

Abstract
The radiosynthesis of [18F]DCFPyL on 2 distinct automated platforms with full regulatory compliant quality control specifications is described. The radiotracer synthesis was performed on a custom-made radiofluorination module and the Sofie Biosciences ELIXYS. The radiofluorination module synthesis was accomplished in an average of 66 minutes from end of bombardment with an average specific activity at end of synthesis (EOS) of 4.4 TBq/μmol (120 Ci/μmol) and an average radiochemical yield of 30.9% at EOS. The ELIXYS synthesis was completed in an average of 87 minutes with an average specific activity of 2.2 TBq/μmol (59.3 Ci/μmol) and an average radiochemical yield of 19% at EOS. Both synthesis modules produced large millicurie quantities of [18F]DCFPyL while conforming to all standard US Pharmacopeia Chapter <823> acceptance testing criteria.
Keywords: fluorine-18, molecular imaging, prostate imaging, radio synthesis
1 | INTRODUCTION
With an estimated incidence of over 1 million cases per year and an estimated mortality of 307 000 men per year, prostate cancer is the most common cancer in men and one of the most prevalent cancers worldwide.1
In the United States alone, there are well over 200 000 new cases diagnosed annually.2 Owing in part to serum diagnostic tests for expression of the prostate-specific antigen in developing prostate cancer, with proper diagnosis and treatment, the 5-year survival is nearly 99% (seer.cancer.gov). With greater frequency, the proper diagnosis and monitoring of treatment involve noninvasive molecular imaging. A recent comprehensive review1 of prostate-specific membrane antigen (PSMA) positron emission tomography (PET) imaging for prostate cancer describing the current state of the art in this area has recently been published and provides a superb assessment of the radiotracers developed to date including [11C]choline-, [18F]fluorocholine-, [68Ga]-, and [18F]-labeled low-molecular- weight PSMA inhibitors including DCFBC3 and DCFPyL.4
The successful use of [18F]DCFPyL4,5 and its favorable distribution and imaging characteristics compared with other PSMA targeting radiotracers6 have led to increased demand for this radiotracer necessitating a reliable radiochemical synthesis with higher radiochemical yield.
For the past 2 years, our PET/computed tomographic (CT) studies utilized 110 preparations of [18F]DCFPyL synthesized from an adaptation of the published multistep synthesis involving the radiofluorination of a prosthetic group and coupling to a urea using an automated radiochemical synthesis module. Along with removing protective ester groups and purification, the adapted automated synthesis of this tracer involved 2 reactors, multiple individual synthesis steps utilizing 2 precursors, and 90 minutes of synthesis time and produced a final product of low to moderate radiochemical yield. This paper describes an improved synthesis of [18F]DCFPyL from a single precursor with an automated synthesis performed on both an in-house custom-build synthesis module and a commercial radiochemistry module involving a single reactor, 5 operational steps, and 65 to 87 minutes of synthesis time with an increased radiochemical yield. While this paper was in preparation, another group published the simplified synthesis using similar conditions.7 In this paper, a radiopharmacy production level synthesis is achieved, and full compliance with all US Pharmacopeia (USP) <823> acceptance criteria is confirmed and presented.
2 | EXPERIMENTAL
All chemicals and solvents were American Chemical Society or high-performance liquid chromatography (HPLC) purity and were purchased through Sigma-Aldrich Chemical Company (St Louis, Missouri) or Fisher Scientific (Waltham, Massachusetts) except where noted. The synthesis of ((2S)-2-[[(1S)-1-carboxy-5-[(6-fluoranylpyridine-3-carbonyl)-amino]-pentyl]-carbamoyl-amino]pentanedioic acid, the DCFPyL standard, ((2S)-2-[[(1S)-1-t-butylcarboxylate- 5-[(6-fluoranylpyridine-3-carbonyl)-amino]-pentyl]-carbamoyl- amino]-di-t-butyl pentanedioate, the fluorinated protected intermediate standard, the formate salt 2-[3-[1-t- butylcarboxylate-(5-aminopentyl)]-uriedo]-di-t-butyl pentandioate (1), and N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)-pyridin-2-aminium trifluoromethanesulfonate (2) were synthesized as described previously.4,8,9
The custom-made radiofluorination module (RFM) was constructed and controlled in a similar fashion to a previously described microwave radiosynthesis module10 with the cavity. The ELIXYS (Sofie Biosciences, Inc, Culver City, California) module is a commercially available automated multireactor radiosynthesizer.11
For routine quality control (QC) HPLC analyses, the chemical and radiochemical identities of [18F]DCFPyL were determined using an Agilent 1260 Infinity System (Santa Clara, California) incorporating a quaternary pump, HiP ALS autosampler, and DAD UV detector with a Max-Light flow cell set to 264 nm (λmax for DCFPyL) plus a Bioscan Flow-Count interface with a NaI radioactivity detector (Eckert & Ziegler, Berlin, Germany). Chromatographic data were acquired and analyzed on an Agilent OpenLAB chromatography data system (Rev. A.04.05). The following chromatographic conditions were used: an Atlantis T3 C18 5 μm 4.6 × 150 mm (Waters Corp, Milford, Massachusetts) column eluted with a mixture of 10:90 acetonitrile (MeCN): triethylamine (TEA)/phosphate buffer (pH 3.2) at a flow rate of 2 mL/min and UV set at 264 nm.
To examine the possibility of residual lipophilic starting materials, we performed gradient HPLC analysis using the same Agilent HPLC system and a Waters Corp Atlantis dC18 5 μm 2.1 × 100 mm column initially equilibrated with solvent A (10:90 MeCN: TEA/phosphate buffer pH 3.2) at a flow rate of 1 mL/min. Solvent A (100%) was flowed from time of injection until 2.5 minutes when solvent B (100% MeCN) linearly increased from 0% to 85% until the end of the chromatogram at 10 minutes.
Analyses of residual solvent levels in [18F]DCFPyL batches were conducted using an Agilent 7890 A gas chromatograph, Agilent OpenLAB chromatography data system for data acquisition and analysis, and a WAX (polyethylene glycol phase: USP G16, G20) 30 m, 0.25 mm ID, 0.25 μm film column.
2.1 | Synthesis of 5-(((S)-6-(tert-butoxy)-5-(3-((S)-1,5-di- tert-butoxy-1,5-dioxopentan-2-yl)ureido)-6-oxohexyl) carbamoyl)-N,N,N-trimethylpyridin-2-aminium trifluoromethanesulfonate (3)
1 (0.303 g, 0.57 mmol) was dissolved in 6 mL dichloromethane. To it was added 0.158 mL TEA and 2 (0.272 g, 0.57 mmol). The reaction mixture was stirred at room temperature for 1 hour. The solvent was then removed under a stream of N2, followed by drying under vacuum. The mixture was dissolved in acetonitrile and diethyl ether added with stirring. The mixture was kept at room temperature for 30 minutes, and a semisolid was formed. The ether layer was removed, and the semisolid redissolved in acetonitrile. Diethyl ether was added with stirring to produce a semisolid. The mixture was kept at room temperature for 30 minutes, and the ether layer was removed. The semisolid was dried under vacuum and purified on C-18 Sep-Pak Vac 35 cc (Waters Corp) using acetonitrile/water (1:9 to 5:5 [v: v]). Fractions were collected and lyophilized to give white solid (0.322 g, 71%). 1H-NMR (500 MHz, MeOD) δ 9.04 (d, J = 2.3 Hz, 1H), 8.54 (dd, J1 = 2.3 Hz, J2 = 8.7 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 4.17 (m, 2H), 3.68 (s, 9H), 3.43 (m, 2H), 2.32 (m, 2H), 2.03 (m, 1H), 1.80 (m, 2H), 1.67 (m, 3H), 1.45 to 1.50 (m, 29H). 13C-NMR (500 MHz, MeOD) δ 174.0, 173.9, 173.6, 166.0, 160.1, 159.7, 149.5, 141.4, 134.3, 121.9 (q, J = 317.9 Hz), 115.8, 83.0, 82.8, 81.9, 56.0, 54.9, 54.3, 41.1, 33.5, 32.6, 29.9, 29.1, 28.5, 28.4, 24.2. Elemental analysis: calcd for C34H56F3N5O11S; C, 51.05; H, 7.06; N, 8.76; found C, 50.58; H, 6.95; N, 8.49. HR-MS calcd for C33H56N5O8+8: 650.4123, found, 650.4138 [M]+.
2.1.1 | Production of [18F]fluoride
Oxygen-18 enriched water (98%, Huayi Isotopes, Jiangsu, China, approximately 2 mL) was loaded into a niobium body, high-yield [18F]fluoride target of a General Electric Medical Systems (GEMS, Uppsala, Sweden) PETtrace cyclotron. The target was irradiated with a proton beam of 55 μA for 30 minutes to produce approximately 61 GBq (1.65 Ci) of aqueous [18F]fluoride ion by the 18O(p,n)18F nuclear reaction.
2.1.2 | Radiosynthesis of [18F]DCFPyL using the radiofluorination module
After all chemicals and components were loaded into the RFM, [18F]fluoride ion was delivered to a Chromafix 30-PS-HCO3 solid-phase extraction (SPE) cartridge (ABX GmbH, Radeberg, Germany) earlier preconditioned by washing with 1 mL high-purity water (Fluka). [18O]Water was collected for recycling. Under RFM computer control (National Instruments LabVIEW, Austin, Texas), the resin cartridge was eluted with an aqueous solution of tetrabutylammonium hydrogen carbonate (TBABC) (600 μL, 0.075M, ABX GmbH, Germany) into a 5-mL reaction vial sealed with a multiport cap; the vials were cleaned with dilute nitric acid, washed with HPLC water, and dried at 80°C overnight prior to the synthesis. After rinsing the cartridge with MeCN (250 μL), the solution was dried at 110°C with controlled nitrogen flow (325 mL/min) for 150 seconds in a standard thermal heating block. To further dry the [18F]fluoride ion, we heated 2 separate additions of MeCN (250 μL each) for 150 and 180 seconds, respectively.
The vial was cooled using compressed air (flow approximately 6 L/min) to a temperature of 50°C. A solution of the DCFPyL precursor (3) (5 mg, 6.25 μmol) in MeCN (500 μL) was added to the reaction vial containing the dried [18F]fluoride ion. The solution was heated at 50°C for 6 minutes. This was followed without cooling by the addition of phosphoric acid (85%, 350 μL). The vial was maintained at 45°C for an additional 6 minutes. A mixture of sodium hydroxide (2M, 2 mL) and sodium dihydrogen phosphate buffer (10mM, pH 2.1, 1 mL) was added to quench the reaction and buffer the reaction mixture to a pH of 2 to 2.5.
The crude reaction mixture was remotely injected onto a Phenomenex Gemini C18 5 μm 10 × 150 mm column (Torrance, California) eluted with a mixture of 15:85 methanol (MeOH): 0.01M sodium dihydrogen phosphate (pH 2.1) at flow rate of 10 mL/min. [18F]DCFPyL (RT = 18 min, k′ = 15.4) was collected in a reservoir of HPLC water (70 mL). The collected fraction was pushed by nitrogen through a C-18 Sep-Pak Plus Long cartridge (Waters Corp), and the cartridge was rinsed to waste with HPLC water (10 mL). The radiotracer product was eluted from the cartridge with absolute ethanol (1 mL) followed by sterile saline (10 mL) through a 0.2-μm sterile Millipore FG filter (25 mm; Merck KGaA, Darmstadt, Germany) into a sterile product vial preloaded with sterile saline (4 mL).
2.1.3 | Radiosynthesis of [18F]DCFPyL using ELIXYS
After all chemicals and components were loaded onto the ELIXYS synthesis cassette, the [18F]fluoride ion was delivered to a 5-mL V-vial in a dose calibrator. The automated ELIXYS synthesis sequence was started. The [18F]fluoride ion was pushed with nitrogen through a Chromafix 30-PS-HCO3 SPE cartridge (ABX GmbH, Germany) previously preconditioned by washing with 1-mL high-purity water (Fluka). [18O]Water was collected for recycling. The resin cartridge was eluted with a solution of TBABC (300 μL, 0.075M, ABX GmbH, Germany) into the 5-mL reactor V-vial with glass stir bar in the ELIXYS reactor. After rinsing the cartridge with MeCN (600 μL), the solution was dried at 110°C under vacuum with nitrogen flow for 270 seconds with stirring. Two separate additions of MeCN (600 μL) were heated under vacuum with nitrogen flow for 90 and 105 seconds, respectively.
The vial was cooled to a temperature of 45°C. A solution of the DCFPyL precursor (3) (5 mg, 6.25 μmol) in MeCN (500 μL) was added to the reaction vessel containing the dried [18F]fluoride. The solution was heated with stirring at 50°C for 6 minutes. Phosphoric acid (85%, 350 μL) was added to the reaction vial. The reaction vial continued to be heated with stirring for an additional 6 minutes at 45°C. A mixture of 2 mL of sodium hydroxide (2M, 2 mL) and sodium dihydrogen phosphate (10mM, pH 2.1, 1 mL) in 2 equal volume aliquots was added with stirring to quench the reaction and buffer the reaction mixture to a pH of 2 to 2.5.
The purification and formulation of the [18F]DCFPyL were the same as described for the RFM radiosynthesis above.
2.1.4 | Quality control procedures
2.1.4.1 | Visual inspection
Using remote handling equipment and appropriate radiation shielding (leaded glass), the vial containing the [18F]DCFPyL product was visually inspected under bright light. The product met this acceptance specification if it was clear and colorless with no evidence of foreign matter.
2.1.4.2 | Radiochemical identity
A reference standard solution (10 μL sample of 0.0074 nmol/μL) was injected on the analytical HPLC to establish the suitability of the system conditions (confirming a match to a standard curve examining retention time and mass). To determine the radiochemical identity, we mixed an aliquot (50 μL) of the final injection matrix of [18F]DCFPyL with an aliquot of the reference standard solution. The product met this acceptance specification if the retention time of the reference material, as determined by UV detector, was consistent with the retention time of the [18F]DCFPyL, as determined by radiation detector, with appropriate correction for the offset between the 2 detector systems.
2.1.4.3 | Radiochemical purity
Using the same HPLC system described for the radiochemical identity test, an appropriate volume (50 μL) of [18F] DCFPyL was injected at a quantity injected that avoids uncorrected dead-time loss (for main peak) in the radioactive detection system. The percent radiochemical purity of [18F] DCFPyL was determined by dividing the radioactivity associated with the [18F]DCFPyL peak by total activity assayed in the chromatogram multiplied by 100. The product met this acceptance specification if the radiochemical purity was greater than or equal to 95%. A sample QC chromatogram is shown in Figure 6 (DCFPyL - RT = 7.6 min, k′ = 8.7).
FIGURE 6.
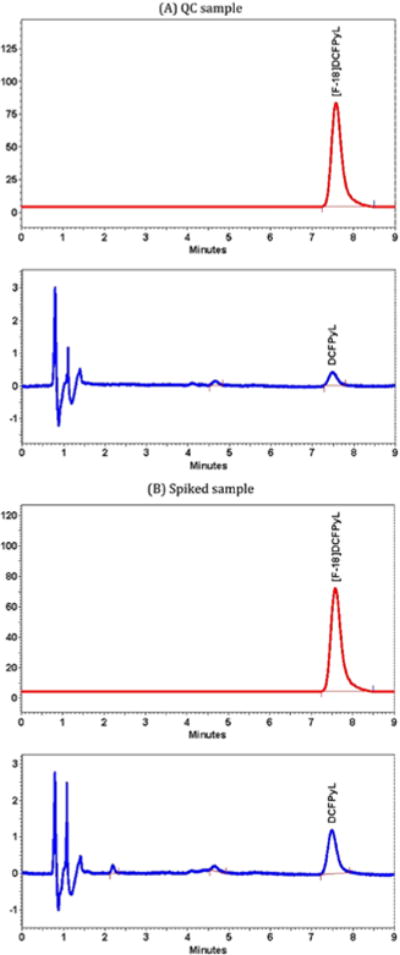
A, Quality control (QC) chromatogram of [18F]DCFPyL. A mass of 0.0134 nmol for the carrier DCFPyL. B, Carrier-added chromatogram of [18F]DCFPyL. Addition of a standard solution of DCFPyL increases the mass of to 0.0384 nmol
2.1.4.4 | Specific activity
The specific activity of [18F]DCFPyL was calculated by dividing the assayed radioactivity of a calibrated aliquot of [18F]DCFPyL (millicurie per milliliter at end of synthesis [EOS]) by the mass concentration of carrier DCFPyL measured by HPLC-UV (micromole of DCFPyL per milliliter) as interpreted from the standard mass calibration curve. The product met this acceptance specification if the specific activity was greater than or equal to 1000 mCi/μmol (a somewhat arbitrary minimum set in the protocol).
2.1.4.5 | Chemical purity
At high specific activity, the use of simple UV peak ratios as an indicator of chemical purity is woefully inadequate as masses are typically diminishingly small. For a successful synthesis of [18F]DCFPyL, not less than 99.5% of the starting precursor must be removed during the synthesis; thus, there may be not more than 0.5% of the precursor or its by-products remaining in the final product matrix. All other UV absorbing HPLC that are not attributed to the matrix must also be less than the same permitted residual precursor concentration. Using the same HPLC system described for the radiochemical identity test, the carrier mass of [18F]DCFPyL was determined. After the initial HPLC column void volume, all other UV peaks were summed and attributed to by-products. The product met this acceptance specification if the mass concentration of these by-products were less than or equal to 1.5 μg/mL.
2.1.4.6 | Residual solvent analysis
An aliquot of a standard solution of 25 mL of HPLC water to which was added 1675 μL of absolute ethanol (6.7%), 12.7 μL of acetonitrile (400 ppm), and 94.7 μL of methanol (3000 ppm) was analyzed to determine system suitability. An aliquot of the final [18F]DCFPyL product matrix was injected, and the levels of residual solvent were calculated by comparison with standards as if from a single-point curve. The product met this acceptance specification if the level of acetonitrile was less than or equal to 400 ppm, the level of methanol was less than or equal to 3000 ppm, and the ethanol level was less than or equal to 10%.12
2.1.4.7 | pH
A drop of the [18F]DCFPyL final product matrix was applied to pH indicator paper (ColorpHast-Indicator strip, pH 2-9; sensitivity of 0.3 to 0.5 units, EMD Chemicals Inc, Gibbstown, New Jersey). The strip color was matched to an indicator chart. The product met this acceptance specification if the pH was between 4.5 and 8.5.
2.1.4.8 | Sterile filter integrity test
The sterile microfilter from the [18F]DCFPyL terminal filtration step was washed with 5 mL of absolute ethanol, left wetted, and attached to a calibrated pressure gauge (Millipore Corp) and air pressure source. The distal end of the filter was placed in a liquid reservoir, and the gas pressure was slowly increased. The product met the acceptance specification for the Millipore Millex FG filter integrity if a pressure of greater than or equal to 13 psi was reached without seeing a stream of bubbles.
2.1.4.9 | Radionuclidic identity
The radioactivity content (millicurie) in an aliquot of the [18F]DCFPyL final product matrix was determined in a Capintec CRC-15R Radioisotope Dose Calibrator (Ramsey, New Jersey) at time (0 min; A) and again 15 minutes later (B). The half-life was calculated using the formula below. The product met this acceptance specification if the calculated half-life was between 105 and 115 minutes.
2.1.4.10 | Radionuclidic purity
Using a suitable gamma-ray spectrometer, an appropriate aliquot of the injection was assayed for a period sufficient to obtain a gamma spectrum. The resultant gamma spectrum was analyzed for the presence of identifiable photopeaks that were not characteristic of 18F emission. The product met this acceptance specification if not less than 99.5% of the total observed gamma emissions corresponded to 0.511 and 1.022 MeV for the radionuclidic purity. Although conducted for all verification productions, this test is considered to be a periodic quality indicator test and is performed annually thereafter.
2.1.4.11 | Endotoxin testing
Endotoxin levels in batches of the [18F]DCFPyL final product matrix were analyzed using a Charles River Laboratories EndoSafe Portable Testing System (Endosafe PTS Reader, Integrated Software Version 7.10, Service Pack 2.0, and printer; Wilmington, Massachusetts). The product met this acceptance specification if the endotoxin level was less than or equal to 11 endotoxin units per milliliter.
2.1.4.12 | Sterility testing
In a laminar flow hood, samples of the [18F]DCFPyL final product matrix (approximately 100 μL each) were added to fluid thioglycolate media and soybean casein digest medium (Becton, Dickinson and Company). The media were incubated at 32.5°C ± 2.5°C and 22.5°C ± 2.5°C, respectively, and observed daily for any turbidity indicative of positive growth. The product met this acceptance specification if no growth was observed during the 14-day incubation period.
2.1.4.13 | Retesting at radiotracer expiry
A subset of the above-listed acceptance tests was performed 360 minutes after the end of the radiotracer synthesis to demonstrate the stability of the radiotracer product upon storage under ambient conditions.
3 | RESULTS AND DISCUSSION
For our human PET/CT studies, the published manual radiosynthesis of [18F]DCFPyL4 was adapted for routine use on an automated, dual reactor synthesis platform (an old Nuclear Interface “Double FDG Synthesis Module”). As part of the original qualification of the radiotracer synthesis for human use, we produced a moderate amount of the final radiotracer product (2.3 GBq; 62 mCi average) with a specific activity at EOS of 192 GBq/μmol (5.2 Ci/μmol). Table 1 shows the original acceptance specifications and results for qualifying productions. Over time, the procedure produced a somewhat variable radiochemical yield of approximately 3% (not corrected for decay, based upon estimated average [18F]fluoride target yields) with frequently lower on average specific activities at EOS for over 110 radiochemical syntheses performed over a 2-year period.
TABLE 1.
QC acceptance specifications: comparison of original synthesis of [18F]DCFPyL with new synthesis on RFM and ELIXYS modules
| Acceptance Specifications in Original Application for First-in-Human Studies | Change in Revised Specification | Revised Acceptance Specifications for this Synthesis (2016) | |||||
|---|---|---|---|---|---|---|---|
| Test | Specification | Average Original (n = 3) | Test | Specification | Average RFM (n = 3) | Average ELIXYS (n = 3) | |
| Initial appearance | Clear, colorless solution, no visible particulate matter | Conforms | No change | Initial appearance | Clear, colorless solution, no visible particulate matter | Conforms | Conforms |
| Appearance, 240 min after EOS | Clear, colorless solution, no visible particulate matter | Conforms | Expiry extended to 360 min | Appearance, 360 min after EOS | Clear, colorless solution, no visible particulate matter | Conforms | Conforms |
| Initial radiochemical purity, % | ≥95% | 100% | No change | Initial radiochemical purity, % | ≥95% | 99.9% | 100% |
| Expiry radiochemical purity, % | ≥95% | 100% | Expiry extended to 360 min | Expiry radiochemical purity, % | ≥95% | 100% | 100% |
| pH, initial | 4.5–8.5 | 5 | No change | pH, initial | 4.5–8.5 | 5 | 5 |
| pH, initial | 4.5–8.5 | 5 | No change | pH, expiry | 4.5–8.5 | 5 | 5 |
| Chemical purity | DCFPyL | 0.33 ± 0.06 μg/mL | No change | Chemical purity | DCFPyL | 0.14 ± 0.03 μg/mL | 0.19 ± 0.08 μg/mL |
| All other impurities ≤ 1.5 μg/mL | 0.05 ± 0.05 μg/mL | All other impurities ≤ 1.5 μg/mL | 0.08 ± 0.08 μg/mL | 0.10 ± 0.09 μg/mL | |||
| Yield | ≥20 mCi [18F]DCFPyL (referenced to assay recorded at end of filtration) | 62.4 ± 10.4 mCi | No change | Yield | ≥ 20 mCi [18F]DCFPyL (referenced to recorded at end of filtration) | 561.7 ± 91.3 mCi | 372.0 ± 199.1 mCi |
| Specific activity | ≥1000 mCi/μmol of [18F]DCFPyL (referenced to end of filtration) | 5210 ± 728 mCi/μmol | No change | Specific activity | ≥1000 mCi/μmol of [18F]DCFPyL (referenced to end of filtration) | 119 950 ± 9165 mCi/μmol | 59 335 ± 12 417 mCi/μmol |
| Residual solvent analysis | Acetonitrile ≤ 400 ppm | 0 ppm | Triethylamine and t-butanol no longer used; methanol added | Residual solvent analysis | Acetonitrile ≤ 400 ppm | 0 ppm | 0 ppm |
| Triethylamine ≤ 400 ppm | 0 ppm | Methanol ≤ 3000 ppm | 17.7 ± 0.6 ppm | 19.6 ± 18.7 ppm | |||
| t-Butanol ≤ 400 ppm | 0 ppm | ||||||
| Radionuclidic identity | t1/2 = 105–115 min | 110.6 ± 3.0 min | No change | Radionuclidic identity | t1/2 = 105–115 min | 109.5 ± 2.2 min | 110.8 ± 2.0 min |
| Radionuclidic purity | 99.5% associated with 18F (0.511 and 1.022 MeV) | Conforms | No change | Radionuclidic purity | 99.5% associated with 18F (0.511 and 1.022 MeV) | Conforms | Conforms |
| Identity (HPLC) | HPLC retention time matches reference standard | Conforms | No change | Identity (HPLC) | HPLC retention time matches reference standard | Conforms | Conforms |
| Filter integrity | Bubble point ≥ 13 psi | 17.3 ± 0.6 psi | No change | Filter integrity | Bubble point ≥ 13 psi | 18.0 ± 1.0 psi | 18.0 ± 1.0 psi |
| Endotoxin | ≤11 EU/mL | <5 EU/mL | No change | Endotoxin | ≤11 EU/mL | <5 EU/mL | <5 EU/mL |
| Kryptofix | ≤50 μg/mL | <50 μg/mL | Kryptofix no longer used | Kryptofix | Specification not needed for this revision. | No Kryptofix is used in this synthesis | No Kryptofix is used in this synthesis |
| Sterility | No growth observed | Conforms | No change | Sterility | No growth observed | Conforms | Conforms |
Abbreviations: EOS, end of synthesis; HPLC, high-performance liquid chromatography; QC, quality control; RFM, radiofluorination module.
All tests except yield, specific activity, and filter integrity were repeated at 360-min expiry and agreed with testing results performed at EOS.
The variable yields and only low to moderate specific activities observed in our original automated synthesis led us to examine an alternative route to the radiotracer. The goal of the work was a [18F]DCFPyL synthesis of higher radiochemical yield and increased specific activity while meeting or exceeding all previously established QC criteria. This involved examining alternative precursors for the radiotracer synthesis, alternative synthesis platforms (including developing our own in-house custom made system), and modifying reaction and purification conditions to achieve a larger-scale radiopharmacy-level production of the radiotracer.
The first step was synthesizing a new precursor. A successful chemical synthesis of the protected trimethylammonium precursor (3) (Figure 1) allowed the radiotracer synthesis of [18F]DCFPyL to evolve. The precursor, 5-(((S)-6-(tert-butoxy)-5-(3-((S)-1,5-di-tert-butoxy-1,5- dioxopentan-2-yl)ureido)-6-oxohexyl)-carbamoyl)-N,N,N- trimethylpyridin-2-aminium trifluoromethanesulfonate (3), was synthesized from the coupling of the triflate salt of the trimethylammonium nicotinic ester (2) and the uriedo compound (1) in suitable yield.
FIGURE 1.
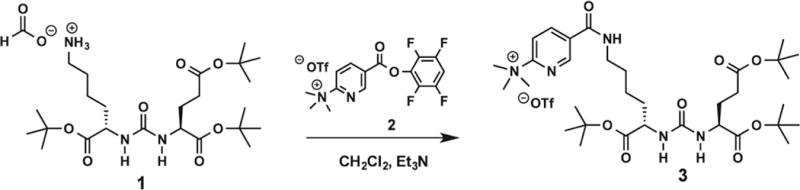
Synthesis of the DCFPyL precursor, 5-(((S)-6-(tert-butoxy)-5-(3-((S)-1,5-di-tert-butoxy-1,5-dioxopentan-2-yl)ureido)-6-oxohexyl)carbamoyl)-N, N,N-trimethylpyridin-2-aminium trifluoromethanesulfonate (3)
Radiofluorination of the trimethylammonium precursor (3) at 50°C produced a clean reaction profile in high yield with the TBABC base. While other bases (eg, potassium bicarbonate or potassium acetate with Kryptofix 2.2.2) were examined, these typically resulted in lower yields. Increasing the quantity of precursor used in the synthesis above 5 mg did not provide an increased radiofluorination yield (data not presented). On the RFM, increasing the amount of TBABC from 11.25 to 22.5 to 45 μmol increased the yield from 10% to 30%. Using gradient HPLC (conditions described in Section 2), an 81% yield of the radiofluorinated protected intermediate was observed (Figure 2A, RT = 8.9 min). Superimposed on the gradient HPLC is the UV trace of the trimethylammonium precursor and nonradioactive fluorinated protected intermediate4 as references (Figure 2B).
FIGURE 2.
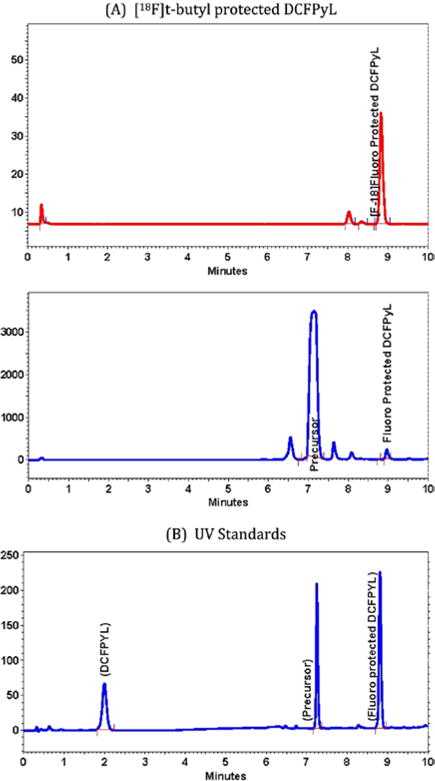
A, Gradient high-performance liquid chromatography (HPLC) of the [18F]t-butyl protected DCFPyL. B, Gradient UV HPLC of DCFPyL, trimethylammonium precursor, and fluoro-protected intermediate standards
After radiofluorination, the deprotection of the ester moieties with a number of different acids was attempted resulting in low yields and by-product formation. Phosphoric acid (pKa1 = 2.12) was found to be sufficiently acidic to remove the butyl groups in a suitable yield13 but not form major radiochemical by-products at 45°C. A gradient HPLC of the final crude deprotected product (RT = 2.0 min) showed a good yield with no radiofluorinated protected intermediate or trimethylammonium precursor remaining in solution (Figure 3).
FIGURE 3.
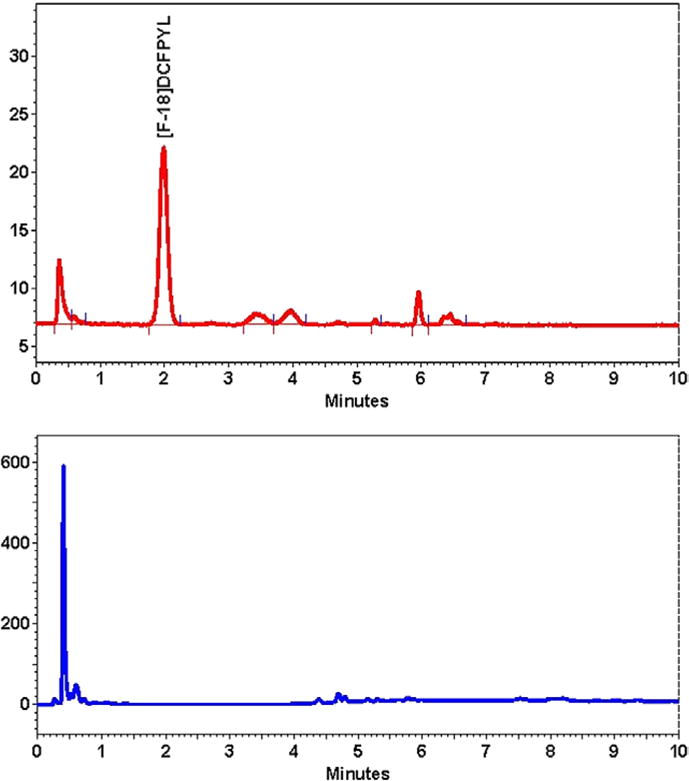
Gradient high-performance liquid chromatography of the final crude [18F]DCFPyL prior to preparative purification
With a radiofluorination synthesis of [18F]DCFPyL that was similar in many ways to some of the standard [18F]-labeled radiotracers previously synthesized (ie, drying radioactive fluoride ion, reaction with an appropriate precursor, deprotection, and some method of purification; like [18F] FDG or [18F]FMISO), we turned our attention to appropriate platforms for performing the synthesis under automation. The in-house, custom-made RFM (a thermal heating adaptation of the microwave synthesis module described previously for making the α7-nicotinergic ligand, [18F] ASEM at high specific activity16) was constructed. The RFM was built with components (valves, tubings, etc) that were free of fluorine in their manufacturing to minimize any fluorine contamination in the synthesis and designed to have fluid pathways that minimize transfer losses and transfer times from upright-positioned, small-volume reagent vessels. The RFM was controlled using National Instruments LabVIEW software.
The radiochemical synthesis was easily transferred to the Sofie Biosciences ELIXYS module. ELIXYS is a 3-reactor system that utilizes replaceable cassettes that provide its fluid pathways and allow the use of one or any combination of the 3 reactors and cassettes in a synthesis sequence.11 For this synthesis, only a single cassette and reactor were needed. The software has an interface that is simple and easily modified for method development or adaptation of existing syntheses. Although potential sources of fluorine contamination have not been rigorously eliminated on ELIXYS, [18F] DCFPyL was obtained in suitable yield at high specific activity. To decrease drying time, less aqueous TBABC solution was utilized, and this may have lowered somewhat the final yield. With the unique integration of robotics for reagent transfers and moveable reactors, ELIXYS synthesis times were longer than those of the RFM.
The final radiosynthesis scheme of [18F]DCFPyL on the custom-made RFM and the Sofie ELIXYS radiosynthesis modules is outlined in Figure 4. With the exception of the amount of TBABC in water that is added (600 μL for the RFM vs 300 μL for the ELIXYS) and the split addition of reaction diluent solution for the ELIXYS prior to HPLC purification vs the single larger volume addition for the RFM, the radiochemical syntheses are identical processes.
FIGURE 4.

Radiosynthesis of [18F]DCFPyL
In both modules, the [18F]fluoride ion was trapped on a Chromafix 30-PS-HCO3 cartridge, eluted from that cartridge with a solution of TBABC in water, and azeotropically dried with heating and additional acetonitrile. An acetonitrile solution of the DCFPyL trimethylammonium precursor (3) was added to the reaction vial, and the vial was heated. Phosphoric acid was added to remove the t-butyl protecting groups with heating. The pH of the crude [18F]DCFPyL solution was adjusted to 2 to 2.5 with the addition and thorough mixing of the sodium hydroxide and sodium dihydrogen phosphate buffer. Injection onto the semipreparative HPLC column produced a typical chromatogram as displayed in Figure 5. The [18F]DCFPyL peak was collected in a water reservoir, and an automated SPE formulation was performed resulting in a final product solution of 1 mL ethanol and 14 mL normal saline.
FIGURE 5.
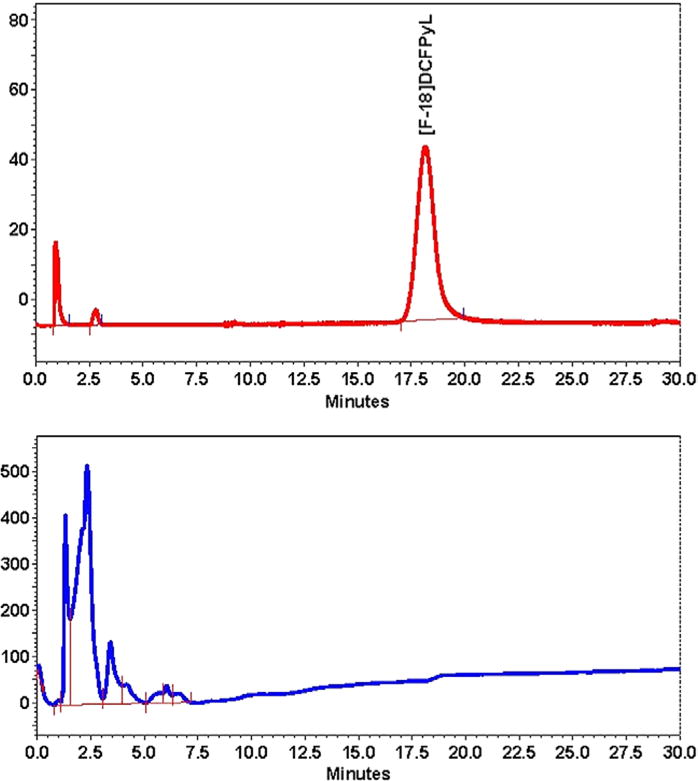
Radioactivity and UV chromatogram of the preparative high-performance liquid chromatography of [18F]DCFPyL
The final sterile solution obtained from the RFM radiosynthesis contained an average of 20.8 ± 3.4 GBq (562 ± 91 mCi; n = 3) of [18F]DCFPyL with an average specific activity of 4.4 ± 0.3 TBq/μmol (120 ± 9.2 Ci/μmol EOS, not corrected for decay). The average EOS non–decay-corrected yield was 30.9% ± 3.0% in an average synthesis time of 66 minutes. The final sterile solution obtained from the ELIXYS radiosynthesis contained an average of 13.8 ± 7.4 GBq (372 ± 199 mCi; n = 3) of [18F]DCFPyL with an average specific activity of 2.2 ± 0.5 TBq/μmol (59.3 ± 12.4 Ci/μmol EOS, not corrected for decay). The average EOS non–decay-corrected yield was 19.4% ± 7.8% in an average synthesis time of 87 minutes. Both of these platforms vastly improved the millicurie yield and specific activity compared with our original multistep synthesis of [18F]DCFPyL that produced an average of 2.3 GBq (62 mCi) with an uncorrected specific activity at EOS of 193 GBq/μmol (5.2 Ci/μmol) in approximately 90 minutes. For comparison, a recent paper using the same precursor with somewhat different reaction and purification conditions7 produced an uncorrected yield of 16% (calculated from the reported decay-corrected yield of 23%) of 3 GBq (81 mCi) of [18F]DCFPyL with a specific activity of 81 to 100 GBq/μmol (2.2-2.7 Ci/μmol) in 55 minutes.
Quality control procedures for [18F]DCFPyL, based upon the current requirements for radiotracers set forth in the USP,14 are summarized in Section 2. Complete QC data, for both radiosynthesis modules, of the initial 3 verification runs of [18F]DCFPyL produced using the methods disclosed herein are summarized in Table 1. These newly reported results meet or exceed all previously stated acceptance specifications. The only changes were the vastly improved amounts of [18F]DCFPyL made with significantly improved radiotracer quality (as demonstrated by the large increases in the final specific activity of the radiotracer product). Two solvents previously used were no longer present in the synthesis, and Kryptofix 2.2.2 was no longer used and thus did not require a final acceptance limit and acceptance specification.
A typical QC chromatogram for chemical and radiochemical purity determinations and a coinjection (“spiked authentic”) chromatogram for the purpose of chemical identity are shown in Figure 6. Carrier mass determinations were performed from a calibration curve generated from 7 mass levels of nonradioactive DCFPyL (6 replicate injections per mass level) relating mass to UV absorbance prepared spanning 0.0046 and 0.2967 nmol with an average goodness of fit (r2) of 0.999985. The lower limit (0.0046 nmol) was established as the limit of quantitation with a signal to noise ratio of 6:1. The limit of detection was determined to be approximately one-half that amount (0.0023 nmol).15 A gradient HPLC of the final formulated solution of [18F]DCFPyL showed no radiofluorinated protected intermediate or trimethylammonium precursor present (Figure 7).
FIGURE 7.
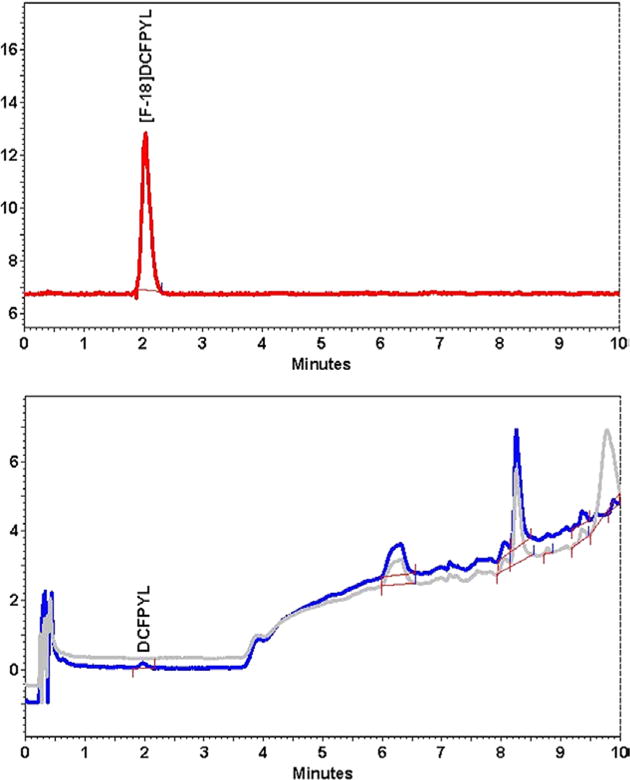
Gradient high-performance liquid chromatography of the final formulated [18F]DCFPyL. Note: The lower chromatogram presents the UV trace of the final product in blue and an overlay of a blank injection of saline in grey to show the gradient trace with no final product present
Our original adaption of the manual synthesis to an automated synthesis platform involved 10 operational steps (trapping fluoride, releasing and drying fluoride, reaction with first precursor, intermediate purification of first precursor, reaction with second precursor, evaporation of reaction solvent, deprotection of t-butyl protected radiofluorinated intermediate with trifluoroacetic acid, removing the acid, buffering for preparative chromatography, purification by HPLC, and formulation of final product). The original synthesis used a module with 2 separate reactors. In this modified synthesis, there are fewer operational steps in part because of the use of a single precursor and deprotection of the penultimate product without the need for solvent evaporation or removing acid prior to preparative HPLC purification. Without the inherent losses of radioactivity during solution transfers, evaporations, and intermediate purification, and the added benefit of shorter reaction times resulting in less radioactivity decay, the current synthesis shows a higher radiochemical yield at EOS compared with the original synthesis.
Finally, to illustrate the potential for large-scale (multi-Curie) production of [18F]DCFPyL, a synthesis using the RFM module produced 83.6 GBq (2.26 Ci) of [18F]DCFPyL from 213 GBq (5.77 Ci) of starting [18F]fluoride representing a 39.2% yield (EOS). The specific activity (EOS) was 4.7 TBq/μmol (127.3 Ci/μmol) with a radiochemical purity of 97.6%. At EOS, all other QC data (as described in Section 2) were within the acceptance specifications set in Table 1. However, a reexamination of this large-scale radiotracer product at the established 6-hour expiry showed the product to be only 51.1% radiochemically pure. While our previous highest product yield validation run was 1.6 GBq/mL (44 mCi/mL) and was confirmed to meet all QC acceptance specifications at the 6-hour expiry, at 5.7 GBq/mL (153 mCi/mL), considerable radiotracer degradation was observed. A subsequent large-scale synthesis using the RFM module in which sodium ascorbate was added to the HPLC collection reservoir and the saline solution used for eluting the product from the Sep-Pak after ethanol elution produced 67.7 GBq (1.83 Ci) of [18F]DCFPyL from 222 GBq (6.0 Ci) of starting [18F]fluoride representing a 30.5% yield (EOS). The specific activity (EOS) was 5.9 TBq/μmol (159.3 Ci/μmol) with a radiochemical purity of 99.7%. At EOS, all other QC data (as described in Section 2) were within the acceptance specifications set in Table 1. The radiochemical purity of this 4.5 GBq/mL (122 mCi/mL) solution of [18F]DCFPyL contained added sodium ascorbate at 3, 4, and 6 hours post EOS were 98.8%, 98.5%, and 98.2%, respectively. It appears in this preliminary examination that there will be some cause for concern when producing highly concentrated [18F]DCFPyL product solutions without some means of moderating the potential for radiolytic decomposition.
4 | CONCLUSION
[18F]DCFPyL was prepared in moderate to high radiochemical yield at very high specific activity. Full regulatory acceptance specifications were described and met for each batch of radiotracer synthesized. This synthesis using either of these automated modules easily provides a sufficient amount of high-quality [18F]DCFPyL radiotracer product for up to 6 PET/CT scans (at 10 mCi per dose) a day injected and imaged 1 hour apart.
Acknowledgments
One of the authors (D.P.H.) was the scientist who designed and built the custom RFM used in this chemistry. The authors would like to thank Jay Burns of the Johns Hopkins School of Medicine Biomedical Engineering Machine Shop for the custom work performed for the RFM. The authors acknowledge CA134675 and CA183031 for partial financial support.
Footnotes
DISCLOSURES
Under a licensing agreement between Progenics and the Johns Hopkins University, Drs Chen, Mease, and Pomper are entitled to royalties on an invention described in this article. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
References
- 1.Maurer T, Eiber M, Schwaiger M, Gschwend JE. Current use of PSMA-PET in prostate management. Nat Rev Urol. 2016;13:226–235. doi: 10.1038/nrurol.2016.26. [DOI] [PubMed] [Google Scholar]
- 2.Seigel R, Ma J, Zhou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Mease RC, Dusich CL, Foss CA, et al. N-[N-[(S)-1.3-dicarboxypropyl]-4- [18F]fluorobenzyl-L-cysteine, [18F]DCFBC: a new imaging probe for prostate cancer. Clin Can Res. 2008;14:3036–3043. doi: 10.1158/1078-0432.CCR-07-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Pullambhatia M, Foss CA, et al. 2-(3-{1-carboxy-5-[(6-[18F]fluoropyridine-3-carbonyl)-amino]-pentyl}-uriedo)-pentanedioic acid, [18F]DCFPyL, a PSMA-based PET imaging agent for prostate cancer. Clin Can Res. 2011;17:7645–7653. doi: 10.1158/1078-0432.CCR-11-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabo Z, Mena E, Rowe SP, et al. Initial evaluation of [18F]DCFPyL for prostate-specific membrane antigen (PSMA)-targeted PET imaging of prostate cancer. Mol Imaging Biol. 2015;17:565–574. doi: 10.1007/s11307-015-0850-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dietlein M, Kobe C, Kuhnert G, et al. Comparison of [18F]DCFPyL and [68Ga]Ga-PSMA-HBED-CC for PSMA-PET imaging in patients with relapsed prostate cancer. Mol Imaging Biol. 2015;17:575–584. doi: 10.1007/s11307-015-0866-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouvet V, Wuest M, Jans HS, et al. Automated synthesis of [18F]DCFPyL via direct radiofluorination and validation in preclinical prostate cancer models. Eur J Nucl Med Mol Imag. 2016;6:40–54. doi: 10.1186/s13550-016-0195-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee SR, Foss CA, Castanares M, et al. Synthesis and evaluation of technetium-99m- and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA) J Med Chem. 2008;51:4504–4517. doi: 10.1021/jm800111u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olberg DE, Arukee JM, Grace D, et al. One step radiosynthesis of 6-[18F] fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]F-Py-TFP): a new prosthetic group for efficient labeling of biomolecules with fluorine-18. J Med Chem. 2010;53:1732–1740. doi: 10.1021/jm9015813. [DOI] [PubMed] [Google Scholar]
- 10.Ravert HT, Holt DP, Dannals RF. A microwave radiosynthesis of the 4-[18F]- fluorobenzyltriphenylphosphonium ion. J Label Compd Radiopharm. 2014;57:695–698. doi: 10.1002/jlcr.3241. [DOI] [PubMed] [Google Scholar]
- 11.Lazari M, Collins J, Shin B, et al. Fully automated production of diverse 18F-labeled PET tracers on the ELIXYS multireactor radiosynthesizer without hardware modification. J Nucl Med Tech. 2014;42:1–8. doi: 10.2967/jnmt.114.140392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline Impurities: Guideline for Residual Solvents. 1997 [Google Scholar]
- 13.Li B, Berliner M, Buzon R, et al. Aqueous phosphoric acid as a mild reagent for deprotection of tert-butyl carbamates, esters, and ethers. J Org Chem. 2006;71:9045–9050. doi: 10.1021/jo061377b. [DOI] [PubMed] [Google Scholar]
- 14.U.S. Pharmacopeia Chapter <823>. Radiopharmaceuticals for Positron Emission Tomography—Compounding. 2009. (USP 32-NF29). [Google Scholar]
- 15.USP (1225) Validation of Compendial Methods
- 16.Ravert HT, Holt DP, Gao Y, Horti AG, Dannals RF. Microwave-assisted radiosynthesis of [18F]ASEM, a radiolabeled α7-nicotinic acetylcholine receptor antagonist. J Label Compd Radiopharm. 2015;58:180–182. doi: 10.1002/jlcr.3275. [DOI] [PubMed] [Google Scholar]