

Abstract
Objective
Extracellular matrix (ECM) proteinases are implicated in the pathogenesis of calcific aortic valve disease (CAVD). The ADAMTS5 (A Disintegrin and Metalloproteinase with Thrombospondin motifs 5) enzyme is secreted, matrix-associated metalloendopeptidase, capable of degrading ECM proteins, particularly matrilin2. We sought to determine the role of the ADAMTS5/matrilin2 axis in mediating the phenotype transition of valvular interstitial cells (VICs) associated with CAVD.
Approach and Results
Levels of ADAMTS5, matrilin2 and α-smooth muscle actin (α-SMA) were evaluated in calcified and normal human aortic valve tissues and VICs. Calcified aortic valves have reduced levels of ADAMTS5 and higher levels of matrilin2 and α-SMA. Treatment of normal VICs with soluble matrilin2 caused an increase in α-SMA level through Toll-like receptor 2 and 4, which was accompanied by upregulation of Runt-related transcription factor 2 and alkaline phosphatase. In addition, ADAMTS5 knockdown in normal VICs enhanced the effect of matrilin2. Matrilin2 activated NF-κB and nuclear factor of activated T cells complex 1 (NFATc1), and induced the interaction of these two nuclear factors. Inhibition of either NF-κB or NFATc1 suppressed matrilin2’s effect on VIC phenotype change. Knockdown of α-SMA reduced and over-expression of α-SMA enhanced the expression of pro-osteogenic factors and calcium deposit formation in human VICs.
Conclusions
Matrilin2 induces myofibroblastic transition and elevates pro-osteogenic activity in human VICs via activation of NF-κB and NFATc1. Myofibroblastic transition in human VICs is an important mechanism of elevating the pro-osteogenic activity. Matrilin2 accumulation associated with relative ADAMTS5 deficiency may contribute to the mechanism underlying CAVD progression.
Keywords: Aortic valve, extracellular matrix protein, signal transduction, α-smooth muscle actin, pro-osteogenic activity
Subject codes: Valvular heart disease, Mechanisms, Cell Signaling/Signal Transduction
Introduction
Calcific aortic valve disease (CAVD) is the most common valvular heart disease in the western countries and increasingly affects the elderly. Approximately 2.8% of people over 75 years old have aortic valve calcification, and 25% of adults over 65 have aortic valve sclerosis1. Currently, there is no effective pharmacological therapy for CAVD other than surgical or interventional valve replacement.
In the past decade, increasing evidence suggests that aortic valve calcification is a regulated pathological process mediated by the phenotype changes of valvular interstitial cells (VICs), the most abundant cell type in valve tissue, and chronic stresses are thought to cause prolonged activation of VICs, which may result in myofibroblastic as well as osteoblast-like phenotypic transition2, 3.
Metalloproteinases, including matrix metalloproteinases, a disintegrin and metalloproteinase (ADAMs) or a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), play pivotal roles in cardiovascular remodeling by degrading matrix or non-matrix substrates4, 5. In contrast to the extensive investigation of metalloproteinases in valvular remodeling, little is known about their role in valvular calcification. Previous study demonstrated that altered proteoglycan cleavage in ADAMTS5 deficient mice results in myxomatous cardiac valves6, suggesting a potential role of ADAMTS5 in congenital valve malformation. However, the role of ADMATS5 in CAVD has not been investigated. Matrilin2 is a novel member of filament-forming oligomeric adaptor protein family that is involved in the development and homeostasis of the extracellular matrix network. The levels of soluble matrilin2 are remarkably increased during liver cirrhosis, osteoarthritis and other inflammation-associated diseases7, 8. Besides its role in modulating extracellular matrix (ECM) network, matrilin2 induces a pro-inflammatory response in macrophages/microglia via Toll-like receptor (TLR)-mediated signaling pathways9. Our previous studies demonstrate that activating TLR2 or TLR4 elevates the pro-osteogenic activity in human VICs10–12 and induces aortic valve lesions in mice13. It is likely that matrilin2 induces VIC pro-osteogenic phenotypic transition through TLR4/2. Matrilin2 is also identified as a shared gene in osteoblast progenitor cells and developing heart valves14. Recently, matrilin2 is recognized as a substrate of ADAMTS515. However, it remains unclear whether matrilin2 accumulation contributes to the pathogenesis of valvular calcification.
We hypothesized that matrilin2 accumulation as a result of relative ADAMTS5 deficiency contributes to valve calcification. The purpose of this study is to determine: (1) whether ADMATS5 deficiency and matrilin2 accumulation are associated with valvular calcification, (2) the effect of soluble matrilin2 on VIC phenotype change and pro-osteogenic activity, (3) the mechanism by which soluble matrilin2 exerts its effect.
Materials and Methods
Materials and Methods are available in the online-only Supplement.
Results
Calcified human aortic valves have lower levels of ADAMTS5 and increased levels of matrilin2
We analyzed gene expression profiles of VICs isolated from 5 normal aortic valves and VICs from non-calcified areas of 5 calcified aortic valves (Valve donor demographics in Supplemental Table I) by gene microarray. The expression of 377 genes were altered (fold change >1.3 and P<0.05; Supplemental Tables II and III) in VICs from calcified aortic valves in comparison to VICs from normal aortic valves. Among them, 21 displayed a change greater than 2.0 fold (Figure 1A). ADAMTS5 was one of the 16 with greater than 2.0 fold down-regulation (Figure 1A). In contrast, levels of mRNA encoding TLR2, TLR4, matrilin2, NFATC1 and NF-κB were comparable between normal and diseased VICs.
Figure 1. Calcified human aortic valves have reduced levels of ADAMTS5 and elevated levels of matrilin2 and α-smooth muscle actin (α-SMA).
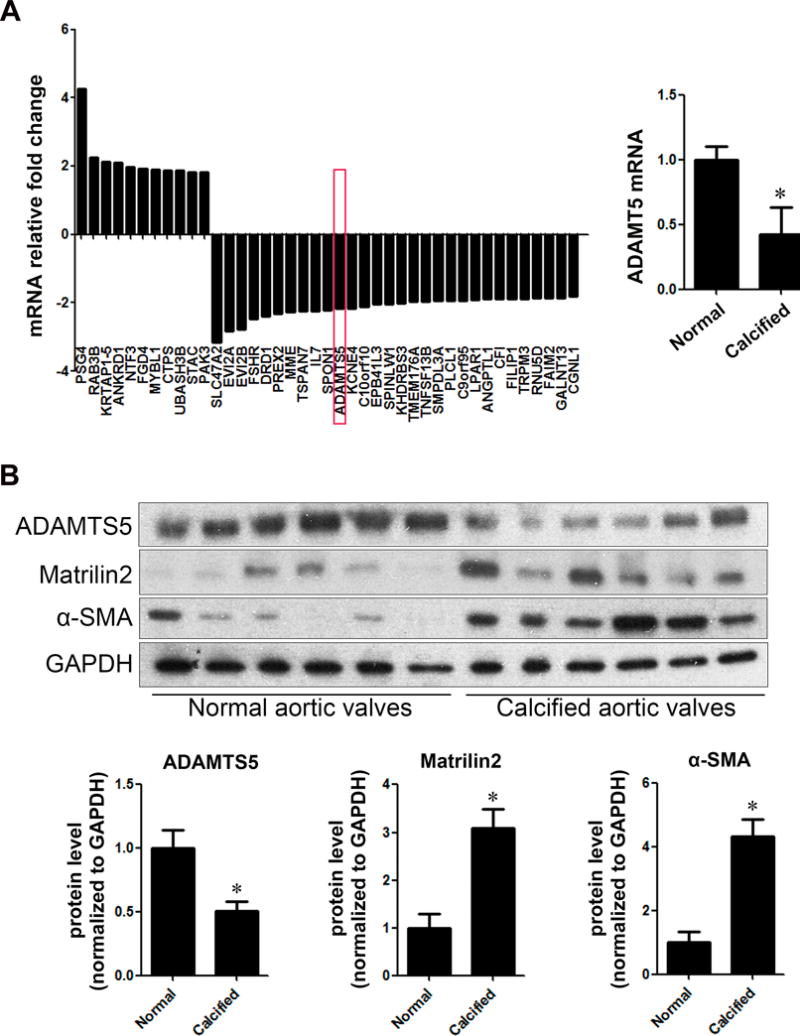
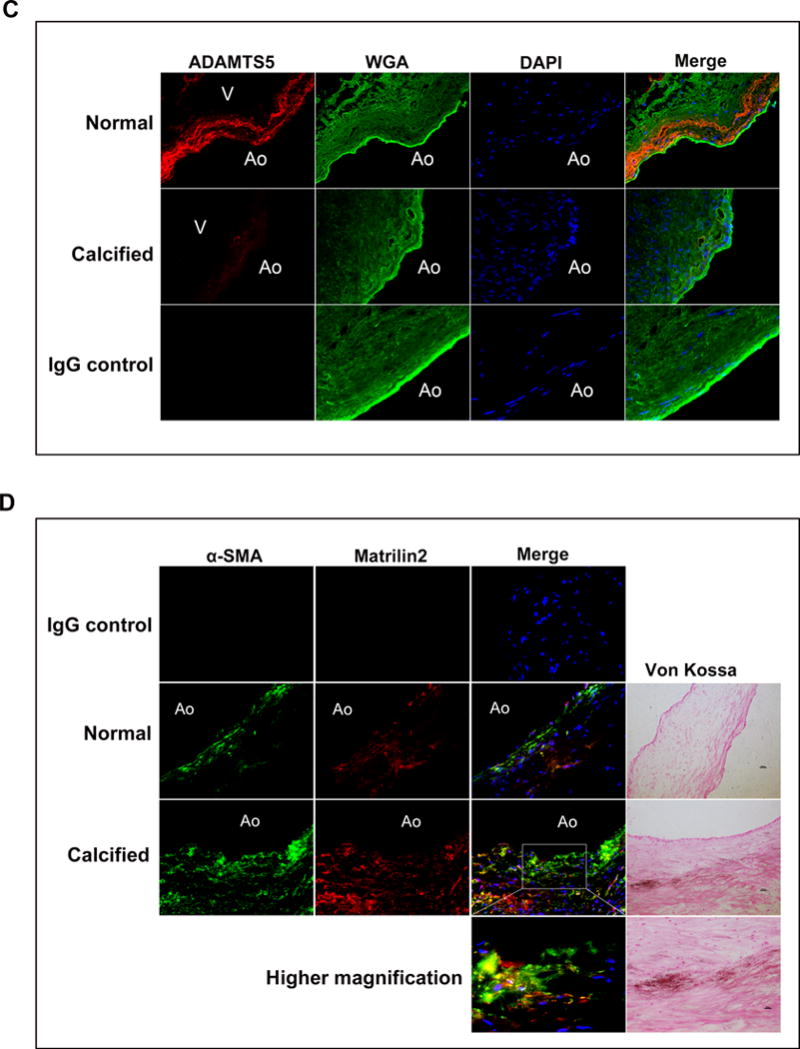
A. Valvular interstitial cells (VICs) were isolated from normal and calcified aortic valves, and gene expression was analyzed by Affymetrix array. Data show markedly altered expression of genes in VICs from calcified aortic valves compared to VICs of normal valves. ADAMT5 mRNA level was significantly decreased in VICs from calcified aortic valves. Mean ± SE; n=5 per group; *P<0.05 vs. normal. B. Immunoblots show that in calcified human aortic valve tissues, ADAMTS5 protein levels were significantly lower, whereas the protein levels of matrilin2 and α-SMA were higher. Mean ± SE; n=6 per group; *P<0.05 vs. normal. C. Representative images of immunofluorescence staining show ADAMTS5 (red) in normal and calcified human aortic valve tissues. Alexa 488-tagged wheat germ agglutinin (WGA) was used to outline plasma membrane (green). 4′, 6-diamidino-2-phenylindole (DAPI) was used for nuclear counterstaining (blue). Ao indicates the aortic side and V the ventricular side of the valve leaflets. Original magnification, 40× objective. D. Representative images of dual immunofluorescence staining of matrilin2 (red) and α-SMA (green) and Von Kossa staining in normal and calcified human aortic valve tissues show that matrilin2 was co-localized with α-SMA and in areas surrounding calcification nodule in calcified aortic valves. Original magnification, 40× objective.
To determine whether ADAMTS5 and matrilin2 levels are altered in calcified aortic valves, we analyzed protein levels of ADMATS5 and matrilin2 in normal valve tissues and tissues from non-calcified areas of calcified aortic valves by immunoblotting and immunofluorescence staining. As shown in Figures 1B–1D, calcified human aortic valves had reduced levels of ADAMTS5 in area without massive calcification, and matrilin2 levels were markedly increased in the same area. Movat’s pentachrome staining confirmed that ADAMTS5 primarily located in the fibrosa layer of normal aortic valve although it was also present in the ventricularis layer (Supplemental Figure I).
VIC transition to myofibroblast is a critical step in valvular pathology associated with CAVD16. We examined myofibroblast by detecting its phenotypic biomarker, α-smooth muscle actin (α-SMA). The protein levels of α-SMA were higher in both tissues and cells from calcified human aortic valves than those from normal valves (Figure 1B and Supplemental Figure II). Dual immunofluorescence staining and Von Kossa staining demonstrated that matrilin2 accumulation was co-localized with α-SMA as well as calcification nodules in calcified aortic valves (Figure 1D). These data suggest that relative ADAMTS5 deficiency and associated matrilin2 accumulation in calcified aortic valves may play a role in VIC myofibroblastic transition and aortic valve calcification.
Matrilin2 induces myofibroblastic transition and upregulates pro-osteogenic activity in VICs
To determine the role of the ADAMTS5/matrilin2 axis in VIC phenotype change, VICs from normal human aortic valves were treated with recombinant matrilin2 (0, 0.5, 1.0 and 2.0 μg/ml) for 48 h. We then examined the expression of myofibroblastic markers, α-SMA and collagen I, as well as pro-osteogenic markers, Runt-related transcription factor 2 (Runx2) and alkaline phosphatase (ALP). As shown in Figures 2A–2C, stimulation of VICs with recombinant matrilin2 resulted in a dose-dependent increase in protein levels of α-SMA, collagen I, Runx2 and ALP. Moreover, VICs exhibited abundant α-SMA-containing stress fibers following matrilin2 treatment (Figure 2B). To determine whether matrilin2 stimulation induces calcium deposition in VICs, we stimulated cells in a conditioning medium (growth medium supplemented with 10 mmol/L β-glycerophosphate, 10 nmol/L dexamethasone, 4 μg/ml cholecalciferol and 8 mmol/L CaCl2) with matrilin2 for 21 days. Alizarin red S staining revealed that prolonged exposure to matrilin2 in the conditioning medium significantly increased the formation of calcium deposits (Figure 2D). In contrast, the conditioning medium alone caused much lower levels of calcium deposit formation. Thus, matrilin2 not only induces myofibroblastic transition, but also elevates the pro-osteogenic activity in VICs.
Figure 2. Matrilin2 induces myofibroblastic transition and upregulates pro-osteogenic activity in valvular interstitial cells (VICs).
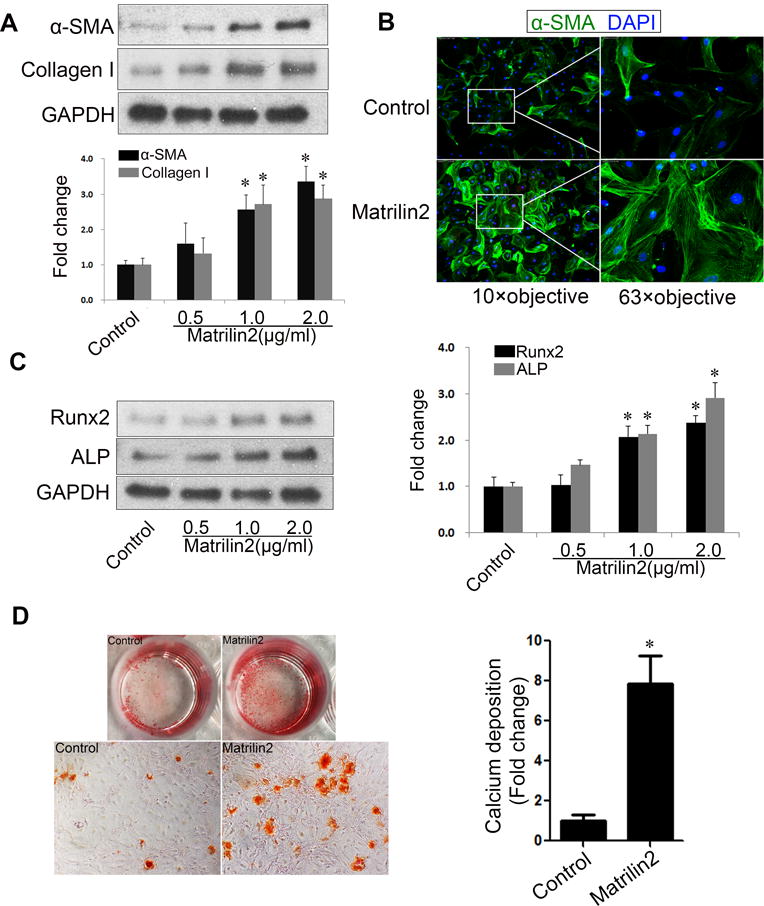
A. VICs from normal human aortic valves were treated with recombinant matrilin2 (2.0 μg/ml) for 48 h. Matrilin2 induced myofibroblastic transition of VICs, as characterized by enhanced expression of α-SMA and collagen I. B. VICs had greater amounts of α-SMA-containing stress fibers (green) following matrilin2 stimulation. 4′, 6-diamidino-2-phenylindole (DAPI) was used for nucleus counterstaining (blue). C. Matrilin2 increased the expression of Runx2 and ALP in VICs in a dose-dependent manner. D. VICs were incubated with matrilin2 for 21 days in conditioning medium (containing 10 mmol/L β-glycerophosphate, 10 nmol/L dexamethasone, 4.0 μg/ml cholecalciferol and 8 mmol/L CaCl2) to examine calcium deposit formation. Images of Alizarin red S staining (10× objective for the high power images) and spectrophotometric analysis show that prolonged stimulation with matrilin2 increased calcium deposit formation. Mean ± SE; n=5 separate experiments using distinct cell isolates; *P<0.05 vs. control.
Matrilin2 induces VIC phenotypic transition through TLR4 and TLR2
Matrilin2 was recently identified as an endogenous damage-associated molecule pattern that can elicit a pro-inflammatory response in macrophage via the TLR signaling pathways9, 17. We examined whether TLR signaling is involved in matrilin2-induced VIC phenotypic transition. VICs were treated with recombinant matrilin2 of varying concentrations for different time periods. We observed that matrilin2 increased cellular levels of TLR4 and TLR2 in a dose-dependent and time-dependent manner (Figure 3A–3C). To determine the role of these 2 innate immune receptors in mediating the effect of matrilin2, we applied siRNAs to knock down TLR4 and TLR2 (Figure 3D). Matrlin2-induced upregulation of α-SMA and collagen I was inhibited by knockdown of either TLR4 or TLR2 (Figure 3E). Similarly, the levels of pro-osteogenic factors Runx2 and ALP were also reduced by knockdown of TLR4 or TLR2 (Figure 3F). In addition, neutralizing antibody against TLR4 or TLR2 also reduced the effect of matrilin2 on the expression of α-SMA, Runx2 and ALP (Supplemental Figure III). Therefore, matrilin2 upregulates TLR4 and TR2, and VIC phenotypic transition induced by matrilin2 is mediated by these two innate immunoreceptors.
Figure 3. Toll-like receptors (TLRs) 4 and 2 mediate matrilin2-induced valvular interstitial cell (VIC) VIC phenotypic transition.
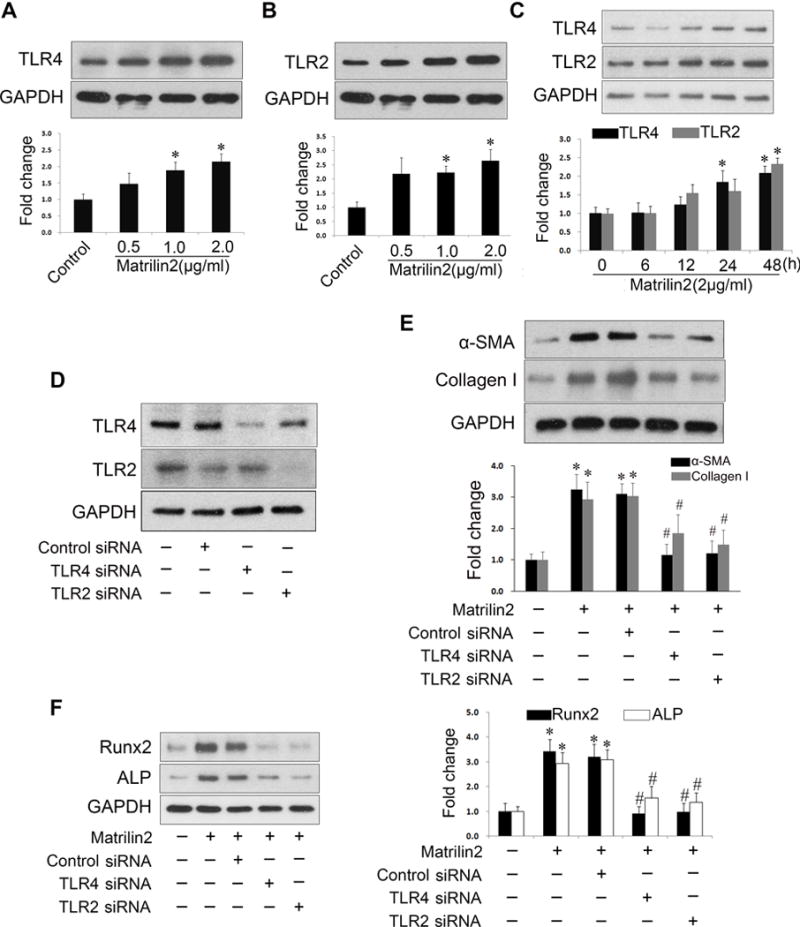
A–C.VICs from normal human aortic valves were treated with recombinant matrilin2 (2.0 μg/ml) for 48 h. Representative immunoblots and densitometric data show that matrilin2 increased the levels of TLR4 and TLR2 in a dose- and time-dependent manner. D. VICs were treated with TLR4 siRNA (100 nM) and TLR2 siRNA (100 nM) for 72 h. TLR4 siRNA and TLR2 siRNA reduced the levels of TLR4 and TLR2, respectively. E and F. VICs with knockdown of TLR4 or TLR2 were treated with matrilin2 (2.0 μg/ml) for 48 h. Knockdown of either TLR4 or TLR2 suppressed the upregulation of α-SMA, collagen I, Runx2 and ALP by matrilin2. Mean ± SE; n=4 separate experiments using distinct cell isolates; *P<0.05 vs. control; #P<0.05 vs. matrilin2 + control siRNA.
NF-κB plays an important role in mediating matrilin2-induced VIC phenotypic transition
To identify the signaling pathway involved in mediating the response to matrilin2, we examined NF-κB activation. We observed a marked increase in NF-κB p65 phosphorylation following matrilin2 treatment, reaching a maximum after 2 h (Figure 4A). Phosphorylation of NF-κB p65 was inhibited by neutralizing antibodies against TLR4 and TLR2 (Figure 4B). Furthermore, immunofluorescence staining confirmed intranuclear translocation of NF-κB p65 following matrilin2 treatment and the inhibitory effect of TLR4 and TLR2 blockades on NF-κB p65 translocation (Figure 4C).
Figure 4. NF-κB plays an important role in matrilin2-induced valvular interstitial cell (VIC) phenotypic transition.
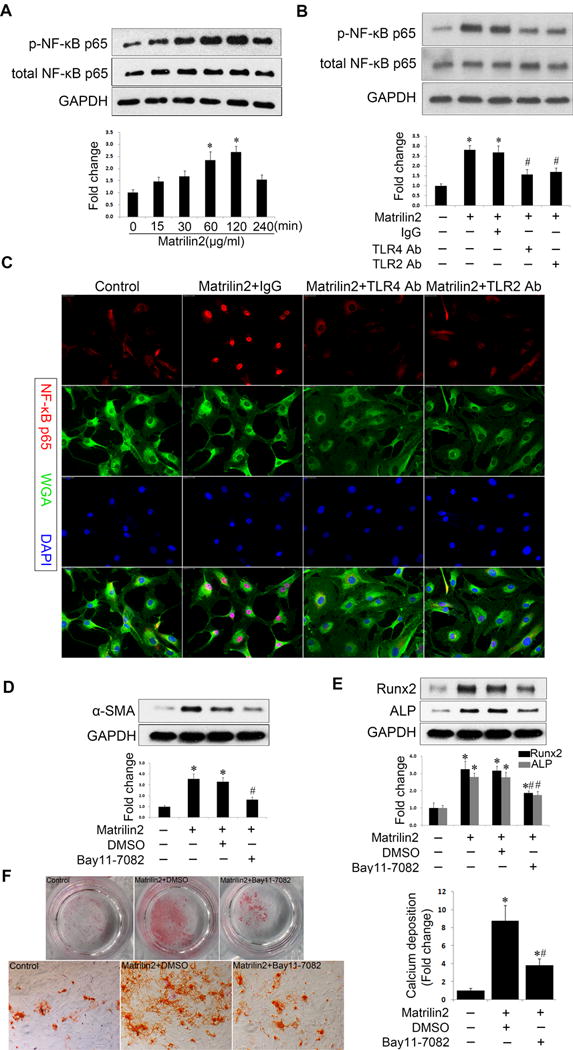
A. VICs from normal human aortic valves were treated with matrilin2 (2.0 μg/ml) for 15 min to 4 h to examine the time course of NF-κB phosphorylation. Phosphorylation of NF-κB p65 occurred mainly at 1 and 2 h following matrilin2 treatment. B and C. VICs were treated with matrilin2 (2.0 μg/ml) for 2 h in the presence or absence of neutralizing antibodies against TLR4 (10 μg/ml) and TLR2 (10 μg/ml). Immunoblots show that neutralizing antibodies against TLR4 and TLR2 suppressed NF-κB p65 phosphorylation following matrilin2 stimulation. Representative images of immunofluorescence staining of NF-κB p65 (red) show that NF-κB p65 intranuclear translocation induced by matrilin2 was reduced by neutralizing antibodies against TLR4 and TLR2. Alexa 488-tagged wheat germ agglutinin (WGA) was used to outline cells (green). 4′, 6-diamidino-2-phenylindole (DAPI) was used for nuclear counterstaining (blue). Original magnification, 40× objective. D and E. NF-κB inhibitor, Bay 11-7082 (10 μM), reduced the levels of α-SMA, Runx2 and ALP in cells exposed to matrilin2 (2.0 μg/ml) for 48 h. F. To examine calcium deposit formation, VICs in the conditioning medium were treated with matrilin2 (2.0 μg/ml) for 21 days in the presence or absence of Bay 11-7082 (10 μM). Alizarin red S staining shows that NF-κB inhibition reduced the formation of calcium deposits in cells treated with matrilin2 (10× objective for the high power image). Mean ± SE; n=4 separate experiments using distinct cell isolates; *P<0.05 vs. control; #P<0.05 vs. matrilin2 + non-immune IgG or matrilin2 + DMSO.
We applied Bay11-7082, a specific inhibitor of NF-κB, to verify the role of NF-κB activation in matrilin2-induced VIC phenotypic transition. As shown in Figure 4D, inhibition of NF-κB significantly reduced the expression of α-SMA after matrilin2 stimulation. Similar reduction was observed in pro-osteogenic markers Runx2 and ALP (Figure 4E). Furthermore, NF-κB inhibition attenuated calcium deposition in VICs exposed to matrilin2 for a prolonged period in the conditioning medium (Figure 4F). Collectively, these results reveal that TLR-dependent NF-κB signaling plays an important role in matrilin2-induced VIC phenotypic transition.
Nuclear factor of activated T cells complex 1 (NFATc1) is involved in VIC phenotypic change induced by matrilin2
NFATc1 is a transcription factor, and its level is found to correlate with osterix and Runx2 levels in calcified human aortic valves18. It has been reported that TLR2 modulates osteoclastogenesis and upregulates NFATc119. We examined the effect of matrilin2 on NFATc1 level and activity. Matrilin2 evoked an increase in NFATc1 expression in a dose- and time-dependent fashion (Figure 5A, 5B). Immunofluorescence analysis confirmed increased NFATc1 levels and intranuclear translocation of this nuclear factor after matrilin2 stimulation (Figure 5C). Neutralization antibodies against TLR4 or TLR2 repressed the increase in NFATc1 (Figure 5D). To investigate whether NFATc1 activation is involved in VIC phenotypic transition, we used both lentiviral NFATc1 shRNA and a specific inhibition peptide, VIVIT. As shown in Figure 5E, lentiviral NFATc1 shRNA reduced the levels of NFATc1. Inhibition of NFATc1 by shRNA or VIVIT markedly suppressed matrilin2-induced upregulation of α-SMA, Runx2 and ALP (Figure 5F, Supplemental Figure IV). Moreover, NFATc1 knockdown attenuated VIC mineralization induced by prolonged stimulation with matrilin2 in the conditioning medium (Figure 5G). Together, these data show that NFATc1 is also involved in the mechanism of VIC phenotypic change induced by matrilin2.
Figure 5. Nuclear factor of activated T cells complex 1 (NFATc1) is involved in valvular interstitial cell (VIC) phenotypic change induced by matrilin2.
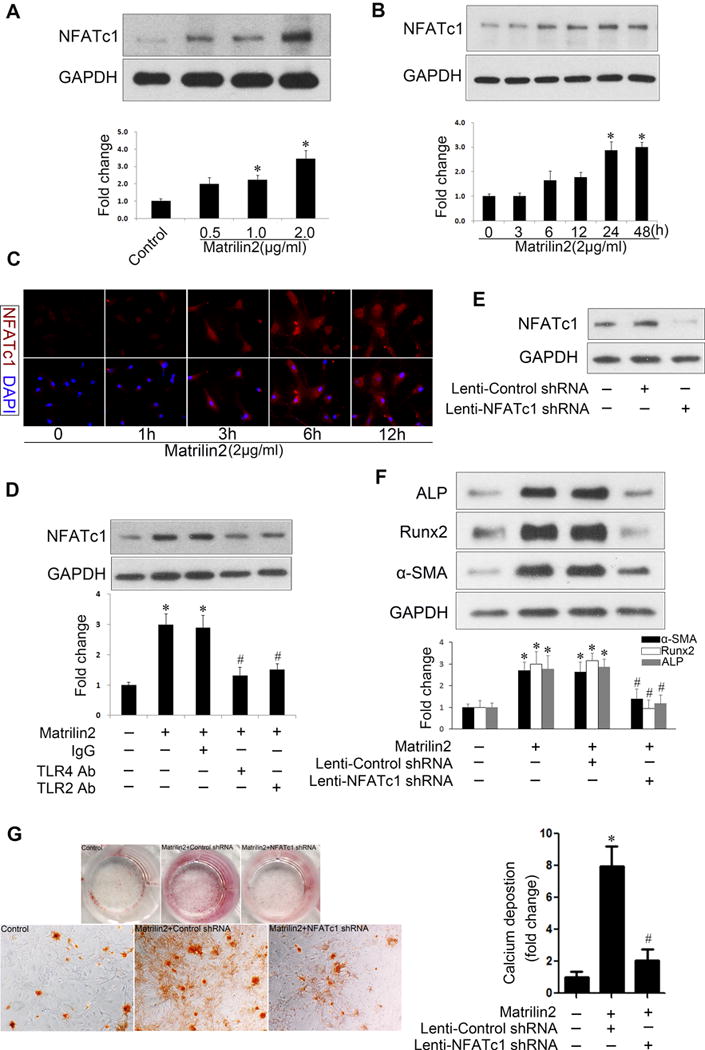
A and B.VICs from normal aortic valves were treated with varying concentrations of matrilin2 for 48 h or with matrilin2 (2.0 μg/ml) for varied time. Matrilin2 enhanced the expression of NFATc1 in a dose- and time-dependent manner. C. VICs were incubated with matrilin2 (2.0 μg/ml) for varied time. Immunofluorescence images show increased NFATc1 immune reactivity (red) in VICs stimulated with matrilin2 for 3 h or longer. Original magnification, 40× objective. D. Neutralizing antibodies against TLR4 (10 μg/ml) and TLR2 (10 μg/ml) suppressed NFATc1 upregulation by matrilin2. E. VICs were transduced with lentivirus expressing control shRNA (100 nM) or NTAT1c1 shRNA (100 nM) for 72 h. NFATc1 shRNA inhibited the expression of NFATc1. F. VICs were transduced with lentivirus expressing NFATc1 shRNA for 72 h prior to matrilin2 stimulation (2.0 μg/ml) for 48 h. Knockdown of NFATc1 attenuated the upregulation of α-SMA, Runx2 and ALP by matrilin2. G. VICs transduced with lentiviral NFATc1 shRNA were treated with matrilin2 (2.0 μg/ml) for 21 days in the conditioning medium to examine calcium deposit formation. Alizarin red S staining and spectrophotometric analysis reveal that NFATc1 knockdown reduced matrilin2-induced calcium deposition (10× objective for the high power images). Mean ± SE; n=4 separate experiments using distinct cell isolates; *P<0.05 vs. control; #P<0.05 vs. matrilin2 + non-immune IgG or matrilin2 + Lenti-control shRNA.
NF-κB modulates NFATc1 activation
Previous studies indicate that NF-κB and NFATc1 interact with each other and that NF-κB modulates the expression of NFATc1 in lymphocyte, macrophage and cardiomyocyte20–22. To examine whether NFATc1 is downstream of NF-κB in matrilin2-induced cellular signaling in human VICs, we pretreated VICs with two distinct NF-κB inhibitors, Bay 11–7082 and SN50, prior to matrilin2 stimulation. However, inhibition of NF-κB with either inhibitor had no effect on the upregulation of NFATc1 protein levels by matrilin2 (Figure 6A, 6B). Interestingly, inhibition of NF-κB suppressed matrilin2-induced NFATc1 activation (Figure 6C). Then, we examined if NFATc1 interacts with NF-κB in human VICs using co-immunoprecipitation. The results in Figure 6D show that NFATc1 is co-immunoprecipitated with NF-κB p65. Conversely, immunoprecipitation of NFATc1 resulted in the co-precipitation of NF-κB p65. Moreover, enhanced binding of the two transcription factors was observed in cells exposed to matrilin2 (Figure 6C). Therefore, NF-κB appears to interact with NFATc1 to modulate its activity.
Figure 6. NF-κB controls the activation of nuclear factor of activated T cells complex 1 (NFATc1).
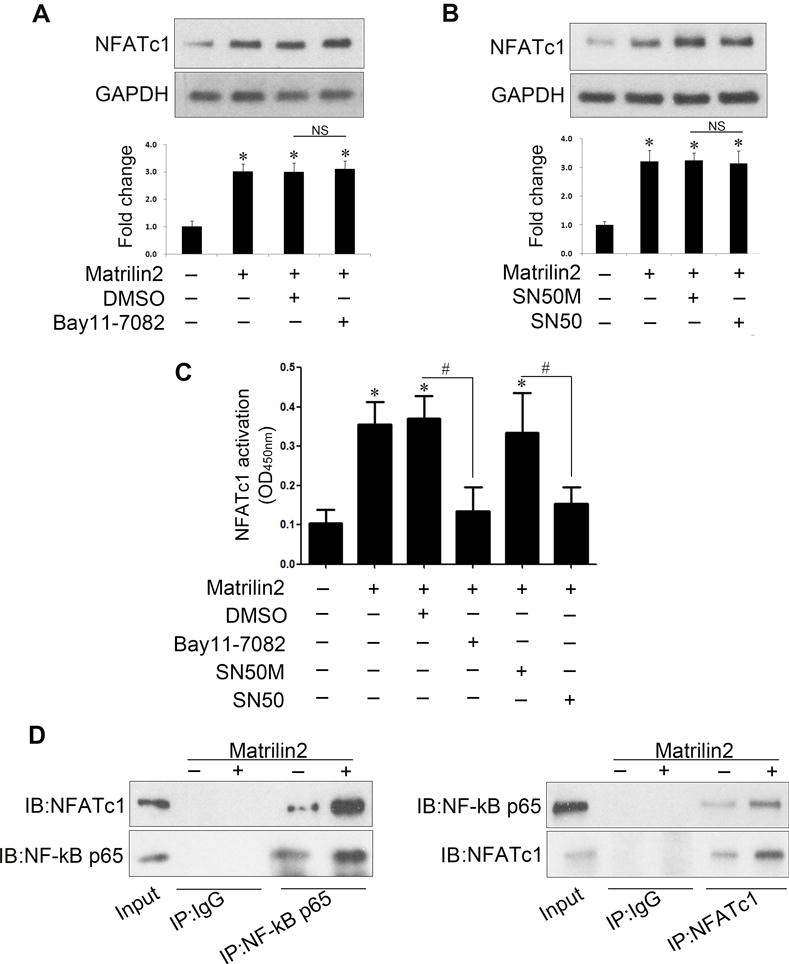
A–C VICs from normal aortic valves were treated with Bay11-7082 (10 μM) or SN50 (100 μg/ml) 1 h prior to matrilin2 (2.0 μg/ml) stimulation for 48 h. Immunoblots and densitometric data show that either Bay11-7082 or SN50 failed to suppress matrilin2-induced expression of NFATc1 (A and B). NFATc1 activity data show that inhibition of NF-κB by Bay11-7082 or SN50 attenuated NFATc1 activation induced by matrilin2 (C). D. Immunoblots for NF-κB p65 and NFATc1 after immunoprecipitation with anti-p65 antibody, anti-NFATc1 antibody or non-immune IgG demonstrate interaction of these two nuclear factors. IP, immunoprecipitation; IB, immunoblotting. Mean ± SE; n=4 separate experiments using distinct cell isolates; *P<0.05 vs. control; #P<0.05 vs. matrilin2 + DMSO or matrilin2 + SN50M; NS = non-significant.
ADAMTS5 deficiency enhances the effect of matrilin2 on VIC phenotypic transition
We treated cells from calcified aortic valves with recombinant human ADMATS5 protein and found that basal levels of α-SMA, Runx2 and ALP in VICs of calcified aortic valves were markedly decreased by this treatment (Figure 7A). In VICs from normal aortic valves, recombinant ADAMTS5 also attenuated the upregulation of α-SMA, Runx2 and ALP induced by matrilin2 (Figure 7B). In addition, we performed loss-of-function experiments to determine whether ADAMTS5 deficiency augments the effect of matrilin2 on normal VICs. As shown in Figure 7C, specific siRNA reduced the levels of ADMATS5. ADMATS5 knockdown resulted in an increase in α-SMA, Runx2 and ALP in VICs of normal aortic valves (Figures 7D and 7E). Interestingly, a combination of ADMATS5 knockdown and matrilin2 stimulation resulted in significantly higher levels of α-SMA, Runx2 and ALP expression as compared with matrilin2 stimulation alone (Figures 7D and 7E). Thus, relative ADAMTS5 deficiency elevates the expression of α-SMA, Runx2 and ALP in human VICs and augments the effect of matrilin2.
Figure 7. ADAMTS5 knockdown enhances the effect of matrilin2 on valvular interstitial cell (VIC) VIC phenotypic change.
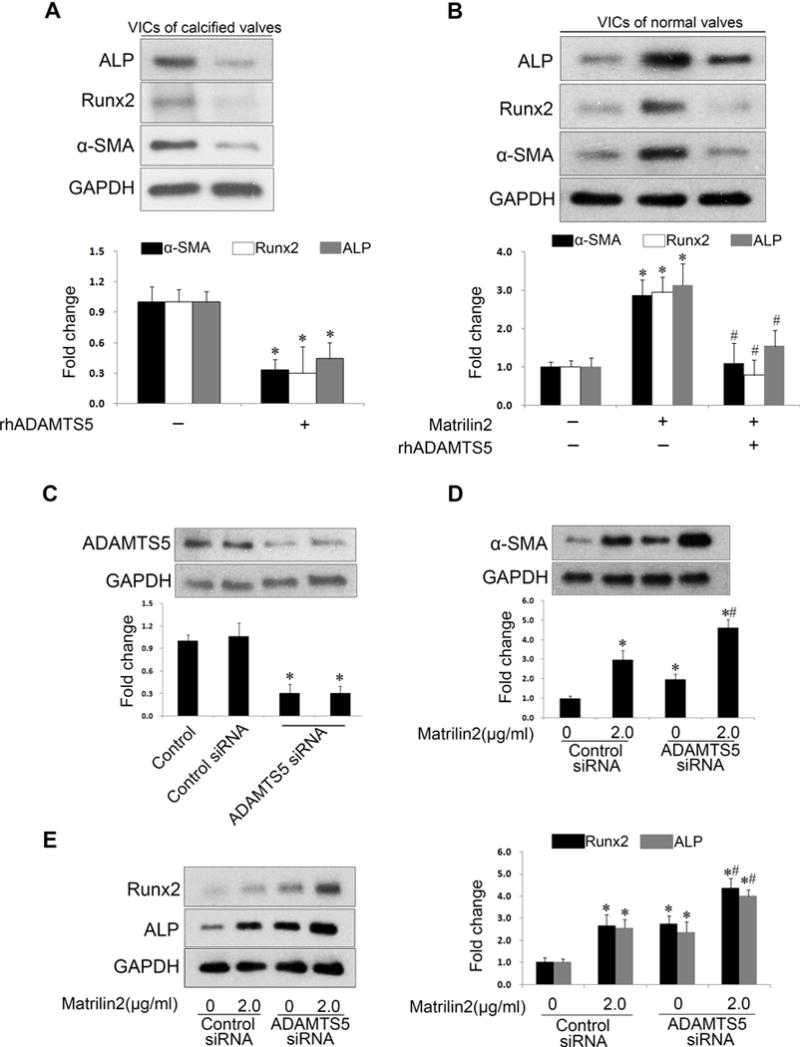
A. VICs from calcified aortic valves were treated with recombinant human ADMATS5 (rhADMATS5, 5.0 μg/ml) for 48 h. Immunoblots and densitometric data show that ADAMTS5 decreased the baseline levels of α-SMA, Runx2 and ALP. B. VICs from normal aortic valves were treated with rhADMATS5 (5.0 μg/ml) 1 h prior to matrilin2 (2.0 μg/ml) stimulation for 48 h. ADMATS5 attenuated the upregulation of α-SMA, Runx2 and ALP by matrilin2. C. ADAMTS5 siRNA reduced ADAMTS5 levels in VICs from normal aortic valves. D and E. VICs from normal aortic valves were transfected with ADAMTS5 siRNA 72 h prior to matrilin2 stimulation for 48 h. Immunoblots and densitometric data show that ADAMTS5 knockdown augmented the upregulation of α-SMA, Runx2 and ALP by matrilin2. Mean ± SE; n=4 separate experiments using distinct cell isolates; *P<0.05 vs. untreated control and control siRNA-treated; #P<0.05 vs. matrilin2 alone or matrilin2 + control siRNA.
Myofibroblastic transition elevates pro-osteogenic activity in VICs
Previous studies suggest a link between myofibroblasts and calcium nodule formation in aortic valves23. However, it remains unclear whether myofibroblasts are the so-called “osteoblast-like” cells. Immunofluorescence microscopy revealed that myofibroblastic biomarker α-SMA and pro-osteogenic biomarker Runx2 were co-localized in VICs from calcified aortic valves as well as in normal VICs exposed to matrilin2 (2.0 μg/ml) for 48 h (Figures 8A and 8B). To determine whether myofibroblast have elevated pro-osteogenic activity, VICs were transduced for 72 h or 96 h with lentivirus expressing α-SMA. The levels of α-SMA level were markedly increased by over-expression (Supplemental Figure V). Over-expression of α-SMA in VICs from normal aortic valves resulted in the upregulation of Runx2 and ALP (Figure 8C). In addition, VICs that over-express α-SMA exhibited greater calcium deposition (Figure 8D). Thus, it appears that myofibroblasts are “osteoblast-like” cells in human aortic valves.
Figure 8. Myofibroblastic transition elevates pro-osteogenic activity in valvular interstitial cells (VICs).
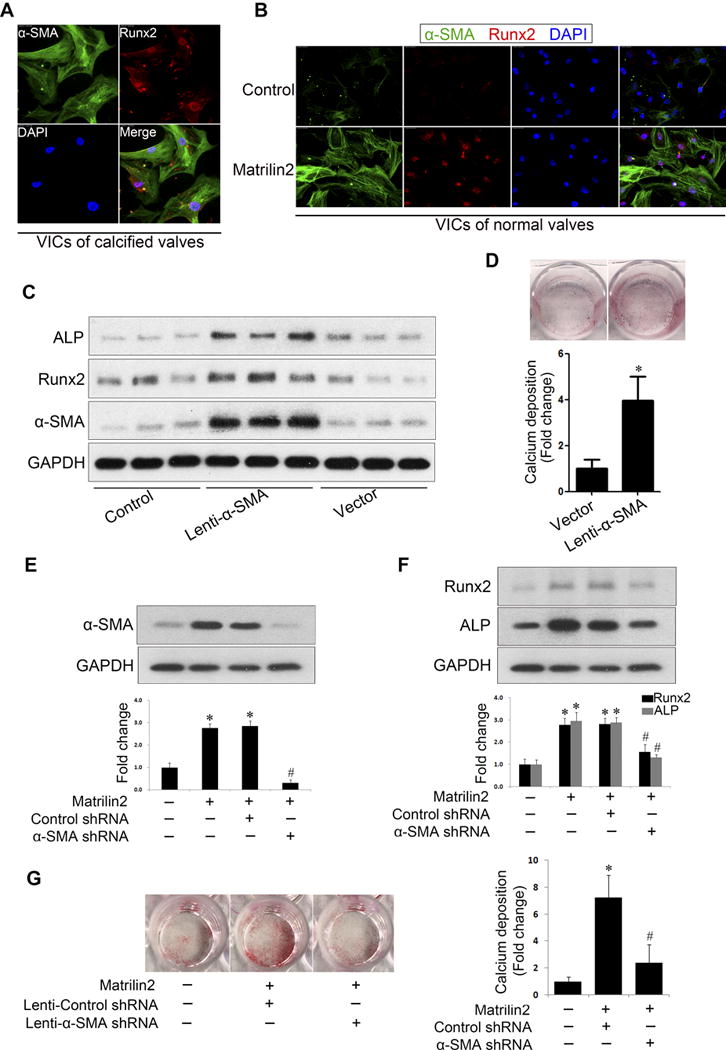
A. Dual immunofluorescence staining was applied to localize α-SMA (green) and Runx2 (red) in VICs from calcified valves. These two proteins were identified in the same cells in the absence of a stimulation. Original magnification, 40× objective. B. In VICs from normal aortic valves, α-SMA and Runx2 were identified by dual immunofluorescence staining after stimulation with matrilin2 (2.0 μg/ml) for 48 h. Original magnification, 40× objective. C. VICs from normal aortic valves were transduced with lentivirus expressing α-SMA for 96 h. Immunoblots show that α-SMA over-expression increased the protein levels of Runx2 and ALP. n=3 separated experiments using distinct cell isolates. D. VICs from normal aortic valves were transduced with lentivirus expressing α-SMA for 96 h and then incubated in the conditioning medium for 21 days to examine calcium deposit formation. Alizarin red S staining shows that α-SMA over-expression increased calcium deposition. Mean ± SE; n=4 separated experiments using distinct cell isolates; *P<0.05 vs. vector control. E and F. VICs were transduced with lentiviral α-SMA shRNA 72 h prior to matrilin2 (2.0 μg/ml) stimulation for 48 h. Lentiviral α-SMA shRNA reduced α-SMA levels and abrogated the upregulation of Runx2 and ALP by martrilin2. Mean ± SE; n=4 separated experiments using distinct cell isolates; *P<0.05 vs. control; #P<0.05 vs. matrilin2 + control shRNA. G. VICs from normal aortic valves were pre-treated with lentiviral α-SMA shRNA or lentiviral control shRNA, and then treated with matrilin2 (2.0 μg/ml) for 21 days in the conditioning medium to examine calcium deposit formation. Images of Alizarin red S staining and spectrophotometric data show that α-SMA knockdown reduced calcium deposition in VICs following prolonged matrilin2 stimulation. Mean ± SE; n=4 separated experiments using distinct cell isolates; *P<0.05 vs. control; #P<0.05 vs. matrilin2 + control shRNA; vector = lentiviral control vector.
To determine whether over-expression of α-SMA (myofibroblastic transition) is responsible for the elevation of pro-osteogenic activity, we transduced cells with lentiviral shRNA to inhibit α-SMA expression. As shown in Figure 8E, matrilin2 failed to increase the protein levels of α-SMA in VICs transduced with lentiviral α-SMA shRNA. Further, the expression of Runx2 and ALP in response to matrilin2 stimulation was greatly reduced in VICs transduced with lentiviral α-SMA shRNA compared to cells transduced with scramble shRNA (Figure 8F). More importantly, α-SMA knockdown significantly reduced calcium deposit formation following prolonged stimulation with matrilin2 in the conditioning medium (Figure 8G). Taken together, these results suggest that matrilin2 induces myofibroblastic transition to elevate the pro-osteogenic activity in VICs and that myofibroblasts are “osteoblast-like” cells in human aortic valves.
Discussion
Numerous studies support the concept that valvular calcification is an active process involving multiple mechanisms, including abnormal calcium or phosphate metabolism, valvular inflammation, pro-osteogenic reprogramming of VICs and ECM remodeling16. Currently, little is known about ADAMTS5 involvement in valvular calcification. In the present study, we identified relative ADAMTS5 deficiency and matrilin2 accumulation in calcified human aortic valves. Stimulation of VICs from normal human aortic valves with soluble matrilin2 induces myofibroblastic and “osteoblast-like” changes through a mechanism dependent of TLR4 and TLR2. Treatment of VICs of calcified aortic valves with recombinant ADAMTS5 reduces levels of myofibroblastic and pro-osteogenic biomarkers. Conversely, knockdown of ADAMTS5 augments the upregulation of myofibroblastic and pro-osteogenic biomarkers by matrilin2 in normal VICs. Mechanistically, NF-κB and NFATc1 signaling pathways play important roles in mediating the effect of matrilin2 on VIC phenotype transition, and NF-κB modulates NFATc1 activity through molecular interaction. Furthermore, we found that myofibroblastic transition is required for the elevated pro-osteogenic activity in VICs exposed to matrilin2. The present study provides the initial evidence that relative ADAMTS5 deficiency in human aortic valves elevates VIC pro-osteogenic activity through matrilin2-mediated cell phenotypic transition.
ADAMTS family is comprised of 19 secreted, ECM-degrading enzymes that can be sub-grouped on the basis of their substrates24. Although much interest has focused on matrix metalloproteinases, pathobiological role of ADAMTS has recently been recognized. Aberrant expression or malfunction of ADAMTS members have been implicated in several conditions, such as arthritis, cancer and atherosclerosis25–27. Previous studies have demonstrated that ADAMTS7, through processing of cartilage oligomeric matrix protein, mediates vascular smooth muscle cell migration and neointimal formation in response to vascular injury28, 29. Matrilins appear as oligomeric adaptor proteins and mediate the interaction between collagen fibrils and other ECM components to form supramolecular networks. Among the four family members, matrilin2, a substrate of ADAMTS5, is identified in cartilage and many other types of connective tissue8. Currently, it is unknown whether the ADMATS5-matrilin2 axis is involved in the mechanism of valvular calcification.
The ADAMTS5-matrilin2 axis plays an important role in VIC phenotypic transitions
We revealed that the levels of ADAMTS5 mRNA and protein are lower in VICs and valvular tissue of patients with CAVD. Interestingly, ADAMTS5 protein is primarily, but not exclusively, localized in the aortic side of normal aortic valves. Interestingly, significantly lower levels of ADAMTS5 are detected in calcified aortic valves. The localization pattern of ADAMTS5 indicates a role of ADAMTS5 in valvular calcification since calcification frequently begins on the aortic side of the valve leaflets30–32. In correlation to relative ADAMTS5 deficiency, matrilin2 levels are markedly higher in the same areas of calcified aortic valves. As matrilin2 is a substrate of ADAMTS5, matrilin2 accumulation in calcified aortic valves is also indicative for ADAMTS5 deficiency. It appears that lower levels of ADAMTS5 in calcified aortic valves result in matrilin2 accumulation. More importantly, elevated levels of matrilin2 are co-localized with α-SMA as well as the calcification nodules in calcified aortic valves.
Previous studies found that VICs from calcified aortic valves produce higher levels of α-SMA and pro-osteogenic biomarkers, including Runx2 and ALP12, 33. However, the underlying molecular mechanism is incompletely understood. We observed that ADAMTS5 deficiency and matrilin2 accumulation are associated with over-expression of myofibroblastic and pro-osteogenic biomarkers in calcified aortic valves. Four lines of evidence from the present study indicate a link between the ADAMTS5-matrilin2 axis and the production of α-SMA and pro-osteogenic biomarkers in VICs. First, lower levels of ADAMTS5 and elevated levels of matrilin2 are co-localized with α-SMA as well as calcification nodules in calcified aortic valves. This provided in vivo evidence that the ADAMTS5-matrilin2 axis is associated with aortic valve calcification. Second, recombinant ADAMTS5 down-regulates the levels of myofibroblastic and pro-osteogenic biomarkers in VICs from calcified aortic valves as well as in matrilin2-stimulated VICs from normal aortic valves. Third, soluble matrilin2 upregulates the expression of α-SMA, collagen I, Runx2 and ALP in normal VICs cultured in growth medium, and prolonged treatment with matrilin2 in the conditioning medium (containing higher calcium and phosphate concentrations) promotes calcium deposit formation. Lastly, knockdown of ADAMTS5 augments the expression of myofibroblastic and pro-osteogenic biomarkers in VICs exposed to matrilin2. Collectively, these data demonstrate that matrilin2 is capable of upregulating the expression of osteogenic biomarkers (Runx2 and ALP) in human AVICs independent of the pro-osteogenic medium and promoting calcium deposit formation in the presence of the pro-osteogenic medium. Matrilin2 accumulation associated with relative ADAMTS5 deficiency appears to be responsible, at least in part, for the upregulation of production of biomarkers for myofibroblastic and osteoblastic-like cells in human VICs. Therefore, it is likely that the ADAMTS5-matrilin2 axis contributes to the mechanism of VIC phenotypic transition associated with CAVD.
However, myxomatous valve disease is found in ADAMTS5−/− mice6. This distinct pathology may be due to total absence of ADAMTS5, rather than a relative deficiency, and the impact of genetic defect on valve development. The discrepancy between our observation and the study on ADAMTS5−/− mice may be explained by the difference between mice and human, as well as the difference between in vivo valve development abnormalities and isolated VICs from diseased aortic valve tissue that has complex pathology. It should also be noted that ADAMTS5 deficiency is likely a contributing factor for CAVD that involves multifactorial pathogenesis.
The NF-κB and NFATc1 signaling pathways mediate the effect of matrilin2
Several ECM proteins, such as fibronectin, hyaluronan-derived oligosaccharides, lumican and biglycan are now recognized as endogenous ligands of the pattern recognition receptors6, 33, 34. A previous study has shown that matrilin2 activates pro-inflammatory signaling pathways in macrophages via TLRs9. In the present study, we found increased expression of both TLR4 and TLR2 in normal human VICs after matrilin2 stimulation. The potency of matrilin2 for the induction of α-SMA, collagen I, Runx2 and ALP expression is attenuated by knockdown of TLR4 or TLR2. These observations suggest that both TLR2 and TLR4 are involved in mediating the responses to matrilin2. It is possible that TLR2 and TLR4 interact. Namely, TLR2 activation affects TLR4 activity and verse versa. Alternatively, these two innate immune receptors may dimerize or synergize in response to matrilin2 stimulation. In this regard, interaction between TLR2 and TLR4 has been reported35. Nevertheless, matrilin2 induces phenotypic transition in human VICs through a TLR-dependent mechanism. This finding is consistent with our previous work that VICs isolated from calcified aortic valves express greater levels of TLR4 and TLR2 than normal VICs, and stimulation of TLR4 and TLR2 in human VICs promotes Runx2 expression and elevates ALP activity11.
Activation of TLR4 or TLR2 initiates several downstream signaling cascades. The data revealed that matrilin2 induces the activation of NF-κB and NFATc1 in human VICs through TLR4 and TLR2. Our previous study revealed that NF-κB signaling elevates the pro-osteogenic activity in human VICs exposed to bacterial or endogenous agents12. Similarly, matrilin2-induced VIC phenotypic transition involves TLR4/2-mediated NF-κB activation since the effects of matrilin2 are attenuated by inhibition of IκB kinase with Bay11-7082. More interestingly, the mechanism of matrilin2 action on human VICs also involves upregulation and activation of NFATc1. NFATc1 is a nuclear factor originally described in immune cells, and it has been implicated in vascular remodeling36. Previous studies found that calcineurin/NFATc1 upregulates Runx2 expression in osteoblasts in response to mechanical stretching37. While NFATc1 has been identified in calcified aortic valves, its role in VIC phenotypic transition and valvular calcification remains unknown. Our data show that matrilin2 increases NFATc1 levels and activity in a TLR4/2-dependent fashion. Knockdown of NFATc1 essentially abrogates the upregulation of α-SMA, Runx2 and ALP, as well as calcium deposition in VICs exposed to matrilin2. These data highlight a novel role of NFATc1 in human VIC transition to myofibroblasts and “osteoblast-like” cells.
Although endothelial cells and immune cells are involved in the pathogenesis of aortic valve calcification, VICs, as the major cell type of aortic valve, and their phenotype transition play a critical role in the development and progression of aortic valve calcification38. Thus, it is important to investigate the mechanism underlying their phenotypic transition. Our data reveal that human VIC express TLR2, TLR4 and NFATc1 and that matrilin2 induces phenotypic transition in human VICs through a TLR-NF-κB/NFATc1 cascade. Thus, the present study identified an endogenous factor and the signaling mechanism by which this factor modulates VIC phenotype transition.
It has been reported that NF-κB and NFATc1 interact, and NF-κB modulates the expression of NFATc1 in several cell types20–22. Whereas inhibition of NF-κB has no effect on NFATc1 upregulation by matrilin2 in human VICs, inhibition of NF-κB suppresses matrilin2-induced activation of NFATc1. Further, we found that p65 interacts with NFATc1 following matrilin2 stimulation in human VICs. Therefore, NF-κB and NFATc1 interact and coordinately mediate VIC myofibroblastic and “osteoblast-like” transition.
Myofibroblastic transition is required for matrilin2-induced elevation of pro-osteogenic activity
VICs comprise a diverse and highly plastic population of resident cells responsible for remodeling and integrity of ECM. In normal aortic valves, VICs are mostly quiescent fibroblast-like cells, and myofibroblasts only account for a small proportion. In calcified valves, however, the VIC population shifts to be mainly activated myofibroblasts displaying increased α-SMA expression, contractility, and collagen production2. Thus, the appearance of increased myofibroblast density is considered as an early pathobiological event in CAVD39. Another major pathogenic phenotype present in calcified valves is the “osteoblast-like” cells that express osteoblast biomarkers, such as Runx2, osterix as well as ALP, and are responsible for the active deposition of calcium40. Like myofibroblasts, these “osteoblast-like” cells might also be derived from VICs in response to pathological cues. However, it remains unclear whether the “osteoblast-like” cells and myofibroblasts are distinct cells or the same identity. Soluble matrilin2 induces the expression of biomarkers for both myofibroblasts and “osteoblast-like” cells in VICs. Elevated levels of Runx2 and α-SMA are co-localized in a population of normal VICs exposed to matrilin2, and co-appearance of increased α-SMA levels and greater Runx2 expression in the same cell is characteristic for a population of VICs of calcified aortic valves. In addition, VICs that over-express α-SMA exhibited increased expression of Runx2 and ALP as well as calcium deposition. Furthermore, by application of a knockdown approach, we demonstrated that upregulation of expression is required for matrilin2-induced expression of pro-osteogenic biomarkers (Runx2 and ALP) and calcium deposit formation. These data led us to speculate that the “osteoblast-like” phenotypic transition involves a fibroblast-myofibroblast-osteoblast process. In this regard, conflicting findings have been reported. A previous study found that over-expression of α-SMA increases VIC contractility and formation of calcification nodules in vitro23. An in vivo study also demonstrated that valvular myofibroblasts exhibit osteoblastic activity in the early stage of aortic valve stenosis41. However, decreased Runx2 expression is associated with enhanced α-SMA expression in Notch1-deficient VICs42. While further work is needed to establish the relationship of myofibroblasts and the “osteoblast-like” cells in calcified aortic valves, our findings suggest that myofibroblasts with greater levels of osteogenic biomarkers are either the “osteoblast-like’ cells or in the intermediate stage of transition to osteoblasts.
In summary, the present study demonstrates a novel role of the ADAMTS5-matrilin2 axis in modulating the pathological phenotype change in human VICs. Relative ADAMTS5 deficiency in calcified human aortic valves is associated with accumulation of matrilin2. Matrilin2 induces myofibroblastic transition and elevates pro-osteogenic activity in VICs via TLR2/4-dependent activation of NF-κB and NFATc1 (Graphic Abstract). Furthermore, myofibroblastic transition elevates VIC pro-osteogenic activity. The results of this study suggest that targeting the ADAMTS5-matrilin2 axis may have therapeutic potential for suppression of VIC myofibroblastic transition and for prevention of CAVD progression.
Supplementary Material
Highlights.
Matrilin2 induces myofibroblastic transition and elevates pro-osteogenic activity in human VICs via TLR2/4-dependent activation of NF-κB and nuclear factor of activated T cells complex 1 (NFATc1).
NF-κB interacts with NFATc1 and modulates its activity in human VICs.
Myofibroblastic transition is responsible for matrilin2-induced elevation of pro-osteogenic activity in human VICs.
Matrilin2 accumulation associated with relative ADAMTS5 deficiency may contribute to the mechanism underlying CAVD progression.
Acknowledgments
Source of funding
This study was supported by National Institutes of Heart, Lung and Blood Grants HL106582 and HL121776. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Fei Li was a participant of the Scientific Research Training Program for Young Talents sponsored by Union Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Non-standard Abbreviations and Acronyms
- CAVD
calcific aortic valve disease
- VICs
valvular interstitial cells
- ECM
extracellular matrix
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs
- α-SMA
α-smooth muscle actin
- Runx2
Runt-related transcription factor 2
- ALP
alkaline phosphatase
- TLR
Toll-like receptor
- NFATc1
nuclear factor of activated T cells complex 1
Footnotes
Disclosures
None.
References
- 1.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: A population-based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 2.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113:198–208. doi: 10.1161/CIRCRESAHA.113.300155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irwin CL, Guzman RJ. Matrix metalloproteinases in medial arterial calcification: Potential mechanisms and actions. Vascular. 2009;17(Suppl 1):S40–S44. doi: 10.2310/6670.2008.00086. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P, Shen M, Fernandez-Patron C, Kassiri Z. Adams family and relatives in cardiovascular physiology and pathology. J Mol Cell Cardiol. 2016;93:186–199. doi: 10.1016/j.yjmcc.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 6.Dupuis LE, McCulloch DR, McGarity JD, Bahan A, Wessels A, Weber D, Diminich AM, Nelson CM, Apte SS, Kern CB. Altered versican cleavage in adamts5 deficient mice; a novel etiology of myxomatous valve disease. Dev Biol. 2011;357:152–164. doi: 10.1016/j.ydbio.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hintermann E, Bayer M, Pfeilschifter JM, Deák F, Kiss I, Paulsson M, Christen U. Upregulation of matrilin-2 expression in murine hepatic stellate cells during liver injury has no effect on fibrosis formation and resolution. Liver Int. 2015;35:1265–1273. doi: 10.1111/liv.12604. [DOI] [PubMed] [Google Scholar]
- 8.Zhang S, Peng J, Guo Y, Javidiparsijani S, Wang G, Wang Y, Liu H, Liu J, Luo J. Matrilin-2 is a widely distributed extracellular matrix protein and a potential biomarker in the early stage of osteoarthritis in articular cartilage. BioMed Res Int. 2014;2014:986127. doi: 10.1155/2014/986127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonas A, Thiem S, Kuhlmann T, Wagener R, Aszodi A, Nowell C, Hagemeier K, Laverick L, Perreau V, Jokubaitis V. Axonally derived matrilin-2 induces proinflammatory responses that exacerbate autoimmune neuroinflammation. J Clin Invest. 2014;124:5042–5056. doi: 10.1172/JCI71385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng X, Ao L, Song Y, Babu A, Yang X, Wang M, Weyant MJ, Dinarello CA, Cleveland JC, Fullerton DA. Expression of functional toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol. 2008;294:C29–C35. doi: 10.1152/ajpcell.00137.2007. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Fullerton DA, Su X, Ao L, Cleveland JC, Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 12.Zeng Q, Song R, Ao L, Weyant MJ, Lee J, Xu D, Fullerton DA, Meng X. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of erk1/2 and nuclear factor-κb activationsignificance. Atertioscler Thromb Vasc Biol. 2013;33:1580–1590. doi: 10.1161/ATVBAHA.112.300912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng Q, Song R, Fullerton DA, Ao L, Zhai Y, Li S, Ballak DB, Cleveland JC, Reece TB, McKinsey TA, Xu D, Dinarello CA, Meng X. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc Natl Acad Sci USA. 2017;114:1631–1636. doi: 10.1073/pnas.1619667114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty S, Cheek J, Sakthivel B, Aronow BJ, Yutzey KE. Shared gene expression profiles in developing heart valves and osteoblast progenitor cells. Physiol Genomics. 2008;35:75–85. doi: 10.1152/physiolgenomics.90212.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Luo J, Iwamoto S, Chen Q. Matrilin-2 is proteolytically cleaved by adamts-4 and adamts-5. Molecules. 2014;19:8472–8487. doi: 10.3390/molecules19068472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD. Calcific aortic valve disease: Not simply a degenerative process. Circulation. 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chi Y, Chai J, Xu C, Luo H, Zhang Q. The extracellular matrix protein matrilin-2 induces post-burn inflammatory responses as an endogenous danger signal. Inflamm Res. 2015;64:833–839. doi: 10.1007/s00011-015-0867-0. [DOI] [PubMed] [Google Scholar]
- 18.Alexopoulos A, Bravou V, Peroukides S, Kaklamanis L, Varakis J, Alexopoulos D, Papadaki H. Bone regulatory factors nfatc1 and osterix in human calcific aortic valves. Int J Cardiol. 2010;139:142–149. doi: 10.1016/j.ijcard.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Su L, Xu Q, Katz J, Michalek SM, Fan M, Feng X, Zhang P. Il-1r/tlr2 through myd88 divergently modulates osteoclastogenesis through regulation of nuclear factor of activated t cells c1 (nfatc1) and b lymphocyte-induced maturation protein-1 (Blimp1) J Biol Chem. 2015;290:30163–30174. doi: 10.1074/jbc.M115.663518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–1086. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muhammad K, Alrefai H, Marienfeld R, Pham DAT, Murti K, Patra AK, Avots A, Bukur V, Sahin U, Kondo E. NF-kB factors control the induction of NFATc1 in B lymphocytes. Eur J Immunol. 2014;44:3392–3402. doi: 10.1002/eji.201444756. [DOI] [PubMed] [Google Scholar]
- 22.Yarilina A, Xu K, Chen J, Ivashkiv LB. TNF activates calcium–nuclear factor of activated t cells (nfat) c1 signaling pathways in human macrophages. Proc Natl Acad Sci USA. 2011;108:1573–1578. doi: 10.1073/pnas.1010030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benton JA, Kern HB, Leinwand LA, Mariner PD, Anseth KS. Statins block calcific nodule formation of valvular interstitial cells by inhibiting α-smooth muscle actin expression. Aterioscler Thromb Vasc Biol. 2009;29:1950–1957. doi: 10.1161/ATVBAHA.109.195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porter S, Clark IM, Kevorkian L, Edwards DR. The adamts metalloproteinases. Biochem J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Didangelos A, Mayr U, Monaco C, Mayr M. Novel role of adamts-5 protein in proteoglycan turnover and lipoprotein retention in atherosclerosis. J Biol Chem. 2012;287:19341–19345. doi: 10.1074/jbc.C112.350785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Du C, Wang H, Zhang C. Increased serum ADAMTS-4 in knee osteoarthritis: A potential indicator for the diagnosis of osteoarthritis in early stages. Genet Mol Res. 2014;13:9642–9649. doi: 10.4238/2014.November.14.9. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Huang J, Yang Z. The roles of adamts in angiogenesis and cancer. Tumour Biol. 2015;36:4039–4051. doi: 10.1007/s13277-015-3461-8. [DOI] [PubMed] [Google Scholar]
- 28.Du Y, Gao C, Liu Z, Wang L, Liu B, He F, Zhang T, Wang Y, Wang X, Xu M. Upregulation of a disintegrin and metalloproteinase with thrombospondin motifs-7 by mir-29 repression mediates vascular smooth muscle calcification. Arterioscler Thromb Vasc Biol. 2012;32:2580–2588. doi: 10.1161/ATVBAHA.112.300206. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Zheng J, Bai X, Liu B, Liu C-j, Xu Q, Zhu Y, Wang N, Kong W, Wang X. Adamts-7 mediates vascular smooth muscle cell migration and neointima formation in balloon-injured rat arteries. Circ Res. 2009;104:688–698. doi: 10.1161/CIRCRESAHA.108.188425. [DOI] [PubMed] [Google Scholar]
- 30.Balachandran K, Sucosky P, Jo H, Yoganathan AP. Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner. Am J Pathol. 2010;177:49–57. doi: 10.2353/ajpath.2010.090631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease. Circulation. 2005;111:3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 32.Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, Chester AH, Butcher JT. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am J Pathol. 2013;182:1922–1931. doi: 10.1016/j.ajpath.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song R, Zeng Q, Ao L, Jessica AY, Cleveland JC, Zhao K-s, Fullerton DA, Meng X. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via toll-like receptor-2. Arterioscler Thromb Vasc Biol. 2012;32:2711–2720. doi: 10.1161/ATVBAHA.112.300116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song R, Ao L, Zhao K-s, Zheng D, Venardos N, Fullerton DA, Meng X. Soluble biglycan induces the production of icam-1 and mcp-1 in human aortic valve interstitial cells through TLR2/4 and the ERK1/2 pathway. Inflamm Res. 2014;63:703–710. doi: 10.1007/s00011-014-0743-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Lizarbe S, Montesinos J, Guerri C. Ethanol induces tlr4/tlr2 association, triggering an inflammatory response in microglial cells. J Neurochem. 2013;126:261–273. doi: 10.1111/jnc.12276. [DOI] [PubMed] [Google Scholar]
- 36.Meloche J, Paulin R, Courboulin A, Lambert C, Barrier M, Bonnet P, Bisserier M, Roy M, Sussman MA, Agharazii M. Rage-dependent activation of the oncoprotein pim1 plays a critical role in systemic vascular remodeling processes. Aterioscler Thromb Vasc Biol. 2011;31:2114–2124. doi: 10.1161/ATVBAHA.111.230573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalagiorgou G, Piperi C, Georgopoulou U, Adamopoulos C, Basdra EK, Papavassiliou AG. Mechanical stimulation of polycystin-1 induces human osteoblastic gene expression via potentiation of the calcineurin/nfat signaling axis. Cell Mol Life Sci. 2013;70:167–180. doi: 10.1007/s00018-012-1164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J-H, Yip CYY, Sone ED, Simmons CA. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am J Pathol. 2009;174:1109–1119. doi: 10.2353/ajpath.2009.080750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 40.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik A, Bonow RO. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Ryzhova LM, Sewell-Loftin M, Brown CB, Huppert SS, Baldwin HS, Merryman WD. Notch1 mutation leads to valvular calcification through enhanced myofibroblast mechanotransductionsignificance. Aterioscler Thromb Vasc Biol. 2015;35:1597–1605. doi: 10.1161/ATVBAHA.114.305095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.