

Abstract
Increased sympathetic nerve activity and the activation of the central renin-angiotensin system are commonly associated with cardiovascular disease states such as hypertension and heart failure, yet the precise mechanisms contributing to the long-term maintenance of this sympatho-excitation are incompletely understood. Due to the established physiological role of neurotrophins contributing towards neuroplasticity and neuronal excitability along with recent evidence linking the renin-angiotensin system and brain-derived neurotrophic factor (BDNF) along with its receptor (TrkB), it is likely the two systems interact to promote sympatho-excitation during cardiovascular disease. However, this interaction has not yet been fully demonstrated, in vivo. Thus, we hypothesized that central angiotensin II (Ang II) treatment will evoke a sympatho-excitatory state mediated through the actions of BDNF/TrkB. We infused Ang II (20 ng/min) into the right lateral ventricle of male Sprague-Dawley rats for twelve days with or without the TrkB receptor antagonist, ANA-12 (50 ng/h). We found that ICV infusion of Ang II increased mean arterial pressure (+40.4 mmHg), increased renal sympathetic nerve activity (+19.4% max activity), and induced baroreflex dysfunction relative to vehicle. Co-infusion of ANA-12 attenuated the increase in blood pressure (−20.6 mmHg) and prevented the increase in renal sympathetic nerve activity (−22.2% max) and baroreflex dysfunction relative to Ang II alone. Ang II increased thirst and decreased food consumption, and Ang II + ANA-12 augmented the thirst response while attenuating the decrease in food consumption. We conclude that TrkB signaling is a mediator of the long-term blood pressure and sympathetic nerve activity responses to central Ang II activity. These findings demonstrate the involvement of neurotrophins such as BDNF in promoting Ang II-induced autonomic dysfunction and further implicate TrkB signaling in modulating presympathetic autonomic neurons during cardiovascular disease.
Keywords: BDNF, blood pressure, baroreflex, neurotrophins
1. Introduction
Increased central renin-angiotensin system (RAS) activation is a hallmark of many cardiovascular diseases such as hypertension and heart failure (Biancardi et al., 2014; Francis, 1985; Guyenet, 2006; Zucker et al., 2012). Although the contribution of the RAS to increased sympathetic tone is well established, the mechanisms contributing to the maintenance and establishment of long-term changes to sympathetic activation remain less well understood. One potential mechanism contributing to this long-term maintenance of sympathetic tone and elevation of blood pressure is neuroplasticity in cardiovascular control centers that potentiate pro-sympathetic pathways driven by neurotrophic factors (Johnson et al., 2015). Neurotrophic factors not only contribute to synaptic connectivity but also mediate changes in neuronal excitability (Benarroch, 2015; Blum et al., 2005; Cao et al., 2010; Rose et al., 2004). To this end, recent studies have investigated the contribution of the ubiquitous neurotrophin brain-derived neurotrophic factor (BDNF) toward promoting autonomic imbalance during RAS activation (Becker et al., 2016; Becker et al., 2015; Clayton et al., 2014; Erdos et al., 2015a; Schaich et al., 2016).
In a recent study where BDNF was overexpressed in the paraventricular nucleus of the hypothalamus (PVN) of rats, an increase in mean arterial pressure (MAP) and an increased hemodynamic (pressor and tachycardia) response to acute stressors such as water and restraint stress was shown (Erdos et al., 2015a). The authors demonstrated that BDNF overexpression augments angiotensin II (Ang II) signaling through the angiotensin type 1 receptor (AT1R). Unfortunately, the role of the BDNF receptor, the Tyrosine receptor kinase B (TrkB) in Ang II signaling was not investigated. BDNF/TrkB signaling may be typical of cardiovascular disease states where central RAS activity may be elevated (Biancardi et al., 2014; Francis, 1985; Zucker et al., 2012). In addition to BDNF, TrkB can bind to neurotrophin 4 (NT4) (Huang et al., 2003; Minichiello, 2009), although the role of NT4/TrkB signaling in sympathetic control of blood pressure is even less well understood than BDNF/TrkB signaling.
Central Ang II evokes hypertension and increased sympathetic nerve activity (SNA) (Camara et al., 1998; Gao et al., 2014; Gao et al., 2005; Osborn et al., 1997) and arterial baroreflex dysfunction (Gao et al., 2008; Pan et al., 2007). Central Ang II has also been shown to modulate metabolic function through increased thirst (Fitzsimons, 1998) and promotion of cachexia, a catabolic state (Brink et al., 2001; Brink et al., 1996; Yoshida et al., 2012). Many of these effects are mediated by Ang II signaling through the AT1R in presympathetic areas of the brainstem and hypothalamus (Gao et al., 2005; Gao et al., 2008; Zucker et al., 2009) and involve reactive oxygen signaling (Chan et al., 2005; Sheh et al., 2007; Yin et al., 2010; Zimmerman et al., 2004). However, little is understood about how AT1R activation by Ang II induces long-term changes relating to neuronal excitability and synaptic plasticity of presympathetic centers, which ultimately results in long-term sympatho-excitation. Signaling mechanisms such as activation of mitogen-activated protein kinase have been implicated in both AT1R (Chan et al., 2005; Gao et al., 2010; Xiao et al., 2013) and TrkB signaling (Becker et al., 2015; Kim et al., 2012; Revest et al., 2014) suggesting a potential convergent signaling mechanism between the two pathways.
Here we aimed to demonstrate, in vivo, the connection between central Ang II and TrkB signaling pathways in mediating sympatho-excitation. Based on recent literature, we propose TrkB signaling as a potential mechanism by which Ang II causes long-term, persistent changes to central autonomic control centers that promote sympatho-excitation. As we have previously demonstrated in vitro Ang II treatment causes an increase in BDNF expression and results in perturbations to K+ currents (Becker et al., 2015), here we hypothesize that central Ang II treatment will evoke a sympatho-excitatory state mediated through the actions of TrkB signaling. We tested our hypothesis by antagonizing TrkB with ANA-12, a specific TrkB antagonist (Cazorla et al., 2011), during ICV Ang II infusion and evaluated hemodynamic, metabolic, and autonomic responses.
2. Materials and Methods
2.1 Animal Model
For these experiments, male Sprague-Dawley rats with starting weights ranging between 350 and 400 g were used. Starting weights between groups were not different (p = 0.15 via one-way ANOVA). The experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center and were carried out under the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. A total of 23 animals were used for these studies, 1 Ang II + ANA-12 animal was excluded due to early death on day 5 of infusion.
2.2 Animal Preparation and ICV Infusions
Blood pressure was measured in conscious, freely moving rats using radiotelemetry as previously described (Haack et al., 2014). In brief, animals were anesthetized with 2–4% isoflurane via inhalation. The left femoral artery was dissected and a DSI telemetry unit (TA11PA-40, Data Science International, Minneapolis, MN) catheter was inserted and advanced into the abdominal aorta. The telemetry unit was placed in a pocket formed under the abdominal skin and tied in place. Immediately following telemetry implantation, anesthetized rats were positioned in a stereotaxic apparatus. The skull was exposed and Bregma was identified. A small hole was bored in the skull at 0.8 mm caudal to Bregma and 1.75 mm lateral to the midline. An Alzet (Cupertino, CA) Brain Infusion Kit 2 cannula was inserted through the hole, and the tip of the cannula was positioned 4 mm deep to the surface of the skull into the right lateral ventricle. The cannula was fixed to the skull with dental acrylic with jewler’s screws positioned both rostrally and caudally to the cannula for support. The cannula was attached to a catheter connected to a subcutaneous osmotic minipump (Alzet 2002; 0.5 μl/h 14 day capacity) containing vehicle. The skin was closed over the skull, and the animal was allowed to recover.
After one week of recovery, rats were placed in metabolic cages for 48 h to acclimate. Following acclimation, baseline metabolic and hemodynamic recordings were collected for seven days. Rats were then anesthetized with 2–4% isoflurane a small incision was made close to the osmotic minipump, and pumps were removed and exchanged with a pump (Alzet 2002) containing either Ang II, Ang II + ANA-12, ANA-12 alone, or vehicle.
Ang II (catalog # A9525) and ANA-12 (N-[2-[[(Hexahydro-2-oxo-1H-azepin-3-yl)amino]carbonyl]phenyl]-benzo[b]thiophene-2-carboxamide; catalog # SML0209) were purchased from Sigma-Aldrich. An effective ICV dose of 20 ng/min Ang II was used based on a survey of the literature (Camara et al., 2001; Camara et al., 1998; Clayton et al., 2014; Fitzsimons, 1998) and through preliminary dose-response studies (data not shown). ANA-12 was infused ICV at 50 ng/h alone or in combination with Ang II. The dosage of ANA-12 was determined by separate preliminary experiments. All drugs were dissolved in vehicle composed of a 1:1 solution of aCSF to DMSO. Rats were then followed over the subsequent 11 days as described below before acute experiments were completed on day 12 in order to operate within the infusion period (2 weeks) of the minipumps.
2.3 Conscious Hemodynamic and Metabolic Measurements
Blood pressure telemetry recordings were collected in freely-moving conscious animals and were recorded between the hours of 11 am to 3 pm using LabChart® software (AD Instruments, Colorado Springs, CO) at a sampling rate of 1 kHz (Bhatia et al., 2010). Periods of 1.5 hr duration without excessive movement artifacts were extracted from the total recordings and used for analysis. Mean arterial pressure (MAP) and heart rate (HR) were extracted from the telemetry pulse wave. Heart rate variability (HRV) measurements were analyzed by the HRV analysis module in LabChart® extracted from the pulse wave after passing through a 45 Hz low-pass filter. Normal to normal intervals were triggered by the maximal derivative of the pulse wave as this best correlates the interval extrapolated from the pulse wave to intervals generated by electrocardiogram (Pellegrino et al., 2014). Standard deviation of normal to normal (SDNN), root mean squared of the successive differences (RMSSD), and the low frequency (LF) (0.2 – 0.6 Hz) to high frequency (HF) (1.0–3.0 Hz) ratio (Erdos et al., 2015b) (LF/HF) were calculated using LabChart® software. The pulse wave recordings were exported to the freely available HemoLab Software (Iowa City, IA) for processing of spontaneous baroreflex sensitivity (BRS). Traces were filtered through a Butterworth low-pass filter with a corner frequency of 20 Hz and the baroreflex gain was determined by the sequence method of BRS (Bertinieri et al., 1985; Stauss et al., 2006). In brief, baroreflex sequences were defined by a minimum of three consecutive beat-to-beat intervals of either increasing or decreasing systolic pressure associated with increasing or decreasing pulse interval, respectively. No time delay was applied between pressure and pulse interval changes, and no threshold for pressure changes was used. Only sequences with a correlation coefficient > 0.8 between pressure and pulse interval were used with the slope of the resultant linear correlation equation set as the baroreflex gain.
Metabolic cage measurements of water and food intake and urine and feces excretion were made gravimetrically once every 24 h at approximately 3 PM following completion of the day’s telemetry recordings.
2.4 Acute Animal Preparation
For the acute, terminal experiments, rats were prepared as previously described (Becker et al., 2016). In brief, animals were anesthetized with urethane (800 mg/kg ip) and α-chloralose (40 mg/kg ip), and appropriate level of anesthesia was determined by absence of reflexive withdrawal from painful stimulus such as foot pinch prior to beginning surgery. The trachea was cannulated, and a Millar catheter (SPR 524; size, 3.5-Fr; Millar Instruments, Houston, TX) was advanced through the right carotid artery into the aorta and left in place to record arterial pressure. Heart rate was derived from the arterial pressure pulse with a PowerLab® model 16S (ADInstruments, Colorado Springs, CO) using LabChart® software. The right jugular vein was cannulated for intravenous injections, and the rats were then paralyzed with pancuronium bromide (1 mg/kg iv, 0.1 mg/kg thereafter as needed) and ventilated artificially with room air supplemented with 100% oxygen. Supplemental doses of α-chloralose (20 mg/kg, iv) were administered every 1.5 h thereafter to maintain an appropriate level of anesthesia or if blood pressure increased by more than 10 mmHg following a strong painful stimulus such as toe pinch. Body temperature was maintained at ∼37°C with an animal temperature controller (ATC1000; World Precision Instruments).
2.5 Renal Sympathetic Nerve Recording
Renal sympathetic nerve activity (RSNA) was recorded as previously described (Becker et al., 2016; Wang et al., 2014). Generally, the left kidney, renal artery, and nerves were exposed through a left retroperitoneal flank incision. Sympathetic nerves running on or beside the renal artery were identified. The renal nerve was cut distally to avoid recording afferent activity. The renal sympathetic nerves were placed on a pair of platinum-iridium recording electrodes and then were covered with a fast-setting silicone (Kwik-Sil; World Precision Instruments). Nerve activity was amplified (×10000) and filtered (bandwidth: 100 to 3000 Hz) using a Grass P55C preamplifier. The nerve signal was monitored on an oscilloscope (model 121 N; Tektronix, Beaverton, OR). The signal from the oscilloscope was displayed on a computer where it was rectified, integrated, sampled (1 kHz), and converted to a digital signal by the PowerLab® data acquisition system. Maximal nerve activity was observed shortly after euthanasia after stopping the heart with a 1 ml iv infusion of air, and the background noise for sympathetic nerve activity was recorded 15–20 min after the rat was euthanized. For description of resting nerve activity (Figure 3 and 4), nerve activity was normalized for each individual rat by setting the maximal activity to 100% and subtracting out the noise floor from the integrated signal. Resting activity was then described as a % of max (Wang et al., 2010).
Figure 3.
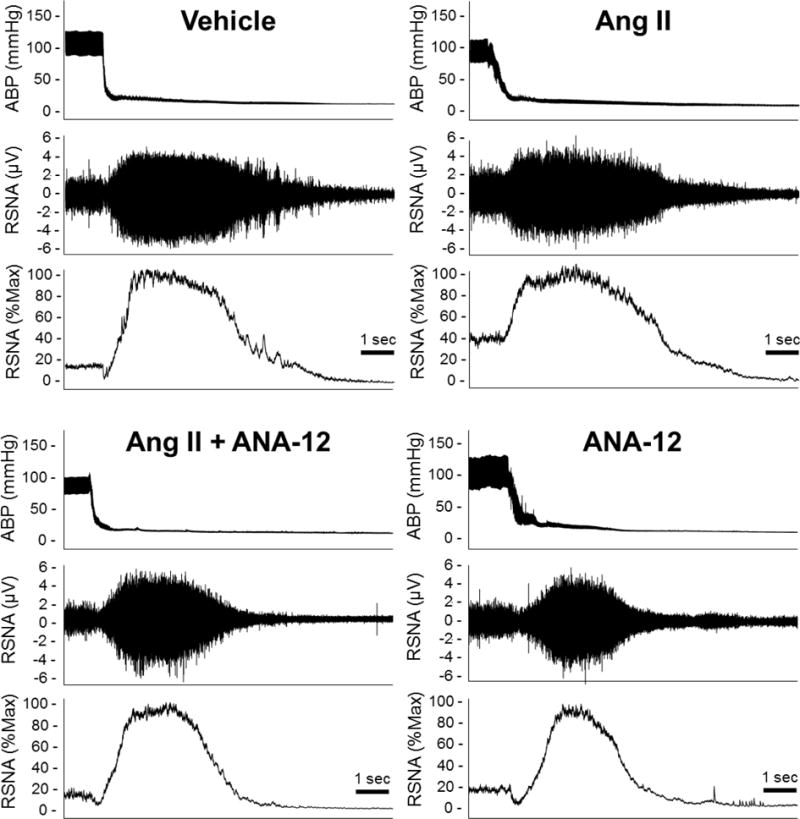
Representative traces of arterial blood pressure (ABP), raw renal sympathetic nerve activity (RSNA), and rectified and integrated channel displayed as percent max RSNA elicited following euthanasia from acute experiments performed on day 12 of ICV infusion.
Figure 4.
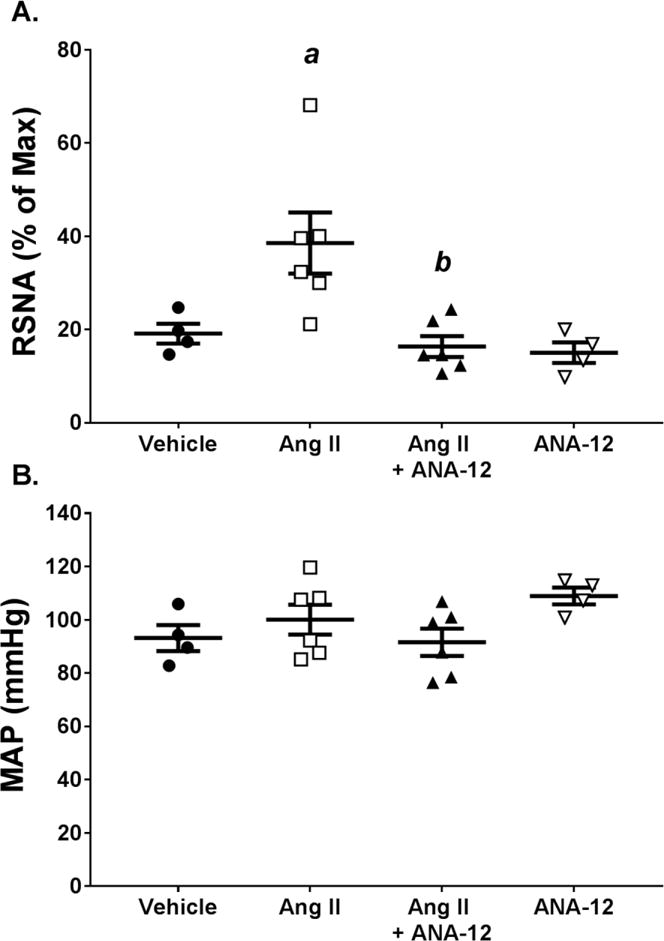
Infusion of Ang II ICV increases renal sympathetic nerve activity (RSNA), which is attenuated by co-infusion of ANA-12. A) RSNA quantified as percent of max elicited following euthanasia. B) MAP measured under anesthesia at same time as RSNA. a, p < 0.05 vs. Vehicle; b, p < 0.05 vs. Ang II.
2.6 Arterial Baroreflex Curves
Baroreflex curves were generated by measuring the HR and RSNA responses to decreases and increases in arterial pressure by intravenous administration of sodium nitroprusside (25 μg) followed by phenylephrine (10 μg) as previously described (Gao et al., 2005; Pan et al., 2007; Wang et al., 2014). The RSNA response (Figure 5 and Table 2) was normalized as a percent of baseline by designating the nerve activity prior to infusion of sodium nitroprusside as 100%. A sigmoid logistic function was fit to the data using a nonlinear regression program (SigmaPlot version 8.0). Four parameters were derived from the following equation: %RSNA or HR = A/1 + exp[B(MAP − C)]} + D; where A is the RSNA or HR range, B is the slope coefficient, C is the pressure at the midpoint of the range (BP50), and D is minimum RSNA or HR. The peak slope [or maximum gain (Gainmax)] was determined by taking the first derivative of the baroreflex curve described by the equation (Kent et al., 1972).
Figure 5.
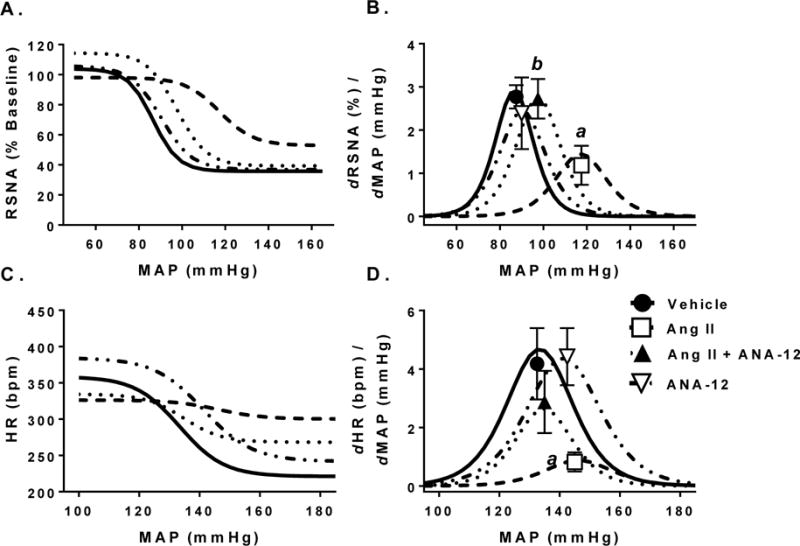
Baroreflex curves of renal sympathetic nerve activity (RSNA) and heart rate (HR) changes to increased mean arterial pressure (MAP) in response to administration of sodium nitroprusside and phenylephrine. A) RSNA response to increases in MAP quantified as a % of baseline where 100% represents the activity before infusion of sodium nitroprusside. B) Gain of RSNA baroreflex across MAP. Symbols represent max gain of baroreflex. C) HR response to increases in MAP. D) Gain of HR baroreflex across MAP. Symbols represent max gain of baroreflex. a, p < 0.05 vs. Vehicle; b, p < 0.05 vs. Ang II.
Table 2.
Heart Rate and RSNA Baroreflex Parameters
| n | a, range (bpm or %) |
x0, BP50 (mmHg) |
y0, min (bpm or %) |
Max Gain (bpm or %/mmHg) |
|
|---|---|---|---|---|---|
| MAP-HR | |||||
| ICV Vehicle | 4 | 137.5 ± 21.7 | 133.4 ± 11.8 | 221.2 ± 15.8 | 4.18 ± 1.41 |
| ICV Ang II | 7 | 26.1 ± 6.3a | 146.2 ± 10.2 | 300.2 ± 16.2a | 0.82 ± 0.36a |
| ICV Ang II + ANA-12 | 6 | 66.2 ± 9.9ab | 134.8 ± 6.4 | 268.2 ± 12.9 | 2.87 ± 1.17 |
| ICV ANA-12 | 4 | 142.8 ± 21.9b | 141.3 ± 2.1 | 241.7 ± 11.4 | 4.42 ± 1.13 |
| MAP-RSNA | |||||
| ICV Vehicle | 4 | 68.1 ± 7.1 | 86.5 ± 7.3 | 35.8 ± 6.7 | 2.77 ± 0.27 |
| ICV Ang II | 7 | 45.2 ± 8.7 | 117.3 ± 3.0a | 52.9 ± 0.2 | 1.18 ± 0.18a |
| ICV Ang II + ANA-12 | 6 | 75.0 ± 9.9 | 97.6 ± 5.4 | 39.3 ± 6.2 | 2.72 ± 0.50b |
| ICV ANA-12 | 4 | 68.9 ± 2.9 | 90.3 ± 9.3b | 36.9 ± 4.4 | 2.39 ± 0.96 |
Baroreflex parameters were recorded in anesthetized rats. Values are mean ± SEM. a is the RSNA or HR range, x0 is the pressure at the midpoint of the range (BP50), y0 is minimum RSNA or HR;
p < 0.05 relative to ICV Vehicle;
p < 0.05 relative to ICV Ang II
2.7 Data Analysis and Statistics
Hemodynamic and metabolic responses to ICV infusion were split into two phases for analysis. The early phase represents the average change from baseline mean (days −3 to −1) of days 3–5 of infusion, and the late phase represents the average change from baseline mean of days 9 to 11 of infusion.
Group and time interactions were compared using a one or two-way ANOVA with a Tukey’s post-hoc test. A p value < 0.05 was considered statistically significant. All data was analyzed using Graphpad Prism 7 software.
3. Results
3.1 Hemodynamic and Sympathetic Responses to ICV Ang II
Rats administered 20 ng/min Ang II ICV demonstrated an immediate and sustained increase in MAP with a peak increase of approximately 50 mmHg (Figure 1A–B). Co-infusion of ANA-12 with Ang II also increased MAP relative to baseline with a peak increase of approximately 20 mmHg (Figure 1A–B); however, this increase was attenuated as compared to Ang II alone (Figure 1B). Heart rate increased soon after infusion in Ang II treated animals as evidenced by an increase in HR relative to baseline during the early period that was absent in the Ang II + ANA-12 treated groups (Figure 1C–D). However, HR was similarly increased relative to baseline in the late period in both Ang II and Ang II + ANA-12 treated groups (Figure 1C–D). Infusion of vehicle or ANA-12 alone had no effect on MAP or HR during either early or late phase.
Figure 1.
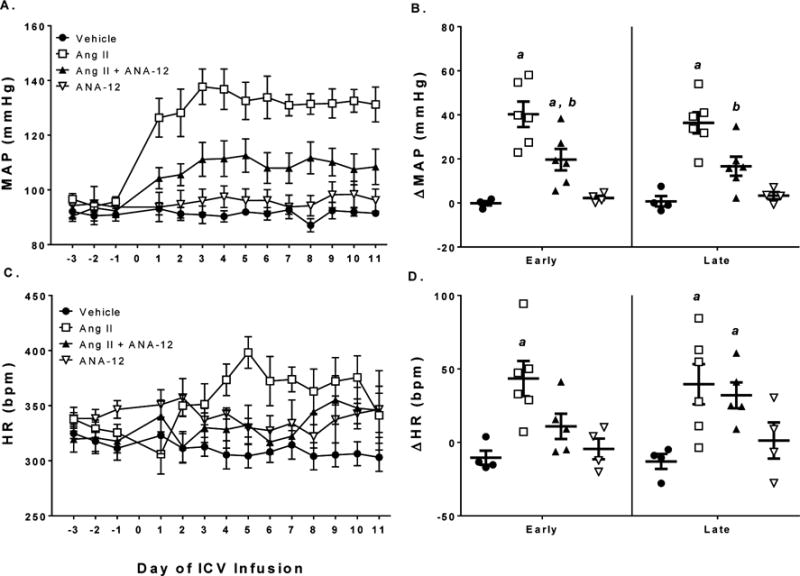
Co-infusion of ANA-12 attenuated the increase in mean arterial pressure following ICV Ang II. A) Mean arterial blood pressure as measured by telemetry over the course of the experiment. B) Change in MAP relative to baseline (days −3 to −1) in early (days 3–5) and late (days 9–11) periods. C) Heart rate over the course of the experiment. D) Change in HR relative to baseline in early and late periods of ICV infusion. a, p < 0.05 vs. Vehicle; b, p < 0.05 vs. Ang II.
None of the treatments had an effect on LF/HF or SDNN in either the early or late phases relative to baseline nor was there an effect between treatments (2-way ANOVA, LF/HF pTreatment = 0.92; SDNN pTreatment = 0.98) (Table 1). Treatment with Ang II decreased RMSSD in both early and late phases, this decrease was not observed in rats co-treated with Ang II and ANA-12 (Table 1). Only Ang II reduced spontaneous BRS relative to baseline during both early (p = 0.0001) and late (p = 0.002) periods (Figure 2). Although it did not reach statistical significance, there was a strong trend toward decreased BRS in rats infused with Ang II compared to vehicle controls, which was not observed in animals co-infused with ANA-12 or ANA-12 alone (Figure 2).
Table 1.
Change in Heart Rate Variability Parameters
| Vehicle | Ang II | Ang II + ANA-12 | ANA-12 | ||
|---|---|---|---|---|---|
| n | 4 | 7 | 6 | 4 | |
| ΔLF/HF | |||||
| Early | 0.04 ± 0.11 | −0.02 ± 0.10 | −0.21 ± 0.19 | −0.12 ± 0.04 | |
| Late | 0.00 ± 0.08 | 0.01 ± 0.11 | 0.07 ± 0.10 | −0.02 ± 0.11 | |
| ΔSDNN (ms) | |||||
| Early | −0.31 ± 1.05 | 0.35 ± 1.54 | 1.36 ± 0.97 | 0.03 ± 0.82 | |
| Late | 0.32 ± 0.80 | 0.29 ± 2.25 | 0.40 ± 0.44 | 1.05 ± 0.76 | |
| ΔRMSSD (ms) | |||||
| Early | −0.56 ± 0.32 | −1.71 ± 0.59a | −0.46 ± 0.49 | 1.37 ± 1.34b | |
| Late | −0.18 ± 0.36 | −1.73 ± 0.47a | −0.63 ± 0.50 | 1.02 ± 0.78b |
Values are changes from baseline (−3 to −1) in Early (3 to 5) and Late (9 to 11) days following initiation of ICV infusions in conscious rats. LF = Low Frequency; HF = High Frequency; SDNN = Standard Deviation of N to N Interval; RMSSD = Root Mean Squared of Successive Differences;
p < 0.05 vs. baseline;
p < 0.05 vs. Ang II
Figure 2.
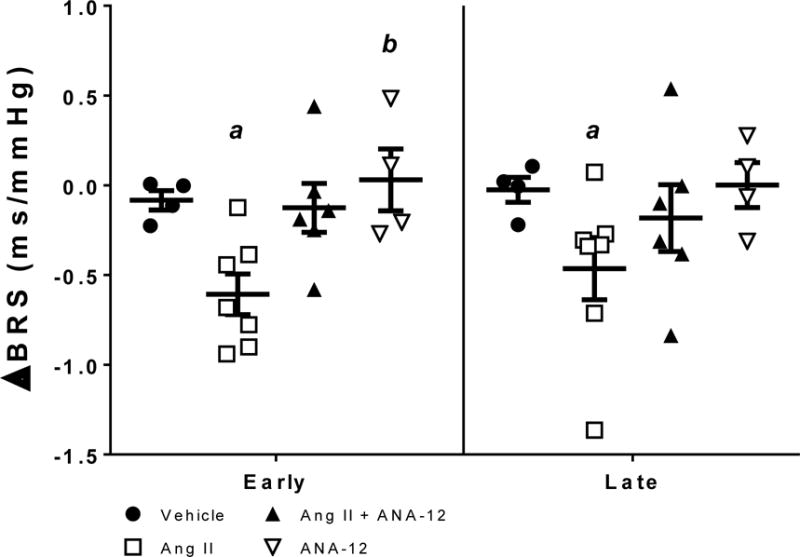
Change in spontaneous baroreflex sensitivity (BRS) relative to baseline (days −3 to −1) in early (days 3–5) and late (days 9–11) of ICV infusion. a, p < 0.05 vs. baseline within group; b, p < 0.05 vs. Ang II.
At the end of the ICV infusion protocol (day 12 or 13) rats were anesthetized and instrumented for measurement of RSNA and baroreflex sensitivity. Resting RSNA levels were significantly elevated in ICV Ang II rats compared to vehicle controls. Co-infusion of ANA-12 with Ang II attenuated this increased RSNA. Vehicle or ANA-12 alone had no effect on RSNA as compared to control (Figures 3 and 4A). The MAP recorded under anesthesia at the same time as the RSNA measurements were not different across groups (Figure 4B).
Baroreflex control of RSNA exhibited a trend toward a blunted response in range, max gain, and midpoint were significantly altered in Ang II treated animals compared to vehicle controls. Ang II + ANA-12 trended to restore RSNA range and max gain compared to Ang II alone but were not statistically significant. ANA-12 alone had no effect compared to vehicle controls (Table 2 and Figure 5A–B). Rats given Ang II ICV exhibited baroreflex dysfunction with a blunted HR range compared to vehicle controls and max gain (Table 2 and Figure 5C–D). Ang II + ANA-12 attenuated the blunted HR baroreflex range and max gain. Although there was a trend toward a rightward shift in the midpoint of the HR baroreflex curve to higher blood pressures following Ang II, this did not reach statistical significance. ANA-12 alone had no effect compared to vehicle (Table 2 and Figure 5C–D).
3.2 ICV Ang II and ANA-12 Alter Metabolic Balance
Following ICV Ang II, daily water intake dramatically increased over the course of the two weeks of infusion to levels approximately 4–5 times the baseline daily intake during the late period (Figure 6A–B). Concurrent with the dipsogenic response, daily urine output also increased by the late period of infusion (Figure 6C–D). Co-infusion of ANA-12 with Ang II resulted in an immediate increase in water intake and urine excretion during the early period of infusion that converged. During the late period, co-infusion of Ang II with ANA-12 was similar as Ang II infusion alone (Figure 6A–D). Rats receiving ICV Ang II also displayed appetite suppression as seen by a decrease in daily food intake and fecal output (Figure 6E–H). Co-infusion of ANA-12 with Ang II attenuated the suppression of appetite as compared to IVC Ang II alone (Figure 6E–F). Infusion of ANA-12 alone or vehicle had no effect on water intake, urine output, food intake, or fecal output over the course of the ICV infusion relative to baseline (Figure 6).
Figure 6.
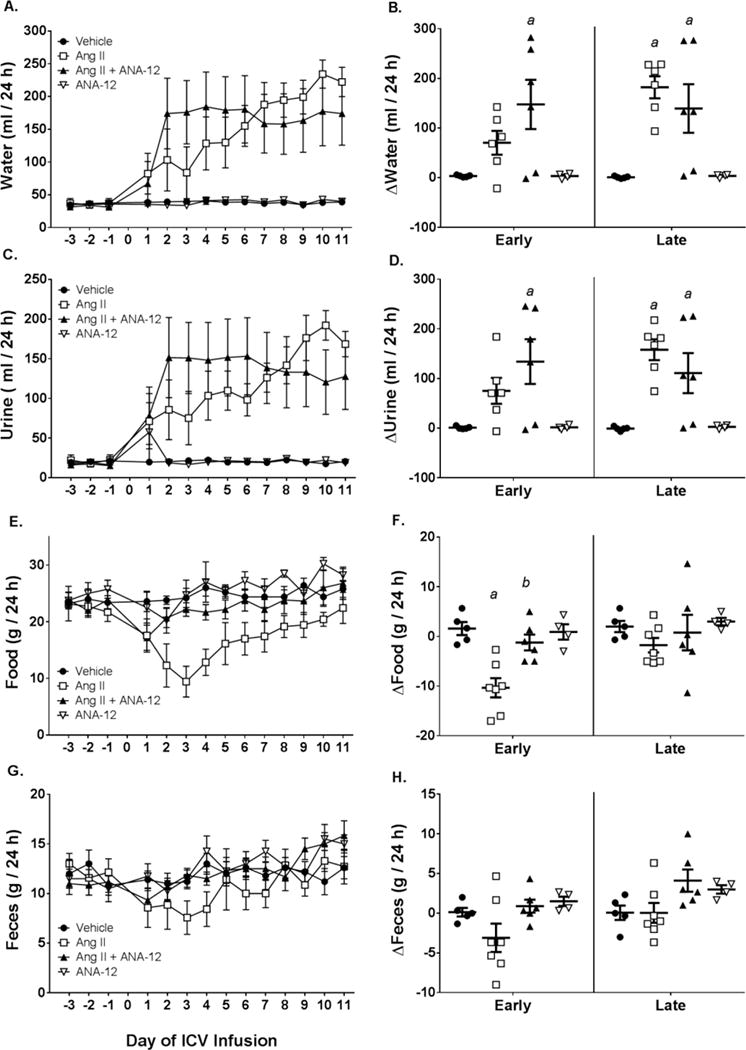
Metabolic parameters following ICV infusion of Ang II, Ang II + ANA-12, ANA-12, or Vehicle. A) Twenty-four hour water intake across the treatment period. B) Change in water intake relative to baseline (days −3 to −1) in early (days 3–5) and late (days 9–11) periods. C) Twenty-four hour urine output across the treatment period. D) Change in urine output relative to baseline in early and late periods. E) Twenty-four hour food intake across the treatment period. F) Change in food intake relative to baseline in early and late periods. G) Twenty-four hour fecal ouput across the treatment period. H) Change in fecal output relative to baseline in early and late periods. a, p < 0.05 vs. Vehicle; b, p < 0.05 vs. Ang II.
Concomitant with the decrease in appetite, rats receiving ICV Ang II had lower final body weight compared to vehicle controls (Figure 6E; Table 3). Although animals receiving Ang II + ANA-12 did not exhibit an appreciable decrease in food intake (Figure 6E–F), final body weight was significantly lower than vehicle controls. Due to the decrease in body weight of Ang II and Ang II+ANA-12 animals, organs such as the heart, wet lung, and kidneys also displayed reductions in absolute weight or when normalized to tibia length. When organ weights were normalized relative to body weight, differences between groups were mostly absent.
Table 3.
Final Body and Organ Weight
| Vehicle | Ang II | Ang II + ANA-12 | ANA-12 | |
|---|---|---|---|---|
| n | 5 | 7 | 6 | 4 |
| Body weight (g) | 480.0 ± 25.8 | 295.7 ± 22.5a | 390.0 ± 16.5b | 502.5 ± 15.6bc |
| Tibia (cm) | 5.88 ± 0.03 | 5.63 ± 0.08 | 5.73 ± 0.07 | 5.85 ± 0.07 |
| Heart weight (mg) | 1571.7 ± 115.1 | 1112.6 ± 78.4a | 1296.1 ± 61.8 | 1555.5 ± 14.6b |
| HW/BW (mg/g) | 3.3 ± 0.2 | 3.8 ± 0.1 | 3.3 ± 0.1 | 3.1 ± 0.1b |
| HW/TL (mg/cm) | 267.0 ± 18.8 | 197.0 ± 12.1a | 225.8 ± 9.2 | 266.0 ± 2.0b |
| Wet Lung (mg) | 2089.4 ± 89.4 | 1517.5 ± 103.1a | 1539.9 ± 41.5a | 1776.0 ± 25.2 |
| WL/BW (mg/g) | 4.5 ± 0.4 | 5.2 ± 0.3 | 4.0 ± 0.1b | 3.6 ± 0.1b |
| WL/TL (mg/cm) | 355.6 ± 16.5 | 268.6 ± 15.6a | 268.6 ± 6.2a | 303.7 ± 4.8 |
| Liver (g) | 17.7 ± 0.9 | 10.9 ± 0.8a | 12.2 ± 0.8a | 18.7 ± 0.8bc |
| LW/BW (mg/g) | 34.4 ± 1.2 | 37.0 ± 1.5 | 31.4 ± 1.8 | 37.2 ± 1.3 |
| LW/TL (g/cm) | 3.0 ± 0.2 | 1.9 ± 0.1a | 2.1 ± 0.1a | 3.2 ± 0.2bc |
| Left Kidney (mg) | 1695.5 ± 17.0 | 1373.6 ± 103.1 | 1651.6 ± 106.4 | 2008.0 ± 63.7 |
| Right Kidney (mg) | 1660.9 ± 24.0 | 1426.4 ± 94.3 | 1644.9 ± 89.6 | 2043.6 ± 72.9 |
BW = body weight; HW = heart weight; TL = tibia length; LW = liver weight;
p < 0.05 vs. Vehicle,
p < 0.05 vs. Ang II;
p < 0.05 vs.
Ang II + ANA-12; one-way ANOVA
4. Discussion
The major findings of the current study are that TrkB antagonism by ANA-12 attenuated the increased MAP, RSNA, and baroreflex dysfunction caused by central Ang II signaling. Other important findings include a potentiated dipsogenic and polyuric response to ICV Ang II by co-infusion of ANA-12 and decreased appetite and cachexia caused by central Ang II infusion. These in vivo observations implicate the importance of endogenous BDNF/TrkB signaling in Ang II-induced sympatho-excitation and suggest potentially disparate signaling mechanisms involved in sympathetic control vs. thirst/metabolic balance following central Ang II.
The finding that BDNF/TrkB signaling is important in mediating the sympatho-excitation following ICV Ang II is in agreement with our previous in vitro observations that Ang II increases BDNF expression and induces BDNF-mediated decrease of K+ currents (Becker et al., 2015). Because antagonizing TrkB with ANA-12 attenuates the increased MAP (Figure 1) and completely prevents the increased RSNA (Figures 3 and 4) following Ang II, we conclude that BDNF/TrkB signaling is an important component in Ang II-mediated sympatho-excitation. A previous study has indicated that overexpression of BDNF in the PVN is sufficient to induce hypertension (Erdos et al., 2015a) implicating BDNF signaling in presympathetic centers of the brain as being hypertensive. Furthermore, acute injections of BDNF into presympathetic areas such as the RVLM in anesthetized rat preparations increases MAP (Wang et al., 2002). However, in vivo interactions between BDNF and Ang II in promotion of sympathetic nerve activity are lacking. Here we extend these previous reports suggesting a sympatho-excitatory effect of BDNF and integrate BDNF/TrkB signaling with centrally mediated Ang II-induced sympathetic activity, which itself has long been appreciated (Biancardi et al., 2014; Patel et al., 2012; Xiao et al., 2013; Zimmerman et al., 2004; Zucker et al., 2012).
Recently Schaich et al. (2016) demonstrated that microinjections of BDNF into the PVN result in an acute increase in MAP that are attenuated by prior treatment with the AT1R antagonist losartan in both conscious and anesthetized rats. The authors suggest that there may be interplay between AT1R and TrkB signaling mechanisms in modulating sympathetic neuronal activity. Our present study lends support to this suggestion in a conscious model of central Ang II application. Future work will be instrumental in deciphering the precise molecular interactions and signaling cascades at play in the interaction between BDNF and Ang II.
Although ANA-12 completely prevented the increase in RSNA caused by central Ang II, ANA-12 only partially attenuated the hypertension and had no measurable impact on HR changes from Ang II. In addition, Ang II only affected RMSSD and not LF/HF or SDNN parameters. One potential explanation for some of these disparities could relate to the nature of the measurements as RMSSD is specifically related to parasympathetic tone whereas SDNN and the LF component of HRV are under mixed sympathetic and parasympathetic influence (Billman, 2013; Houle et al., 1999). Alterations to both sympathetic and parasympathetic arms may offset one another and be unnoticed by these measurements. It is also possible that during these conscious measurements of autonomic tone that the parasympathetic arm is affected more. This is suggested by the robust decrease to RMSSD and sBRS following Ang II and the attenuation of these decreases with co-infusions of ANA-12.
The effect of Ang II may also vary across different sympathetic nerve beds. For instance, the renal sympathetic bed may be more directly influenced by an Ang II-BDNF interaction than the cardiac sympathetic bed, which would result in an observable difference in RSNA, but not HR or markers of HRV. Vasomotor sympathetic tone may be under mixed influence from BDNF/TrkB-dependent and BDNF/TrkB-independent presympathetic centers thus antagonism of TrkB may only partially prevent an Ang II-induced hypertensive response. The answers to these questions are beyond the scope of the current study, and future work will be important in exploring the particular autonomic centers impacted by an Ang II-BDNF/TrkB signaling mechanism. In a salt-loaded model, previous studies have found that renal denervation blunts the hypertensive response of ICV Ang II (Osborn et al., 1997). Thus the renal nerves may play a predominant role in mediating central Ang II-induced hypertension, and RSNA may be preferentially activated relative to other sympathetic nerve beds.
Central administration of Ang II is well known to evoke a potent and immediate drinking response (as comprehensively reviewed by Fitzsimons, 1998). This response is a primary, neurogenic dipsogenic response as adrenalectomized, hypophysectomized (Avrith et al., 1980b) or bilaterally nephrectomized rats (Avrith et al., 1980a) still respond to central bolus injections of Ang II. Interestingly, in rats receiving 6 μg/h (100 ng/min) in the lateral cerebral ventricle for 7 days, there was only a transient dipsogenic response that the authors speculated was due to thirst inhibition by the sustained increase in MAP (Dinicolantonio et al., 1982). The dose used in the present study was lower than used by Di Nicolantonio et al. (1982) and evoked a less drastic hypertensive response, which could explain the lack of a complete pressor-mediated suppression of Ang II-induced thirst. Furthermore, in animals receiving coinfusion of ANA-12, there was a more robust and immediate increase in daily water intake following Ang II, suggesting a potential uncoupling of the inhibitory feedback control of Ang II-induced thirst. Previous studies investigating the influence of arterial pressure and baroreflex input on the dipsogenic response of central Ang II found a reciprocal relationship between MAP and central Ang II-induced thirst that was not affected by sino-aortic denervation, indicating the relationship is baroreflex independent (Thunhorst et al., 1993). Similarly, acute hypertension has been shown to inhibit thirst (Stocker et al., 2001). Due to the observation that co-infusion of ANA-12 attenuated the increase in MAP in response to central Ang II, this lower pressure would have reduced negative feedback on Ang II-induced thirst. Hence, it is likely that ANA-12 is predominately attenuating the increase in sympathetic tone following central Ang II and the potentiating effect on thirst is a secondary response to attenuated MAP. Although there was an initial potentiation of thirst by co-infusion of ANA-12 with Ang II, both groups exhibited similar drinking behavior by the end of the infusion protocol. To our knowledge, few studies have investigated this relationship for longer infusion periods as most studies are acute or conclude by day 7 of ICV infusion. The convergence of drinking behavior at the later stages of infusion suggests a withdrawal of the pressure-mediated inhibition of drinking and provides observations warranting further study in long-term, chronically instrumented animals.
We observed changes to baroreflex function in both the Ang II and Ang II + ANA-12 groups. Previous work has implicated central Ang II signaling in altered baroreflex (Gao et al., 2014; Gao et al., 2005; Pan et al., 2007) particularly through its action on AT1R and promotion of sympatho-excitation in presympathetic areas such as the PVN and RVLM. Here we altered the baroreflex through central infusion of Ang II eliciting a neurogenic form of hypertension. Only the Ang II group had an observable decrease in spontaneous BRS in both the early (days 3–5) and late (days 9–11) periods of infusion. Co-infusion of ANA-12 exhibited improvements on a number of baroreflex parameters such as significantly attenuating the decrease in heart rate range and RSNA gain observed in the Ang II alone group. ANA-12 also tended to attenuate the rightward shift of the baroreflex after 12 days of infusion in the acute, anesthetized preparation (Table 2 and Figures 5). These results suggest that the altered baroreflex function following central Ang II is mediated through TrkB signaling.
The direct effects of ANA-12 on modulating the baroreflex through its actions on central autonomic neurons cannot be excluded. As we have previously demonstrated, ANA-12 in the dmNTS inhibits baroreflex function, a finding that at first appears in conflict with the results presented here (Becker et al., 2016). An important distinction between these two studies is the route of ANA-12 delivery. In the previous study, ANA-12 was directly microinjected into the dmNTS, thereby isolating its effects to a specific, sympatho-inhibitory autonomic center. Conversely, in the present study, ANA-12 was infused ICV and would likely affect multiple autonomic centers, particularly those presympathetic areas of the SFO and PVN relatively close to the site of infusion, which are sensitive to factors circulating in the CSF (Buggy et al., 1978; Buggy et al., 1984; Fitzsimons, 1998; Mangiapane et al., 1980). These sites also contain a high concentration of AT1R protein making them likely targets for the action of ICV Ang II (Allen et al., 2000; Lenkei et al., 1997; Song et al., 1992). Therefore, we speculate that the predominant actions of ICV infusion of Ang II and ANA-12 are on control of neuronal activity of presympathetic sites close to infusion. It is less likely through actions on areas such as the NTS that is further from the infusion site. Furthermore, the SFO, PVN, and RVLM are autonomic integration sites that provide excitatory sympathetic output ultimately projecting to the intermediolateral (IML) spinal cord. Thus, even if central Ang II and ANA-12 have an effect on the NTS, the effect may be buffered by the direct effects on these autonomic centers with direct outflow to the peripheral sympathetic nervous system. The method of delivery via ICV infusions is a limitation of our study as we are unable to conclusively pinpoint the involvement of these areas over others. Furthermore, we cannot exclude the possibility that Ang II and ANA-12 are acting on completely separate brain nuclei and that our observed physiological outcomes are the result of an integrative effect of presympathetic neurons. For example, it is possible that Ang II could act predominately through the SFO, and ANA-12 could inhibit neurons in the PVN. This would have similar effects to what we observe without requiring an interaction between Ang II and TrkB signaling in the same individual neurons. Future work will be necessary addressing these areas specifically in order to reach the conclusions speculated here.
Another observation from this study is the marked cachexia and appetite suppression following ICV Ang II. Peripheral Ang II signaling results in cachexia through direct actions on skeletal muscle and activation of neurohumoral factors (Brink et al., 2001; Brink et al., 1996). The central actions of Ang II on metabolic state, appetite, and muscle wasting are less known; however, previous work from the Grobe and Sigmund laboratories has also demonstrated central Ang II signaling in energy balance and thirst (Claflin et al., 2015; Coble et al., 2015; Coble et al., 2014). ICV Ang II infusion also demonstrated a decrease in appetite and severe fat and muscle tissue wasting concurrent with a decrease in orexic peptides such as neuropeptide Y and orexin in the hypothalamus (Yoshida et al., 2012). The present study demonstrates a similar finding with a robust decrease in body mass and food intake following ICV infusion of Ang II. Interestingly, co-infusion with ANA-12 prevented the depressed appetite followingAng II and attenuated the decrease in body mass relative to Ang II alone, but did not normalize body mass to that of vehicle controls. This indicates that BDNF/TrkB signaling may be involved in the appetite suppressant actions of central Ang II, but may only be partially effective in reducing the sympathetic-driven cachexia (Hryniewicz et al., 2003; Laviano et al., 2008). Consistent with a decrease in total body mass, most organ masses were reduced following ICV Ang II or Ang II + ANA-12 (Table 3). When normalized to body mass, most organ masses were similar across groups indicating that the reduction in organ mass was proportional to the reduced body weight.
One limitation of our current study is the use of only male rats. As recent attention has highlighted the importance of sex on cardiovascular function (Blenck et al., 2016; Sandberg et al., 2012), evaluating the effect of sex on Ang II and BDNF/TrkB signaling interactions may provide interesting avenues for clarifying sex differences. However, as the importance of the Ang II, BDNF/TrkB pathway is only recently being described, studies such as ours demonstrating the existence of an interaction in either sex are important first steps toward understanding the pathways involved. Future studies are warranted to further clarify the role of biological sex.
In conclusion, antagonizing BDNF/TrkB signaling with ANA-12 attenuated parameters of sympatho-excitation and hypertension following ICV Ang II infusion further implicating the necessity of TrkB signaling in partially mediating the neuronal effects of Ang II. ANA-12 however did not prevent all effects of ICV Ang II such as cachexia and water balance, suggesting differential control of the Ang II – BDNF/TrkB axis in mediating various organ bed sympathetic tone, thirst, and metabolism.
Acknowledgments
Grants:
This study was supported, in part, by National Heart, Lung, and Blood Institute Grants PO1 HL62222 to I.H.Z; R01 HL116608-01A1 to H.W. and I.H.Z.; and F31 HL126286 to B.K.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
No conflicts of interest, financial or otherwise, are declared by the authors.
Author Contributions:
Conceptualization, B.K.B, H.W. and I.H.Z.; Performed experiments, B.K.B.; Analyzed data, B.K.B., H.W. and I.H.Z; Interpreted results, B.K.B, H.W. and I.H.Z.; Prepared figures, B.K.B.; Drafted manuscript, B.K.B.; Edited and revised manuscript, B.K.B, H.W. and I.H.Z.; Approved final version of manuscript, B.K.B, H.W. and I.H.Z.
References
- Allen AM, Zhuo J, Mendelsohn FA. Localization and function of angiotensin AT1 receptors. Am J Hypertens. 2000;13:31S–38S. doi: 10.1016/s0895-7061(99)00249-6. [DOI] [PubMed] [Google Scholar]
- Avrith DB, Fitzsimons JT. Increased sodium appetite in the rat induced by intracranial administration of components of the renin-angiotensin system. J Physiol. 1980a;301:349–364. doi: 10.1113/jphysiol.1980.sp013210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrith DB, Wiselka MJ, Fitzsimons JT. Increased sodium appetite in adrenalectomized or hypophysectomized rats after intracranial injections of renin or angiotensin II. J Endocrinol. 1980b;87:109–112. doi: 10.1677/joe.0.0870109. [DOI] [PubMed] [Google Scholar]
- Becker BK, Tian C, Zucker IH, Wang HJ. Influence of brain-derived neurotrophic factor-tyrosine receptor kinase B signalling in the nucleus tractus solitarius on baroreflex sensitivity in rats with chronic heart failure. J Physiol. 2016;594:5711–5725. doi: 10.1113/JP272318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker BK, Wang HJ, Tian C, Zucker IH. BDNF contributes to angiotensin II-mediated reductions in peak voltage-gated K+ current in cultured CATH.a cells. Physiol Rep. 2015;3:1–8. doi: 10.14814/phy2.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Brain-derived neurotrophic factor: Regulation, effects, and potential clinical relevance. Neurology. 2015;84:1693–1704. doi: 10.1212/WNL.0000000000001507. [DOI] [PubMed] [Google Scholar]
- Bertinieri G, di Rienzo M, Cavallazzi A, Ferrari AU, Pedotti A, Mancia G. A new approach to analysis of the arterial baroreflex. J Hypertens Suppl. 1985;3:S79–81. [PubMed] [Google Scholar]
- Bhatia V, Rarick KR, Stauss HM. Effect of the data sampling rate on accuracy of indices for heart rate and blood pressure variability and baroreflex function in resting rats and mice. Physiol Meas. 2010;31:1185–1201. doi: 10.1088/0967-3334/31/9/009. [DOI] [PubMed] [Google Scholar]
- Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension. 2014;63:572–579. doi: 10.1161/HYPERTENSIONAHA.113.01743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billman GE. The LF/HF ratio does not accurately measure cardiac sympatho-vagal balance. Front Physiol. 2013;4:26. doi: 10.3389/fphys.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blenck CL, Harvey PA, Reckelhoff JF, Leinwand LA. The Importance of Biological Sex and Estrogen in Rodent Models of Cardiovascular Health and Disease. Circ Res. 2016;118:1294–1312. doi: 10.1161/CIRCRESAHA.116.307509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum R, Konnerth A. Neurotrophin-mediated rapid signaling in the central nervous system: mechanisms and functions. Physiology (Bethesda) 2005;20:70–78. doi: 10.1152/physiol.00042.2004. [DOI] [PubMed] [Google Scholar]
- Brink M, Price SR, Chrast J, Bailey JL, Anwar A, Mitch WE, Delafontaine P. Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology. 2001;142:1489–1496. doi: 10.1210/endo.142.4.8082. [DOI] [PubMed] [Google Scholar]
- Brink M, Wellen J, Delafontaine P. Angiotensin II causes weight loss and decreases circulating insulin-like growth factor I in rats through a pressor-independent mechanism. J Clin Invest. 1996;97:2509–2516. doi: 10.1172/JCI118698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggy J, Fink GD, Haywood JR, Johnson AK, Brody MJ. Interruption of the maintenance phase of established hypertension by ablation of the anteroventral third ventricle (AV3V) in rats. Clin Exp Hypertens. 1978;1:337–353. doi: 10.3109/10641967809068612. [DOI] [PubMed] [Google Scholar]
- Buggy J, Huot S, Pamnani M, Haddy F. Periventricular forebrain mechanisms for blood pressure regulation. Fed Proc. 1984;43:25–31. [PubMed] [Google Scholar]
- Camara AK, Osborn J. Central AT1 and AT2 receptors mediate chronic intracerebroventricular angiotensin II-induced drinking in rats fed high sodium chloride diet from weaning. Acta Physiol Scand. 2001;171:195–201. doi: 10.1046/j.1365-201x.2001.00790.x. [DOI] [PubMed] [Google Scholar]
- Camara AK, Osborn JL. AT1 receptors mediate chronic central nervous system AII hypertension in rats fed high sodium chloride diet from weaning. J Auton Nerv Syst. 1998;72:16–23. doi: 10.1016/s0165-1838(98)00080-0. [DOI] [PubMed] [Google Scholar]
- Cao XH, Byun HS, Chen SR, Cai YQ, Pan HL. Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J Neurochem. 2010;114:1460–1475. doi: 10.1111/j.1471-4159.2010.06863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Premont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97:772–780. doi: 10.1161/01.RES.0000185804.79157.C0. [DOI] [PubMed] [Google Scholar]
- Claflin KE, Grobe JL. Control of energy balance by the brain renin-angiotensin system. Curr Hypertens Rep. 2015;17:38. doi: 10.1007/s11906-015-0549-x. [DOI] [PubMed] [Google Scholar]
- Clayton SC, Zhang Z, Beltz T, Xue B, Johnson AK. CNS neuroplasticity and salt-sensitive hypertension induced by prior treatment with subpressor doses of ANG II or aldosterone. Am J Physiol Regul Integr Comp Physiol. 2014;306:R908–917. doi: 10.1152/ajpregu.00010.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coble JP, Grobe JL, Johnson AK, Sigmund CD. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: importance of the subfornical organ. Am J Physiol Regul Integr Comp Physiol. 2015;308:R238–249. doi: 10.1152/ajpregu.00486.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coble JP, Johnson RF, Cassell MD, Johnson AK, Grobe JL, Sigmund CD. Activity of protein kinase C-alpha within the subfornical organ is necessary for fluid intake in response to brain angiotensin. Hypertension. 2014;64:141–148. doi: 10.1161/HYPERTENSIONAHA.114.03461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinicolantonio R, Mendelsohn FaO, Hutchinson JS, Takata Y, Doyle aE. Dissociation of Dipsogenic and Pressor-Responses to Chronic Central Angiotensin-Ii in Rats. American Journal of Physiology. 1982;242:R498–R504. doi: 10.1152/ajpregu.1982.242.5.R498. [DOI] [PubMed] [Google Scholar]
- Erdos B, Backes I, McCowan ML, Hayward LF, Scheuer DA. Brain-derived neurotrophic factor modulates angiotensin signaling in the hypothalamus to increase blood pressure in rats. Am J Physiol Heart Circ Physiol. 2015a;308:H612–622. doi: 10.1152/ajpheart.00776.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdos B, Clifton RR, Liu M, Li H, McCowan ML, Sumners C, Scheuer DA. Novel mechanism within the paraventricular nucleus reduces both blood pressure and hypothalamic pituitary-adrenal axis responses to acute stress. Am J Physiol Heart Circ Physiol. 2015b;309:H634–645. doi: 10.1152/ajpheart.00207.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev. 1998;78:583–686. doi: 10.1152/physrev.1998.78.3.583. [DOI] [PubMed] [Google Scholar]
- Francis GS. Neurohumoral mechanisms involved in congestive heart failure. Am J Cardiol. 1985;55:15A–21A. doi: 10.1016/0002-9149(85)90791-x. [DOI] [PubMed] [Google Scholar]
- Gao J, Zucker IH, Gao L. Activation of central angiotensin type 2 receptors by compound 21 improves arterial baroreflex sensitivity in rats with heart failure. Am J Hypertens. 2014;27:1248–1256. doi: 10.1093/ajh/hpu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Li Y, Schultz HD, Wang WZ, Wang W, Finch M, Smith LM, Zucker IH. Downregulated Kv4.3 expression in the RVLM as a potential mechanism for sympathoexcitation in rats with chronic heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H945–955. doi: 10.1152/ajpheart.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol Heart Circ Physiol. 2005;288:H2271–2279. doi: 10.1152/ajpheart.00949.2004. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang WZ, Wang W, Zucker IH. Imbalance of angiotensin type 1 receptor and angiotensin II type 2 receptor in the rostral ventrolateral medulla: potential mechanism for sympathetic overactivity in heart failure. Hypertension. 2008;52:708–714. doi: 10.1161/HYPERTENSIONAHA.108.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Haack KK, Marcus NJ, Del Rio R, Zucker IH, Schultz HD. Simvastatin treatment attenuates increased respiratory variability and apnea/hypopnea index in rats with chronic heart failure. Hypertension. 2014;63:1041–1049. doi: 10.1161/HYPERTENSIONAHA.113.02535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle MS, Billman GE. Low-frequency component of the heart rate variability spectrum: a poor marker of sympathetic activity. Am J Physiol. 1999;276:H215–223. doi: 10.1152/ajpheart.1999.276.1.H215. [DOI] [PubMed] [Google Scholar]
- Hryniewicz K, Androne AS, Hudaihed A, Katz SD. Partial reversal of cachexia by beta-adrenergic receptor blocker therapy in patients with chronic heart failure. J Card Fail. 2003;9:464–468. doi: 10.1016/s1071-9164(03)00582-7. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Johnson AK, Zhang Z, Clayton SC, Beltz TG, Hurley SW, Thunhorst RL, Xue B. The roles of sensitization and neuroplasticity in the long-term regulation of blood pressure and hypertension. Am J Physiol Regul Integr Comp Physiol. 2015;309:R1309–1325. doi: 10.1152/ajpregu.00037.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent BB, Drane JW, Blumenstein B, Manning JW. A mathematical model to assess changes in the baroreceptor reflex. Cardiology. 1972;57:295–310. doi: 10.1159/000169528. [DOI] [PubMed] [Google Scholar]
- Kim JH, Roberts DS, Hu Y, Lau GC, Brooks-Kayal AR, Farb DH, Russek SJ. Brain-derived neurotrophic factor uses CREB and Egr3 to regulate NMDA receptor levels in cortical neurons. J Neurochem. 2012;120:210–219. doi: 10.1111/j.1471-4159.2011.07555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviano A, Inui A, Marks DL, Meguid MM, Pichard C, Rossi Fanelli F, Seelaender M. Neural control of the anorexia-cachexia syndrome. Am J Physiol Endocrinol Metab. 2008;295:E1000–1008. doi: 10.1152/ajpendo.90252.2008. [DOI] [PubMed] [Google Scholar]
- Lenkei Z, Palkovits M, Corvol P, Llorens-Cortes C. Expression of angiotensin type-1 (AT1) and types (AT2) receptor mRNAs in the adult rat brain: a functional neuroanatomical review. Front Neuroendocrinol. 1997;18:383–439. doi: 10.1006/frne.1997.0155. [DOI] [PubMed] [Google Scholar]
- Mangiapane ML, Simpson JB. Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. Am J Physiol. 1980;239:R382–389. doi: 10.1152/ajpregu.1980.239.5.R382. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Osborn JL, Camara AK. Renal neurogenic mediation of intracerebroventricular angiotensin II hypertension in rats raised on high sodium chloride diet. Hypertension. 1997;30:331–336. doi: 10.1161/01.hyp.30.3.331. [DOI] [PubMed] [Google Scholar]
- Pan YX, Gao L, Wang WZ, Zheng H, Liu D, Patel KP, Zucker IH, Wang W. Exercise training prevents arterial baroreflex dysfunction in rats treated with central angiotensin II. Hypertension. 2007;49:519–527. doi: 10.1161/01.HYP.0000256955.74461.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KP, Zheng H. Central neural control of sympathetic nerve activity in heart failure following exercise training. Am J Physiol Heart Circ Physiol. 2012;302:H527–537. doi: 10.1152/ajpheart.00676.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino PR, Schiller AM, Zucker IH. Validation of pulse rate variability as a surrogate for heart rate variability in chronically instrumented rabbits. Am J Physiol Heart Circ Physiol. 2014;307:H97–109. doi: 10.1152/ajpheart.00898.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revest JM, Le Roux A, Roullot-Lacarriere V, Kaouane N, Vallee M, Kasanetz F, Rouge-Pont F, Tronche F, Desmedt A, Piazza PV. BDNF-TrkB signaling through Erk1/2 MAPK phosphorylation mediates the enhancement of fear memory induced by glucocorticoids. Mol Psychiatry. 2014;19:1001–1009. doi: 10.1038/mp.2013.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Blum R, Kafitz KW, Kovalchuk Y, Konnerth A. From modulator to mediator: rapid effects of BDNF on ion channels. Bioessays. 2004;26:1185–1194. doi: 10.1002/bies.20118. [DOI] [PubMed] [Google Scholar]
- Sandberg K, Ji H. Sex differences in primary hypertension. Biol Sex Differ. 2012;3:7. doi: 10.1186/2042-6410-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaich CL, Wellman TL, Koi B, Erdos B. BDNF acting in the hypothalamus induces acute pressor responses under permissive control of angiotensin II. Auton Neurosci. 2016;197:1–8. doi: 10.1016/j.autneu.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheh YL, Hsu C, Chan SH, Chan JY. NADPH oxidase- and mitochondrion-derived superoxide at rostral ventrolateral medulla in endotoxin-induced cardiovascular depression. Free Radic Biol Med. 2007;42:1610–1623. doi: 10.1016/j.freeradbiomed.2007.02.019. [DOI] [PubMed] [Google Scholar]
- Song K, Allen AM, Paxinos G, Mendelsohn FA. Mapping of angiotensin II receptor subtype heterogeneity in rat brain. J Comp Neurol. 1992;316:467–484. doi: 10.1002/cne.903160407. [DOI] [PubMed] [Google Scholar]
- Stauss HM, Moffitt JA, Chapleau MW, Abboud FM, Johnson AK. Baroreceptor reflex sensitivity estimated by the sequence technique is reliable in rats. Am J Physiol Heart Circ Physiol. 2006;291:H482–483. doi: 10.1152/ajpheart.00228.2006. [DOI] [PubMed] [Google Scholar]
- Stocker SD, Stricker EM, Sved AF. Acute hypertension inhibits thirst stimulated by ANG II, hyperosmolality, or hypovolemia in rats. Am J Physiol Regul Integr Comp Physiol. 2001;280:R214–224. doi: 10.1152/ajpregu.2001.280.1.R214. [DOI] [PubMed] [Google Scholar]
- Thunhorst RL, Johnson AK. Effects of arterial pressure on drinking and urinary responses to intracerebroventricular angiotensin II. Am J Physiol. 1993;264:R211–217. doi: 10.1152/ajpregu.1993.264.1.R211. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhou XF. Injection of brain-derived neurotrophic factor in the rostral ventrolateral medulla increases arterial blood pressure in anaesthetized rats. Neuroscience. 2002;112:967–975. doi: 10.1016/s0306-4522(02)00085-4. [DOI] [PubMed] [Google Scholar]
- Wang HJ, Pan YX, Wang WZ, Gao L, Zimmerman MC, Zucker IH, Wang W. Exercise training prevents the exaggerated exercise pressor reflex in rats with chronic heart failure. J Appl Physiol (1985) 2010;108:1365–1375. doi: 10.1152/japplphysiol.01273.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HJ, Wang W, Cornish KG, Rozanski GJ, Zucker IH. Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension. 2014;64:745–755. doi: 10.1161/HYPERTENSIONAHA.114.03699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Haack KK, Zucker IH. Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am J Physiol Cell Physiol. 2013;304:C1073–1079. doi: 10.1152/ajpcell.00364.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JX, Yang RF, Li S, Renshaw AO, Li YL, Schultz HD, Zimmerman MC. Mitochondria-produced superoxide mediates angiotensin II-induced inhibition of neuronal potassium current. Am J Physiol Cell Physiol. 2010;298:C857–865. doi: 10.1152/ajpcell.00313.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Semprun-Prieto L, Wainford RD, Sukhanov S, Kapusta DR, Delafontaine P. Angiotensin II reduces food intake by altering orexigenic neuropeptide expression in the mouse hypothalamus. Endocrinology. 2012;153:1411–1420. doi: 10.1210/en.2011-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- Zucker IH, Patel KP, Schultz HD. Neurohumoral stimulation. Heart Fail Clin. 2012;8:87–99. doi: 10.1016/j.hfc.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker IH, Schultz HD, Patel KP, Wang W, Gao L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol Heart Circ Physiol. 2009;297:H1557–1566. doi: 10.1152/ajpheart.00073.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]