

Abstract
Survival analyses based on the Kaplan–Meier estimate have been pervasively used to support or validate the relevance of biological mechanisms in cancer research. Recently, with the appearance of gene expression high-throughput technologies, this kind of analysis has been applied to tumor transcriptomics data. In a ‘bottom–up’ approach, gene-expression profiles that are associated with a deregulated pathway hypothetically involved in cancer progression are first identified and then subsequently correlated with a survival effect, which statistically supports or requires the rejection of such a hypothesis. In this work, we propose a ‘top–down’ approach, in which the clinical outcome (survival) is the starting point that guides the identification of deregulated biological mechanisms in cancer by a non-hypothesis-driven iterative survival analysis. We show that the application of our novel method to a population of ~2,000 breast cancer patients of the METABRIC consortium allows the identification of several well-known cancer mechanisms, such as ERBB4, HNF3A and TGFB pathways, and the investigation of their paradoxical dual effect. In addition, several novel biological mechanisms are proposed as potentially involved in cancer progression. The proposed exploratory methodology can be considered both alternative and complementary to classical 'bottom–up' approaches for validation of biological hypotheses. We propose that our method may be used to better characterize cancer, and may therefore impact the future design of therapies that are truly molecularly tailored to individual patients. The method, named SURCOMED, was implemented as a web-based tool, which is publicly available at http://surcomed.vital-it.ch. R scripts are also available at http://surcomed.sourceforge.net).
Introduction
It is increasingly accepted that cancer is not a single disease but rather a group of molecularly distinct neoplastic disorders,1 with a variety of underlying molecular mechanisms, morphological features, and biomarkers that hinders the design of treatments and disease prognosis. In addition, due to ethical limitations, most current experimental biomedical research is carried out using either in vitro or in vivo models. Interpreting the relevance of findings in such models and relating them to human health remains challenging.
In the last decades, we have witnessed hundreds of oncology studies, including those at the forefront of cancer research, where the relevance of a given cancer-driving mechanism to human disease is normally determined by applying the following three steps: (1) characterize the mechanism in a biological model; (2) identify a marker whose expression changes when the mechanism is perturbed; and (3) show the correlation between the marker presence and a clinical outcome in human patients. This correlation is frequently illustrated by means of a Kaplan–Meier plot,2 which represents a difference in survival between groups of patients with and without the marker previously identified in the corresponding or closest pre-clinical (animal) model.
Here we propose a novel reverse approach that can be considered both alternative and complementary to hypothesis-driven strategies (Figure 1). Unlike the standard approach of identifying markers in a pre-clinical model based on a given hypothesis and checking whether the clinical outcome in patients supports this hypothesis, our approach searches at the tumor transcriptomic level for relevant combinations of markers in a population of patients based on the optimization of the differences in the observed clinical outcome (patient survival) between groups of patients defined by these markers. In other words, these combinations of markers, which are composed by an arbitrary number of genes, allow the stratification of the population of patients into groups with an optimized difference in the clinical outcome. Subsequently, a comparative analysis of the resulting pro- and anti-survival patient groups for each combination of markers allows generating hypotheses regarding the underlying molecular mechanisms of action. The exploration of these results for multiple combinations of survival biomarkers allows the identification of common mechanisms of action associated with survival that apply to the majority of the patients, as well as mechanisms specific to distinct groups of patients.
Figure 1.
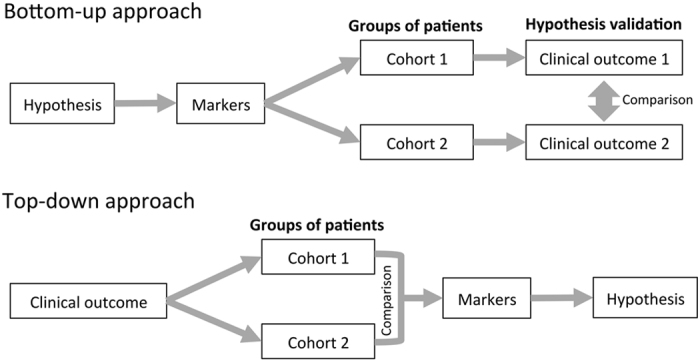
Two strategies to identify relevant cancer-driving mechanisms. In a ‘bottom–up’ or hypothesis-driven approach, gene expression profiles associated with a biological phenotype or deregulated pathway are first identified and then subsequently correlated with the clinical outcome. In a ‘top–down’ approach, cohorts of patients with known clinical outcomes are characterized and compared in order to identify biological mechanisms associated with prognosis without any a priori biological assumption.
It is important to note here that despite the fact that the proposed method fragments the population into groups of patients based on both the gene expression profile and clinical information, it is not intended merely for patient classification or for individual patient prognosis but for hypothesis generation on cancer-driving mechanisms. During the last years a considerable effort has been invested to identify gene-expression signatures for better patient characterization and prognosis. At present, besides the classical identification of single markers and clinical parameters, such as tumor size, grade, cancer stage, or node status, high-throughput technologies allow the measurement of thousands of messenger RNA transcripts in a single experiment and enable identification of multi-gene expression signatures associated with clinical outcomes.1 These multi-gene expression signatures are derived from collections of human tumor transcriptomics data sets and can be used, for example, to classify breast cancer tumors into subtypes reflecting discrete cancer phenotypes.3–5 In general, it has been shown that such signatures are more reliable for predicting clinical outcome than single markers,6,7 and currently several assays are commercially available (MammaPrint, OncotypeDX, Theros, MapQuantDX, Mammostrat and Prosigna).
Instead, rather than attempting to find a comprehensive and predictive classifier for patients, the methodology proposed here is intended to assist in the identification of cancer-driving mechanisms of action, which may coexist in, be shared by or distinguish groups of patients stratified by the abovementioned gene-expression signatures or pathological markers such us ER, PR or Her2. Moreover, the proposed methodology allows investigating biological mechanisms with a dual effect, sometimes protective, sometimes representing risk factors, which hinders the interpretation of their role in cancer progression.
In summary, the algorithm, named SURCOMED (SUrvival COmbined effect-driven cancer MEchanism Discovery), allows the identification of specific mechanisms of action up- or downregulated between subgroups of a given population of cancer patients with a significant survival separation between them (Figure 2). Given that the mechanisms identified by SURCOMED are intrinsically supported by the data in patients, we consider that our approach can help to shorten the process of identification of both cancer-driving mechanisms and therapeutic targets valid in human beings.
Figure 2.
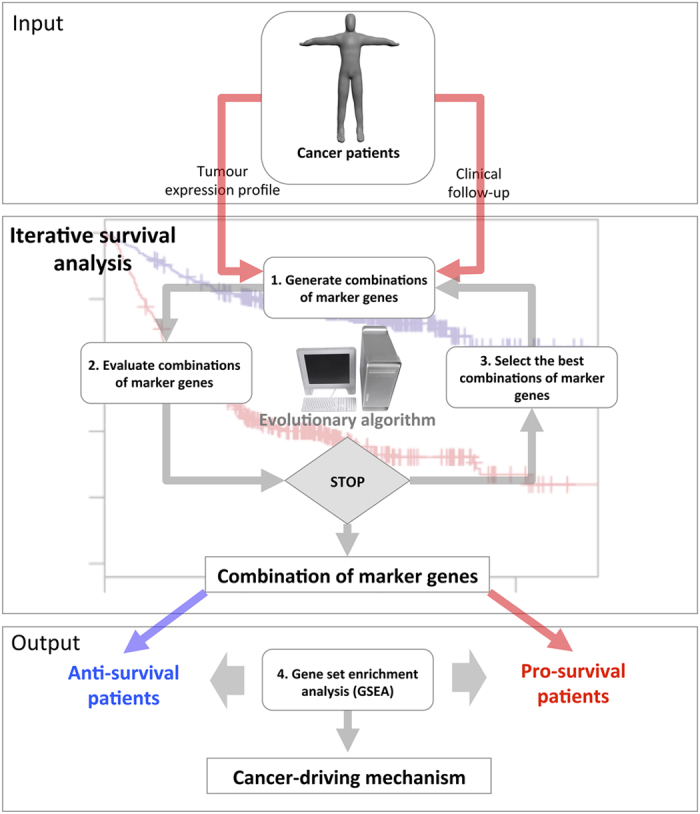
SURCOMED flow chart. SURCOMED takes as input tumor transcriptomics data and the clinical information from the corresponding patients, in particular, the survival time. The output consists of biological processes, molecular mechanisms or pathways up- or downregulated between groups of patients with long and short survival time. These groups of patients are defined by sets of marker genes identified by iterative survival analysis using an evolutionary algorithm. The iterative survival analysis can be described in 3 steps: (1) Generate combinations of marker genes. At the first iteration, the combinations are totally random; in posterior iterations, the generation of new combinations is based on the probability distribution of survival marker genes within the best combinations in the previous iteration; (2) Evaluate combinations of marker genes. This evaluation is based on the difference between the restricted mean survival time between the pro- and anti-survival groups; and (3) Select the best combinations of marker genes. Once the iterative survival analysis finishes, the resulting optimized combinations of marker genes are used to split the population of patients into pro- and anti-survival groups. A gene set enrichment analysis (GSEA) is subsequently applied in order to identify molecular mechanisms, biological processes or pathways for which their constituent genes exhibit concordant differences between pro- and anti-survival groups. This allows the identification of deregulated mechanisms between pro- and anti-survival patients.
Results
In order to illustrate the usage of the proposed methodology, we chose breast cancer because of its relevance to public health and the abundance of publicly available data sets with both tumor transcriptomics and patient clinical follow-up data, which is required for use of the method. We applied SURCOMED to two independent data sets for discovery and validation purposes, with 1,971 and 1,809 breast cancer patients respectively. The ‘discovery’ data set was used to search for processes or pathways potentially relevant for patient survival after tumor resection. To this end and according to our proposed strategy, the first step is to identify genome-wide combinations of marker genes with an optimal survival separation between patients with and without these markers. This search is not biased by prior knowledge and, in principle any gene is equally eligible. It is important to note that in this work we define a survival marker gene not only by the gene itself but also by the expression level (either ‘high’ or ‘low’) within the context of the population of patients (see Materials and Methods). The genome-wide search was performed on a group of 11,849 genes shared by the microarray platforms used in both the ‘discovery’ and the ‘validation’ data sets. The complete lists of genes are included in Supplementary Table S1.
Identification of optimized combinations of survival marker from genome-wide search
In order to perform this search, we used the 1,971 patients of the ‘discovery’ data set, and looked for combinations of 4 survival marker genes. Indeed, in a preliminary analysis, we observed that the survival separations based on 4 genes performed favorably with respect to combinations of smaller and greater number of marker genes in terms of survival separation, but it is worth noting here that the method can be applied to any combination size (see Discussion). After the application of SURCOMED, we selected among 15,000 combinations evaluated by the algorithm (see Materials and Methods) the top 50 for further analysis, 49 of them with a statistically significant survival separation (Benjamini and Hochberg adjusted P value<0.05). Only the combination CENPF::low HTR2C::high HTR4::high CDHA5::high obtained a non-significant adjusted P value. The calculation of adjusted P values is needed because, unlike the classical survival analysis of a single hypothesis, which implies a strong and simple statistical model, here we are considering multiple hypotheses and the resulting P values have to be adjusted according to the number of hypotheses tested.
These combinations were compared with respect to a population of randomly selected combinations (n=1,000) using a t-test, and all of them were found significantly different from the survival values of the random population (P value of the t-test<0.05). The results of the t-test are included in Supplementary Table S2. In summary, taking together the survival analysis of each combination and the comparison with respect to population of combinations randomly selected, we concluded that all selected combinations exhibited not only a statistically significant survival separation but also that the magnitude of this separation is very unlikely to be obtained by chance.
The top 50 combinations of survival markers (Table 1) were identified after the iterative optimization (maximization) of the survival separation between groups of patients with opposite expression profile for a given combination. In other words, patients with a profile for genes A, B, C and D of, for example, A::high, B::low, C::high and D::high are compared (in survival terms) with respect to patients with A::low, B::high, C::low, and D::low (see Materials and Methods section for more details). After several iterations of the algorithm with the evaluation of multiple combinations and selection of the best ones, a final optimized set of combinations of survival markers is obtained. Details of this optimization are included in the Materials and Methods section.
Table 1. Top 50 combinations of survival markers identified by SURCOMED.
| Combinations of survival markers (1–25) | Combinations of survival markers (25–50) | ||
|---|---|---|---|
| ABCG1::high | HSD17B1::high | CDCP1::low | ETV2::low |
| RNASEH2A::low | RPS9::high | HMGCR::low | PLA2G2F::high |
| ACADVL::high | FGL2::high | CDCP1::low | GSTM2::high |
| S100A9::low | SNX1::high | PLOD2::low | YBX2::low |
| ACOXL::low | GSTM2::high | CDCP1::low | MYO7B::high |
| PLOD2::low | YBX2::low | TMEM156::high | TNFRSF4::low |
| ACOXL::low | RSRC1::low | CENPF::low | HTR2C::high |
| SLC2A6::low | STX8::high | HTR4::high | CDHA5::high |
| ACTB::low | CRIM1::high | COL16A1::high | DERL1::low |
| CYP19A1::low | GALNT12::high | TESK1::low | ZNF177::high |
| ADCY3::high | MT1M::high | COL16A1::high | FIBP::low |
| PTTG1::low | TP53BP2::low | IL21R::high | PSMD3::low |
| ADCY3::high | NHLH1::high | COPZ1::low | EGR1::high |
| SERPINE1::low | SORL1::high | HINFP::high | KIF20B::low |
| ANXA2::low | CENPF::low | CSH2::low | GSTT2::high |
| HTR4::high | PCDHA5::high | KL::high | ZNF277::high |
| ANXA2::low | CENPF::low | CYFIP2::high | LUZP4::low |
| PCDHA5::high | TMEM156::high | PHLPP2::low | WDR19::high |
| ANXA2::low | MYO7B::high | DCAF7::low | FAS::low |
| TMEM156::high | TNFRSF4::low | PLAC8::high | PRMT3::low |
| AQP4::high | COPZ1::low | DERL1::low | DPT::high |
| HINFP::high | KIF20B::low | TESK1::low | ZNF177::high |
| AQP4::high | MT1M::high | DLEC1::high | ETV2::low |
| PTTG1::low | TP53BP2::low | HMGCR::low | PLA2G2F::high |
| ATPAF2::high | BMS1::low | DLEC1::high | FIBP::low |
| FBXL14::high | INSR::low | HINFP::high | TNFRSF4::low |
| ATXN3::low | SLC6A6::low | DPT::high | RSRC1::low |
| WDR60::high | ZNF16::high | SLC2A6::low | STX8::high |
| B4GALT7::low | BUB1::low | EGR1::high | NHLH1::high |
| C10orf95::low | CCL3L1::low | SERPINE1::low | SORL1::high |
| BLM::low | CXCL3::low | ELK4::low | GUCY1B2::low |
| NUP133::low | TROAP::low | MBNL1::high | PDCD6IP::high |
| BMPR1B::high | FIBP::low | ETV2::low | PSMD3::low |
| IL21R::high | PSMD3::low | HMGCR::low | MYO7B::high |
| BMPR1B::high | ITM2A::high | GCLC::high | MX2::low |
| OAS3::low | RAB30::high | NQO1::low | SPG20::high |
| BRD7::low | LRPPRC::low | GPX4::high | MRPS28::high |
| SH2B3::low | TRIB3::low | MSR1::low | TROAP::low |
| C16orf70::low | KCTD2::high | HTR2C::high | ITM2A::high |
| PHLDA2::low | SPAST::high | OAS3::low | RAB30::high |
| C17orf53::low | HNRNPA3P1::high | LILRA3::low | NES::low |
| PTPN1::low | TRIM45::high | RRAGA::high | SGCE::high |
| CANT1::low | CXCL9::low | MVD::low | RNASEH2A::low |
| PECR::low | PSME3::high | SLC2A6::high | STK17B::low |
| CATSPER2P1::high | HIST1H2BF::low | NDUFA4L2::low | PDK1::low |
| RPS7::low | SETD1B::low | RGS5::high | UBE2L3::high |
| CBX8::low | FGF17::high | NPY2R::high | PIGV::high |
| FIS1::high | PRKAR1B::low | RPL10L::low | ST5::high |
| CCL20::low | PDLIM1::high | SERPINA7::high | SLC7A6::low |
| STX12::high | WDR19::high | SPG11::high | TMEM186::low |
SURCOMED was applied to a data set of 1,971 breast cancer patients. Combinations with the greatest difference in restricted mean survival time (RMST) between pro- and anti-survival groups of patients are shown in the table.
Validation of the identified combinations of survival marker genes in an independent data set
As we showed, it is very unlikely that combinations of genes randomly selected constitute a good combination of survival markers, but it is possible that some combinations of survival markers identified in our 'discovery' data set cannot be extrapolated to other data sets. To explore this possibility, the identified signatures were validated using an independent 'validation' data set with 1,809 breast cancer patients. Of them, 62% obtained a statistically significant survival separation and similar behavior to that observed in the ‘discovery’ data set. Signatures without statistical support in the ‘validation’ analysis can be considered as over-fitted to the ‘discovery’ data set or to correspond to survival signatures missing in the ‘validation’ data set. The survival curves of DPT::high RSRC1::low SLC2A6::low STX8::high and SERPINA7::high SLC7A6::low SPG11::low STX8::high are shown in Figure 3 as examples of identified validated and non-validated signatures respectively. Complete results comparing the identified signatures in both ‘discovery’ and ‘validation’ data sets are included in Supplementary Table S3.
Figure 3.

Survival analysis of identified signatures in both 'discovery' and 'validation' data sets. (a) Example of validated signature. The survival signature DPT::high RSRC1::low SLC2A6::low STX8::high exhibited a statistically significant survival separation in both ‘discovery’ and ‘validation’ data sets (P value<0.05). (b) Example of non-validated signature. The survival signature SERPINA7::high SLC7A6::low SPG11::low STX8::high was identified in the ‘discovery’ data set, but no significance difference in survival was found in the ‘validation’ data set.
Identification of pathways, biological processes or molecular mechanisms associated with survival
In order to identify mechanisms of action potentially involved in the observed survival effect, for each of the 50 selected combination of markers we applied a gene set enrichment analysis (GSEA), a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences in expression between two biological phenotypes.8,9 These predefined gene sets consist of groups of genes with the same functional annotation (for example, biological processes or pathways) in databases such as GO or KEGG, so they are constant, whereas, for each combination, the biological phenotypes are different and defined by the combination of survival marker genes, which split the population of patients into pro- and anti-survival phenotypes. The results of the GSEA allow not only to detect concordant differences in gene expression between pro- and anti-survival phenotypes but also to determine the sign of the difference or, in other words, whether a given gene set corresponding to a mechanism of action (biological process, pathway or molecular mechanism) is up- or down- regulated in the phenotypes under consideration. This analysis is intended to detect not only those mechanisms that are more common or frequent but also those that are restricted to a reduced subset of the population of patients.
Figure 4 shows the dendrogram of survival combinations organized by hierarchical clustering based on the vector of all biological mechanisms found significant in any of the combinations by GSEA, and a heatmap representing the activity of the biological mechanisms for each survival combination. The exploration of the heatmap in Figure 4 shows both biological mechanisms consistently up- or down- regulated across survival combinations and others that are only up- or down- regulated in a fraction of the survival combinations or even with opposite regulation in different groups of pro-survival patients.
Figure 4.

Heatmap representing molecular mechanisms, biological processes, and pathways identified by SURCOMED in association with 50 different combinations of survival marker genes. Within the heatmap, if a biological mechanism is up- or downregulated, it is represented in red and blue respectively (white if it is not statistically significant). Some of the identified biological mechanisms are consistently up- or downregulated in most of the combinations, whereas others appeared only in some of the combinations. Interestingly, there exist some biological mechanisms with opposite regulation in different groups of pro-survival patients, which reflects the dual effect of some mechanisms, such as the 'Nuclear signaling by ERBB4' (HER4), 'HNF3A (FOXA1) pathway' and 'Transforming growth factor beta (TGFB) signaling pathway'.
Table 2 shows the most frequent up- and down- regulated biological mechanisms across the top 50 combinations of survival markers (complete results in Supplementary Table S4). Table 3 shows 20 frequent biological mechanisms that are either up- or downregulated depending of the survival combinations of markers. Some of these mechanisms are usually associated with a better disease progression, such as the up-regulation of 'T cell activation' or 'T cell differentiation'. However, we found that for some combinations of survival markers, or, in other words, for some subgroups of patients with high survival, these mechanisms are clearly downregulated. Elucidation of the circumstances of such dual mechanisms is a matter for future work.
Table 2. Top 40 survival mechanisms found by SURCOMED in 1,791 breast cancer patients.
| Biological processes, molecular mechanisms and pathways | Occurrences | State |
|---|---|---|
| Reactome nuclear signaling by ERBB4 | 35 | Up |
| Amine binding | 31 | Up |
| Auxiliary transport protein activity | 31 | Up |
| Neuron differentiation | 31 | Up |
| Transmembrane receptor protein serine threonine kinase signaling pathway | 31 | Up |
| Axonogenesis | 30 | Up |
| PID ERA genomic pathway | 30 | Up |
| PID HNF3A pathway | 30 | Up |
| Reactome G alpha S signaling events | 30 | Up |
| Ligand dependent nuclear receptor activity | 29 | Up |
| Neurite development | 29 | Up |
| Reactome phase1 functionalization of compounds | 29 | Up |
| Cellular morphogenesis during differentiation | 27 | Up |
| Channel regulator activity | 27 | Up |
| Kegg drug metabolism cytochrome P450 | 27 | Up |
| Pattern specification process | 27 | Up |
| Biocarta BAD pathway | 26 | Up |
| Neurogenesis | 26 | Up |
| Reactome nuclear receptor transcription pathway | 26 | Up |
| Transforming growth factor beta receptor signaling pathway | 25 | Up |
| Reactome kinesins | 44 | Down |
| Regulation of mitosis | 44 | Down |
| M phase | 43 | Down |
| Reactome cell cycle checkpoints | 43 | Down |
| Transferase activity transferring phosphorus containing groups | 43 | Down |
| M phase of mitotic cell cycle | 42 | Down |
| Mitosis | 42 | Down |
| Organelle organization and biogenesis | 42 | Down |
| Reactome assembly of the pre replicative complex | 42 | Down |
| Reactome cell cycle | 42 | Down |
| Reactome DNA replication | 42 | Down |
| Reactome M-G1 transition | 42 | Down |
| Reactome mitotic M–M–G1 phases | 42 | Down |
| Reactome regulation of mitotic cell cycle | 42 | Down |
| Cell cycle process | 41 | Down |
| Chromosome segregation | 41 | Down |
| PID ATR pathway | 41 | Down |
| Reactome APC C CDH1 mediated degradation of CDC20 and other APC C CDH1 targeted proteins in late mitosis early G1 | 41 | Down |
| Reactome cell cycle mitotic | 41 | Down |
| Cell cycle GO 0007049 | 40 | Down |
The table shows the most frequent up- and downregulated biological mechanisms across the top 50 combinations of survival markers. Highlighted in italic, 'ATR pathway', 'Nuclear signaling by ERBB4' (HER4), 'HNF3A (FOXA1) pathway', and 'Transforming growth factor beta (TGFB) signaling pathway', appeared in 82, 70, 60 and 50% of the survival combinations of markers, respectively. They constitute examples of well-known cancer-driving mechanisms.
Table 3. 20 survival mechanisms that can be either up- or downregulated.
| Biological mechanism | Up | Down | Total |
|---|---|---|---|
| Reactome class A1 rhodopsin like receptors | 8 | 8 | 16 |
| Homeostatic process | 7 | 8 | 15 |
| Reactome G alpha I signaling events | 8 | 7 | 15 |
| Reactome gpcr ligand binding | 11 | 7 | 18 |
| Regulation of developmental process | 6 | 9 | 15 |
| Kegg cytokine cytokine receptor interaction | 6 | 8 | 14 |
| Negative regulation of developmental process | 6 | 6 | 12 |
| Regulation of signal transduction | 6 | 6 | 12 |
| Regulation of multicellular organismal process | 7 | 6 | 13 |
| Reactome peptide ligand binding receptors | 10 | 6 | 16 |
| Regulation of biological quality | 11 | 6 | 17 |
| Positive regulation of immune response | 5 | 12 | 17 |
| T cell activation | 5 | 12 | 17 |
| Reactome hemostasis | 5 | 11 | 16 |
| Regulation of cell proliferation | 5 | 11 | 16 |
| T cell differentiation | 5 | 10 | 15 |
| Biocarta NKT pathway | 5 | 9 | 14 |
| Cytokine activity | 5 | 8 | 13 |
| Cytokine binding | 5 | 8 | 13 |
| Kegg regulation of actin cytoskeleton | 5 | 8 | 13 |
The table shows 20 frequent biological mechanisms across combinations of survival markers that can be either up- or downregulated depending of the particular case.
Among the most frequent up-regulated biological mechanisms, it is worth highlighting 'Nuclear signaling by ERBB4' (HER4), 'HNF3A (FOXA1) pathway' and 'Transforming growth factor beta (TGFB) signaling pathway', which appeared in 70, 60, and 50% of the survival combinations of markers, respectively. These three pathways are particularly interesting because they have been reported to be both positively and negatively associated with a favorable clinical outcome, and their dual behavior has been proposed to be associated with specific subgroups of the population of patients.
In particular, ERBB4 has been suggested to have both oncogenic and tumor suppressive functions.10,11 According to some reports, high expression levels of ERBB4 are associated with favorable clinical outcome.12,13 However, other reports have suggested an association between ERBB4 and a poor prognosis.14,15 Furthermore, it has been reported that, in breast cancer, ERBB4 is associated with favorable prognosis in ER-positive patients, but not in ER-negative patients,16 which may explain the dual effect due to the stratification of different populations of patients.
HNF3A is a downstream target of GATA3 in the mammary gland, and it is highly correlated with low-grade morphology and improved survival in ER-negative breast cancer.17 Interestingly and similarly to the ERBB4 case, in ER-positive breast cancer HNF3A it might be associated with poor clinical outcomes and treatment resistance due to the rapid reprogramming of the ERa signaling.18
Similarly to ERBB4 and HNF3A, TGFB has been proposed to be both positively and negatively associated with the clinical outcomes. In this case, in early stages of breast cancer, TGFB shows tumor suppressive effects with the inhibition of the epithelial cell cycle progression and promotion of apoptosis. However, in late stages it has been associated with an increased tumor progression, higher cell motility, invasiveness and metastasis.19
Given that all of our previously identified combinations of survival markers exhibited either the up-regulation of these three pathways or non-significant deregulation, we decided to investigate their dual effect by performing three new analyses in which the search for combinations of survival markers was constrained by the downregulation of ERBB4, HNF3A, and TGFB1 genes correspondingly. In all of these three new searches, SURCOMED managed to find several combinations of survival markers statistically significant (P value<0.05) and remarkable survival effect (see examples in Figure 5).
Figure 5.
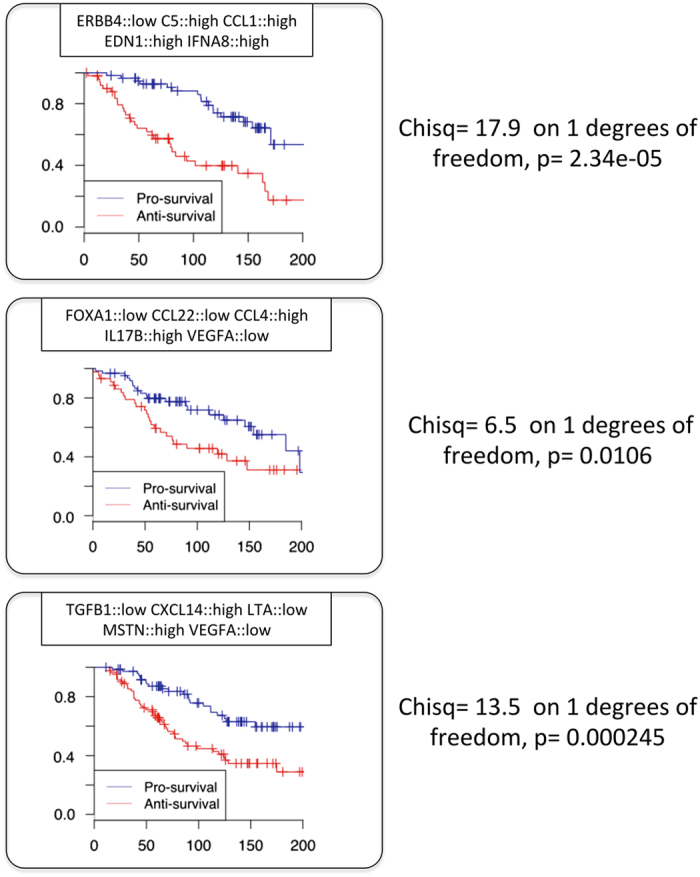
Combinations of survival markers found by SURCOMED with forced downregulation of ERBB4, HNF3A (FOXA1) and TGFB1. All combinations exhibited a clear separation between pro- and anti-survival groups and a P value <0.05.
Concerning the top downregulated biological mechanisms, it is worth noting that some of them were in most of the 50 selected survival combinations, as for example the downregulation of functional categories related to cell division and regulation of mitosis. Among the most frequent downregulated we also found the 'ATR pathway' in 82% of the combinations, which is involved in the regulation of the DNA damage response. The DNA repair pathway, when less efficient, is associated with better survival because it induces the formation of neoantigens. Highly selective small molecule inhibitors of ATR are currently in clinical development for cancer therapy.20
These results highlight the capacity of SURCOMED to identify relevant biological mechanisms that become potential targets for novel therapies and illustrate the utility of SURCOMED to investigate complex paradox behavior of biological mechanisms involved in survival.
It is worth noting that there exist other methods potentially useful to detect biological mechanisms differentially regulated between groups of samples or patients once they are defined by the detected combination of genes. To illustrate an alternative approach to GSEA, we performed a second analysis where a list of genes differentially expressed between pro- and anti-survival groups of patients is analyzed to find statistical overrepresentation of functional annotation terms. Complete results of this analysis and R scripts are provided as Supplementary Information (Supplementary Table S5 and Supplementary Data S2 respectively).
Interestingly, the alternative approach using differential expression analysis between pro- and anti-survival groups of patients and statistical overrepresentation of functional annotation terms, instead of GSEA, also detected ERBB signaling pathway (GO:0038127), TGFB signaling pathway (KEGG: hsa04350) and several terms associated to response to DNA damage (GO:0000077, GO:1902402 and GO:1902230) as statistically significant.
The three examples of paradoxical dual effects emphasize the necessity of novel methodology such as the one proposed in this work in order to disentangle the complexity of cancer mechanisms. It seems clear that different subgroups of patients behave differently and that a single combination of survival markers is not sufficient to capture the variety of biological mechanisms underlying disease progression and survival.
Impact of the data set in the identification of biological mechanisms
In order to investigate the impact of the data set in the identification of biological mechanisms, we applied SURCOMED to the 'validation' data set (acting now as 'discovery') and compared the results from both data sets. Among the 1516 significant biological mechanism identified in this second analysis, 1417 were common to those obtained from the 'discovery' data set. Concerning the top 40 biological mechanisms identified in the 'discovery' data set, 36 of them were also identified in the 'validation' data set. Some of them were very similar in frequency, such as the 'Nuclear receptor transcription pathway' present in 52 and 54% of the signatures in 'discovery' and 'validation' data set respectively, whereas others differ in frequency, such as 'Nuclear signaling by ERBB4', 'ATR pathway' or 'Transmembrane receptor protein serine threonine kinase signaling pathway' (complete results are included in Supplementary Table S6). Regardless of the differences in frequency, which may reflect a different population structure or some kind of bias in the patient recruitment for the study, these results stress the robustness of the analysis across different data sets.
Stratification of patients based on multiple combinations of survival markers
Despite the methodology proposed here is mainly intended to assist in the identification of cancer-driving mechanisms of action, it can be also applied to case classification. The stratification of the population of patients is the other side of the coin of personalized therapy; it allows us to determine groups of patients for which targeting a specific cancer-driving mechanism is suitable, once such mechanisms are identified and validated.
To illustrate this idea, the 50 selected combinations of survival marker genes were used to construct a matrix patient-combination, where, there exists a Boolean vector of 50 elements for each patient that take values of '1' if the combination of survival markers is present in the patient and '0' if it is absent (Supplementary Table S7). These vectors allowed us to compare and cluster patients in different groups based on the entire collection of 50 combinations of survival markers at the same time. The results of this analysis were summarized in a dendrogram of patients organized by hierarchical clustering based on the occurrence of the 50 combinations of survival markers, and the corresponding heatmap precisely representing these occurrences (Figure 6).
Figure 6.

Heatmap representing the occurrence of 50 combinations of survival markers found by SURCOMED in breast cancer patients. The dendrogram on the top corresponds to the hierarchical clustering of patients based on these combinations. This figure illustrates that multiple survival signatures can be used to structure the population of patients and potentially to classify new patients in specific groups. The figure also shows that different combinations of survival markers can coexist in the same patient, either because they are associated with the same underlying biological mechanism or because multiple mechanisms are present in the same patient.
It is also worth noting that the construction of such a matrix also allowed us to explore the co-occurrence of different combinations of survival markers, ranging from 0 up to 20 combinations in the same patient (Supplementary Table S8). Despite some of these signatures possibly corresponding to different markers of the same underlying biological mechanisms, which is perfectly possible as it is shown in Figure 4, others may produce an additive or synergistic survival effect. To illustrate this idea, we performed a survival analysis (Figure 7) comparing the two tails (~5% or 100 patients) of the patients list ranked based on number of combinations of survival markers present in a given patient. In one of the tails, we have patients with only 1 survival signature, whereas the other tail combinations of survival markers range between 10 and 20 per patient. Results showed a clear survival separation of these two groups of patients, which supports the idea that survival can operate through different and sometimes independent mechanisms with additive or synergistic effect when they are together in the same patient.
Figure 7.
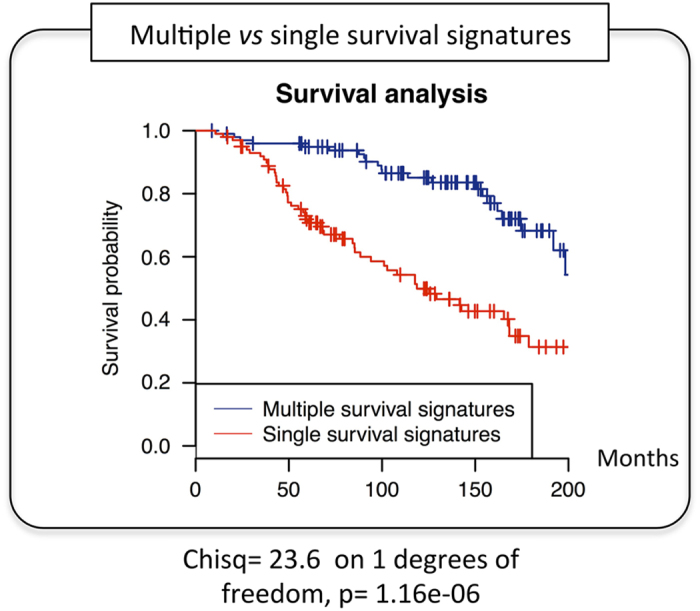
Survival analysis comparing patients with multiple and single survival signatures. The blue curve corresponds to 100 patients with a number of combinations of survival markers (survival signatures) ranging between 10 and 20, whereas the red curve corresponds to 100 patients with only 1 signature. Results showed a clear separation between the two curves, with a P value=1.16e−06, which supports the presence of multiple and independent underlying mechanisms with an additive or synergistic effect when they coexist in the same patient.
The list of coexisting combinations of survival markers for each patient is included in Supplementary Table S8. The complete deconvolution of the underlying dependent and independent survival mechanisms is beyond the scope of this article and a matter of future work.
Discussion
In this work, we propose a novel method, designated SURCOMED, in which patient survival is the starting point that guides the identification of molecular mechanisms of action involved in cancer progression. This method conceptually differs from the regular use of the Kaplan–Meier estimate to validate or support a given hypothesis and can be considered both an alternative and complementary to ‘bottom–up’ approaches to elucidate cancer progression mechanisms.
The main strength of SURCOMED is that it is not biased by prior knowledge; we report here that SURCOMED can be applied in a blind manner to discover biological mechanisms involved in disease progression, which can lead to unexpected findings. The blindfolded application of SURCOMED allowed us to identify several deregulated pathways well known as involved in breast cancer progression, such as the up- and downregulation of ERBB4, HNF3A and TGFB and ATR pathways, respectively. In addition, SURCOMED can also be applied under certain constrains in order to investigate specific questions. We illustrated this capability in three specific cases by searching for combinations of survival markers accompanying the downregulation of ERBB4, HNF3A and TGFB genes. In the three cases, SURCOMED managed to find combinations of survival markers under these constraints, which is consistent with the previously reported dual effect of these genes on disease progression.
The main limitation of SURCOMED is that it operates only on existing expression profiles within the population of patients, and only these existing expression profiles can be compared and selected as optimal combinations of survival markers; there is no prediction or projection to anticipate the behavior of expression profiles that are not already present in the population of patients. This means that a greater variety of comparisons can be performed on large, heterogeneous data sets, leading to better rank combinations of survival markers, whereas results will be more limited for smaller, more homogeneous data sets. Similarly, the way in which SURCOMED intersects groups of patients to calculate the combined survival effect makes this calculation only possible for large data sets, and this problem is exacerbated as the number of genes in the combination is increased. We empirically observed that, for combinations of 4 genes, SURCOMED performs reasonably well with data sets as small as 100 patients randomly selected from larger populations, and with results consistent with those obtained using greater number patients.
We have shown that SURCOMED can be used for patient stratification based on multiple combinations of survival signatures, which suggest potential translational applications. However, we believe that the capacity to classify a case in a predefined group is more useful if the group is well characterized in terms of biological mechanisms, which ultimately would lead to the development of more personalized therapies.
Moreover, it sounds reasonable to classify patients based on the solvency of the different biological mechanisms underlying disease progression, and treat them according to their special demands. It is precisely this mechanism-centered vision of patient classification that is the main motivation of this work, and the identification of combinations of genes that make the difference at the mechanism level (summarized as the clinical outcome in terms of patient survival) is the core of the proposed methodology. The deconvolution of the contributing biological mechanisms to cancer progression is by no means simple, but we consider that this work constitutes a step forward in this direction. Of course, a final mechanism-centered patient classification would require the experimental validation of the identified biological mechanisms, which constitutes a monumental challenge beyond the scope of this work and motive of future directions.
Finally, it is worth mentioning that, in this study, we have used breast cancer as an illustrative example, but SURCOMED is by no means restricted to this disease, and we plan to apply SURCOMED to a variety of solid tumors and other diseases or biological processes beyond cancer in the coming years.
Materials and methods
Iterative survival analysis: search of optimal combinations of survival marker genes
The algorithm takes as the input a collection of transcriptomics data derived from resected tumors and the clinical information of the corresponding patients, more specifically, the survival time. As the output, the algorithm delivers one or several combinations of highly or lowly expressed genes with an optimized survival separation associated with their expression levels. The exhaustive search of optimal combinations of marker genes is a huge combinatorial problem that we addressed with a strategy that explores the vast search space by iterative optimization of the clinical outcome, i.e., the observed survival effect. To this end, the algorithm uses an evolutionary optimization based on an estimation of distribution algorithm, a well-known strategy commonly used in bioinformatics.21
During the search process, there are several rounds of optimization or iterations (we used 50 iterations in this work) in which new combinations of genes are generated based on the probability distribution of gene states calculated in the subset of combinations with the best survival separation in the previous iteration. The optimization continues until the maximum number of iterations (defined by the user) is reached. One or several combinations with an optimized survival separation constitute the final output.
The algorithm for the iterative search of combinations of marker genes can be described in 3 steps (Figure 2)—(1) generate combinations of marker genes; (2) evaluate combinations of marker genes; and (3) select the best combinations of marker genes.
Generate combinations of marker genes. In the first iteration, the algorithm creates a population of combinations by random selection of a given number of genes with their corresponding states, i.e., either ‘higher’ or ‘lower’ (see ‘Combined survival analysis’ section for details). The specific number of genes is defined by the user and is the same as the number of marker genes in the final optimized combinations; in the analysis included in this work, we used combinations of four genes because we empirically observed that the survival separations based on four genes performed reasonably well with respect to smaller and greater combinations, but the method is applicable to any number of genes. In the second and later iterations, new combinations of marker genes are selected by randomly sampling the set of genes based on the probability distribution of genes and their corresponding expression levels (discretized to either ‘high’ or ‘low’) within the subset of combinations with the best survival separation in the previous iteration. In this work, we used populations of 30 combinations and selected at each iteration the top 15 combinations with the best survival separation. Of note, SURCOMED allows the user to force or to censor the presence or absence of specific gene states during the search.
- Evaluate combinations of marker genes. The algorithm evaluates the combined survival signature for each combination of genes by calculating the restricted mean survival time (RMST) of two groups of patients. The first group corresponds to those patients who fulfill the expression profile defined by the combination, and the second group corresponds to the opposite expression profile (see ‘Combined survival analysis’ section). The RMST is defined as the area under the curve of the survival function, and it corresponds to the average time that patients will survive. The difference between the RMST of the first and second groups constitutes the score of the combination:
Select the best combinations of marker genes. Once all combinations have been scored and ranked, only the best combinations will be used to calculate the probability distribution of all gene states. In this work, we used a selection number that equals half of the population number (n=15). Within the subset of combinations with the best survival separation, the algorithm calculates the probability distribution of gene states. That means that for every selected combination, SURCOMED calculates gene-by-gene how many patients have a ‘higher’ and a ‘lower’ expression value in the group of patients with an expression profile consistent with the combination (Figure 8). The criterion to decide whether a given expression value is ‘higher’ or ‘lower’ is based on the median of the expression value of the gene in the entire population of patients. Consequently, expression values above and below the median will be considered to be in the ‘higher’ and in the ‘lower’ group, respectively. The total number of occurrences for each gene state will be divided by the total number of patients in the group with an expression profile consistent with the combination. These frequencies are calculated for each gene and combination. Once all combinations are calculated, SURCOMED computes the average frequency across combinations for each gene. The resulting frequency will become the weight for each gene state. The greater the weight, the higher the probability that the gene in a given state is present when generating the next population of combinations. To sample the new set of combinations SURCOMED uses the R function sample() with the vector of average frequencies as argument (prob argument).
Figure 8.
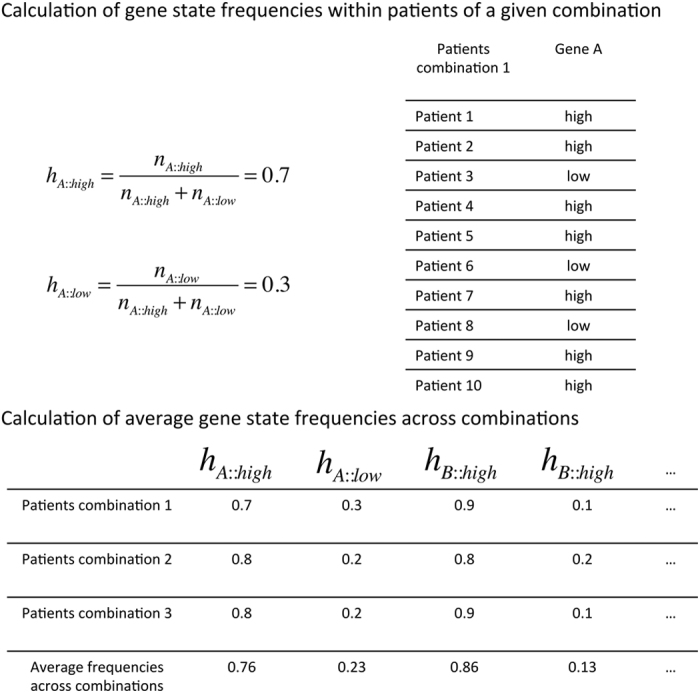
Calculation of gene state frequencies in patients of a given combination and across combinations. The frequencies of gene A in states of 'high' and 'low' expression in the subset of patients are defined by the expression profile of a given combination. These 'high' or 'low' values are assigned when the expression levels are respectively above or below the median of the expression for this gene in the entire population of patients. Once this frequency has been calculated for each of the selected combinations of genes, the average value of these frequencies across combinations are used as weights to sample a new set of combinations. Those gene states with higher average values across combinations have a better chance to be selected in the new set of combinations generated in the next iteration of the algorithm.
In this work, we ran SURCOMED 10 times with 50 iterations of the algorithm and collected the top 5 combinations from each run, which resulted in the selected 50 combinations that we used for further analysis. Given that at each iteration of the algorithm 30 combinations were evaluated, a total of 15,000 combinations were assessed across runs and iterations.
It is worth mentioning that we decided to work with a population size of 30 combinations and 50 iterations after some preliminary tests. The optimization may be benefited from greater population sizes but with a computational time cost. We strongly suggest users planning to apply SURCOMED to a different data set to perform some preliminary runs with different population size and small number of iterations before the actual analysis in order to find a suitable configuration of the algorithm, which ultimately depends on the underlying structure of the population of patients.
Implementation of the SURCOMED algorithm
The algorithm is implemented as a web application written with the AngularJS and d3js frameworks for the front-end part and with PHP and R on the server side. It performs three types of analysis: first, the search for combinations of genes; second, the test of specific combinations; and third, results from these two previous analyses can be redirected to investigate deregulated mechanisms between pro- and anti-survival groups. Both intermediate and final results can be retrieved and downloaded or visualized online. The web-based display of the results enables the user to explore the survival curve of the top combinations, χ2, and P value of the log-rank test and the number of patients considered in each specific comparison, as well as to explore the underlying mechanisms that distinguish pro- and anti-survival groups.
The user can define the search space by providing a list of gene symbols or probes, or by selecting a genome-wide search. Once the analysis is complete, an email will be sent to the user with a link to the web-based display of the results, which can be visualized or downloaded.
The analysis can also be constrained by means of two entry-boxes where the user can introduce a list of ‘forced’ and ‘censored’ gene states that will be taken into account during the search; ‘forced’ refers to gene states that will always be included in any combination, whereas ‘censored’ refers to gene states that will be systematically avoided.
We also provide R scripts for the search, and mechanisms detection, which can be run locally from the command line without graphical interface (Supplementary Data File S2 and http://surcomed.sourceforge.net). In addition, a user’s guide is available as Supplementary Data File S3 and http://surcomed.sourceforge.net.
Combined survival analysis
In survival analysis based on the expression of a single gene, the population of patients is divided into two groups based on a given threshold (for example, the median of the expression values), and the corresponding survival curves derived from these groups are compared. The combined survival analysis is performed by subsetting from the population of patients two groups for comparison: those patients who fulfill a given expression profile and those who fulfill the opposite profile. To this end, for each individual gene the algorithm splits the population of patients into two groups based on the median of the expression value of this gene in the entire population of patients. The first group corresponds to those patients with an expression value above the median (‘higher’); the second group corresponds to those patients below the median (‘lower’). Subsequently, the resulting groups of patients are intersected according to the gene states in the combination in order to obtain the final two groups for the survival comparison (Figure 9). It may happen that there are patients in the population that do not fulfill the combination profile or the opposite profile for all of the genes; consequently, they are not taken into account for the comparison.
Figure 9.

Intersection of patients to perform a combined survival analysis. In this example, the combination evaluated is A::high-B::low-C::high (pro-survival group) against A::low-B::high-C::low (anti-survival group). Red and black blocks refer to the subsets of patients with expression values higher and lower than the median respectively. The separation between the pro- and anti-survival curves (in blue and red respectively) is optimized by SURCOMED to search for optimal combinations of marker genes.
All survival analyses were performed using the R package ‘Survival’ (http://cran.r-project.org/web/packages/survival/index.html). The statistical significance of these analyses was determined based on the log-rank test with a critical value calculated using a χ2 with a degree of freedom equal to 1. Only analyses with a P value<0.05 were considered as statistically significant and further evaluated using the RMST difference between pro- and anti-survival groups. In the case that the survival separation is not statistically significant, the score of the combination is equal to 0.
It is worth noting that more stringent thresholds than the median, such as first and third quartiles or 10th percentile, could be potentially applied to split the population of patients, but such thresholds would decrease the number of patients available for the intersections and even prevent from finding patients with an expression profile consistent with the combination under evaluation. We admit that patients right above and below the median may be very similar, and that there is not strong evidence to support that they have to be classified in different categories. However, as we have shown in this work, the noise potentially included by wrong patient classification does not prevent the optimization algorithm from finding groups of patients with a clear survival separation. Consequently, for the sake of general applicability, we decided to use the median as a general threshold, which takes advantage of all the patients in the data set.
GSEA
GSEA allows the determination of whether an a priori defined set of genes shows statistically significant, concordant differences in expression between pro- and anti-survival groups of patients. To this end, we used the GSEA software8,9 and as gene sets the collections C2 and C5 from the Molecular Signatures Database (MSigDB).9 In order to obtain the statistical significance for each gene set, we ran 1,000 permutations of the GSEA for each optimized combination of marker genes with a random assignment of phenotype for each patient (either pro- or anti-survival) and considered only those gene sets identified with a P value<0.05 with respect to the permutations. Alternatively, using the SURCOMED interface users can generate and download the files required for running the GSEA analysis locally.
Data sets
We used two different data sets in this work, namely, the ‘discovery’ and ‘validation’ sets. The ‘discovery’ data set corresponds to the collection of patients from METABRIC (Molecular Taxonomy of Breast Cancer International Consortium).22 It includes microarray expression data and clinical information from 1,971 breast cancer patients; the platform used for the transcriptional profiling was Illumina HT-12 v3. The ‘validation’ data set includes 1,809 patients. It includes microarray expression data and clinical information from 1,809 breast cancer patients.23 The platform used was either on HG-U133A (GPL96) or HG-U133 Plus 2.0 (GPL570).
Availability of data and materials
The discovery data set is available at the European Genome-phenome Archive (www.ebi.ac.uk/ega). The ‘validation’ data set is publicly available at the KM plotter repository (www.kmplot.com). The algorithm for the iterative survival analysis was implemented as a web-based tool, which is publicly available at http://surcomed.vital-it.ch. R scripts for the search test of combinations, and mechanisms detection are available at http://surcomed.sourceforge.net).
Acknowledgments
We thank Dr Brian Stevenson and Dr Mark Ibberson for careful and critical reading of the manuscript. This project has been funded with support from Vital-IT-SIB (Swiss Institute of Bioinformatics) at the University of Lausanne; the MEDIC Foundation, and the Swiss National Science Foundation (SNSF), grant CR32I3_156915 (MAD), as well as the Ludwig Institute for Cancer Research. (GC).
Footnotes
Supplementary Information accompanies the paper on the npj Systems Biology and Applications website (http://www.nature.com/npjsba)
The authors declare no conflict of interest.
References
- Sotiriou, C. & Pusztai, L. Gene-expression signatures in breast cancer. N. Engl. J. Med. 360, 790–800 (2009). [DOI] [PubMed] [Google Scholar]
- Goel, M. K., Khanna, P. & Kishore, J. Understanding survival analysis: Kaplan-Meier estimate. Int. J. Ayurveda Res. 1, 274 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA 98, 10869–10874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou, C. M. et al. Molecular portraits of human breast tumours. Nature 406, 747–752 (2000). [DOI] [PubMed] [Google Scholar]
- Van't Veer, L. J. et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536 (2002). [DOI] [PubMed] [Google Scholar]
- Wang, Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 365, 671–679 (2005). [DOI] [PubMed] [Google Scholar]
- Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullick, W. J. c‐erbB‐4/HER4: friend or foe? J. Pathol. 200, 279–281 (2003). [DOI] [PubMed] [Google Scholar]
- Junttila, T. T., Sundvall, M., Määttä, J. A. & Elenius, K. Erbb4 and its isoforms: selective regulation of growth factor responses by naturally occurring receptor variants. Trends Cardiovasc. Med. 10, 304–310 (2000). [DOI] [PubMed] [Google Scholar]
- Pawlowski, V., Révillion, F., Hebbar, M., Hornez, L. & Peyrat, J.-P. Prognostic value of the type I growth factor receptors in a large series of human primary breast cancers quantified with a real-time reverse transcription-polymerase chain reaction assay. Clin. Cancer Res. 6, 4217–4225 (2000). [PubMed] [Google Scholar]
- Witton, C. J., Reeves, J. R., Going, J. J., Cooke, T. G. & Bartlett, J. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J. Pathol. 200, 290–297 (2003). [DOI] [PubMed] [Google Scholar]
- Bièche, I. et al. Prognostic value of ERBB family mRNA expression in breast carcinomas. Int. J. Cancer 106, 758–765 (2003). [DOI] [PubMed] [Google Scholar]
- Lodge, A. et al. Type 1 growth factor receptor expression in node positive breast cancer: adverse prognostic significance of c-erbB-4. J. Clin. Pathol. 56, 300–304 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila, T. T. et al. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 65, 1384–1393 (2005). [DOI] [PubMed] [Google Scholar]
- Albergaria, A. et al. Expression of FOXA1 and GATA-3 in breast cancer: the prognostic significance in hormone receptor-negative tumours. Breast Cancer Res. 11, 1–15 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes, C. S. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzynska, J. M. Two faces of TGF-beta1 in breast cancer. Mediat. Inflamm. 2014, 141747 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, A. M. & Ryan, A. J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 149, 124–138 (2015). [DOI] [PubMed] [Google Scholar]
- Armañanzas, R. et al. A review of estimation of distribution algorithms in bioinformatics. BioData Mining 1, 6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györffy, B. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The discovery data set is available at the European Genome-phenome Archive (www.ebi.ac.uk/ega). The ‘validation’ data set is publicly available at the KM plotter repository (www.kmplot.com). The algorithm for the iterative survival analysis was implemented as a web-based tool, which is publicly available at http://surcomed.vital-it.ch. R scripts for the search test of combinations, and mechanisms detection are available at http://surcomed.sourceforge.net).