

Abstract
An important subset of asymmetric synthesis is dynamic kinetic resolution, dynamic kinetic asymmetric processes and stereoablative transformations. Initially, only enzymes were known to catalyze dynamic kinetic processes but recently various synthetic catalysts have been developed. This review summarizes major advances in non-enzymatic, transition metal promoted dynamic asymmetric transformations reported between 2005 and 2015.
Keywords: Dynamic kinetic resolution, dynamic kinetic asymmetric transformation, stereoablative reactions, transition-metal catalysis, asymmetric catalysis, cooperative catalysis
1. Introduction
One of the ultimate challenges in the field of organic synthesis is the development of methods for the construction of enantioenriched, carbon-based molecules. The vast majority of processes currently in existence toward this goal proceed through the selective construction of a stereocenter where one previously did not exist. An alternative strategy involves subjecting a racemic mixture of enantiomers to an enantioselective catalytic transformation, wherein the chiral catalyst undergoes preferential reaction with only one of the two enantiomers. This phenomenon is known as kinetic resolution, and while highly useful, kinetic resolutions suffer from a major practical limitation – they cannot produce enantioen-riched products in greater than 50% yield.1,2,3,4,5,6
If a mechanism for the rapid interconversion of enantiomers can be established in the presence of a chiral catalyst, however, the concept of kinetic resolution can lead to full conversion of a racemic mixture to a single, enantioenriched product, a concept known generally as stereoconvergence. It is the focus of this review to highlight the developments of stereoconvergent transformations, with a particular emphasis on transition metal-catalyzed processes. Within this realm there are three subclasses: Stereoablative transformations, Dynamic Kinetic Resolutions (DKRs), and Dynamic Kinetic Asymmetric Transformations (DyKATs). Each subclass contains its own section, and is carefully defined in the corresponding section’s introduction.
During the preparation of this review, we happened upon a number of asymmetric transformations that were conceptually similar to DKR and DyKAT processes; however, the chiral information was contained on the substrate, rather than the catalyst. Strictly speaking, these transformations do not involve asymmetric catalysis and therefore can be considered neither a DKR nor a DyKAT. While few in number, we found these transformations intellectually stimulating, and included them in a final section under the title Dynamic Substrate-Directed Resolutions (DSDRs).
The final goal of this review is to instruct readers as to the proper use of these terms. Due to the inherent similarity of these processes, particularly with respect to DKRs and DyKATs, some of the transformations described herein have been incorrectly classified by their authors. We have re-classified these examples into their proper categories according to the definitions presented in this review. It is our hope that this review will serve as a guide to the reader as to the proper use of these terms.
2. Stereoablative Transformations
In the last decade the synthesis of enantiomerically enriched molecules via stereoablative processes has received much attention.7 A stereoablative process is one in which a key reactive intermediate is formed via the irreversible destruction of a stereocenter; this prochiral species then interacts with a catalyst to form a new stereocenter selectively. As shown in Figure 1, both enantiomers of starting material, (R)-A and (S)-A, undergo a reaction that irreversibly destroys a stereocenter – a process termed “stereoablation” – to produce prochiral intermediate B. Interaction of B with a chiral catalyst can lead preferentially to one enantiomer of product [(R)-C in this case]. Importantly, stereoablative enantioconvergent catalytic systems involve identical or nearly identical rates of stereoablation (i.e. kracR ≈ kracS) but display substantially different rates of product formation (i.e. kR ≫ kS). Stereoablative processes differ from traditional DKR or DyKAT processes in that there is no discernable dynamic or reversible nature to the process with respect to the organic stereogenicity.8
Figure 1.
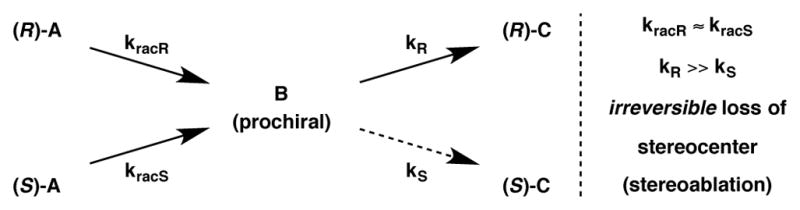
Stereoablative enantioconvergent catalysis.
While the remainder of this article remains focused on transition metal-catalyzed processes, due to the relative scarcity of truly stereoablative transformations, we chose to include transformations that operate through the use of chiral organic catalysts. We hope that this review will inspire the development of novel stereoablative transformations catalyzed by chiral transition metal catalysts.
2.1 Decarboxylative Processes
2.1.1 Enantioselective Allylic Alkylation
Perhaps the most prevalent and commonly utilized stereoablative process is the enantioselective allylic alkylation9 pioneered by Stoltz and coworkers (Scheme 1).10 In 2004, the authors introduced a palladium/PHOX-based catalytic system for the formation of α-ketone quaternary stereocenters from cyclohexanone-based allyl enol carbonates. One year later, they adapted this protocol for the use of chiral racemic α-quaternary β-ketoesters substrates 1.11 This process represents a substantial advance in the construction of carbonyl α-quaternary stereocenters through a three-step process: carboxylation of a ketone enolate with allyl cyanoformate, alkylation of the resultant β-ketoester, and finishing with the decarboxylative allylic alkylation to set the aforementioned fully substituted tertiary or all-carbon quaternary stereocenter.
Scheme 1.
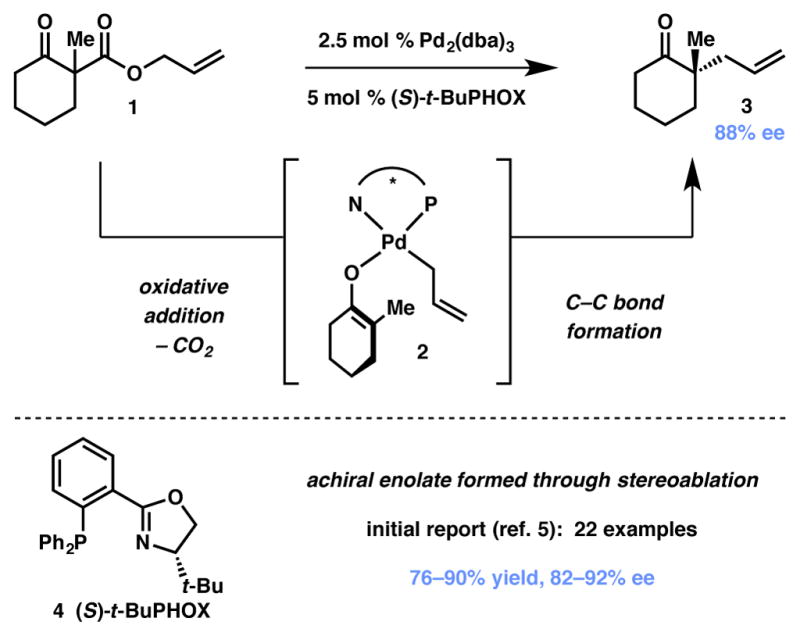
Stoltz’s stereoablative allylic alkylation.
Substantial effort has resulted in a detailed mechanistic understanding of this stereoablative process.12,13,14 Preliminary studies (Scheme 1) suggested that an internal mechanism (i.e. reductive elimination) is a lower-energy pathway than the corresponding external mechanism involving attack of the enolate onto an η3-allyl complex; it was later discovered that η1-allylpalladium carboxylate 5 was found to be the resting state of the catalyst and that decarboxylation was likely rate-limiting.15,16,17 The lowest-energy pathway for carbon–carbon bond formation occurs through a seven-membered, Claisen-like transition state (6) similar to that originally proposed by Echavarren,18 in which the chiral ligand imparts facial selectivity of the allylic alkylation. The sigmatropic character of the transition state likely accounts for the high efficiency with which these sterically hindered quaternary centers are formed, as sigmatropic rearrangements remain a preeminent method for their construction.19 Crucially, these mechanistic studies found that palladium enolate 2 is the reactive intermediate that proceeds to the enantioenriched products. The chiral racemic allyl β-ketoester starting material is converted to this achiral intermediate through catalytic resting state 5, and the catalyst-controlled allylic alkylation event provides the product in an enantioenriched fashion. Furthermore, an allyl β-ketoester, allyl enol carbonate, or a combination of silyl enol ether and fluoride can be used as the enolate precursor to give almost identical yields ee’s, thus confirming the stereoablative nature of the transformation.2,20
Since their development of an enantioselective allylic alkylation, the Stoltz group has increased the scope of this transformation substantially to include, 1,3-dioxan-5-one- (9),21 β-thiocyclohexenone-(10),22,23 3-ketal- (11),24 β-alkoxy-cycloheptenone- (12),25,26 5-alkyl- and 5-alkoxy- (13), β-aminocyclohexenone- (14), 1-alkoxypiperidine-2,6-dione- (15), dihydropyridin-4(1H)-one- (16),27 cyclobutanone- (17),28 valerolactam and 2-piperazinone- (18),29 2-aminomethylcyclohexanone- (19),30 4-oxazolidinone- (20), morpholin-3-one- (21), 1,2-oxazepan-3-one- (22),31 and cyclopentanone-based (23)32 allyl β-ketoesters (Figure 2). Notably, the use of oxygen- and nitrogen-based heterocycles allows for the facile synthesis of quaternary stereocenter-bearing polyketide and pharmaceutical-type fragments in a straightforward manner.
Figure 2.

Substrate scope in Stoltz’s allylic alkylation.
2.1.2 Enantioselective Allylic Alkylation in Total Synthesis
Given the value of ketones bearing enantioenriched α-quaternary stereocenters, it is not surprising that this chemistry has found substantial application to the total synthesis of biologically active natural products. Perhaps most notable is the total synthesis of cyanthiwigin F (31) by Stoltz in 2008 (Scheme 3).33,34 Bis-β-ketoester 24 was produced in two steps from diallyl succinate as a mixture of racemic and meso diastereomers (1:1). Treatment of 24 with the Pd/PHOX catalyst (vide supra) provided a 4.4:1 mixture of syn:meso diallylated products 25 with the major product produced in 99% ee. The stereoablative nature of this transformation simplifies the process of setting two quaternary stereocenters by the clever recognition of latent C2 symmetry. As shown in Scheme 4, after a desymmetrizing vinyl triflate formation/alkyl Negishi coupling sequence to produce tetraene 26 the synthesis is completed in four steps: tandem ring-closing/cross metathesis with vinyl boronate and its concomitant oxidation to aldehyde 29, radical hydroacylation with a polarity reversal thiol catalyst to yield ketone 30,35,36 and a challenging vinyl triflate formation/alkyl Kumada coupling sequence. Overall, the total synthesis of cyanthiwigin F (31) was completed in just nine linear steps utilizing the enantioselective allylic alkylation.
Scheme 3.

Double allylic alkylation leads to excellent ee.
Scheme 4.
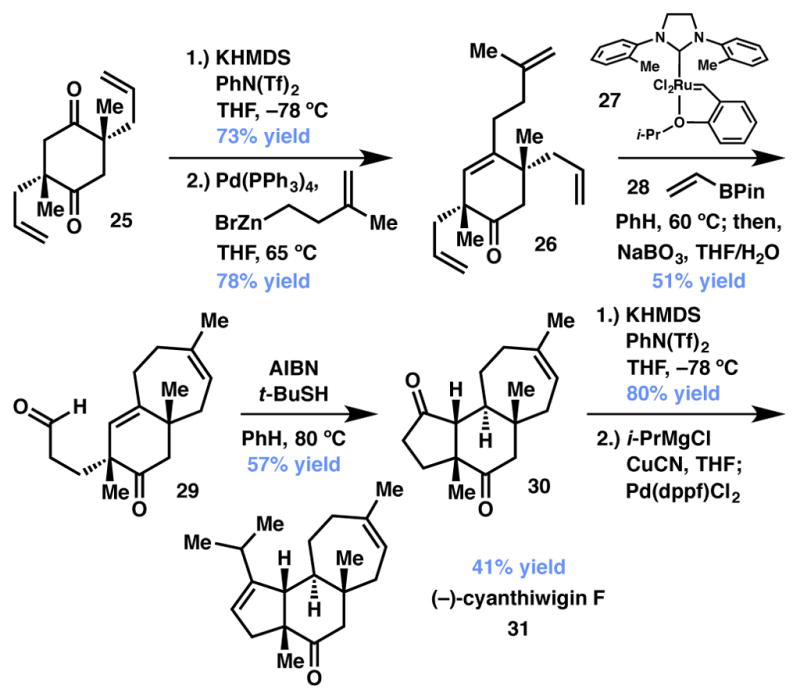
Completion of the synthesis of (−)-cyanthiwigin F.
More recently, the synthetic community has recognized the power of the allylic alkylation for setting such crucial stereocenters and as such has responded with a variety of total syntheses (Figure 3). In 2009, Stoltz applied the β-ketoester-derived from α-methyl-β-phenylthiocyclohexenone to the total syntheses of (+)-carissone (32)13a and (+)-cassiol (33)13b utilizing the stereoablative allylation chemistry coupled with Stork–Danheiser-type cyclohexenone manipulations. Later, the Stoltz and Grubbs groups collaborated on an allylic alkylation/ring-closing metathesis strategy for a general synthesis of the chamigrene natural products including (+)-elatol (35).37 In 2013, Lupton38 and Shao39 reported the formal and total syntheses of (+)-kopsihainanine A (38), respectively, along with the total synthesis of (−)-aspidospermidine (39) by Shao. More recently, in 2015 Zhu and coworkers reported the total synthesis of (−)-isoschizogamine (41) that utilized a stereoablative, enantioselective cyclopentanone allylic alkylation.40 Given the power of this method for the construction of valuable all-carbon quaternary stereocenters, it is likely that we will continue to witness its use in natural product total synthesis for years to come.
Figure 3.

Total syntheses using allylic alkylation.
2.2 Enantioselective Protonation
In 2006, Stoltz and coworkers applied this stereoablative concept to the enantioselective protonation of trisubstituted ketone enolates (Scheme 5, conditions A).41 By employing an allyl β-ketoester with Pd(OAc)2 and a chiral ligand, a similar palladium enolate as 2 (Scheme 1) was formed; however, instead of allylation, the authors reported that they could induce enantioselective protonation using formic acid and 4Å molecular sieves. The authors noted that substantial optimization was required for each substrate; as a result, they also developed a fully homogenous variant of this reaction shortly thereafter (Scheme 5, conditions B).42 In this report, Meldrum’s acid served a dual purpose as both the proton source and as an allyl group scavenger. The latter conditions offered improved generality and scalability. Although only one antipode of the catalyst was used for these studies, the authors noted that the enolates were not always protonated from the same face. Despite the synthetic utility of this reaction, the mechanism of protonation remains unclear.
Scheme 5.
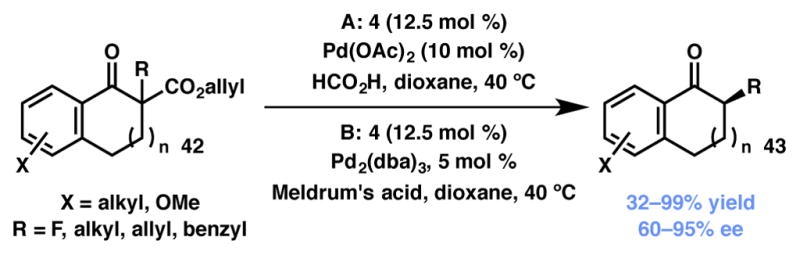
Stoltz’s stereoablative protonation reaction.
2.3 Cross-Coupling Reactions
2.3.1 Couplings with Organometallic Nucleophiles
In 2005 Fu and coworkers disclosed the use of a chiral nickel catalyst capable of performing enantioselective Negishi couplings of racemic alkyl halides (Figure 4).43,44 These reports provide efficient procedures of constructing relatively remote tertiary stereocenters that possess three alkyl units. Following the initial disclosures the authors have demonstrated the exceptionally wide scope of the transformation, with efficient couplings of primary and secondary alkyl chlorides, bromides, iodides, carbonates, and sulfonates with alkyl-, vinyl-, and arylzinc nucleophiles (Figure 4).45,46,47,48,49,50,51,52,53,54 Furthermore, the authors have also developed conditions for enantioselective Suzuki,55,56,57,58,59,60 Hiyama,61 Kumada,62 and zirconium-Negishi53,63 couplings with a variety of alkyl electrophiles.
Figure 4.

Selected scope of Fu’s stereoablative couplings.
In each of the above reports, Fu and coworkers designed a catalyst that could overcome the inherent challenges associated with this type of coupling, most notably the use of alkyl electrophiles and nucleophiles without any competing β-hydride elimination or isomerization.65,66,67,68,69 Furthermore, in all cases racemic alkyl electrophiles were employed, yet the products are produced in high enantiomeric excess. Recent mechanistic studies have elucidated the operating catalytic cycle (Scheme 6).70 The cycle begins with a halide abstraction from 49 by in situ-generated nickel(I) species 48 to yield prochiral alkyl radical 50 and nickel(II) complex 51. Transmetalation between 51 and an arylzinc reagent occurs, generating arylnickel(II) species 52 – the catalytic resting state. Radical addition to 52 is facile, and at this stage the chiral ligand controls facial selectivity of the prochiral radical addition to the metal, generating transient nickel(III) species 53. Subsequent reductive elimination furnishes the enantio-enriched cross-coupled product 54.
Scheme 6.
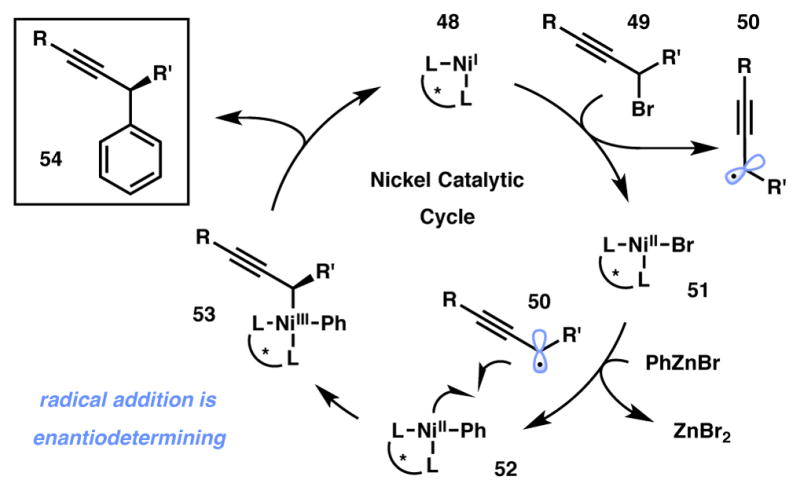
Simplified Nickel Catalytic Cycle.64
2.3.2 Cross-Electrophile Coupling
In recent years, there has been a great deal of interest in cross-electrophile coupling.71,72,73,74 In particular, nickel catalysts have displayed exceptional reactivity and selectivity for cross-coupling processes, rather than simply providing statistical mixtures of products. Extensive mechanistic studies by Weix have led to an understanding of the relevant catalytic cycles in these couplings (Scheme 7).75 sp2-Hybridized electrophiles 55 undergo oxidative addition selectively and rapidly in the presence of nickel(0) species such as 56, thereby producing nickel(II) complex 57. Bimetallic oxidative addition76 of alkyl halide 58 leads to transient nickel(III) species 59. Rapid reductive elimination then produces nickel(I) intermediate 60 and the Csp3-Csp2 product 61; 60 is then reduced to the active nickel(0) catalyst 56 by a low-valent metal reducing agent to complete the catalytic cycle.
Scheme 7.
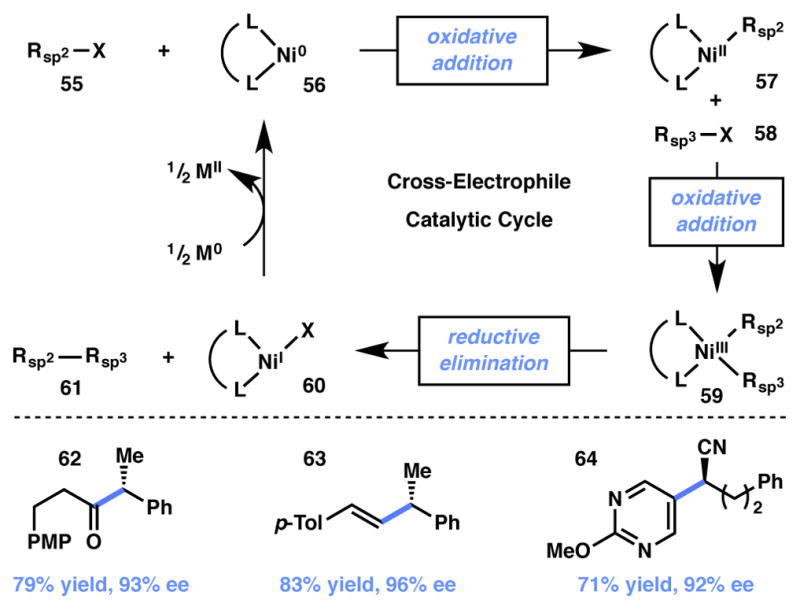
Reisman’s cross-electrophile coupling reactions.
In 2013, Reisman and coworkers reported an enantioselective cross-electrophile coupling of acyl chlorides and racemic benzyl chlorides (Scheme 7, product 62).77 The authors propose a mechanism similar to that proposed by Fu70 wherein enantiodiscrimination occurs upon addition of a prochiral radical to nickel(II) species 57. Since this original report, the authors have also disclosed the enantioselective coupling of vinyl halides with racemic benzyl chlorides (product 63)78 and that of heteroaryl halides with racemic α-chloronitriles (product 64).79
2.4 Enantioselective Oxindole Functionalization
2.4.1 Copper Catalysis
3,3-Disubstituted oxindoles are prominent pharmacophores and as such methods for their construction are in high demand. In 2009 Stoltz and coworkers introduced an enantioselective protocol for their construction from substituted 3-bromooxindoles using a copper-bisoxazoline catalyst.80,81 In the presence of an amine base, the bromide-bearing stereocenter at the 3-position of 65 is ablated, generating o-azaxylylene 66 – an extended π-electrophile – that is attacked by the Lewis acid-bound enolate. Chirality is transferred from the ligand to generate a variety of enantioenriched 3,3-disubstituted oxindole motifs (68). The Stoltz group has utilized this strategy for the syntheses of communesin F82 and perophoramidine.83
2.4.2 Brønsted Acid Catalysis
Since the initial report on the use of stereoablation as a handle for the efficient construction of 3,3-disubstituted oxindoles, a number of groups have disclosed similar strategies. Intriguingly, no other group has reported the use of copper-bisoxazoline catalysts, opting instead to employ cinchona alkaloid-based hydrogen-bonding catalysts.84 Yuan and coworkers reported in 2012 the use of cinchona alkaloid 73 for the O-alkylation of aldoximes with oxindole-based electrophiles (product 69).85 Their system was later modified to allow for the use of ketone enolates as π-nucleophiles (product 70).86 More recently, Jing and coworkers developed cinchona catalyst 74 that bears a squaramide hydrogen-bond donor for the O-alkylation of α-nitrophosphonates under similar conditions (product 71).87
2.5 Miscellaneous Reactions
Terada and coworkers disclosed an enantioselective aza-Petasis–Ferrier reaction in 2009 (Scheme 9).88 The authors found that treatment of racemic O-vinyl-N,O-acetals 76 with chiral phosphoric acid (CPA) 77 (Figure 5) promoted the cleavage of these labile fragments to their corresponding enolate-iminium ion pair (78, Scheme 9), the collapse of which provided the corresponding Mannich adducts 79 after reductive workup. Not surprisingly, the authors noted a strong dependence of the enantioselectivity of the transformation on the geometry of the latent enolate (a product of the olefin geometry of the O-vinyl group). This observation led the authors to develop a set of novel, bulky bisphosphine-nickel complexes for the convenient preparation of the starting materials from the readily accessible O-allyl analogues 78.
Scheme 9.
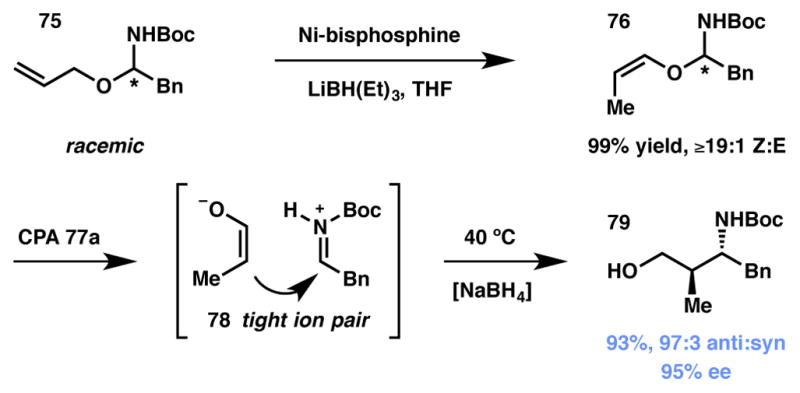
Asymmetric aza-Petasis–Ferrier rearrangement.
Figure 5.
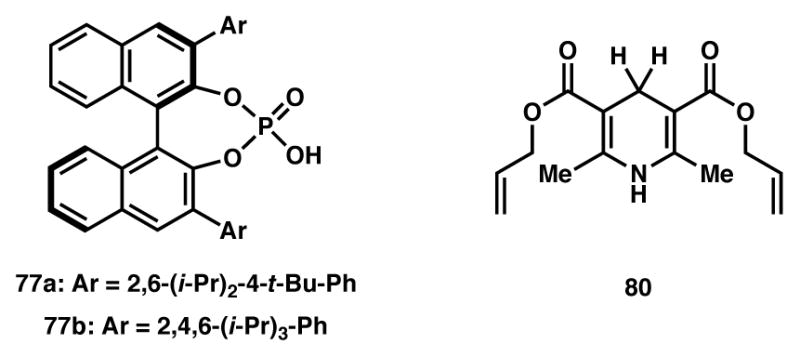
In contrast to many enantioselective catalysis protocols, the authors found that the enantioselectivity of the reaction increased with increasing temperature. The authors later disclosed a thorough mechanistic evaluation to further probe this reaction and account for the anomalous temperature effect.89 They found that the CPA catalyst serves two purposes, acting both as a hydrogen-bond donor to the N-Boc-imine and a hydrogen-bond acceptor from the transient enol. This scaffolding bifunctionality is responsible for both the large degree of anti-selectivity as well as the high enantioselectivities the authors typically observed. Additionally, through an elegant crossover experiment they elucidated the underlying cause of the temperature effect: at high temperatures the two ions produced from the fragmentation of the N,O-acetal dissociate fully and the catalyst-controlled addition of the enol to the protonated imine is the dominant mechanism, resulting in high anti-selectivity and high enantioselectivity. At low temperature, however, dissociation of the two ions is incomplete and a non-catalyst-controlled pathway becomes competitive, thus leading to diminished enantioselectivity.
As a final example, in 2013 Terada disclosed a CPA catalytic system coupled with Hantzsch ester hydride for the enantioselective 1,4-reduction of 1-benzopyrylium ions (Scheme 10).90 In this case, racemic benzannulated lactols 81 are converted to achiral benzopyrylium ions 82 through the action of the CPA catalyst (77). This intermediate is then reduced in a 1,4-fashion with diallyl Hantzsch ester 80 to provide enantioenriched 4-aryl-4H-chromenes 83 with high yields and enantioselectivities. To our knowledge, this is the only report of a stereoablative process wherein the stereocenter created is not that which was originally destroyed.
Scheme 10.
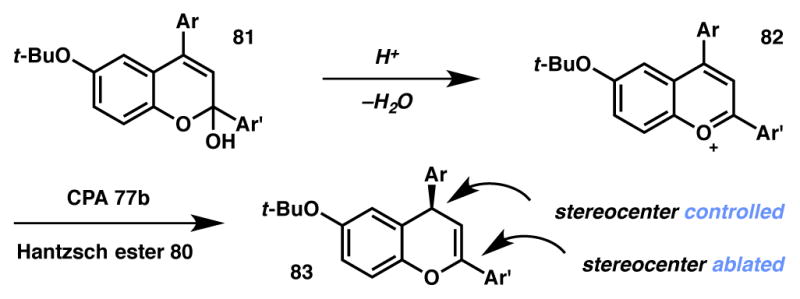
CPA-catalyzed synthesis of 4H-chromenes.
2.6 Concluding Remarks – Stereoablative Transformations
In conclusion, stereoablative enantioselective catalysis is a field that has burgeoned over the past decade, seeing advances in palladium, nickel, copper, and organocatalysis during that span. In contrast to DKR and DyKAT systems that often feature reversible racemization pathways, true stereoablative processes involve irreversible racemization and enantioselective reaction of the prochiral intermediate thereafter. Given the substantial amount of growth in this field over the past decade, we can expect to see even more in the years to come.
3. Dynamic Kinetic Resolutions
Introduction
In contrast to Stereoablative Transformations, Dynamic Kinetic Resolutions involve the reversible racemization prior to the selective reaction of one enantiomer with the chiral catalyst. The first requirement that must be fulfilled to achieve efficient and selective DKR is that the interconversion of enantiomers must be rapid and independent of the catalyst [the equilibration of (R)-A and (S)-A with a high krac, as shown in Scheme 11]. The second requirement is that the reaction of one enantiomer of substrate with the chiral catalyst must occur with a significantly higher rate than that of the other enantiomer (i.e. kR ≫ kS) to provide the enantioenriched product [(R)-B in this case]. As one enantiomer of substrate A reacts with the catalyst, the equilibrium between (R)-A and (S)-A shifts according to Le Châtelier’s principle, such that all of the racemic starting material is eventually funneled through a single enantiomer by the chiral catalyst. As a result, the maximum theoretical yield for a DKR process is 100%.
Scheme 11.
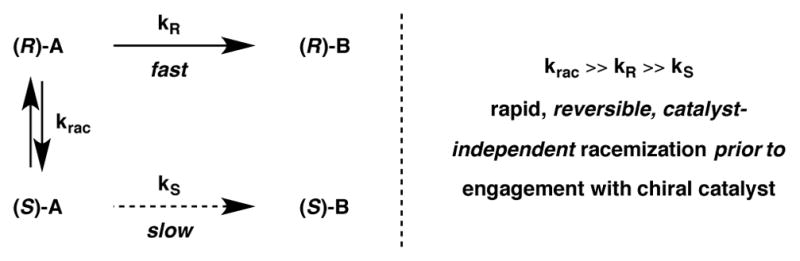
Dynamic kinetic resolutions.
3.1 Annulation Reactions
Asymmetric Spiroannulation
Luan and coworkers devised a novel approach to affect axial-to-central chirality transfer via Pd-catalyzed DKR (Scheme 12).91 In the event, racemic biaryl phenolic substrates (84) were efficiently converted to enantioenriched spirocyclic products (86) in good to excellent yields and enantioselectivities. It is proposed that the catalyst system comprising of Pd(OAc)2 and chiral NHC ligand 87 (Figure 6) complex can preferentially undergo oxidative addition with one of the rapidly interconverting atropisomers of the brominated biaryl phenol substrate. The alkyne partner then intercepts the Pd-bound intermediate and following a phenolic dearomatization event, the sprioannulated product is formed. The methodology is quite general and a wide variety of substitutions are tolerated on the bromide-bearing (hetero)aromatic ring as well as on the alkyne unit. Interestingly, the positional isomer of substrate 84 where the phenolic –OH group is at the 3-position, affords the expected product albeit as a racemate. This observation can be explained by taking into account the detrimental effect of the –OH functionality on facile rotation along the biaryl bond. Although unsymmetrical alkynes react smoothly giving excellent yields, regioselectivities were modest (1.3:1–2:1), slightly favoring the product with the smaller substituent proximal to the quaternary spirocenter.
Scheme 12.

Luan’s dearomative spirocyclization via DKR.
Figure 6.
Chiral ligands from Schemes 12–14 and 45.
Asymmetric Heck Cyclizations
In 2006 the Stephenson group examined the mechanistic aspects of the asymmetric Heck reaction methodology previously laid out by Overman and coworkers.92,93,94,95 The Overman group had found that intramolecular Heck cyclization of 2-iodoanilides produced either enantiomers of 3,3-disubstituted oxindoles using a single enantiomer of chiral bisphosphine ligand. In further studies by the Stephenson et al.96 over a range of temperatures, a distinct pattern took place (Scheme 13): reaction profiles at low conversions saw low ee’s, while at high conversions high ee’s were observed. This observation could not be explained simply by the interconversion of the rotameric forms of the starting material leading to DKR induced enantioenriched formation of the product. Instead, the authors used a variety of x-ray crystal structures to propose a pathway between the Pd-bound oxidative addition complexes of the P and M helices opened in the presence of silver phosphate. Whereas the transformation from the M starting material to its corresponding Pd-bound complex is faster than that from the P starting material, the conversion of the (M)-Pd-bound complex to the (P)-Pd-bound complex is faster than the reverse, thereby leading to preferential access to the (S)-(−) form of the product and provides a suitable explanation for the change in ee based on conversion.
Scheme 13.

Asymmetric Heck reaction of helical amide rotamers.
Ozeki and Yamashita recently reported an interesting example of Pd-SYNPHOS-catalyzed asymmetric Heck reaction that proceeds via DKR (Scheme 14).97 The hindered rotation about the aryl–alkenyl bond results in atropisomerism in triflate 92 and, at temperatures above 60 °C, equilibrium is attained between the atropisomers. Recognizing the potential of an intramolecular Heck reaction via DKR, the authors subjected triflate 92 to Pd(OAc)2 and (R)-SYNPHOS (89, Figure 6) and obtained tricyclic product 93 with excellent yield and high enantioselectivity. The authors postulated that the chiral Pd complex readily differentiates between the two enantiotopic faces of the cyclohexenyl ring, so as to selectively produce the favored enantiomer. A detailed mechanistic discussion, including DFT calculations on the various possible transition states, can be found in the original reference.
Scheme 14.

Asymmetric Heck reaction controlled by restricted rotation.
3.2 Enolate Alkylation
Jacobsen and coworkers reported a Cr(salen)-catalyzed method for enantioselective alkylation of acyclic α,α-disubstituted tin enolates (Scheme 15).98 It is known that tin enolates readily undergo tautomerization in solution,99 leading to the dynamic interconversion of the E and Z isomers. The Cr(salen) complex 97 (Figure 7) is able to selectively react with one of the rapidly equilibrating geometric isomers and leads to the enantioselective construction of quaternary carbon stereocenters.100,101
Scheme 15.
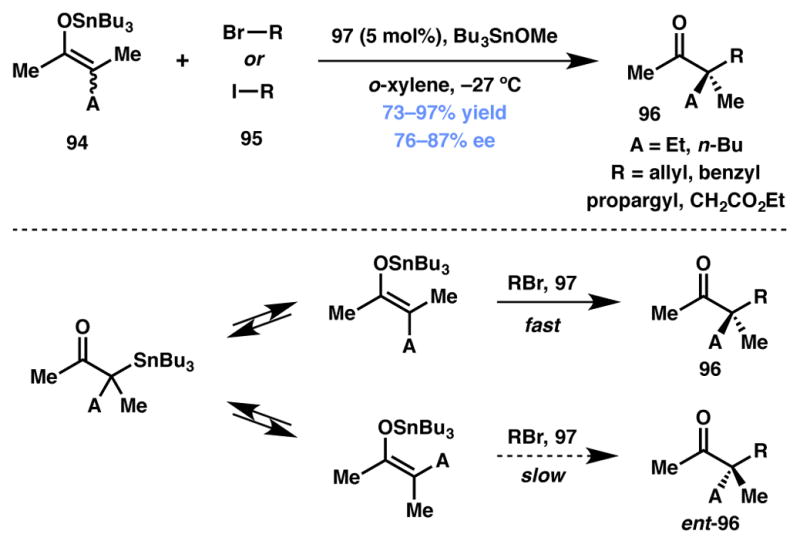
Jacobsen’s enantioselective enolate alkylation.
Figure 7.
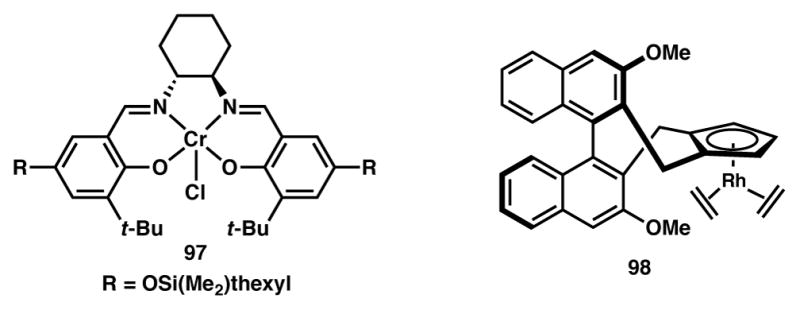
Catalysts from Schemes 15 and 16. Changed
3.3 C–H Activation
In recent years, DKRs proceeding via C–H activation have attracted attention from researchers around the globe.102,103,104 In one example, the You group described a dehydrogenative Heck coupling that occurred via the Rh-catalyzed C–H activation of biaryl substrates 99 to produce corresponding alkenylated biaryls 100 with high efficiency and enantioselectivity (Scheme 16).105 The atroposelectivity the authors observed is believed to originate from the recognition by the chiral catalyst complex of only one atropisomer of either the substrate or the reactive intermediate. This methodology is the first instance in the literature where a chiral CpRh complex (98, Figure 7) is utilized in an oxidative coupling reaction. Although the substrate scope remains limited, this methodology is a promising lead that can spawn more general applications.
Scheme 16.
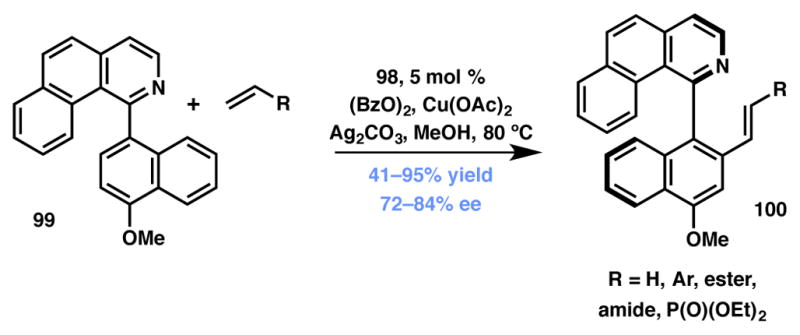
You’s atroposelective C–H activation strategy.
3.4 Amination
Zhao and coworkers recently disclosed a useful protocol for the conversion of racemic secondary alcohols to enantio- and diastereopure secondary amines (Scheme 17).106 The transformation utilizes cooperative catalysis involving chiral Ir complex 103 and CPA catalyst 104 (Figure 8). Impressively, a mixture of four isomers of alcohol 101 can be converted to predominantly a single diastereomer of acyclic amine 102 through a “borrowing hydrogen,” redox-neutral pathway. The racemic alcohol substrate is first dehydrogenated to ketone by the Ir-catalyst (103), followed by CPA-promoted enantioselective protonation, resulting in the formation of an imine intermediate. This species is then reduced diastereoselectively by the [IrH2] complex to afford the amine product in high enantio-and diastereopure fashion. In addition to providing facile access to chiral secondary amines, the Zhao group has also shown that chiral amino alcohols (R2 = OTBS) can be obtained using this methodology. As the racemization is not affected by the catalyst responsible for establishing the initial point of enantioselectivity, this example is classified as a DKR, rather than a DyKAT (vide infra).
Scheme 17.
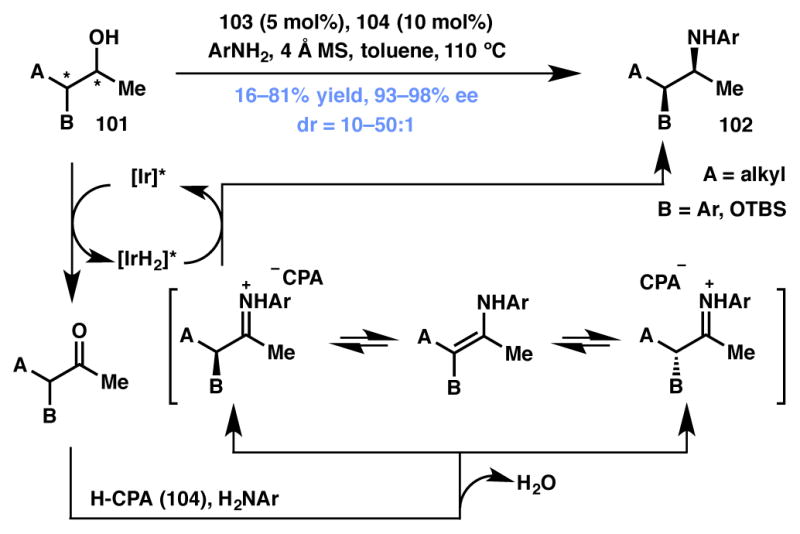
Zhao’s borrowing-hydrogen DKR strategy.
Figure 8.
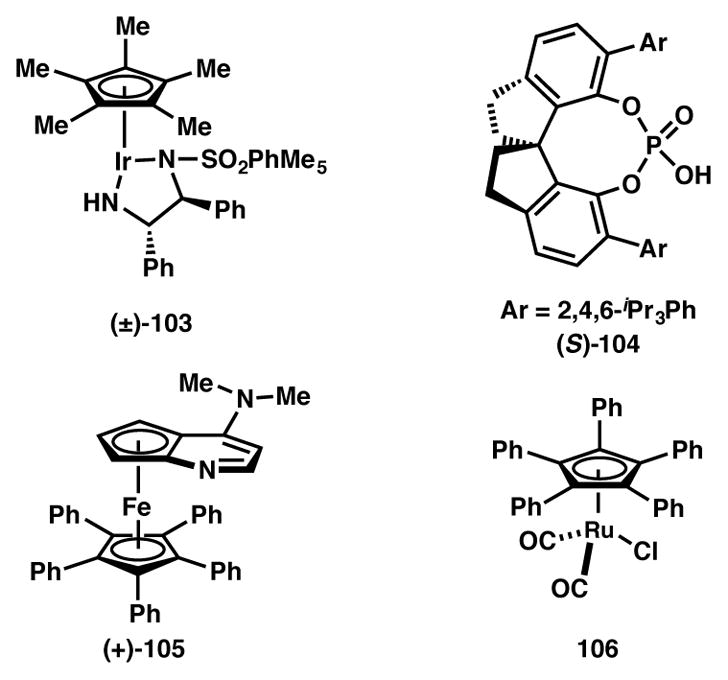
Catalysts from Schemes 17 and 18.
3.5 Acylation
In 2012, the Fu group reported the first example of a non-enzymatic DKR of aryl alkyl carbinols via enantioselective acylation (Scheme 18).107 Previously, these authors had established a classical kinetic resolution for the acetylation of secondary alcohols using planar-chiral DMAP derivative 105 (Figure 8).108 Unfortunately, direct application of these conditions to a dynamic variant by simply adding a Ru-based racemization catalyst (106) did not prove fruitful.109 It was observed that the Ru complex was being acylated by Ac2O, thereby inhibiting the racemization catalyst. To circumvent this problem, less electrophilic acyl carbonates were explored as alternative acylation agents. Of the acyl carbonates examined, acetyl isopropyl carbonate gave the best yields and enantioselectivities. Using a mixture of toluene and t-amyl alcohol as solvent, the DKR of a variety of aryl alkyl carbinols was achieved in both high yield and enantioselectivity. Of note, this methodology applies to branched alkyl substrates, a current limitation of the analogous enzymatic methodologies. Substrates with electron-rich and electron-poor as well as ortho-, meta-, or para-substituted aromatic groups, extended π-systems and allylic alcohols were well tolerated. Mechanistic studies reveal the reversible nature of the N-acylation of the chiral DMAP catalyst (105) and the rate-determining step as the acyl transfer from 105 to the alcohol facilitated by carbonate anion.
Scheme 18.
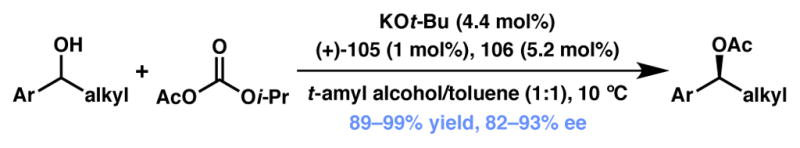
Fu’s non-enzymatic acylation via DKR.
3.6 Asymmetric Reduction
The Noyori group laid the foundations in the field of asymmetric hydrogenation with their pioneering research in the Ru-catalyzed asymmetric hydrogenation of β-ketoesters via DKR.110 Since then, asymmetric hydrogenation and asymmetric transfer hydrogenation have become the most extensively studied and utilized class of transition-metal-catalyzed-DKR transformations. The huge numbers of reports on this concept cover a broad substrate scope and are routinely used on milligram-scale in research laboratories to multikilogram-scale in industrial settings. Due to the large number of examples reported in the literature covering asymmetric reduction via DKR, this review will only cover a select few and the reader is directed to references for the rest.
3.6.1 Asymmetric Hydrogenation
In the present context, DKR–asymmetric hydrogenation processes have been a popular method to reduce aldehydes, ketones, and carbon–carbon double bonds. The typical catalyst system includes a transition-metal (usually Ru, Rh, Ir, Ni, Pd, and Pt) precatalyst, a chiral 1,2-diamine ligand, and in some cases a chiral bisphosphine ligand. The chiral catalyst reacts preferentially with one enantiomer of the substrate. For aldehydes, ketones and other enolizable substrates, the reaction is performed in the presence of a base, which facilitates the substrate racemization via enolate formation, thus rendering the overall transformation dynamic kinetic resolution.
Aldehydes
Zhou and List independently reported the Ru(II) catalyzed asymmetric reduction of racemic, α-branched aldehydes via DKR (Scheme 19). While the Zhou group enlisted SDP as the chiral ligand (111, Figure 9) to obtain moderate to good selectivity,111,112 List and coworkers found that DM-BINAP (116, Figure 9) affords the alcohol product (110) with excellent enantioselectivity.113 The List group has also reported reductive amination via DKR of the same substrate class.114
Scheme 19.

Zhou’s and List’s aldehyde reduction via DKR. changed
Figure 9.
Chiral ligands used in Schemes 19, 20, and 32.
Ketones
Within the past few years, a number of groups have developed methods for the asymmetric hydrogenation of various α-functionalized ketones via DKR.115,116,117 In 2009 the Zhou group developed a chiral RuCl2-SDP/DPEN-catalyzed asymmetric hydrogenation of racemic α-amino aliphatic ketones to their corresponding chiral amino alcohols with two adjacent stereocenters of anti configuration (Scheme 20a).118 Both alkyl and aryl substitution alpha to the ketone and on the nitrogen atom are tolerated, as are secondary amines. In 2010 these authors extended this methodology to α-aryloxydialkyl ketones (Scheme 20b).119 Again using a chiral RuCl2-SDP/DPEN catalyst, the substrates were converted to their corresponding chiral β-aryloxy alcohols with two adjacent stereocenters with anti configuration. The substrate scope could be extended to α-aryl substitution, as well as to α-heteroaryloxy substitution. In 2009 the Hamada group also employed a DKR process to access anti amino alcohols (Scheme 20c).120 In contrast to Zhou’s approach, Hamada utilized nickel catalysis to effect the asymmetric hydrogenation of aromatic α-aminoketone hydrochlorides to their corresponding β-aminoalcohols, again with anti stereochemistry. In 2013, the Zhang group utilized ruthenium catalysis to convert α-amido-β-ketophosphonates to their corresponding β-hydroxy-α-amido phosphonates with syn stereochemistry (Scheme 20d).121 The scope of this transformation extends to alkyl, aryl and heteroaryl ketones with secondary amines of acyl or Cbz protection. Similarly, in 2010 Ratovelomanana-Vidal, Genêt and coworkers achieved the ruthenium-catalyzed asymmetric hydrogenation of α-chloro β-ketophosphonates to their corresponding α-chloro-β-hydroxyphosphonates with syn stereochemistry (Scheme 20e).122
Scheme 20.

Enantioselective hydrogenative DKRs. Changed
β-Ketoesters
In addition to ketones, several examples have also been reported on the asymmetric hydrogenation via DKR of racemic β-ketoesters,123,124,125 especially with amino groups at the α-position.126,127,128,129,130 A representative example on this topic described by the Hamada group is depicted in Scheme 21.131 An Ir(I)–(S)-MeOBIPHEP (123, Figure 10) catalyst system is used in the hydrogenation of racemic α-amino-β-ketoester hydrochlorides 121 to obtain anti-β-hydroxy-α-amino acid derivatives 122 in high yield, high enantioselectivity and with excellent diastereoselectivity.
Scheme 21.

Hamada’s α-amino-β-ketoester hydrogenative DKR.
Figure 10.
Indoles
In 2010, Zhou and coworkers reported an asymmetric hydrogenation protocol of unprotected indoles that proceeds via DKR.132,133,134 Both mono- and disubstituted indoles are readily reduced in the presence of a Brønsted acid promoter and Pd(II) and (R)-H8-BINAP (125, Figure 10) as the catalyst (Scheme 22). The reaction is believed to proceed through the equilibrating iminium species 129a and 129b, which are generated through reversible, non-selective protonation of the indole 3-position. The iminium ions thus produced are then selectively reduced to afford the desired indoline (130) in high yield and enantioselectivity.
Scheme 22.
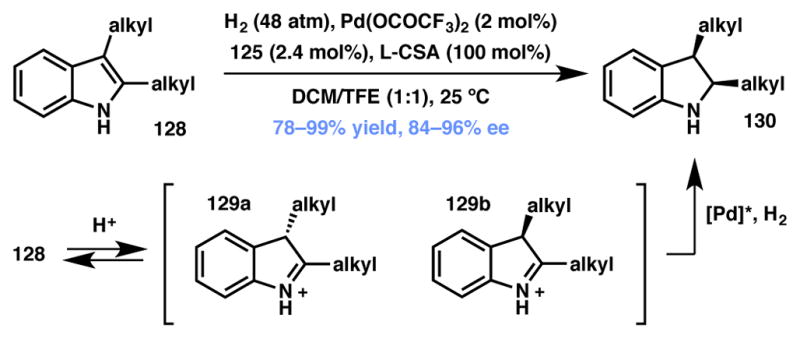
Enantioselective indole protonation/reduction. Changed
3.6.2 Asymmetric Transfer Hydrogenation
In contrast to the examples discussed above where a number of transition-metal catalysts have been developed, Ru-based catalysts almost exclusively catalyze the corresponding asymmetric transfer hydrogenation (ATH) transformations. Additionally, the reducing agent is generated in situ using a number of recipes, most common being a 5:2 cocktail of formic acid and triethylamine. As was the case in the asymmetric hydrogenation reactions that proceed via DKR, the asymmetric transfer hydrogenations of enolizable substrates are also performed under conditions that are conducive to enolization.
Ketones
Fernández and Lassaletta have made significant contribution in the field of Ru-catalyzed hydrogenation of ketones and imines. For example, in 2005 they reported a highly enantio- and diastereoselective reduction protocol for α-substituted cyclic imines (Scheme 23a), with catalyst loadings as low as 0.2 mol %.135 These authors also made another interesting breakthrough in the field by developing a method for the reduction of α-halo ketones to the corresponding vicinal halohydrins (Scheme 23b)136 without the concomitant reduction of the alkyl halide, a common problem with this type of substrate.
Scheme 23.

ATH-based DKR on 5-membered ring ketones and ketimines. changed
Through the endeavors of a number of research groups all over the world, a wide range of functionalities is now tolerated at the α-position of carbonyl compounds.137,138,139,140,141,142 In 2009, Zhang and coworkers disclosed the asymmetric transfer hydrogenation DKR strategy for the reduction of cyclic and acyclic β-ketosulfones under mild conditions with a broad substrate scope (Scheme 24).143 Along similar lines, β-formyl-sulfones can also be reduced efficiently to the corresponding primary alcohols.144
Scheme 24.

ATH-based DKR on β-ketosulfones.
A substantial impact in the ATH-based DKR arena can be attributed to the Johnson group. They have developed a robust methodology for the reduction of α-acyl phosphonates (133) that provides access to α-hydroxyalkylphosphonates (134) in excellent yield along with very high enantio- and diastereocontrol (Scheme 25a).145 Another noteworthy accomplishment from Johnson and coworkers is the reduction of β-aryl α-ketoester via DKR followed by cyclization onto a pendent malonate to form lactone products (Scheme 25b).146 This methodology provides access to highly functionalized γ-butyrolactones (136) that possess three contiguous stereocenters and, not surprisingly, has found applications in natural product synthesis and is discussed in Section 3.6.4 of this review.
Scheme 25.

Johnson’s ATH-based DKR strategies.
Liu and coworkers have demostrated that this technology can also be used to control diastereoselectivity in a 1,3-fashion in the reduction of β-ketophthalides (137) to obtain β-hydroxy isobenzofuranones (138) (Scheme 26).147 The reaction only requires very low catalyst loading and affords desired product in excellent yield and enantioselectivity albeit with modest to good diastereoselecitvity.
Scheme 26.

ATH-based DKR on β-ketophthalides.
β-Ketoesters and β-Ketoamides
Transfer hydrogenation-based DKR has been extended to β-ketoesters and β-ketoamides, including those with heteroatom substitution at the α-position (Scheme 27). The products thus obtained are valuable chiral building blocks. For example, the Ratovelomanana-Vidal group has developed a protocol that delivers mono-differentiated syn-1,2-diols with high enantio- and diastereocontrol via the reduction of racemic α-alkoxy-β-ketoesters (Scheme 27a).148,149 Seashore-Ludlow, Somfai and coworkers employ a similar catalyst system to effectively reduce racemic α-NHBoc-β-ketoesters to anti-β-hydroxy-α-amino acid derivatives in aqueous media (Scheme 27b).150 A team of researchers from Merck led by Limanto and Krska has found that α-alkyl β-ketoamides to be excellent substrates for Ru-catalyzed ATH-DKR that afford syn-α-alkyl β-hydroxyamides with high enantio- and diastereoselectivity (Scheme 27c).151
Scheme 27.

ATH-based DKR on β-ketoesters and -amides. Changed
3.6.3 Reduction with Hydride
In 2008 Yamada extended Bringmann’s DKR-based synthesis152 of axially chiral biaryl compounds via the reduction of biaryl lactones (Scheme 28).153 With the β-ketoiminatocobalt(II) catalyst ((S,S)-141, Figure 11) various substituted optically active biaryl compounds were synthesized in good yields and high ee’s. Biaryl lactones required an increase in temperature to 50 °C for facile racemization of atropisomers. These new conditions allowed for the synthesis of chiral biaryls in high yields and enantioselectivity.
Scheme 28.

Cobalt-based ATH for chiral biaryl synthesis.
Figure 11.
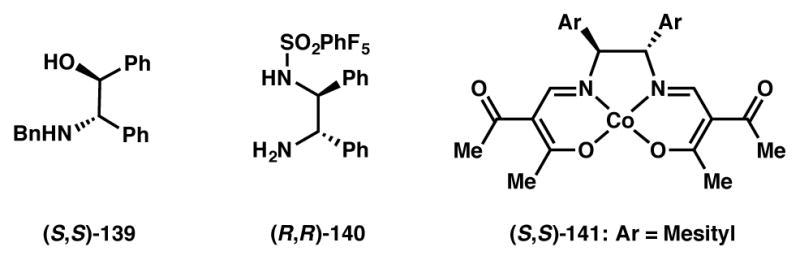
Chiral ligands used in Schemes 27 and 28.
3.6.4 Application of Asymmetric Reduction in Complex Molecule Synthesis
Asymmetric Transfer Hydrogenation
Megaceratonic acid & Shimobashiric acid
The Johnson group recently reported the first asymmetric total synthesis of megacerotonic acid (145) and shimobashiric acid (146) that utilizes their group’s methodology for the asymmetric construction of g-butyrolactones using a Ru-catalyzed, ATH-based DKR strategy (Scheme 29).154 The racemic substrates were prepared in one step via an NHC-catalyzed Stetter reaction of β-aryl enones 147 and 148 with ethyl glyoxylate. The crude product was subjected to the key ATH-based DKR with Rumonosulfonamide 127 (Figure 10) as the catalyst. The requisite alcohol products 150 and 151 were obtained with over 90% yield with high enantio- and diastereoselectivity. With an efficient route to these key intermediate, the asymmetric synthesis of megacerotonic acid (145) and shimobashiric acid (146) was accomplished in nine and eleven steps, respectively.
Scheme 29.

Johnson’s total syntheses of megacerotonic acid and shimobashiric acid.
(−)-epi-Cytooxazone
A useful addition to the scope of ATH via DKR methodology is the asymmetric reduction of prochiral cyclic sulfamate imines that was pioneered by Lee and workers (Scheme 30).155,156,157 The methodology provides an enantioselective technique to synthesize cyclic sulfamates that can be valuable chiral building blocks for the synthesis of complex molecules. In their initial efforts, substrates bearing aryl and alkyl groups at the 4-position were studied and enantioenriched products were obtained, however only with a maximum of 75% ee, while substrates bearing alkyl groups at the 5-position afforded products in 98% ee. In order to improve the results, the authors hypothesized that by increasing the acidity of the hydrogen at the 5-position, rapid racemization would be facilitated and improve the stereoselectivity of the transformation. Excellent results were obtained with electron withdrawing groups at the 5-position. In a representative example, sulfonyl imine 152 was treated with a chiral rhodium catalyst to obtain sulfamate 153 in near perfect yield, enantio-, and diastereoselectivity, and was used to synthesize (−)-epi-cytoxazone (154).156
Scheme 30.
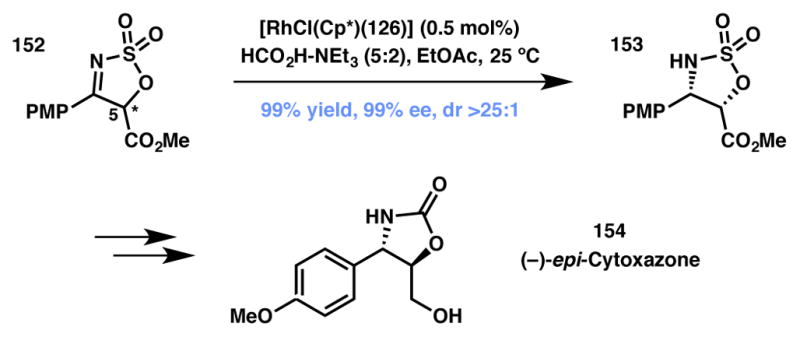
Lee’s total synthesis of (−)-epi-cytoxazone. changed
Reboxetine
Another interesting application of DKR-based ATH from the Lee laboratory describes the construction of two contiguous stereocenters in one step to obtain various 2-substituted morpholine analogs (Scheme 31).158 In the event, subjecting racemic morpholin-3-one 155 to ATH conditions cleanly affords the reduction product 156 with 98% yield and 99% ee. The method provides access to the pharmaceutically relevant 2-substituted morpholine benzyl alcohols in enantioenriched form. Alcohol 156 was transformed to the known antidepressant (S,S)-reboxetine (157) in a few straightforward steps.
Scheme 31.
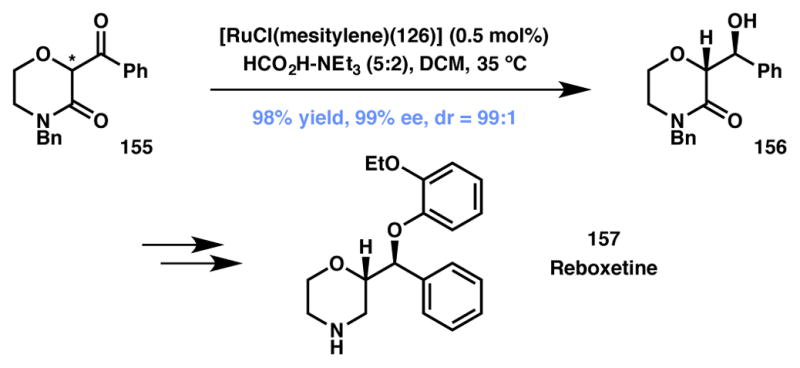
Lee’s ATH-based synthesis of Reboxetine. changed
Other noteworthy examples of ATH-based DKRs in complex molecules syntheses that are not discussed presently are depicted in Figure 12.159,160,161,162
Figure 12.

Other total syntheses involving ATH-based DKR.
Asymmetric Hydrogenation
(+)-γ-Lycorane
In a series of publications, Xie and Zhou have disclosed their group’s efforts on the application of Ru-catalyzed asymmetric hydrogenation involving DKR for the total synthesis of a number of natural products.163,164,165,166 Racemic a-substituted cyclic ketones substrates were processed via this technology to establish two or three contiguous stereocenters. This approach has several advantages: the reaction is carried out on readily accessible substrates, tolerates a number of functional groups, and works well with sterically hindered substrates. The most impressive application of this methodology is showcased in the total synthesis of (+)-γ-lycorane (162).155 α,α′-Disubstituted ketone 163 was subjected to asymmetric hydrogenation to afford alcohol 164 thus creating three stereocenters via DKR in a single step (Scheme 32). With facile access to enantiopure 164, Xie, Zhou and coworkers completed the asymmetric synthesis of 162 in three additional steps.
Scheme 32.

Xie and Zhou’s total synthesis of (+)-γ-lycorane.
PF-00951966
Lall and coworkers reported the use of β-ketopyrrolidinone 165 as the substrate for asymmetric hydrogenation-based DKR (Scheme 33).167 In a reaction performed on multi-gram scale, 165 was reduced via a ruthenium-catalyzed process that afforded β-hydroxylactam 166 in good yield and excellent selectivity. Alcohol 166 was then transformed to furnish fluoroquinolone antibiotic 167 (PF-00951966) in ten steps.
Scheme 33.
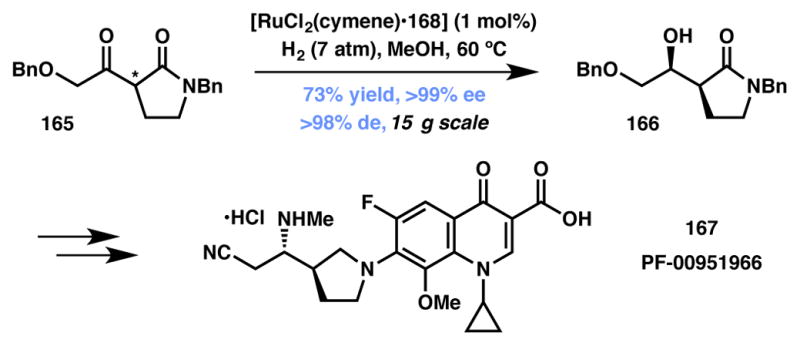
Pfizer’s DKR-based synthesis of PF-00951966.
Glucagon Receptor Antagonist
A tour de force application of asymmetric hydrogenation–DKR transformation was developed at Merck, with the key reaction performed on an impressive 110 kg scale en route to the synthesis of 172, a glucagon receptor antagonist (GRA) (Scheme 34), which has been recognized as potential candidates in the treatment of type-2 diabetes.168 In the event, ketone 173 was reduced in the presence of Ru-SEGPHOS (168, Figure 13) catalyst to afford alcohol 174 in 94% and greater than 98% ee and dr. In five subsequent steps, alcohol 174 was processed to the target molecule.
Scheme 34.
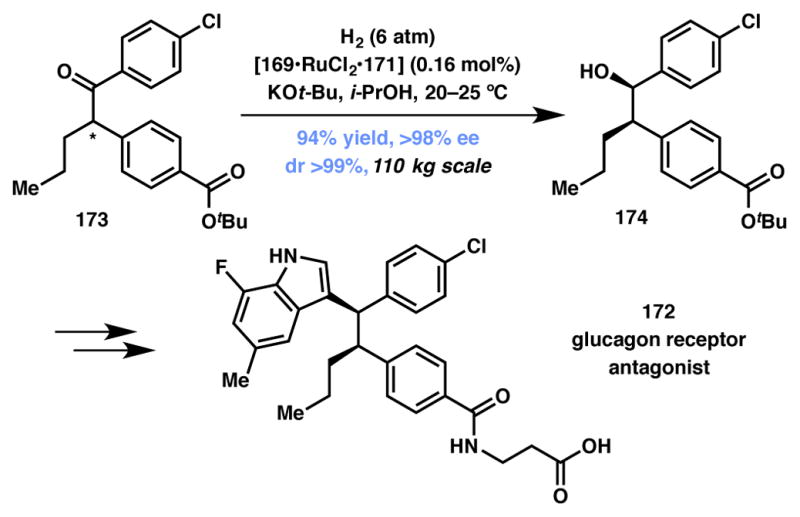
Merck process’ synthesis of GRA 172.
Figure 13.
Chiral ligands used in Schemes 33–34, 41, and 51.
Reduction with Hydride
Eupomatilone-3
In 2005 Buchwald and co-workers completed the first asymmetric total synthesis of eupomatilone-3 (173, Scheme 35).169 All members of the family possess a highly oxygenated biaryl moiety appended to a cis-4,5-disubstituted butyrolactone which, for eupomatilone-3, was accessed via the asymmetric conjugate reduction via DKR of the intermediate 3-methyl-4-aryl butenolide. Using MeO-BIPHEP (123, Figure 10) as the chiral ligand, PMHS as the hydride source, and an excess of base at room temperature, the corresponding cis-4,5-disubstituted lactone was obtained as a single diastereomer in 85% yield and 93% ee. The scope of this DKR was successfully extended to other 3-methyl-4-aryl butenolides; however, the extension of these conditions to γ-alkyl lactones proved unsuccessful. Nonetheless, this work represents the first copper-catalyzed DKR of an unsaturated lactone, enabling the asymmetric total synthesis of eupomatilone-3 in six steps and in 48% overall yield.
Scheme 35.
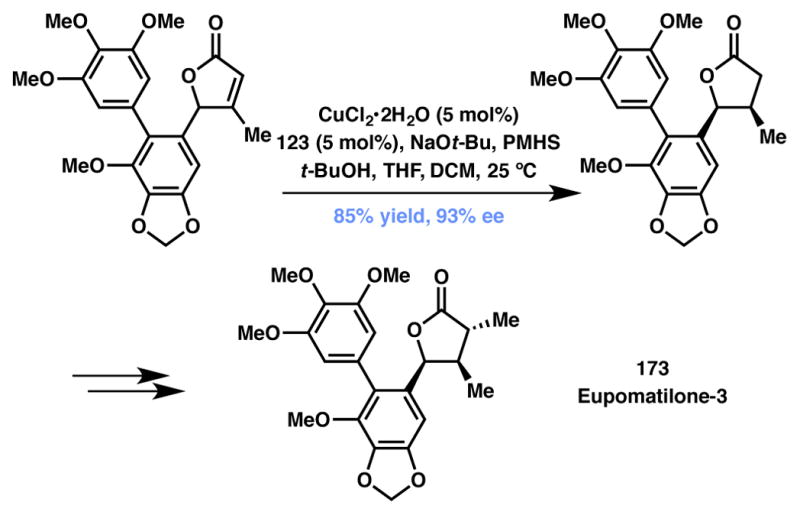
Buchwald’s total synthesis of eupomatilone-3.
The examples discussed above were chosen to give the reader an idea of the versatility of the asymmetric hydrogenation-DKR strategy in complex molecules synthesis. Other examples that were not covered are depicted in Figure 14.170,171,172,173,174,175,176
Figure 14.

Other natural products and pharmaceuticals synthesized via DKR.
3.7 Concluding Remarks – Dynamic Kinetic Resolutions
DKRs have become a vital piece in the organic chemists toolkit, particularly with respect to asymmetric hydrogenation and transfer hydrogenation. The possibility of funneling all material through a single enantiomer brings incredible value to DKRs, especially when compared to classical kinetic resolution. It is our hope that this important research area will continue to grow in the coming years.
4. Dynamic Kinetic Asymmetric Transformations
The third section in this review belongs to Dynamic Kinetic Asymmetric Transformations, or DyKATs.177 Similar to DKRs, DyKATs also involve an equilibration of substrate enantiomers; however, they differ in that a chiral catalyst is responsible for this equilibration. Furthermore, DyKATs can be divided into two types. Type I DyKATs involve the binding of both enantiomers of the substrate to the catalyst to provide a mixture of diastereomeric substrate-catalyst pairs {cf. (R)-A•[cat] vs. (S)-A•[cat], Scheme 36a}. These pairs are then rapidly equilibrated, often through a prochiral intermediate B (B•[cat]), with one of the two reacting to form the product [(R)-C in this case] with a much higher rate than the other (i.e. kP-R ≫ kP-S, Scheme 36a). Type I DyKATs resemble DKRs in that the rate of enantiomeric equilibration krac must be faster than the rate of product formation kP-R, with the notable difference that the interconversion of enantiomers is catalyst-mediated. In contrast, Type II DyKATs involve the loss of the substrate’s chiral center during its interaction with the chiral catalyst to form a prochiral substrate B bound to the chiral catalyst. Selectivity in the overall transformation is achieved when one enantiomer of the product [(R)-C in this case] is produced with a significantly higher rate than the other (i.e. kP-R ≫ kP-S, Scheme 36b). Type II DyKATs bear similarity to stereoablative transformations in that the rate of racemization of each enantiomer must be similar, and that both must be faster than the rate of product formation [i.e., kracR ≈ kracS ≫ kP-R (or kP-S)]. Crucially, the loss of chirality in Type II DyKATs is both reversible and catalyst-mediated, distinguishing them from stereoablative transformations (Section 2).
Scheme 36.

Schematic representation of Type I and Type II DyKATs.
4.1. Carbon–Carbon Bond Forming Reactions
4.1.1 Asymmetric Allylic Alkylation (AAA)
Recognized as one of the most general and reliable transformations, the AAA reactions come in many flavors including DyKAT. This particular subset has found numerous applications and has been reviewed periodically.178,179,180,181 As such, a limited number of examples are presented here for illustration and the reader is advised to consult previous reviews dedicated to this topic for more information.
In 2009 Trost and coworkers disclosed the allylic alkylation of benzylic nucleophiles generated from the deprotonation of BF3-bound 2-alkyl pyridine units.182 A mixture of BF3·OEt2, LiHMDS, and n-BuLi was needed to affect this challenging deprotonation, while a Pd-ANDEN (173, Figure 15) catalyst system was used to activate the racemic cyclic-pivalate electrophile. This combination makes for a highly efficient and selective method for the construction of vicinal tertiary stereocenters, a stereodyad whose construction is certainly not trivial.
Figure 15.
Chiral ligands used in Schemes 37–39.
The Fletcher group disclosed an important breakthrough in this area of non-stabilized AAA reactions with their report on the copper-catalyzed AAA between alkyl zirconium reagents and racemic allylic chloride substrates (Scheme 38).183,184 The organozirconium species can be conveniently generated in situ from alkenes and Schwartz reagent (Cp2ZrHCl). Interestingly, neither Pd- nor Ir-based catalysts delivered the desired product. While it is not entirely clear as to how the chiral catalyst system interacts with the substrate and the alkylzirconium species to afford the product, the authors believe a rapid, copper catalyst-promoted syn-SN2′ mechanism to be the reason behind the observed dynamic behavior. The most practical aspect of this transformation is that readily available terminal alkenes can be added to allylic halides to obtain products in good to excellent yield and enantioselectivity. A minor drawback of this strategy, however, is that the presence of Schwartz reagent in the reaction may limit the overall functional group tolerance of the transformation. Thus, there is room for expanding the substrate scope for this methodology.
Scheme 38.

Fletcher’s copper-catalyzed AAA reaction (Type II DyKAT).
4.1.2 Cross–Coupling Reactions
Suzuki–Miyaura Cross-Coupling
Fernández and Lassaletta demonstrated that the DyKAT strategy could be applied to generate axial chirality via C–C bond construction in racemic biaryl substrates (Scheme 39).185 Using Pd(0) and TADDOL-derived phosphoramidite ligand 175 (Figure 15) as the catalyst system, a Suzuki–Miyaura coupling between racemic 1-aryl-2-triflyloxynaphthalenes and triaryl boroxines effected the asymmetric synthesis of atropisomeric heterocycles.186,187,188,189,190,191 It is postulated that the palladium intermediate generated upon the oxidative addition of a racemic 1-aryl-2-triflyloxynaphthalenese 176 to Pd(0) can rotate freely around the biaryl bond, allowing for the facile interconversion of enantiomers. There are two possible mechanisms for enantiodiscrimination: enrichment of one of the two possible oxidative addition products prior to reductive elimination (Type I DyKAT), or a relatively equal ratio of the two in solution with different rates of reductive elimination leading to enantioenriched products (Type II DyKAT). The authors demonstrated that this strategy was effective when a number of 2-substituted pyridines and isoquinolines as well as 4-substituted quinazolines were utilized, obtaining the corresponding products with good to high yield and enantioselectivity.
Scheme 39.

Fernández and Lassaletta’s asymmetric biaryl synthesis.
Nickel-Catalyzed Cross-Couplings
In 2015, Molander and Kozlowski disclosed mechanistic insights into the asymmetric cross-coupling reactions between a racemic secondary alkyltrifluoroborate (178) and three aryl bromides (179) (Scheme 40).192 The catalyst system for the transformation comprises of an Ir(III) photoredox catalyst (187), Ni(COD)2, and biox ligand 188 (Figure 16). The iridium(III) photocatalyst 187 plays a dual role in the overall transformation, generating carbon-centered radicals from the alkyltrifluoroborate via single electron transfer (SET) and by reducing Ni(I) species 186 to regenerate the Ni(0) catalyst. DFT calculations suggest that enantioselectivity arises via reversible association and dissociation of the stabilized radical to the Ni(II) intermediate. The authors propose that the diastereomeric Ni(III) complexes display different rates of reductive elimination, providing modestly enantioneriched products through a Type II DyKAT process. The authors did not report the yields of these coupling reactions.
Scheme 40.

Molander’s SET-based coupling (Type II DyKAT).
Figure 16.
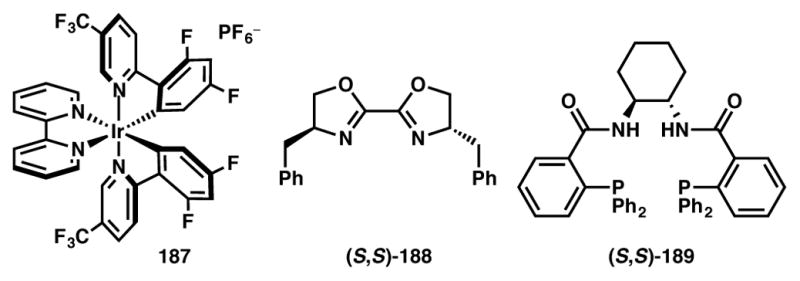
Catalyst and chiral ligands used in Schemes 40 and 42.
4.1.3 Cycloadditions
[3+2] Cycloadditions
In 2013, the Davies group reported the first example of DyKAT in carbenoid chemistry in the formation of cyclopentene derivatives from the gold(I)-catalyzed formal [3+2] cycloaddition of enol ethers 190 and vinyldiazoacetates 191 (Scheme 41).193 This reaction delivered highly functionalized spirocyclic cyclopentene products in high yield and in greater than 90% ee. The reaction generates three contiguous stereocenters in a single step and, remarkably, the products are formed as a single diastereomer. It is proposed that both enantiomers of the product are accessed via rapid, gold-promoted equilibration of E- and Z-enol isomers via the diastereomeric complex 190-[Au]. Intriguingly, the chiral center itself is inert to the transformation, but this equilibration of enol isomers results in scrambling of its R and S identity. Subsequent to this equilibration, only the enantiomer that is matched for the reaction at the Re face of the DTBM-SegPhos [(R)-170, Figure 13] gold-vinylcarbene undergoes cycloaddition via initial attack at the vinylogous position of the vinylcarbene intermediate.
Scheme 41.
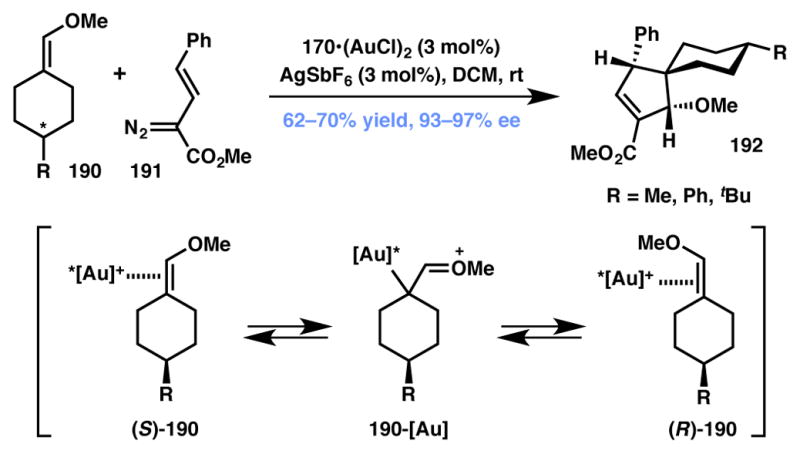
Gold-catalyzed [3+2] cycloaddition featuring a gold-mediated interconversion of enantiomers (Type II DyKAT).
Reactions involving donor–acceptor cyclopropanes (DACs) have recently found widespread use.194,195,196,197,198,199 The Trost group reported a Pd-catalyzed formal [3+2] cycloaddition between racemic vinyl cyclopropane 193 and alkylidene azalactones 194 via a DyKAT process to afford spirocyclic cycloadduct 195 (Scheme 42).200 Impressively, the reaction produces mainly one of the four possible diastereomers as the major product in good yield and excellent enantioselectivity. Mechanistically, the reaction initiates through the non-selective ionization of the vinyl cyclopropane to generate intermediates 196 and 197. The unique “wall and flap” steric environment created by ligand 189 is able to funnel the equilibrating mixture to thermodynamically favored intermediate 196. The malonate anion in 196 attacks the azalactone in a 1,4-fashion and the resulting azalactone enolate traps the π-allylpalladium moiety to produce the desired product. The methodology can be applied to a broad substrate scope and provides access to highly functionalized products.
Scheme 42.

Trost’s formal [3+2] cycloaddition (Type I DyKAT).
Xu, Shi and coworkers developed a Pd-catalyzed formal [3+2] cycloaddition reaction of vinyl cyclopropanes with β,γ-unsaturated-α-ketoesters to obtain highly functionalized cyclopentane derivatives 198 bearing three contiguous stereocenters (Scheme 43).201 The reaction tolerates a variety of substitution on the enone substrate and delivers products with excellent yields and high enantio- and diastereoselectivities.202 The mechanistic rationale for the observed selectivity is expected to be similar to that observed in Trost’s methodology (Scheme 42).
Scheme 43.
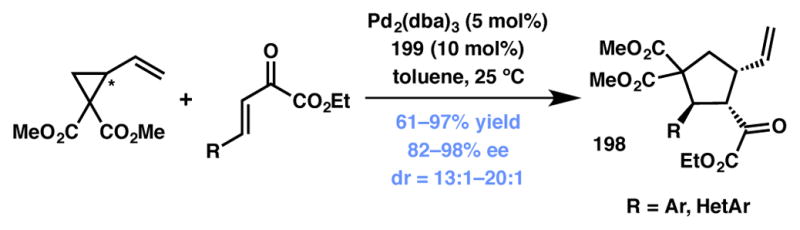
Xu’s and Shi’s formal [3+2] cycloaddition (Type I DyKAT). changed
Tang and coworkers have reported a DyKAT involving [3+2] annulation of cyclic silyl enol ethers and racemic DACs catalyzed by a copper(II)/200 system (Scheme 44a).203 More recently, Waser and coworkers reported [3+2] cycloaddition via DKR that utilizes amino-DACs and aldehydes or enol ethers as annulation partners to afford the tetrahydrofuran or cyclopentane products respectively, in good to excellent yields, enantio- and diastereoselectivity (Scheme 44b).204 Readily available copper/t-Bu-Box (201, Figure 17) complex was used as the chiral catalyst. The authors propose that the facial selectivity of the formal [3+2] cyclization event is dictated by catalyst system and that the dynamic process proceeds via reversible cyclopropane ring opening/closing, which may also be mediated by the copper catalyst.
Scheme 44.

Copper-catalyzed formal [3+2] cycloadditions from Tang and Waser (Type II DyKAT).
Figure 17.
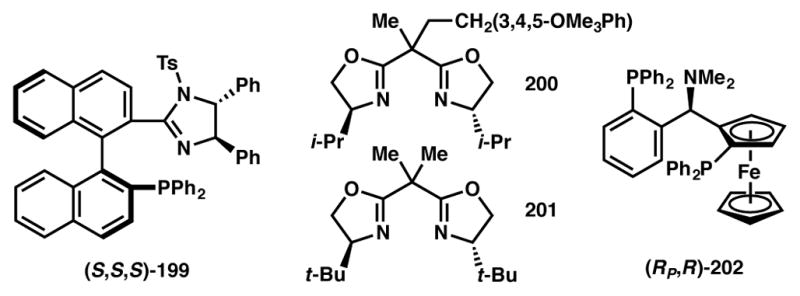
Chiral ligands used in Schemes 43, 44, and 46.
[3+2] Annulation of racemic allenes with aryl ketimines
Cramer and coworkers reported a useful method for the selective construction of substituted indanylamine building blocks (Scheme 45).205 The Rh(I)-BINAP (88, Figure 6) catalyzed transformation can tolerate a broad substrate scope using readily available precursors. Importantly, the good to excellent chemical yield, enantio- and diastereoselectivity obtained makes this technology a particularly attractive route to access complex scaffolds. The reaction is believed to proceed through a rhodium-catalyzed, ketimine directed C–H activation and is followed by coordination and insertion of the allenic moiety. A dynamic system is established as a result of isomerization of the diastereomeric allyl rhodium intermediates (206) as shown in Scheme 45. The isomerization occurs faster than the ultimate addition across the imine fragment. It was observed that the reaction stereochemistry is controlled entirely by the chiral catalyst system and the axial chirality of the allene component has no effect on the product stereochemistry. Moreover, submitting enantioenriched allene to the rhodium complex in the absence of imine led to complete racemization, providing further evidence for a catalyst-mediated interconversion of enantiomers.
Scheme 45.
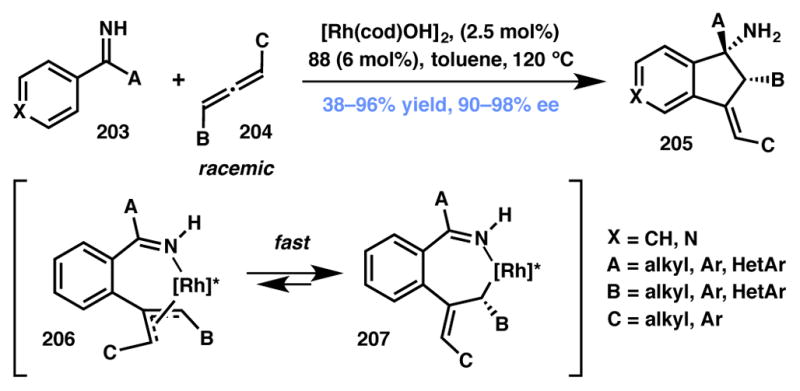
Cramer’s C–H activation via Type II DyKAT.
4.1.4 Hydroacylation
Willis and coworkers have used allenes as substrates in rhodium–catalyzed dynamic kinetic asymmetric hydroacylation reactions (Scheme 46).206 Preliminary reports focused on the use of aliphatic and aromatic aldehydes bearing a thiomethyl moiety at the β-position, presumably to facilitate metal coordination and subsequent insertion in the aldehyde C–H bond. Under Rh(I)/Me-DuPhos catalysis (211, Figure 18), various 1,3-disubstituted allenes smoothly reacted with aldehydes to afford the corresponding β,γ-enone products in good to excellent yields and enantioselectivities. The authors also carried out mechanistic studies that support a DyKAT mechanism responsible for the observed selectivities. Catalyst control was observed when an enantiomerically enriched allene was employed in the reaction. Moreover, the ee of the recovered allene was found to be significantly reduced, strengthening the proposal of a DyKAT mechanism.
Scheme 46.

Willis’ rhodium-catalyzed allene hydroacylation.
Figure 18.
Chiral ligands used in Schemes 46 and 49.
4.2 Carbon–Heteroatom Bond Forming Reactions
4.2.1 Carbon–Nitrogen Bond-Forming Reactions
Amination
In 2005, Trost and coworkers reported the Pd-catalyzed dynamic kinetic asymmetric addition of secondary amines to racemic allenyl acetates (Scheme 47).207 The reaction efficiently produced allenamine 213 via the rapid interconversion of vinyl-Pd(II) intermediates 214 and 215. In addition to secondary amines, this methodology also tolerates malonate nucleophiles.208
Scheme 47.

Trost’s C–N bond-forming Type I DyKAT on allenyl acetates.
The Widenhoefer group reported a gold-catalyzed enantioselective intramolecular hydroamination209,210,211 of γ-amino allenes 216 to form 2-vinyl pyrrolidine products 217 (Scheme 48).212, The cationic gold complex participates in the racemization of enantiomers 218 and 219 selectively reacts with one enantiomer of the substrate, thus qualifying as a Type I DyKAT system.213 Under these conditions, disubstituted allenes deliver the corresponding product ent-217 with poor enantioselectivity.214
Scheme 48.
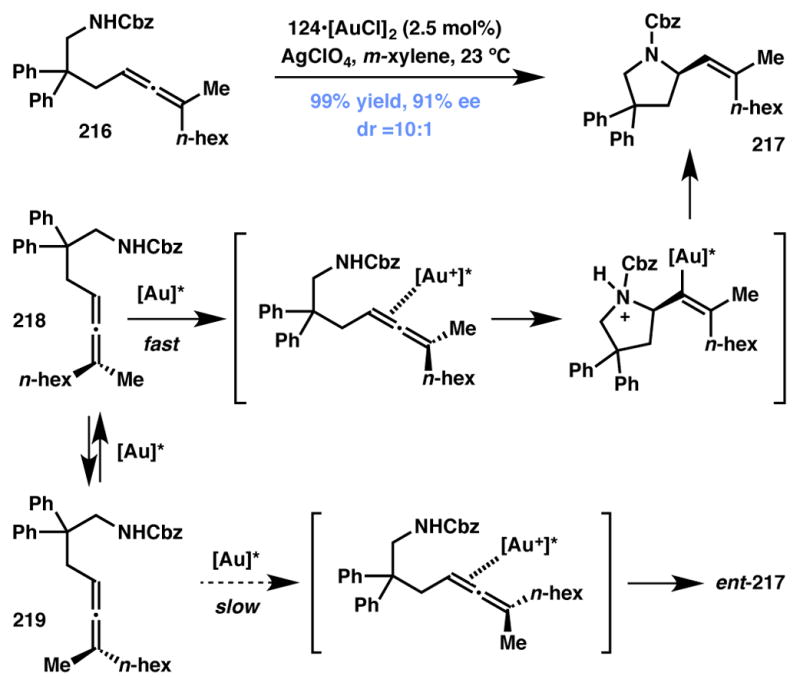
Widenhoefer’s vinylative pyrrolidine formation via Type I DyKAT.
Kawatsura, Itoh and coworkers developed an interesting approach for accessing acyclic, chiral CF3-bearing amines from unsymmetrical 1,3-disubstituted allylic acetates and carbonates via a Pd-catalyzed DyKAT (Scheme 49). The desired α-product (221) was obtained with good to excellent enantioselectivity along with minor amounts of nearly racemic γ-product (222). It was found that the presence of silver additive was critical in achieving the observed enantioselectivity in this dynamic process.
Scheme 49.
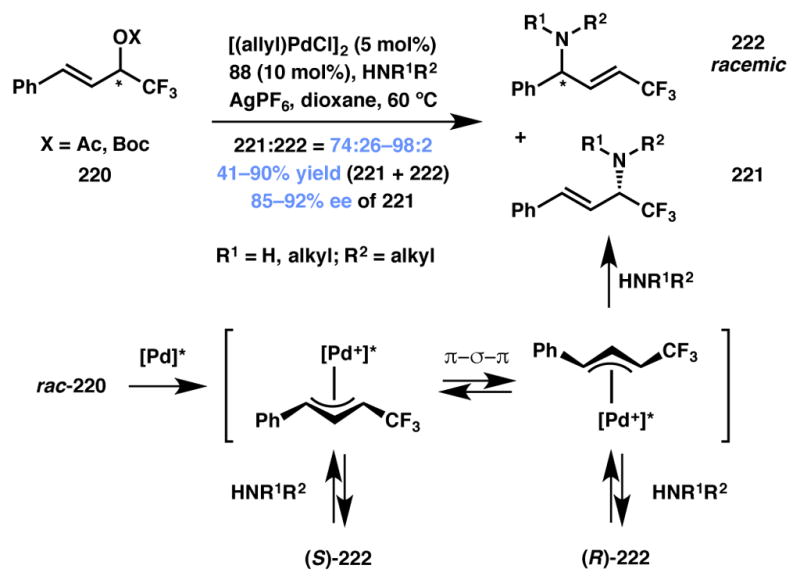
Reversible addition of amine to benzylic site leads to efficient Type I DyKAT.
In 2012 the Nguyen group developed a rhodium-catalyzed regio- and enantioselective amination via DyKAT of racemic tertiary allylic trichloroacetimidates with anilines (Scheme 50).215 Prior research in their group on racemic allylic amination216 had indicated that the oxidative addition of trichloroimidates 226 and 228 occurred with different rates, suggesting a kinetic resolution was at play. The authors also noted, however, that isomerization of the diastereomeric π-allylrhodium intermediates appeared to be facile, which could be taken advantage of in a DyKAT. The authors hypothesized that the use of an appropriate ligand that would slow the rate of aniline addition could allow more time for a π-σ-π intercon-version of the diastereomeric π-allylrhodium intermediates. If this ligand was chiral, such a DyKAT could be realized. A broad ligand evaluation showed that diarylbicyclo[2.2.2]octadiene 230 (Figure 19) provided the desired product in both high yield and enantioselectivity. Consistent with the authors’ observations, electron-rich anilines formed adducts with lower enantioselectivity than their electron-deficient counterparts. This trend likely stems from the correspondingly higher rates of addition of these more nucleophilic coupling partners.
Scheme 50.
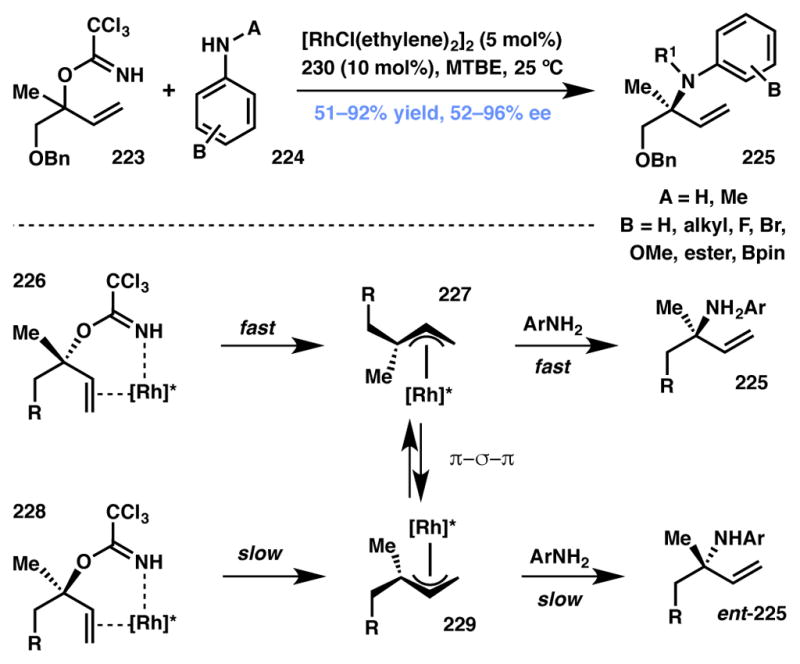
Nguyen’s Type I DyKAT-based synthesis of allylic amines.
Figure 19.
Chiral ligands used in Schemes 50 and 52.
These authors have also developed a closely related Ir-catalyzed dynamic kinetic asymmetric fluorination of racemic, secondary allylic trichloroacetimidates with Et3N·3HF as the fluoride source.217
Kitagawa and coworkers reported the use of a DyKAT process for the synthesis of axially chiral 1,2-biaryl indoles via a Pd-catalyzed C–N bond-forming 5-endo-hydroamination (Scheme 51).218,219 The authors recognized that the (2-t-butylphenyl)indoles have a high rotational barrier and can be accessed via an atroposelective, intramolecular cyclization starting from ethynylaryl anilines 232. Of the numerous chiral ligands screened, (R)-SEGPHOS 168 (Figure 13) was found to affect the cyclization with highest enantioselectivity. Best results were obtained with substrates in which the alkyne moiety was capped with 2-susbtituted phenyl groups (ring A). It is believed that in the enantiodeterming step, axial chirality is generated due to the presence of a substituent at the 2-position of ring A. As a result, the conformation with minimum steric clash between the R group on ring A, the t-butyl moiety on ring B and the chiral ligand on the catalyst is favored. Thus, for R = H, the indole product was obtained in 60% ee whereas for R = Br the ee was determined to be 83%, correlating agreeably with the hypothesis. Replacing the tert-butyl group on ring B with the smaller isopropyl or phenyl groups was detrimental to the observed enantioselectivities.220,221,222
Scheme 51.
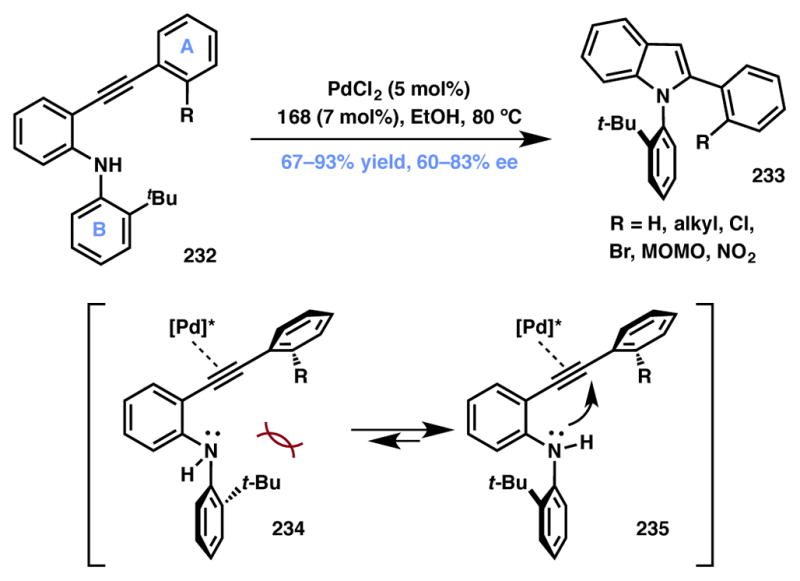
Kitagawa’s Type II DyKAT method for atroposelective indole synthesis.
4.2.2 Carbon–Oxygen Bond-Forming Reactions
Trost and coworkers reported a Pd-catalyzed AAA–DyKAT approach for the synthesis of tetrahydropyran (THP) moieties from racemic Baylis–Hillman-type adducts bearing a tethered alcohol as the nucleophile (Scheme 52).223 High yield and enantioselectivity was observed for both ester and nitrile substrates, although the two required slightly different conditions to achieve the highest levels of efficiency and enantioselectivity. The dynamic system is set up through π-allyl equilibration and enantioselectivity is achieved when this process is faster than the attack by the pendant alcohol. Interestingly, a highly selective kinetic resolution process is observed when the reaction is carried out at 23 °C, indicating that higher temperature is required for the π-allyl equilibration. Kitamura disclosed a similar transformation utilizing allylic alcohols, rather than acetates, and showed that one of their substrates provides enantioselective products through a Type I DyKAT.224
Scheme 52.
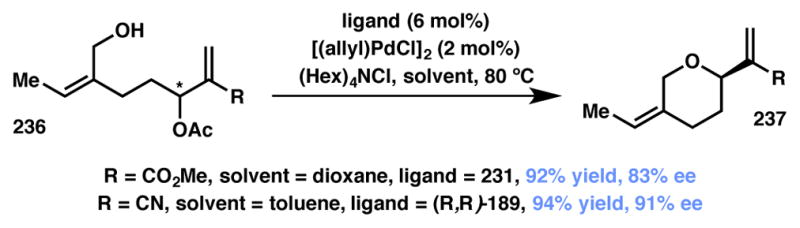
Trost’s Type I DyKAT-based method for THP synthesis.
Zhang and coworkers have reported an interesting extension to the Pd-catalyzed allylic substitution by O-nucleophiles (Scheme 53).225 Their approach utilizes racemic vinyl-substituted ethylene carbonates as substrates that undergo CO2 extrusion upon exposure to catalytic Pd2(dba)3·CHCl3 and (S,S,S)-phosphoramidite 174 (Figure 20), leading to a rapidly interconverting dynamic system comprising of diastereomeric π-allylpalladium intermediates. Formaldehyde then reversibly captures the Pd-alkoxide intermediate, generating a new pair of diastereomeric Pd-allyl complexes, which is much slower to equilibrate. The DyKAT is realized when due to rapid reductive elimination of one of the two glycolates undergoes reductive elimination, affording methylene acetal-protected tertiary vinylglycols in excellent yield and enantioselectivity.
Scheme 53.
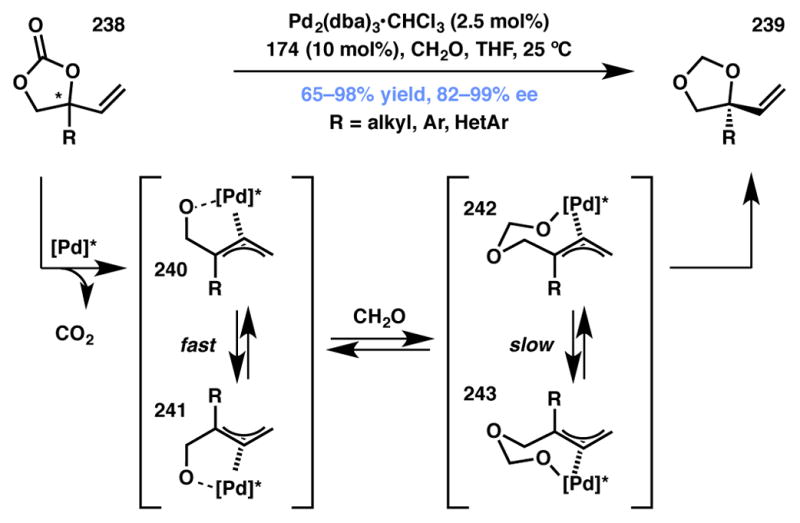
Zhang’s decarboxylative Type I DyKAT for chiral dioxolane synthesis.
Figure 20.
Chiral ligands from Schemes 53 and 54.
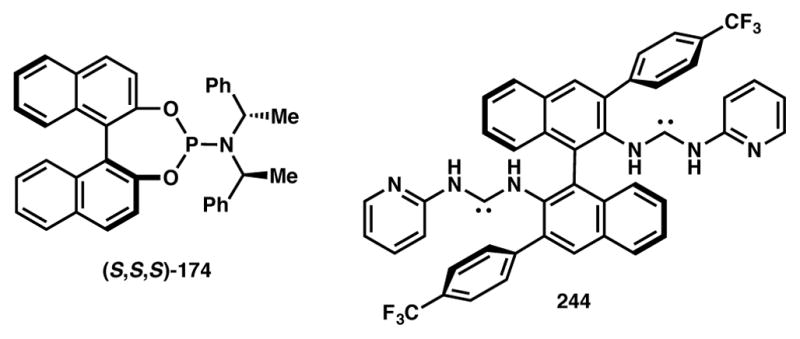
The Toste group has demonstrated the use of a chiral cationic Au(I)–carbene complex as catalyst for the asymmetric synthesis of highly substituted chromene analogs from suitably functionalized propargylic esters (Scheme 54).226 A variety of chiral phosphines and NHC ligands were evaluated, and ligand 244 (Figure 20) was identified to be the optimum candidate that delivered the desired product in high yield and selectivity. Both free phenol and benzyl aryl ethers may be used as substrates. The reaction initiates with Au-catalyzed formal [3,3] sigmatropic rearrangement of propargylic ester substrate to generate a gold-bound allene. The allene–gold interaction results in scrambling of axial chirality and stereoselectivity is achieved via a 6-endo-trig attack of the phenolic oxygen to construct the chromene skeleton, followed by either proton or benzyl group transfer to the insipient cation. Mechanistic studies indicate that one of the enantiomers of the substrate reacts faster while the unreacted isomer undergoes racemization via the aforementioned gold-allene pathway.
Scheme 54.
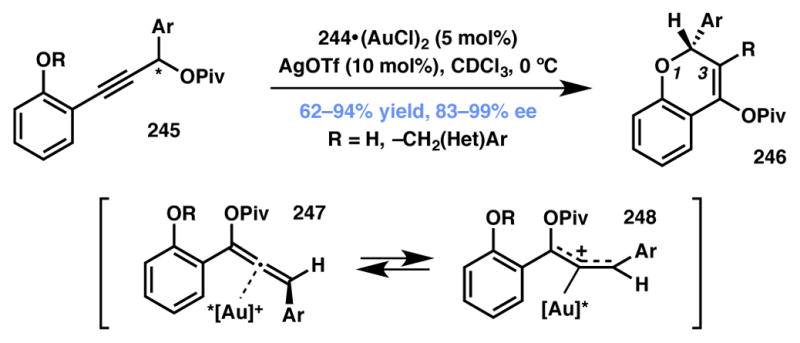
Toste’s Type II DyKAT-based strategy for asymmetric chromene synthesis.
4.2.3 Carbon–Phosphorous Bond-Forming Reactions
Glueck and coworkers pioneered Pd-catalyzed C–P cross-couplings involving DKR to obtain chiral phosphines.227,228 Since then, this field has evolved substantially and reviewed in the recent literature.229,230 As such, only a couple of representative examples will be discussed here. In 2007 Bergman and Toste designed a Pd-catalyzed arylation of tertiary racemic silylphosphines as a means to synthesize P-stereogenic phosphines (Scheme 55).231 After extensive screening it was discovered that ortho-benzamide substituents improved the enantioselectivity of the C–P coupling. The authors found that the one-carbon arylacetamide homologs provided decreased ee’s, implying that a five-membered palladacycle was crucial for obtaining the highest enantioselectivity. The source of DyKAT stems from the low barrier to epimerization of Pd(II)-phosphide intermediates (due to the facile pyramidal inversion of metal phosphido complexes) which occurs faster than the reductive elimination. Exploration of substrate scope around iodobenzamides revealed that changes in electronics para- to the iodide or to the amide have very little effect on the ee. Electron-rich iodides required longer reaction times but also show excellent enantioselectivities. Other competent variations include substrates with extended conjugation, electron-rich heteroarenes, thiophenyl substrates, and sterically congested amides. Exploration of substrate scope around phosphines revealed that electron-poor substrates decrease ee. Tolerated phosphine variations include less sterically congested alkyl groups as well as oxygenated alkyl groups.
Scheme 55.

Bergman and Toste’s Type I synthesis of enantioenriched P-stereogenic phosphines. changed
Stoltz and Virgil reported a palladium-catalyzed, atroposelective DyKAT for the asymmetric synthesis of the chiral ligand QUINAP (258, Scheme 56).232 Using a Pd(0)/Josiphos (118, Figure 9) catalyst system, racemic triflate precursor 257 was phosphinated to afford QUINAP 258 in good yield and enantioselectivity. It is believed that the diastereomeric arylpalladium intermediates 259 and 261, produced after a nonselective oxidative addition event, undergo racemization to preferentially form one atropisomer that upon phosphination yields QUINAP with high selectivity. Interestingly, replacing triflate with bromide on the substrate results in a kinetic resolution process that provides both QUINAP and the unreacted bromide in high yield and enantioselectivity.
Scheme 56.

Pd-mediated Type I DyKAT for the synthesis of QUINAP.
4.3 Asymmetric Reduction
Dong and coworkers have developed a Rh-catalyzed asymmetric hydrogenation proceeding through a DyKAT process on allylic sulfoxides, an entirely different substrate class than those discussed above.233,234 This interesting transformation provides a complementary route to chiral sulfoxide products (Scheme 57), which are usually accessed via the oxidation of sulfides. Racemic allylic sulfoxides of the type 262 were transformed into enantioenriched sulfoxides 263 under a hydrogen atmosphere with Rh(cod)BF4-(S,S)-Ph-BPE (255, Figure 21) as the catalyst. The authors postulate a pathway wherein the Rh-catalyst is involved in both the racemization of the allylic sulfoxide via reversible C–S bond cleavage-recombination, in addition to the selective hydrogenation of one of the resulting substrate enantiomers to deliver the desired product in moderate to good yield and enantioselectivity. The reaction optimization process revealed several noteworthy aspects: a relatively low hydrogen pressure (0.1 atm) ensures that the rate of hydrogenation is slow compared to the rate of racemization, and the use of polar solvents, such as methanol, favors the intermediacy of polar intermediates during racemization.
Scheme 57.
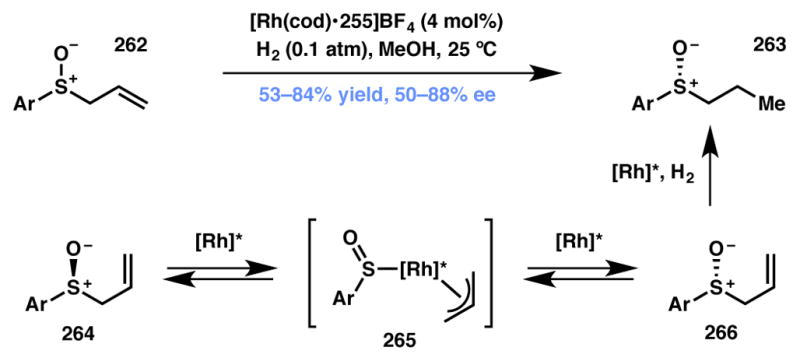
Dong’s reductive chiral sulfoxide synthesis via a rhodium-catalyzed Type II DyKAT.
Figure 21.
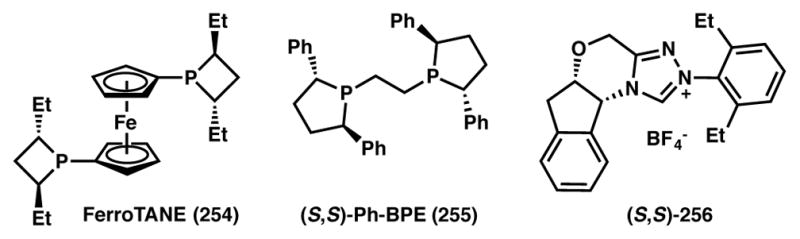
Chiral ligands used in Schemes 55, 57, and 58.
4.4 Miscellaneous Reactions
Wang and coworkers recently disclosed the first example of an intermolecular DKR of α-ketoesters through the synergistic combination of NHC and Lewis acid-catalysis (Scheme 58).235 The reaction between α-ketoesters (267) and β-methylenals (268) occurred to produce δ-lactone products (269) in good to excellent chemical and stereochemical efficiency. The methodology tolerates a wide substrate scope and furnishes products that have numerous functional group handles for further manipulations. The exact role of the Lewis acidic Sc(OTf)3 co-catalyst is not fully understood at this time, however it was required to achieve high enantioselectivity in addition to high yield in the transformation.
Scheme 58.
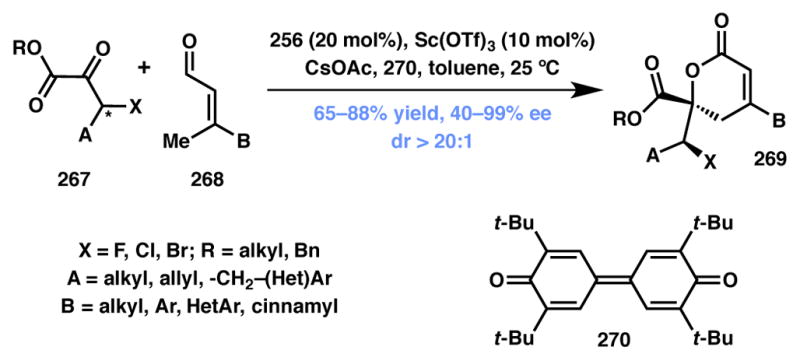
Wang’s Type I DyKAT for the synthesis of unsaturated lactones.
Moriwaki, Liu, Soloshonok and coworkers recently reported a Ni-promoted DKR of unprotected α-amino acids (271, Scheme 59).236 It was postulated that when the enantiomers in the racemic sample were subjected to the reaction conditions, chiral ligand 273 (Figure 22) reacts faster with the R enantiomer of the substrate to make the (S,R) diastereomer (272) as the kinetically favored product. The other (S,S) diastereomer (not shown) forms slowly and converts to the (S,R) diastereomer via a base-catalyzed enolate equilibration. Access to the enantiopure amino acid products can be readily realized via treatment of intermediate 272 with 6N HCl. This also allows for the recovery and recycling of the chiral ligand. Furthermore, similar efficiency of this DKR to the S/R interconversion of α-amino acids was also demonstrated. Structural analyses show that (S)-Ligand 273 creates S-helical chirality of the chelate rings, which gives rise to the R-absolute configuration of the α-amino acid. The substrate scope of this resolution encompasses aliphatic, aromatic, and ω-functionalized amino acids with high yields and diastereoselectivity.
Scheme 59.
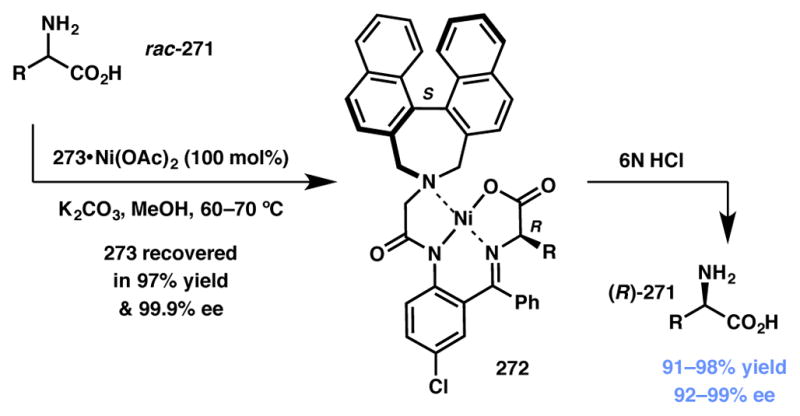
Type I DyKAT-based method for enantioconvergence of racemic α-amino acids.
Figure 22.
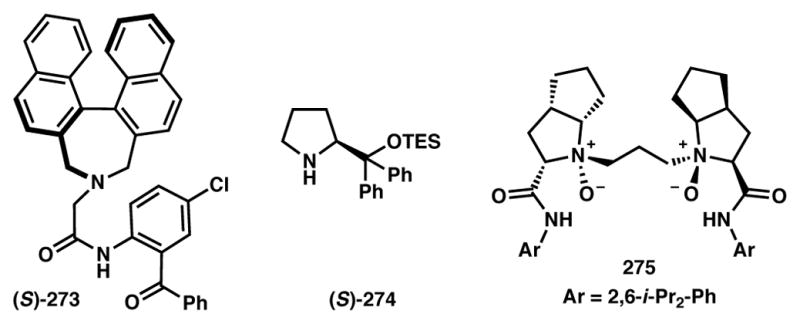
Chiral ligands and catalysts used in Schemes 59–61.
Córdova and coworkers have developed a fascinating one-pot oxa-Michael/cyclization cascade between propargyl alcohols 276 and enals 277 to produce highly substituted dihydrofurans (DHFs) 278 that proceeds under a synergistic combination of chiral amine 274 (Figure 22) and PdCl2 (Scheme 60).237 The authors reasoned that while the addition of propargyl alcohol (PrgOH) to the iminium intermediate was expected to be reversible and non-selective, the participation of palladium via its interaction with the triple bond would force the reaction forward. Irreversible asymmetric induction takes place at this stage of the reaction, as only the sterically favored diastereomeric Pd-alkyne complex 281 can entertain a nucleophilic attack to forge oxacyclopentane 282 that contains an exocyclic olefin at this stage. Thermodynamic equilibration to the fully substituted and conjugated olefin eventually yields 278 as the overall product. The authors were not able to rule out the possibility of Pd(II) acting as a Lewis acid that activates the triple bond toward an enantioselective, and therefore a non-DyKAT, attack by the enamine onto the Pd-alkyne prior to oxa-Michael addition, however. The Cordova group has since expanded this technology to obtain a variety of useful structural motifs.238,239
Scheme 60.

Córdova’s Type II DyKAT for enantioselective DHF synthesis.
Feng and coworkers recently disclosed an iron-catalyzed asymmetric Cannizzaro reaction (Scheme 61).240 A variety of aryl and alkyl glyoxal hydrates reacted smoothly with alcohols to produce α-hydroxyesters with excellent ee. The mechanism for stereoinduction in asymmetric Cannizzaro reactions is subject to an ongoing debate, and where one proposal qualifies as a DyKAT (path A),241,242 another favors an enantioselective addition route (path B).243 It is postulated that under the reaction conditions, substrate 283 dehydrates to form glyoxal 285 that can reversibly form racemic hemiacetals 285. The chiral iron-N,N′-dioxide (275, Figure 22) complex then selectively binds and promotes the suprafacial hydride migration to afford α-hydroxyester product 284. As shown in path B, one can imagine the iron catalyst chelating to the glyoxal (287) and inducing enantioselective alcohol addition. In the present case the authors, based on experimental evidence, believe that both DyKAT (path A) and enantioselective addition of alcohol to glyoxal (path B) to be in synergy and deliver the products in excellent yield and enantioselectivity. Considering that these conditions promote the reverse of the alcohol addition (rendering path B reversible), we feel that path A is likely predominant, and thus classify this transformation as a DyKAT.
Scheme 61.

Feng’s asymmetric Cannizzaro reaction via a Type II DyKAT process. changed
The Dong group reported their initial investigations on the rhodium–catalyzed ring expansion methodology to obtain indanone 292 from benzocyclobutanone 288 (Scheme 62).244 The proposed C–C bond cleavage approach was broadly effective for racemic reactions, however, limited success was achieved in an asymmetric variant catalyzed by Rh(I)/(S)-SEGPHOS (168, Figure 23) with product 292 obtained in a modest 42% ee (Scheme 62). The reaction is believed to be an example of DyKAT and at the moment requires careful optimization for achieving useful levels of yield and selectivity.
Scheme 62.

Dong’s Type II DyKAT for benzocyclobutanone expansion.
Figure 23.

Chiral ligands and catalysts used in Schemes 62–64.
4.5 DyKATs in Complex Molecule Synthesis
Muscone
In 2005 Ikariya and co-workers completed the asymmetric synthesis of muscone (297) establishing the lone stereocenter with a ruthenium-catalyzed asymmetric double bond isomerization of an allylic alcohol precursor (295, Scheme 63).245 Using a chiral catalyst comprising of Ru(I) and L-proline derived ligand 293 (Figure 23) the (±)-(E)-allylic alcohol precursor 295 afforded the (S)-ketone 296 in 74% ee. The authors speculate that the allylic alcohol is reversibly oxidized to enone 298, providing the active hydrogenation catalyst and electronically activating the now-conjugated olefin. Enantioselective reduction delivers ketone 297 via a selective 1,4-addition event.
Scheme 63.

Ikariya’s total synthesis of muscone utilizing a Type I DyKAT.
M58163 and M58169
Saitoh, Mikami and co-workers completed the asymmetric total syntheses of M58163 (303) and M58169 (304), both of which display antithrombotic activity (Scheme 64). Access to their imidazopyrazinone core (301) was achieved using a lanthanum-catalyzed asymmetric cascade cyclization proceeding through racemic aminal 302.246,247 Mechanistic studies revealed that the aminal is formed reversibly, with enantioselection occurring during the La-catalyzed amide bond-forming step. A survey of various lanthanide metal complexes revealed that the La-linked BINOL complex 294 (Figure 23) gave the best yield and selectivity. Despite the modest enantioselectivity, the authors note that this marks the first catalytic enantioselective aminal synthesis.
Scheme 64.

Saitoh’s and Mikami’s synthesis of antithrombotic agents 303 and 304 utilizing a Type II DyKAT.
Hydroxypyrrolidine Natural Products
Trost and coworkers reported a versatile application of the Pd-catalyzed AAA in the total synthesis of (+)-broussonetine G (313) and other structurally related pyrrolidine-containing alkaloids like (+)-dihydroxymethyldihydroxypyrrolidine (DMDP) (311) and (−)-bulgecinine (312, Scheme 65).248 In the event, racemic butadiene monoxide undergoes a Pd-catalyzed DyKAT with phthalimide to afford homoallylic alcohol 307 with excellent yield and enantioselectivity (Scheme 65). 307 was then transformed into oxazolidinone 308, which was used for a second Pd-catalyzed DyKAT with butadiene monoxide to produce alcohol 309, once again with high yield and selectivity. Pyrrolidine scaffold 310 is forged using RCM technology. Thus sequential application of AAA transforms readily available feed-stock material into a flexible intermediate 309 that was further elaborated to accomplish the total synthesis of (+)-broussonetinine G (313), (+)-DMDP (311) and (−)-bulgecinine (312).
Scheme 65.

Trost’s syntheses of some hydroxypyrrolidine natural products utilizing a Type I DyKAT.
Elbasavir
Researchers from Process Chemistry at Merck have recently disclosed a Pd-catalyzed Buchwald–Hartwig C–N coupling to synthesize chiral indoles 317 (Scheme 66).249 Using a high-throughput experimentation approach, palladium(II) acetate, bis-phosphine ligand 315 (Figure 24) and K3PO4 in toluene were identified as the optimum conditions to transform racemic hemiaminals 316 to chiral benzoxazino-indoles in excellent yield and high enantioselectivity. While the exact mechanistic pathway has yet to be confirmed, the authors postulate a base-promoted epimerization to be key in the isomerization of the substrate via its ring-open form (318). The “matched” hemiaminal-Pd complex (320) selectively undergoes reductive amination to forge the C–N bond. The methodology was used as the key step in the efficient synthesis of elbasvir (322), a drug candidate for treating HCV infections.
Scheme 66.

Merck’s Type I DyKAT-based synthesis of Elbasavir.
Figure 24.
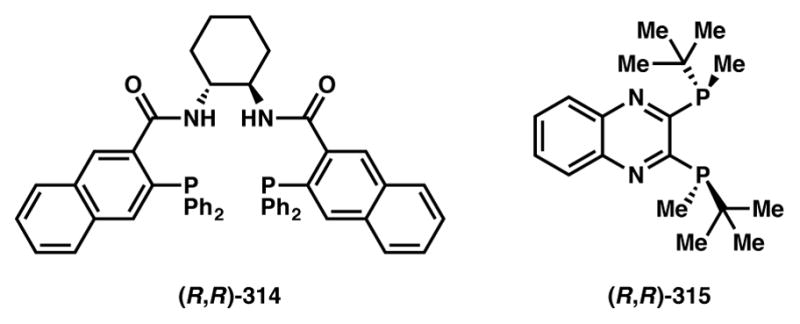
Chiral ligands used in Schemes 65 and 66.
4.6 Concluding Remarks – Dynamic Kinetic Asymmetric Transformations
While DyKAT systems have the added complexity of a catalyst-mediated racemization event, they also offer the possibility of stereoconvergence to substrates that are not typically prone to epimerization. As such they form a fine complement to DKR processes, and as newer and more sophisticated catalytic systems are introduced, we can expect this field to continue its pattern of rapid growth.
5. Dynamic Substrate-Directed Resolutions
As mentioned in the Introduction section, during the preparation of this review we came across a few systems that displayed dynamic behavior, yet were not controlled by a chiral catalyst. While these do not fall under one of the traditional categories of stereoconvergence, we were intrigued by the concept of a Dynamic Substrate-Directed Resolution and thus opted for their inclusion with the hope of inspiring future work in this area.
Azatitanacyclopropane Reductive Cross-Coupling
In 2013, Micalizio and co-workers, observed the epimerization of azatitanacyclopropanes under reductive cross-coupling conditions (Scheme 67).250,251,252,253,254 Application of this phenomenon to the stere-oconvergent reductive cross–coupling of achiral aromatic imines 323 with chiral allylic alcohols 326 provided homoallylic amines 327 in good yields and high enantioselectivity. Treatment of the achiral aromatic imines with Ti(OiPr)4 and c-C5H9MgCl gives a rapidly interconverting mixture of enantiomeric azatitanacyclopropanes, which upon treatment with 1.3–2.0 equivalents of chiral allylic alcohol provides the stereodefined amine product (327). Both TMS- and Bn-substituted aromatic imines can be employed. The methodology has also been applied cyclic and acyclic allylic alcohols, with di- or tri-substituted alkene functionality, including and is tolerant of vinyl bromides.
Scheme 67.

Micalizio’s use of chiral alcohols for the synthesis of enantiopure amines.
Intramolecular Ring-Closing Ene Reaction
In 2005, the Pearson group reported a novel intramolecular ene-type reaction between a diene-Fe(CO)3 complex and an alkene resulting in the stereospecific generation of the corresponding spirolactam (Scheme 68).255 The precursor can be easily synthesized from the corresponding chiral amino ester and the racemic carboxylic acid as a diastereomeric mixture of 328 and 329. Under photolytic conditions, the iron-center loses coordination to the diene, eventually establishing an equilibrium between the two π-faces that allows for the dynamic interconversion of 328 and 329. The two diastereomers can be separated, and when individually subjected to the [6+2] ene-type reaction conditions one generated the spirolactam and the other converted to the reactive diastereomer and then underwent the cyclization. The authors propose that interconversion of diastereomeric precursors 328 and 329 occurs faster than the formation of the putative reactive intermediate 330. Initially, one product is formed but thermal rearrangement gives access to olefin isomers (not shown); however, all isomers converge to a single product after demetalation and hydrogenation. In this process the chiral amide substituent directs the stereospecific formation of a single enantiomer, setting multiple stereocenters in a single step.
Scheme 68.

Pearson’s use of an iron-bound diene for enantioselective lactam synthesis.
Atroposelective Biaryl Vinylation
Soon afterward, Yang and co-workers256 reported the C–H alkenylation of biaryls bearing a chiral phosphinate (Scheme 69).257 Using Pd(OAc)2 and N-acyl glycine as the catalyst system, a wide variety of alkenes were coupled in good yield and diastereoselectivity. The methodology can also be extended to acylation, hydroxylation, acetoxylation and iodination on the same substrates albeit though kinetic resolution.
Scheme 69.

Yang’s enantioselective biaryl vinylation.
Carbon–Oxygen Bond Formation via C–H Activation
A highly diastereoselective Pd-catalyzed acetoxylation of biaryls 335 that contain a chiral sulfoxide auxiliary was disclosed by Wencel-Delord and Colobert and coworkers (Scheme 70).258,259 The C–O coupling was realized using ammonium persulfate in a 1:1 mixture of acetic acid and hexafluoroisopropanol (HFIP) as the solvent. The reaction was performed at ambient temperature and was found to be tolerant to air and moisture, implying that Pd(II), rather than Pd(0), was the key catalytic intermediate in the transformation. Moreover, replacing the persulfate reagent with N-iodosuccinimide affords the corresponding iodinated product with good to excellent yield and diastereoselectivity.
Scheme 70.

Colobert’s atroposelective C–H acetoxylation/iodination.
Application of DSDR in Total Synthesis
Carbohydrates
Guo and Tang recently reported an impressive application of the Achmatowicz rearrangement260,261 for the formal synthesis of several deoxysugars that progress via an iridium-catalyzed dynamic kinetic internal transfer hydrogenation (Scheme 71).262 Alcohol 337, which can be accessed as an enantiopure, 3:1 mixture of diastereomers in two steps via the asymmetric reduction of acetylfuran followed by an Achmatowicz rearrangement. In the key step, subjecting the diastereomeric mixture of 337 to catalytic [Ir(cod)Cl]2 and 2,6-dichlorobenzoic acid (2,6-DCBA) results in a stereoselective internal transfer hydrogenation to generate lactone 338 in 99% ee with complete diastereocontrol. Mechanistically, the reaction is believed to proceed through a rapid acid-catalyzed epimerization of the hemiacetal, followed by stereoselective Ir-catalyzed internal transfer hydrogenation via a dynamic system. Lactone 338 can be processed to a number of deoxysugars by utilizing previously established protocols.263
Scheme 71.

Guo and Tang’s synthesis of deoxysugars.
6. Conclusion
Stereoconvergent methods for the construction of enantioenriched organic molecules remain among the most valuable for the construction of chiral, non-racemic organic molecules. In particular, the ablative or dynamic aspect of these processes is a significant advantage, allowing for the full conversion of racemic starting materials to products of a single enantiomer. We hope that this review has served as an educational tool to not only summarize work in this field, but also instruct readers as to the proper use of these terminologies.
Scheme 2.
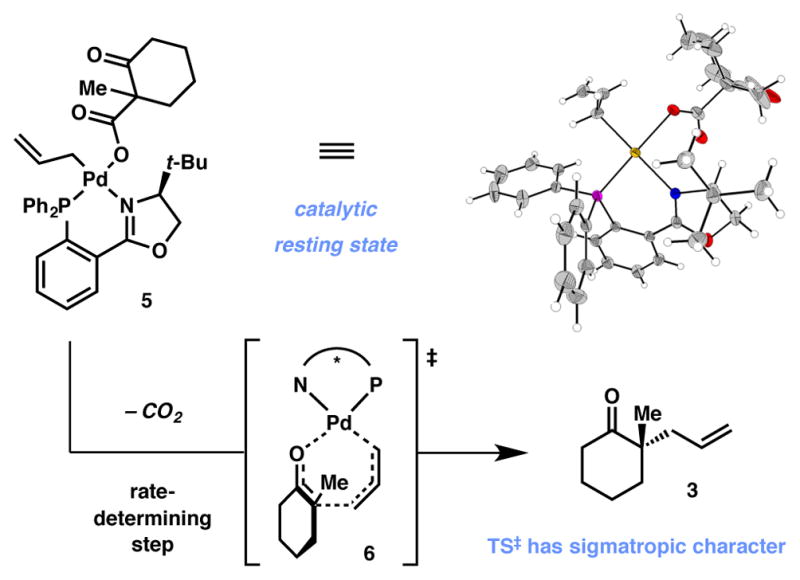
Mechanistic investigation into allylic alkylations.
Scheme 8.

Catalytic stereoablative oxindole synthesis.
Scheme 37.
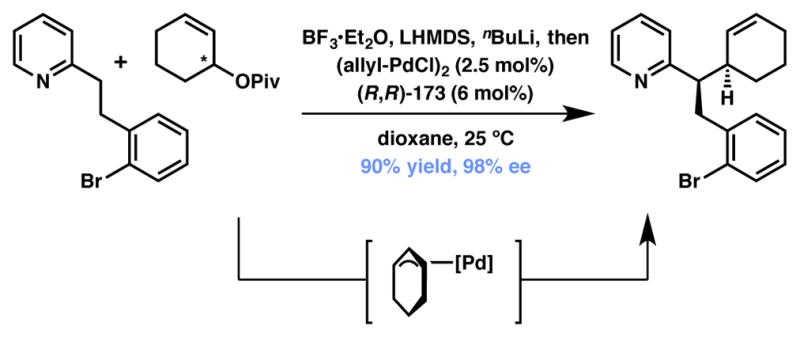
Trost’s AAA reaction (Type I DyKAT) on 2-alkylpyridine systems. changed
Acknowledgments
This work was supported in part by the NIH-NIGMS (Grant R01GM080269), Caltech, the Gordon and Betty Moore Foundation, and the Margaret E. Early Medical Research Trust. E.R.W. was supported by a Postdoctoral Fellowship, PF-16-011-01-CDD, from the American Cancer Society.
ABBREVIATIONS
- 9-BBN
9-borabicyclo[3.3.1]nonane
- AAA
asymmetric allylic alkylation
- acac
acetylacetonate
- Ar
aryl
- ATH
asymmetric transfer hydrogenation
- BArF
tetrakis[3,5-bis(trifluoromethyl)phenyl]borate
- Boc
tert-butoxycarbonyl
- BPE
1,2-di(phospholan-1-yl)ethane
- BPin
(4,4,5,5-tetramethyl-1,3,2-dioxaborolane)-2-yl
- Bz
benzoyl
- cat.
catalyst
- Cbz
carboxybenzyl
- CFL
compact fluorescent lamp
- cod
1,5-cyclooctadiene
- conv.
conversion
- CPA
chiral phosphoric acid
- CPME
cyclopentyl methyl ether
- CSA
camphor sulfonic acid
- DAC
donor-acceptor cyclopropane
- dba
dibenzylideneacetone
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCBA
dichlorobenzoic acid
- DCC
N,N′-dicyclohexylcarbodiimide
- DCM
dichloromethane
- DFT
density functional theory
- DHF
dihydrofuran
- DIBAL-H
diisobutylaluminum hydride
- DKR
dynamic kinetic resolution
- DMA
N,N-dimethylacetamide
- DMAP
4-(dimethylamino)pyridine
- DME
1,2-dimethoxyethane
- DMF
N,N-dimethylformamide
- DMMPh
3,5-dimethyl-4-methoxyphenyl
- DMPU
N,N′-dimethylpropyleneurea
- DMSO
dimethylsulfoxide
- DPEN
diphenylethylenediamine
- dppb
1,4-bis(diphenylphosphino)butane
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- dr
diastereomeric ratio
- DTBM
3,5-di-tert-butyl-4-methoxyphenyl
- DyKAT
dynamic kinetic asymmetric transformation
- ee
enantiomeric excess
- EtOAc
ethyl acetate
- Gly
glycine
- HetAr
heteroaryl
- HFIP
1,1,1,3,3,3-hexafluoro-2-propanol
- HMPA
hexamethylphosphoramide
- Men(−)
(−)-menthyl
- MOM
methoxymethyl
- MS
molecular sieves
- MTBE
methyl tert-butyl ether
- Nap
naphthyl
- nbd
norbornadiene
- NBS
N-bromosuccinimide
- NHC
N-heterocyclic carbene
- NIS
N-iodosuccinimide
- phen
1,10-phenanthroline
- Pin
pinacol; 2,3-dimethyl-2,3-butanediol
- PMB
p-methoxybenzyl
- PMP
p-methoxyphenyl
- Prg
propargyl, 1-propynyl
- SDP
7,7′-Bis(diphenylphosphino)-1,1′-spirobiindane
- TADDOL
α,α,α′,α′-tetraaryl-1,3-dioxolane-4,5-dimethanol
- TASF
tris(dimethylamino)sulfonium difluorotrimethylsilicate
- TBAB
tetrabutylammonium bromide
- TBAF
tetrabutylammonium fluoride
- TBAT
tetrabutylammonium difluorotriphenylsilicate
- TBD
1,5,7-triazabicyclo[4.4.0]dec-5-ene
- TBDPS
tert-butyldiphenylsilyl
- TBS
tert-butyldimethylsilyl
- TEA
triethylamine
- TEMPO
2,2,6,6-tetramethylpiperidine-1-oxyl
- TFE
2,2,2-trifluoroethanol
- thexyl
2,3-dimethyl-2-butyl
- THF
tetrahydrofuran
- THP
tetrahydropyran
- Tf
triflyl, trifluoromethanesulfonyl
- Tol
tolyl, 4-methylphenyl
- Ts
tosyl, p-toluenesulfonyl
Biographies
Vikram Bhat was born in Ajmer, India. He obtained his undergraduate degree at the Indian Institute of Technology, Bombay. He then attended the University of Chicago for graduate studies in natural product synthesis with Professor Viresh Rawal. His dissertation topic was the total synthesis of welwitindolinone alkaloids, which possess a dauntingly complex molecular architecture and interesting biological properties. His work was recognized through the 2011 Reaxys Ph.D. Prize. Upon completion of his doctorate in chemistry, he took a postdoctoral position at the Center for Catalysis and Chemical Synthesis at the California Institute of Technology. There, he developed a novel methodology for the asymmetric synthesis of QUINAP and related P,N ligands. He is currently a Senior Scientist at AbbVie Inc.
Eric R. Welin was born in Columbus, Ohio in 1987. He obtained his B.S. degree in Chemistry in 2010 from the Ohio State University, where he conducted undergraduate research in the laboratory of Professor James P. Stambuli. In the same year, he began his graduate studies at Princeton University under the supervision of Professor David W. C. MacMillan. At Princeton his research focused on developing new methods utilizing photoredox catalysis. He earned his Ph.D. in 2015, and later that year joined the laboratory of Professor Brian M. Stoltz as an American Cancer Society postdoctoral fellow. His current research focuses on the total synthesis of bioactive natural products.
Xuelei Guo received her B.S. in Chemistry from the University of California at Berkeley in 2009. As an undergraduate researcher she worked in the lab of Professor Ahamindra Jain, focusing on the synthesis of menthol derivatives, and in the lab of Professor Richmond Sarpong, focusing on the synthesis of the icetexane and cortistatin families of natural products. Upon graduation, Xuelei worked at the National Institutes of Health under the direction of Dr. Victor Pike on the synthesis of radioactive PET ligands. In 2011 Xuelei received her M.S. in Chemistry from the University of Chicago where she conducted research toward the total synthesis of (+)-catharanthine under the direction of Professor Viresh Rawal. Currently, she is an Associate Scientist at AbbVie Inc.
Brian M. Stoltz was born in Philadelphia, PA, in 1970 and obtained his B.S. degree from the Indiana University of Pennsylvania in Indiana, PA. After graduate work at Yale University in the laboratories of John L. Wood and an NIH postdoctoral fellowship at Harvard with E. J. Corey, he took a position at the California Institute of Technology. A member of the Caltech faculty since 2000, he is currently Professor of Chemistry. His research interests lie in the development of new methodology for general applications in synthetic chemistry.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.For selected examples and reviews, see refs. 1–6: Martin VS, Woodward SS, Katsuki T, Yamada Y, Ikeda M, Sharpless KB. Kinetic Resolution of Racemic Allylic Alcohols by Enantioselective Epoxidation. A Route to Substances of Absolute Enantiomeric Purity? J Am Chem Soc. 1981;103:6237–6240.
- 2.Kagan HB, Flaud JC. Kinetic Resolution. Top Stereochem. 1988;18:249–330. [Google Scholar]
- 3.Somfai P. Nonenzymatic Kinetic Resolution of Secondary Alcohols. Angew Chem, Int Ed. 1997;36:2731–2733. [Google Scholar]
- 4.Spivey AC, Maddaford A, Redgrave AJ. Asymmetric Catalysis of Acyl Transfer by Lewis Acids and Nucleophiles. A Review. Org Prep Proc Int. 2000;32:331–365. [Google Scholar]
- 5.Keith JM, Larrow JF, Jacobsen EN. Practical Considerations in Kinetic Resolution Reactions. Adv Synth Catal. 2001;343:5–26. [Google Scholar]
- 6.Vedejs E, Jure M. Efficiency in Nonenzymatic Kinetic Resolution. Angew Chem, Int Ed. 2005;44:3974–4001. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 7.Mohr JT, Ebner DC, Stoltz BM. Catalytic Enantioselective Stereoablative Reactions: An Unexploited Approach to Enantioselective Catalysis. Org Biomol Chem. 2007;5:3571–3576. doi: 10.1039/b711159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.While the rest of this review has been limited to transition metal-catalyzed processes: due to the relatively nascent state of this field organocatalytic methods were included in this section.
- 9.Trost BM, Van Vranken DL. Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 10.Behenna DC, Stoltz BM. The Enantioselective Tsuji Allylation. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 11.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Deracemization of Quaternary Stereocenters by Pd-Catalyzed Enantioconvergent Decarboxylative Allylation of Racemic β-Ketoesters. Angew Chem, Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 12.Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III The Inner-Sphere Process in the Enantioselective Tsuji Allylation Reaction with (S)-t-Bu-phosphinooxazoline Ligands. J Am Chem Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]
- 13.Behenna DC, Mohr JT, Sherden NH, Marinescu SC, Harned AM, Tani K, Seto M, Ma S, Novák Z, Krout MR, et al. Enantioselective Decarboxylative Alkylation Reactions: Catalyst Development, Substrate Scope, and Mechanistic Studies. Chem–Eur J. 2011;17:14199–14223. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keith JA, Behenna DC, Sherden N, Mohr JT, Ma S, Marinescu SC, Nielsen RJ, Oxgaard J, Stoltz BM, Goddard WA., III The Reaction Mechanism of the Enantioselective Tsuji Allylation: Inner-Sphere and Outer-Sphere Pathways Internal Rearrangements and Asymmetric C–C Bond Formation. J Am Chem Soc. 2012;134:19050–19060. doi: 10.1021/ja306860n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Unusual Allylpalladium Carboxylate Complexes: Identification of the Resting State of Catalytic Enantioselective Decarboxylative Allylic Alkylation Reactions of Ketones. Angew Chem, Int Ed. 2009;48:6840–6843. doi: 10.1002/anie.200902575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma S, Reeves CM, Craig RA, II, Stoltz BM. Palladium-Catalyzed Decarboxylative Allylic Alkylation of Diastereomeric β-Ketoesters. Tetrahedron. 2014;70:4208–4212. doi: 10.1016/j.tet.2014.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohr JT, Krout MR, Stoltz BM. Preparation of (S)-2-Allyl-2-Methylcyclohexanone. Org Synth. 2009;86:194–211. doi: 10.15227/orgsyn.086.0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Intramolecular Coupling of Allyl Carboxylates with Allyl Stannanes and Allyl Silanes: A New Type of Reductive Elimination Reaction? Chem–Eur J. 2002;8:3620–3628. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 19.Hiersemann M, Nubbemeyer U, editors. The Claisen Rearrangement: Methods and Applications. Wiley-VCH; Weinheim; Germany: 2007. [Google Scholar]
- 20.Reeves CM, Behenna DC, Stoltz BM. Development of (Trimethylsilyl)ethyl Ester Protected Enolates and Applications in Palladium-Catalyzed Enantioselective Allylic Alkylation: Intermolecular Cross-Coupling of Functionalized Electrophiles. Org Lett. 2014;16:2314–2317. doi: 10.1021/ol500355z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seto M, Roizen JL, Stoltz BM. Catalytic Enantioselective Alkylation of Substituted Dioxanone Enol Ethers: Ready Access to C(α)-Tetrasubstituted Hydroxy-Ketones, Acids, and Esters. Angew Chem, Int Ed. 2008;47:6873–6876. doi: 10.1002/anie.200801424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine SR, Krout MR, Stoltz BM. Catalytic Enantioselective Approach to the Eudesmane Sesquiterpenoids: Total Synthesis of (+)-Carissone. Org Lett. 2009;11:289–292. doi: 10.1021/ol802409h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrova KV, Mohr JT, Stoltz BM. Enantioselective Total Synthesis of (+)-Cassiol. Org Lett. 2009;11:293–295. doi: 10.1021/ol802410t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDougal NT, Virgil SC, Stoltz; BM. High-Throughput Screening of the Asymmetric Decarboxylative Alkylation Reaction of Enolate-Stabilized Enol Carbonates. Synlett. 2010:1712–1716. doi: 10.1055/s-0030-1258094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong AY, Krout MR, Jensen T, Bennett NB, Harned AM, Stoltz BM. Ring-Contraction Strategy for the Practical, Scalable, Catalytic-Asymmetric Synthesis of Versatile γ-Quaternary Acylcyclopentenes. Angew Chem, Int Ed. 2011;50:2756–2760. doi: 10.1002/anie.201007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett NB, Hong AY, Harned AM, Stoltz BM. Synthesis of γ-Quaternary Cycloheptanones using a Combined Allylic Alkylation/Stork-Danheiser Approach: Preparation of Mono-, Bi-, and Tricyclic Systems. Org Biomol Chem. 2012;10:56–59. doi: 10.1039/c1ob06189e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett NB, Duquette DC, Kim J, Liu WB, Marziale AN, Behenna DC, Virgil SC, Stoltz BM. Expanding Insight into Asymmetric Palladium-Catalyzed Allylic Alkylation of N-Heterocyclic Molecules and Cyclic Ketones. Chem–Eur J. 2013;19:4414–4418. doi: 10.1002/chem.201300030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reeves CM, Eidamshaus C, Kim J, Stoltz BM. Enantioselective Construction of α-Quaternary Cyclo-butanones by Catalytic Asymmetric Allylic Alkylation. Angew Chem, Int Ed. 2013;52:6718–6721. doi: 10.1002/anie.201301815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korch KM, Eidamshaus C, Behenna DC, Nam S, Horne D, Stoltz BM. Enantioselective Synthesis of α-Secondary and α-Tertiary Piperazin-2-ones and Piperazines by Catalytic Asymmetric Allylic Alkylation. Angew Chem, Int Ed. 2015;54:179–183. doi: 10.1002/anie.201408609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Numajiri Y, Pritchett BP, Chiyoda K, Stoltz BM. Enantioselective Synthesis of α-Quaternary Mannich Adducts by Palladium-Catalyzed Allylic Alkylation: Total Synthesis of (+)-Sibirinine. J Am Chem Soc. 2015;137:1040–1043. doi: 10.1021/ja512124c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Numajiri Y, Jiménez-Osés G, Wang B, Houk KN, Stoltz BM. Enantioselective Synthesis of Dialkylated N-Heterocycles by Palladium-Catalyzed Allylic Alkylation. Org Lett. 2015;17:1082–1085. doi: 10.1021/ol503425t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craig RA, II, Loskot SA, Mohr JT, Behenna DC, Harned AM, Stoltz BM. Palladium-Catalyzed Enantioselective Decarboxylative Allylic Alkylation of Cyclopentatones. Org Lett. 2015;17:5160–5163. doi: 10.1021/acs.orglett.5b02376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enquist JA, Jr, Stoltz BM. The Total Synthesis of (–)-Cyanthiwigin F by Means of Double Catalytic Enantioselective Alkylation. Nature. 2008;453:1228–1231. doi: 10.1038/nature07046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enquist JA, Jr, Virgil SC, Stoltz BM. Total Syntheses of Cyanthiwigins B, F, and G. Chem–Eur J. 2011;17:9957–9969. doi: 10.1002/chem.201100425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts BP. Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem Soc Rev. 1999;28:25–35. [Google Scholar]
- 36.Dang H-S, Roberts BP. Radical-Chain Addition of Aldehydes to Alkenes Catalysed by Thiols. J Chem Soc, Perkin Trans 1. 1998:67–75. [Google Scholar]
- 37.White DE, Stewart IC, Seashore-Ludlow BA, Grubbs RJ, Stoltz BM. A General Enantioselective Route to the Chamigrene Natural Product Family. Tetrahedron. 2010;66:4668–4686. doi: 10.1016/j.tet.2010.04.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gartshore CJ, Lupton DW. Enantioselective Palladium-Catalyzed Decarboxylative Allylation of Carbazolones and Indolones: Formal Synthesis of (+)-Kopsihainanine A. Angew Chem, Int Ed. 2013;52:4113–4116. doi: 10.1002/anie.201209069. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Zhang S, Wu S, Shen X, Zou L, Wang F, Li X, Peng F, Zhang H, Shao Z. Enantioselective Palladium-Catalyzed Decarboxylative Allylation of Carbazolones: Total Synthesis of (–)-Aspidospermidine and (+)-Kopsihainanine A. Angew Chem, Int Ed. 2013;52:4117–4121. doi: 10.1002/anie.201209878. [DOI] [PubMed] [Google Scholar]
- 40.Xu Z, Bao X, Wang Q, Zhu J. An Enantioselective Total Synthesis of (–)-Isoschizogamine. Angew Chem, Int Ed. 2015;54:14937–14940. doi: 10.1002/anie.201508150. [DOI] [PubMed] [Google Scholar]
- 41.Mohr JT, Nishimata T, Behenna DC, Stoltz BM. Catalytic Enantioselective Decarboxylative Protonation. J Am Chem Soc. 2006;128:11348–11349. doi: 10.1021/ja063335a. [DOI] [PubMed] [Google Scholar]
- 42.Marinescu SC, Nishimata T, Mohr JT, Stoltz BM. Homogenous Pd-Catalyzed Enantioselective Decarboxylative Protonation. Org Lett. 2008;10:1039–1042. doi: 10.1021/ol702821j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischer C, Fu GC. Asymmetric Nickel-Catalyzed Negishi Cross-Couplings of Secondary α-Bromo Amides with Organozinc Reagents. J Am Chem Soc. 2005;127:4594–4595. doi: 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]
- 44.Arp FO, Fu GC. Catalytic Enantioselective Negishi Reactions of Racemic Secondary Benzylic Halides. J Am Chem Soc. 2005;127:10482–10483. doi: 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]
- 45.Son S, Fu GC. Nickel-Catalyzed Asymmetric Negishi Cross-Couplings of Secondary Allylic Chlorides with Alkylzincs. J Am Chem Soc. 2008;130:2756–2757. doi: 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]
- 46.Smith SW, Fu GC. Nickel-Catalyzed Asymmetric Cross-Couplings of Racemic Propargylic Halides with Arylzinc Reagents. J Am Chem Soc. 2008;130:12645–12647. doi: 10.1021/ja805165y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lundin PM, Esquivias J, Fu GC. Catalytic Asymmetric Cross-Couplings of Racemic α-Bromoketones with Arylzinc Reagents. Angew Chem, Int Ed. 2009;48:154–156. doi: 10.1002/anie.200804888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oelke AJ, Sun J, Fu GC. Nickel-Catalyzed Enantioselective Cross-Couplings of Racemic Secondary Electrophiles that Bear an Oxygen Leaving Group. J Am Chem Soc. 2012;134:2966–2969. doi: 10.1021/ja300031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi J, Fu GC. Catalytic Asymmetric Synthesis of Secondary Nitriles via Stereoconvergent Negishi Arylations and Alkenylations of Racemic α-Bromonitriles. J Am Chem Soc. 2012;134:9102–9105. doi: 10.1021/ja303442q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Binder JT, Cordier CJ, Fu GC. Catalytic Enantioselective Cross-Couplings of Secondary Alkyl Electrophiles with Secondary Alkylmetal Nucleophiles: Negishi Reactions of Racemic Benzylic Bromides with Achiral Alkylzinc Reagents. J Am Chem Soc. 2012;134:17003–17006. doi: 10.1021/ja308460z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Do HQ, Chandrashekar ERR, Fu GC. Nickel/Bis(oxazoline)-Catalyzed Asymmetric Negishi Arylations of Racemic Secondary Benzylic Electrophiles to Generate Enantioenriched 1,1-Diarylalkanes. J Am Chem Soc. 2013;135:16288–16291. doi: 10.1021/ja408561b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang Y, Fu GC. Catalytic-Asymmetric Synthesis of Tertiary Alkyl Fluorides: Negishi Cross-Coupling of Racemic α,α-Dihaloketones. J Am Chem Soc. 2014;136:5520–5524. doi: 10.1021/ja501815p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi J, Martín-Gago P, Fu GC. Stereoconvergent Arylations and Alkenylations of Unactivated Alkyl Electrophiles: Catalytic Enantioselective Synthesis of Secondary Sulfonamides and Sulfones. J Am Chem Soc. 2014;136:12161–12165. doi: 10.1021/ja506885s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang Y, Fu GC. Stereoconvergent Negishi Arylations of Racemic Secondary Alkyl Electrophiles: Differentiating between a CF3 and an Alkyl Group. J Am Chem Soc. 2015;137:9523–9526. doi: 10.1021/jacs.5b04725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saito B, Fu GC. Enantioselective Alkyl-Alkyl Suzuki Cross-Couplings of Unactivated Homobenzylic Halides. J Am Chem Soc. 2008;130:6694–6695. doi: 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- 56.Lundin PM, Fu GC. Asymmetric Suzuki Cross-Couplings of Activated Secondary Alkyl Electrophiles: Arylations of Racemic α-Chloroamides. J Am Chem Soc. 2010;132:11027–11029. doi: 10.1021/ja105148g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Owston NA, Fu GC. Asymmetric Alkyl-Alkyl Cross-Couplings of Unactivated Secondary Alkyl Electrophiles: Stere-oconvergent Suzuki Reactions of Racemic Acylated Halohydrins. J Am Chem Soc. 2010;132:11908–11909. doi: 10.1021/ja105924f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu Z, Wilsily A, Fu GC. Stereoconvergent Amine-Directed Alkyl-Alkyl Suzuki Reactions of Unactivated Secondary Alkyl Chlorides. J Am Chem Soc. 2011;133:8154–8157. doi: 10.1021/ja203560q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zultanski SL, Fu GC. Catalytic Asymmetric γ-Alkylation of Carbonyl Compounds via Stereoconvergent Suzuki Cross-Couplings. J Am Chem Soc. 2011;133:15362–15364. doi: 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilsily A, Tramutola F, Owston NA, Fu GC. New Directing Groups for Metal-Catalyzed Asymmetric Carbon–Carbon Bond-Forming Processes: Stereoconvergent Alkyl–Alkyl Suzuki Cross-Couplings of Unactivated Electrophiles. J Am Chem Soc. 2012;134:5794–5797. doi: 10.1021/ja301612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai X, Strotman NA, Fu GC. Catalytic Asymmetric Hiyama Cross-Couplings of Racemic α-Bromo Esters. J Am Chem Soc. 2008;130:3302–3303. doi: 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- 62.Lou S, Fu GC. Nickel/Bis(oxazoline)-Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic α-Bromoketones. J Am Chem Soc. 2010;132:1264–1266. doi: 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lou S, Fu GC. Enantioselective Alkenylation via Nickel-Catalyzed Cross-Coupling with Organozirconium Reagents. J Am Chem Soc. 2010;132:5010–5011. doi: 10.1021/ja1017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.In ref. 70 the authors find that the nickel(I) species that performs the halide abstraction need not be the nickel(II) species with which the transient radical combines. The cycle shown here involves a single nickel species for simplicity.
- 65.Koga N, Obara S, Kitaura K, Morokuma K. Role of Agostic Interaction in β–Elimination of Pd and Ni Complexes. An ab Initio MO Study. J Am Chem Soc. 1985;107:7109–7116. [Google Scholar]
- 66.Strömberg S, Zetterberg K, Siegbahn PEM. Trends within a Triad: Comparison between σ-Alkyl Complexes of Nickel, Palladium and Platinum with Respect to Association of Ethylene, Migratory Insertion and β-Hydride Elimination. A Theoretical Study. J Chem Soc, Dalton Trans. 1997:4147–4152. [Google Scholar]
- 67.Pudasaini B, Janesko BG. Computational Investigation of Selectivity in Suzuki-Miyaura Coupling of Secondary Alkyl Boranes. Organometallics. 2011;30:4564–4571. [Google Scholar]
- 68.Podasaini B, Janesko BG. Computational Mechanistic Study of Stereoselective Suzuki Coupling of an α-Cyano-Activated Secondary Alkyl. Organometallics. 2012;31:4610–4618. [Google Scholar]
- 69.Pudasaini B, Janesko BG. Agostic Interactions in Nickel(II) Complexes: Trans Influence of Ancillary Ligands on the Strength of the Bond. Organometallics. 2014;33:84–93. [Google Scholar]
- 70.Schley ND, Fu GC. Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J Am Chem Soc. 2014;136:16588–16593. doi: 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Everson DA, Weix DJ. Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J Org Chem. 2014;79:4793–4798. doi: 10.1021/jo500507s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weix DJ. Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc Chem Res. 2015;48:1767–1775. doi: 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ackerman LKG, Lovell MM, Weix DJ. Multimetallic Catalysed Cross-Coupling of Aryl Bromides with Aryl Triflates. Nature. 2015;524:454–457. doi: 10.1038/nature14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang P, Le CC, MacMillan DWC. Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J Am Chem Soc. 2016;138:8084–8087. doi: 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Biswas S, Weix DJ. Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J Am Chem Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Breitenfeld J, Wodrich MD, Hu X. Bimetallic Oxidative Addition in Nickel-Catalyzed Alkyl–Aryl Kumada Coupling Reactions. Organometallics. 2014;33:5708–5715. doi: 10.1021/ja4051923. [DOI] [PubMed] [Google Scholar]
- 77.Cherney AH, Kadunce NT, Reisman SE. Catalytic Asymmetric Reductive Acyl Cross-Coupling: Synthesis of Enantioenriched Acyclic α,α-Disubstituted Ketones. J Am Chem Soc. 2013;135:7442–7445. doi: 10.1021/ja402922w. [DOI] [PubMed] [Google Scholar]
- 78.Cherney AH, Reisman SE. Nickel-Catalyzed Asymmetric Reductive Cross-Coupling Between Vinyl and Benzyl Electrophiles. J Am Chem Soc. 2014;136:14365–14368. doi: 10.1021/ja508067c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kadunce NT, Reisman SE. Nickel-Catalyzed Asymmetric Reductive Cross-Coupling between Heteroaryl Iodides and α-Chloronitriles. J Am Chem Soc. 2015;137:10480–10483. doi: 10.1021/jacs.5b06466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma S, Han X, Krishnan S, Virgil SC, Stoltz BM. Catalytic Enantioselective Stereoablative Alkylation of 3-Halooxindoles: Facile Access to Oxindoles with C3 All-Carbon Quaternary Stereocenters. Angew Chem, Int Ed. 2009;48:8037–8041. doi: 10.1002/anie.200902943. [DOI] [PubMed] [Google Scholar]
- 81.Lee CW, Han SJ, Virgil SC, Stoltz BM. Stereochemical Evaluation of Bis(phosphine) Copper Catalysts for the Asymmetric Alkylation of 3-Bromooxindoles with α-Arylated Malonate Esters. Tetrahedron. 2015;71:3666–3670. doi: 10.1016/j.tet.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han SJ, Vogt F, Krishnan S, May JA, Gatti M, Virgil SC, Stoltz BM. A Diastereodivergent Synthetic Strategy for the Syntheses of Communesin F and Perophoramidine. Org Lett. 2014;16:3316–3319. doi: 10.1021/ol5013263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han SJ, Vogt F, May JA, Krishnan S, Gatti M, Virgil SC, Stoltz BM. Evolution of a Unified, Stereodivergent Approach to the Synthesis of Communesin F and Perophoramidine. J Org Chem. 2015;80:528–547. doi: 10.1021/jo502534g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doyle AG, Jacobsen EN. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 85.Liao YH, Wu ZJ, Han WY, Zhang XM, Yuan WC. Organocatalytic Enantioselective Stereoablative Hydroxylation of 3-Halooxindoles: An Effective Method for the Construction of Enantioenriched 3-Substituted 3-Hydroxy-2-Oxindoles. Chem–Eur J. 2012;18:8916–8920. doi: 10.1002/chem.201201395. [DOI] [PubMed] [Google Scholar]
- 86.Zuo J, Liao YH, Zhang XM, Yuan WC. Organocatalyzed Enantioselective Decarboxylative Stereoablation Reaction for the Construction of 3,3′-Disubstituted Oxindoles Using β-Ketoacids and 3-Halooxindoles. J Org Chem. 2012;77:11325–11332. doi: 10.1021/jo302048v. [DOI] [PubMed] [Google Scholar]
- 87.Xie X, Jing L, Qin D, He W, Wu S, Jin L, Luo G. Regioselective Asymmetric Stereoablative O-Alkylation of α-Nitrophosphonates via O-Azaxylylene Intermediates Generated in situ from 3-Bromooxindoles. RSC Adv. 2014;4:11605–11609. [Google Scholar]
- 88.Terada M, Toda Y. Double Bond Isomerization/Enantioselective Aza-Petasis–Ferrier Rearrangement Sequence as an Efficient Entry to Anti- and Enantioenriched β-Amino Aldehydes. J Am Chem Soc. 2009;131:6354–6355. doi: 10.1021/ja902202g. [DOI] [PubMed] [Google Scholar]
- 89.Terada M, Komuro T, Toda Y, Korenaga T. Mechanistic Studies of Highly Enantio- and Diastereoselective Aza-Petasis–Ferrier Rearrangement Catalyzed by Chiral Phosphoric Acid. J Am Chem Soc. 2014;136:7044–7057. doi: 10.1021/ja5017206. [DOI] [PubMed] [Google Scholar]
- 90.Terada M, Yamanaka T, Toda Y. Chiral Anion Catalysis in the Enantioselective 1,4-Reduction of the 1-Benzopyrylium Ion as a Reactive Intermediate. Chem–Eur J. 2013;19:13658–13662. doi: 10.1002/chem.201302486. [DOI] [PubMed] [Google Scholar]
- 91.Yang L, Zheng H, Luo L, Nan J, Liu J, Wang Y, Luan X. Palladium-Catalyzed Dynamic Kinetic Asymmetric Transformation of Racemic Biaryls: Axial-to-Central Chirality Transfer. J Am Chem Soc. 2015;137:4876–4879. doi: 10.1021/jacs.5b01285. [DOI] [PubMed] [Google Scholar]
- 92.Madin A, Overman LE. Controlling Stereoselection in Intramolecular Heck Reactions by Tailoring the Palladium Catalyst. Tetrahedron Lett. 1992;33:4859–4862. [Google Scholar]
- 93.Ashimori A, Overman LE Catalytic Asymmetric Synthesis of Quarternary Carbon Centers. Palladium-Catalyzed Formation of Either Enantiomer of Spirooxindoles and Related Spirocyclics using a Single Enantiomer of a Chiral Diphosphine Ligand. J Org Chem. 1992;57:4571–4572. [Google Scholar]
- 94.Ashimori A, Matsuura T, Overman LE, Poon DJ Catalytic Asymmetric Synthesis of Either Enantiomer of Physostigmine. Formation of Quaternary Carbon Centers with High Enantioselection by Intramolecular Heck Reactions of (Z)-2-Butenanilides. J Org Chem. 1993;58:6949–6951. [Google Scholar]
- 95.Ashimori A, Bachand B, Calter MA, Govek SP, Overman LE, Poon DJ Catalytic Asymmetric Synthesis of Quaternary Carbon Centers. Exploratory Studies of Intramolecular Heck Reactions of (Z)-α, β-Unsaturated Anilides and Mechanistic Investigations of Asymmetric Heck Reactions Proceeding via Neutral Intermediates. J Am Chem Soc. 1998;120:6488–6499. [Google Scholar]
- 96.McDermott MC, Stephenson GR, Hughes DL, Walkington AJ. Intramolecular Asymmetric Heck Reactions: Evidence for Dynamic Kinetic Resolution Effects. Org Lett. 2006;8:2917–2920. doi: 10.1021/ol0606132. [DOI] [PubMed] [Google Scholar]
- 97.Hosoi S, Ozeki M, Nakano M, Arimitsu K, Kajimoto T, Kojima N, Iwasaki H, Miura T, Kimura H, Node M, Yamashita M. Mechanistic Aspects of Asymmetric Intramolecular Heck Reaction Involving Dynamic Kinetic Resolution: Flexible Conformation of the Cyclohexenylidene–Benzene System. Tetrahedron. 2015;71:2317–2326. [Google Scholar]
- 98.Doyle AG, Jacobsen EN. Enantioselective Alkylation of Acyclic α,α-Disubstituted Tributyltin Enolates Catalyzed by a {Cr(salen)} Complex. Angew Chem, Int Ed. 2007;46:3701–3705. doi: 10.1002/anie.200604901. [DOI] [PubMed] [Google Scholar]
- 99.Kobayashi K, Kawanisi M, Hitomi T, Kozima S. Bonding Isomers of Triorganostannyl Enolates Analyzed by 119Sn NMR Spectroscopy. Chem Lett. 1984;13:497–500. [Google Scholar]
- 100.For a recent review, see: Quasdorf KW, Overman LE. Catalytic Enantioselective Synthesis of Quaternary Carbon Stereocentres. Nature. 2014;516:181–191. doi: 10.1038/nature14007.
- 101.Stoltz BM, Bennett NB, Duquette DC, Goldberg AFG, Liu Y, Loewinger MB, Reeves CM. In: Comprehensive Organic Synthesis II. 2. Knochel P, editor. Elsevier; Amsterdam: 2014. pp. 1–55. [Google Scholar]
- 102.Chen X, Engle KM, Wang DH, Yu JQ. Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew Chem, Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davies HML, Morton D. Recent Advances in C–H Functionalization. J Org Chem. 2016;81:343–350. doi: 10.1021/acs.joc.5b02818. [DOI] [PubMed] [Google Scholar]
- 104.Gensch T, Hopkinson MN, Glorius F, Wencel-Delord J. Mild Metal-Catalyzed C–H Activation: Examples and Concepts. Chem Soc Rev. 2016;45:2900–2936. doi: 10.1039/c6cs00075d. [DOI] [PubMed] [Google Scholar]
- 105.Zheng J, You S-L. Construction of Axial Chirality by Rhodium-Catalyzed Asymmetric Dehydrogenative Heck Coupling of Biaryl Compounds with Alkenes. Angew Chem Int Ed. 2014;53:13244–13247. doi: 10.1002/anie.201408805. [DOI] [PubMed] [Google Scholar]
- 106.Rong ZQ, Zhang Y, Chua RHB, Pan HJ, Zhao Y. Dynamic Kinetic Asymmetric Amination of Alcohols: From A Mixture of Four Isomers to Diastereo- and Enantiopure α-Branched Amines. J Am Chem Soc. 2015;137:4944–4947. doi: 10.1021/jacs.5b02212. [DOI] [PubMed] [Google Scholar]
- 107.Lee SY, Murphy JM, Ukai A, Fu GC. Nonenzymatic Dynamic Kinetic Resolution of Secondary Alcohols via Enantioselective Acylation: Synthetic and Mechanistic Studies. J Am Chem Soc. 2012;134:15149–15153. doi: 10.1021/ja307425g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ruble JC, Latham HA, Fu GC. Effective Kinetic Resolution of Secondary Alcohols with a Planar–Chiral Analogue of 4-(Dimethylamino)pyridine. Use of the Fe(C5Ph5) Group in Asymmetric Catalysis. J Am Chem Soc. 1997;119:1492–1493. [Google Scholar]
- 109.For a review on racemization catalysts see: Ahn Y, Ko S-B, Kim M-J, Park J. Racemization Catalysts for the Dynamic Kinetic Resolution of Alcohols and Amines. Coord Chem Rev. 2008;252:647–658.
- 110.Kitamura M, Ohkuma T, Tokunaga M, Noyori R. Dynamic Kinetic Resolution in BINAP–Ruthenium(II) Catalyzed Hydrogenation of 2-Substituted 3-Oxo Carboxylic Esters. Tetrahedron: Asymmetry. 1990;1:1–4. [Google Scholar]
- 111.Zhou ZT, Xie JH, Zhou QL. Enantioselective Synthesis of Chiral β-Aryloxy Alcohols by Asymmetric Hydrogenation of a-Aryloxy Aldehydes via Dynamic Kinetic Resolution. Adv Synth Catal. 2009;351:363–366. [Google Scholar]
- 112.Xie JH, Zhou ZT, Kong WL, Zhou QL. Ru-Catalyzed Asymmetric Hydrogenation of Racemic Aldehydes via Dynamic Kinetic Resolution: Efficient Synthesis of Optically Active Primary Alcohols. J Am Chem Soc. 2007;129:1868–1869. doi: 10.1021/ja0680109. [DOI] [PubMed] [Google Scholar]
- 113.Li X, List B. Catalytic Asymmetric Hydrogenation of Aldehydes. Chem Commun. 2007:1739–1741. doi: 10.1039/b703977h. [DOI] [PubMed] [Google Scholar]
- 114.Hoffmann S, Nicoletti M, List B. Catalytic Asymmetric Reductive Amination of Aldehydes via Dynamic Kinetic Resolution. J Am Chem Soc. 2006;128:13074–13075. doi: 10.1021/ja065404r. [DOI] [PubMed] [Google Scholar]
- 115.For more examples on asymmetric hydrogenation via DKR of ketone substrates, see refs. 116–117.
- 116.Arai N, Ooka H, Azuma K, Yabuuchi T, Kurono N, Inoue T, Ohkuma T. General Asymmetric Hydrogenation of α-Branched Aromatic Ketones Catalyzed by TolBINAP/DMAPEN–Ruthenium (II) Complex. Org Lett. 2007;9:939–941. doi: 10.1021/ol070125+. [DOI] [PubMed] [Google Scholar]
- 117.Ooka H, Arai N, Azuma K, Kurono N, Ohkuma T. Asymmetric Hydrogenation of Aromatic Ketones Catalyzed by the TolBINAP/DMAPEN–Ruthenium(II) Complex: A Significant Effect of N-Substituents of Chiral 1,2-Diamine Ligands on Enantioselectivity. J Org Chem. 2008;73:9084–9093. doi: 10.1021/jo801863q. [DOI] [PubMed] [Google Scholar]
- 118.Xie JH, Liu S, Kong WL, Bai WJ, Wang XC, Wang LX, Zhou QL. Highly Enantioselective and Diastereoselective Synthesis of Chiral Amino Alcohols by Ruthenium-Catalyzed Asymmetric Hydrogenation of α-Amino Aliphatic Ketones. J Am Chem Soc. 2009;131:4222–4223. doi: 10.1021/ja901058u. [DOI] [PubMed] [Google Scholar]
- 119.Bai WJ, Xie JH, Li YL, Liu S, Zhou QL. Enantioselective Synthesis of Chiral β-Aryloxy Alcohols by Ruthenium-Catalyzed Ketone Hydrogenation via Dynamic Kinetic Resolution (DKR) Adv Synth Catal. 2010;352:81–84. [Google Scholar]
- 120.Hibino T, Makino K, Sugiyama T, Hamada Y. Homogeneous Chiral Nickel-Catalyzed Asymmetric Hydrogenation of Substituted Aromatic α-Aminoketone Hydrochlorides through Dynamic Kinetic Resolution. ChemCatChem. 2009;1:237–240. [Google Scholar]
- 121.Tao X, Li W, Li X, Xie X, Zhang Z. Diastereo- and Enantioselective Asymmetric Hydrogenation of α-Amido-β-keto Phosphonates via Dynamic Kinetic Resolution. Org Lett. 2013;15:72–75. doi: 10.1021/ol303105d. [DOI] [PubMed] [Google Scholar]
- 122.Prévost S, Gauthier S, Caño de Andrade MC, Mordant C, Touati AR, Lesot P, Savignac P, Ayad T, Phansavath P, Ratovelomanana-Vidal V, et al. Dynamic Kinetic Resolution of α-Chloro β-Keto Esters and Phosphonates: Hemisynthesis of Taxotere® through Ru-DIFLUORPHOS Asymmetric Hydrogenation. Tetrahedron: Asymmetry. 2010;21:1436–1446. [Google Scholar]
- 123.For selected examples: see ref. 124–125.
- 124.Szori K, Szollosi G, Bartok M. Dynamic Kinetic Resolution over Cinchona-Modified Platinum Catalyst: Hydrogenation of Racemic Ethyl 2-Fluoroacetoacetate. Adv Synth Catal. 2006;348:515–522. [Google Scholar]
- 125.Li X, Tao X, Ma X, Li W, Zhao M, Xie X, Ayad T, Ratovelomanana-Vidal V, Zhang Z. Dynamic Kinetic Resolution of β′-Keto-β-Amino Esters using Ru–DTBM–Sunphos Catalyzed Asymmetric Hydrogenation. Tetrahedron. 2013;69:7152–7156. [Google Scholar]
- 126.Makino K, Iwasaki M, Hamada Y. Dynamic Kinetic Resolution by a Cationic Iridium Complex in the Synthesis of β-Hydroxy-α-Amino Acid Esters. Org Lett. 2006;8:4573–4576. doi: 10.1021/ol061796v. [DOI] [PubMed] [Google Scholar]
- 127.Makino K, Fujii T, Hamada Y. Rhodium-Catalyzed Asymmetric Hydrogenation through Dynamic Kinetic Resolution: Asymmetric Synthesis of anti-β-Hydroxy-α-Amino Acid Esters. Tetrahedron: Asymmetry. 2006;17:481–485. [Google Scholar]
- 128.Makino K, Goto T, Hiroki Y, Hamada Y. Direct anti-Selective Asymmetric Hydrogenation of α-Amino-β-Keto Esters through Dynamic Kinetic Resolution using Ru-Axially Chiral Phosphine Catalysts–Stereoselective Synthesis of anti-β-Hydroxy-α-Amino Acids. Tetrahedron: Asymmetry. 2008;19:2816–2828. [Google Scholar]
- 129.Hamada Y, Koseki Y, Fujii T, Maeda T, Hibino T, Makino; K. Cataytic Asymmetric Hydrogenation of α-Amino-β-Keto Ester Hydrochlorides using Homogeneous Chiral Nickel-Bisphosphine Complexes through DKR. Chem Commun. 2008:6206–6208. doi: 10.1039/b816524f. [DOI] [PubMed] [Google Scholar]
- 130.Echeverria PG, Férard C, Cornil J, Guérinot A, Cossy J, Phannarath P, Ratovelomanana-Vidal V. Iridium–SYNPHOS-Catalyzed Hydrogenation through Dynamic Kinetic Resolution of α-Amino β-Keto Ester Hydrochlorides. Synlett. 2014;25:2761–2764. [Google Scholar]
- 131.Makino K, Hiroki Y, Hamada Y. Dynamic Kinetic Resolution Catalyzed by Ir Axially Chiral Phosphine Catalyst: Asymmetric Synthesis of anti Aromatic β-Hydroxy-α-Amino Acid Esters. J Am Chem Soc. 2005;127:5784–5785. doi: 10.1021/ja0432113. [DOI] [PubMed] [Google Scholar]
- 132.Wang DS, Chen QA, Li W, Yu CB, Zhou YG, Zhang X. Pd-Catalyzed Asymmetric Hydrogenation of Unprotected Indoles Activated by Brønsted Acids. J Am Chem Soc. 2010;132:8909–8911. doi: 10.1021/ja103668q. [DOI] [PubMed] [Google Scholar]
- 133.Wang DS, Tang J, Zhou YG, Chen MW, Yu CB, Duan Y, Jiang GF. Dehydration Triggered Asymmetric Hydrogenation of 3-(α-Hydroxyalkyl)Indoles. Chem Sci. 2011;2:803–806. [Google Scholar]
- 134.Duan Y, Chen MW, Chen QA, Yu CB, Zhou YG. Pd-Catalyzed Asymmetric Hydrogenation of 3-(Toluenesulfonamidoalkyl)Indoles. Org Biomol Chem. 2012;10:1235–1238. doi: 10.1039/c1ob06777j. [DOI] [PubMed] [Google Scholar]
- 135.Ros A, Magriz A, Dietrich H, Ford M, Fernández R, Lassaletta JM. Transfer Hydrogenation of α-Branched Ketimines: Enantioselective Synthesis of Cycloalkylamines via Dynamic Kinetic Resolution. Adv Synth Catal. 2005;347:1917–1920. [Google Scholar]
- 136.Ros A, Magriz A, Dietrich H, Fernández R, Alvarez E, Lassaletta JM. Enantioselective Synthesis of Vicinal Halohydrins via Dynamic Kinetic Resolution. Org Lett. 2006;8:127–130. doi: 10.1021/ol052821k. [DOI] [PubMed] [Google Scholar]
- 137.Fernández R, Ros A, Magriz A, Dietrich H, Lassaletta JM. Enantioselective Synthesis of cis-α-Substituted Cycloalkanols and trans-Cycloalkyl Amines thereof. Tetrahedron. 2007;63:6755–6763. [Google Scholar]
- 138.Yun W, Zhicong G, Jinjin B, Yawen Z. Synthesis of Chiral 2-Aroyl-1-Tetralols: Asymmetric Transfer Hydrogenation of 2-Aroyl-1-Tetralones via Dynamic Kinetic Resolution. Chin J Chem. 2011;29:1467–1472. [Google Scholar]
- 139.Son SM, Lee HK. Dynamic Kinetic Resolution Based Asymmetric Transfer Hydrogenation of α-Alkoxy-β-Ketophosphonates. Diastereo- and Enantioselective Synthesis of Monoprotected 1,2-Dihydroxyphosphonates. J Org Chem. 2014;79:2666–2681. doi: 10.1021/jo500148j. [DOI] [PubMed] [Google Scholar]
- 140.Villacrez M, Somfai P. Enantioselective Synthesis of anti-β-Amido-α-Hydroxy Esters via Asymmetric Transfer Hydrogenation Coupled with Dynamic Kinetic Resolution. Tetrahedron Lett. 2013;54:5266–5268. [Google Scholar]
- 141.Steward KM, Corbett MT, Goodman CG, Johnson JS. Asymmetric Synthesis of Diverse Glycolic Acid Scaffolds via Dynamic Kinetic Resolution of α-Keto Esters. J Am Chem Soc. 2012;134:20197–20206. doi: 10.1021/ja3102709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goodman CG, Do DT, Johnson JS. Asymmetric Synthesis of anti-β-Amino-α-Hydroxy Esters via Dynamic Kinetic Resolution of β-Amino-α-Keto Esters. Org Lett. 2013;15:2446–2449. doi: 10.1021/ol4009206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ding Z, Yang J, Wang T, Shen Z, Zhang; Y. Dynamic Kinetic Resolution of β-Keto Sulfones via Asymmetric Transfer Hydrogenation. Chem Commun. 2009:571–573. doi: 10.1039/b818257d. [DOI] [PubMed] [Google Scholar]
- 144.Wu G, Zhu J, Ding Z, Shen Z, Zhang Y. Dynamic Kinetic Resolution of Racemic α-Sulfonylaldehydes via Asymmetric Transfer Hydrogenation. Tetrahedron Lett. 2009;50:427–429. [Google Scholar]
- 145.Corbett MT, Johnson JS. Diametric Stereocontrol in Dynamic Catalytic Reduction of Racemic Acyl Phosphonates: Divergence from α-Keto Ester Congeners. J Am Chem Soc. 2013;135:594–597. doi: 10.1021/ja310980q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steward KM, Gentry EC, Johnson JS. Dynamic Kinetic Resolution of α-Keto Esters via Asymmetric Transfer Hydrogenation. J Am Chem Soc. 2012;134:7329–7332. doi: 10.1021/ja3027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cheng T, Ye Q, Zhao Q, Liu G. Dynamic Kinetic Resolution of Phthalides via Asymmetric Transfer Hydrogenation: A Strategy Constructs 1,3-Diastereocentered 3-(2-Hydroxy-2-Arylethyl-Isobenzofuran-1(3H)-one. Org Lett. 2015;17:4972–4975. doi: 10.1021/acs.orglett.5b02394. [DOI] [PubMed] [Google Scholar]
- 148.Cartigny D, Puntener K, Ayad T, Scalone M, Ratovelomanana-Vidal V. Highly Diastereo- and Enantioselective Synthesis of Monodifferentiated syn-1,2-Diol Derivatives through Asymmetric Transfer Hydrogenation via Dynamic Kinetic Resolution. Org Lett. 2010;12:3788–3791. doi: 10.1021/ol101451s. [DOI] [PubMed] [Google Scholar]
- 149.Monnereau L, Cartigny D, Scalone M, Ayad T, Ratovelomanana-Vidal V. Efficient Synthesis of Differentiated syn-1,2-Diol Derivatives by Asymmetric Transfer Hydrogenation–Dynamic Kinetic Resolution of α-Alkoxy-Substituted β-Ketoesters. Chem–Eur J. 2015;21:11799–11806. doi: 10.1002/chem.201501884. [DOI] [PubMed] [Google Scholar]
- 150.Seashore-Ludlow B, Saint-Dizier F, Somfai P. Asymmetric Transfer Hydrogenation Coupled with Dynamic Kinetic Resolution in Water: Synthesis of anti-β-Hydroxy-α-Amino Acid Derivatives. Org Lett. 2012;14:6334–6337. doi: 10.1021/ol303115v. [DOI] [PubMed] [Google Scholar]
- 151.Limanto J, Krska SW, Dorner BT, Vazquez E, Yoshikawa N, Tan L. Dynamic Kinetic Resolution: Asymmetric Transfer Hydrogenation of α-Alkyl-Substituted β-Ketoamides. Org Lett. 2010;12:512–515. doi: 10.1021/ol902715d. [DOI] [PubMed] [Google Scholar]
- 152.Bringmann G, Breuning M, Tasler; S. The Lactone Concept: An Efficient Pathway to Axially Chiral Natural Products and Useful Reagents. Synthesis. 1999:525–558. [Google Scholar]
- 153.Ashizawa T, Tanaka S, Yamada T. Catalytic atropo-Enantioselective Reduction of Biaryl Lactones to Axially Chiral Biaryl Compounds. Org Lett. 2008;10:2521–2524. doi: 10.1021/ol800802c. [DOI] [PubMed] [Google Scholar]
- 154.Krabbe SW, Johnson JS. Asymmetric Total Syntheses of Megacerotonic Acid and Shimobashiric Acid A. Org Lett. 2015;17:1188–1191. doi: 10.1021/acs.orglett.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Han J, Kang S, Lee HK. Dynamic Kinetic Resolution in the Stereoselective Synthesis of 4,5-Diaryl Cyclic Sulfamidates by using Chiral Rhodium-Catalyzed Asymmetric Transfer Hydrogenation. Chem Commun. 2011;47:4004–4006. doi: 10.1039/c0cc05289b. [DOI] [PubMed] [Google Scholar]
- 156.Kim JA, Seo YJ, Kang S, Han J, Lee HK. Stereoselective Synthesis of 4-Substituted-Cyclic Sulfamidate-5-Carboxylates by Asymmetric Transfer Hydrogenation Accompanied by Dynamic Kinetic Resolution and Applications to Concise Stereoselective Syntheses of (−)-epi-Cytoxazone and the Taxotere Side-Chain. Chem Commun. 2014;50:13706–13709. doi: 10.1039/c4cc06395c. [DOI] [PubMed] [Google Scholar]
- 157.Seo YJ, Kim JA, Lee HK. Stereoselective Synthesis of 4-Substituted Cyclic Sulfamidate-5-Phosphonates by Using Rh Catalyzed, Asymmetric Transfer Hydrogenation with Accompanying Dynamic Kinetic Resolution. J Org Chem. 2015;80:8887–8902. doi: 10.1021/acs.joc.5b01434. [DOI] [PubMed] [Google Scholar]
- 158.Son SM, Lee HK. Dynamic Kinetic Resolution-Based Asymmetric Transfer Hydrogenation of 2-Benzoylmorpholinones and its Use in Concise Stereoselective Synthesis of All Four Stereoisomers of the Antidepressant Reboxetine. J Org Chem. 2013;78:8396–8404. doi: 10.1021/jo401102d. [DOI] [PubMed] [Google Scholar]
- 159.Tone H, Buchotte M, Mordant C, Guittet E, Ayad T, Ratovelomanana-Vidal V. Asymmetric Total Synthesis and Stereochemical Revision of Gymnangiamide. Org Lett. 2009;11:1995–1997. doi: 10.1021/ol900184d. [DOI] [PubMed] [Google Scholar]
- 160.Xu F, Zacuto MJ, Kohmura Y, Rosen J, Gibb A, Alam M, Scott J, Tschaen D. Asymmetric Synthesis of Highly Functionalized Tetrahydropyran DPP-4 Inhibitor. Org Lett. 2014;16:5422–5425. doi: 10.1021/ol502661g. [DOI] [PubMed] [Google Scholar]
- 161.Bromhead LJ, Visser J, McErlean CSP. Enantioselective Synthesis of the Strigolactone Mimic (+)-GR24. J Org Chem. 2014;79:1516–1520. doi: 10.1021/jo402722p. [DOI] [PubMed] [Google Scholar]
- 162.Perez M, Echeverria PG, Martinez-Arripe E, Zoubir ME, Touati R, Zhang Z, Genet JP, Phansavath P, Ayad T, Ratovelomanana-Vidal V. An Efficient Stereoselective Total Synthesis of All Stereoisomers of the Antibiotic Thiamphenicol through Ruthenium-Catalyzed Asymmetric Reduction by Dynamic Kinetic Resolution. Eur J Org Chem. 2015;27:5949–5958. [Google Scholar]
- 163.Chen JQ, Xie JH, Bao DH, Liu S, Zhou QL. Total Synthesis of (−)-Galanthamine and (−)-Lycoramine via Catalytic Asymmetric Hydrogenation and Intramolecular Reductive Heck Cyclization. Org Lett. 2012;14:2714–2717. doi: 10.1021/ol300913g. [DOI] [PubMed] [Google Scholar]
- 164.Cheng LJ, Xie JH, Wang LX, Zhou QL. Enantioselective Synthesis of (−)-CP-55940 via Ruthenium-Catalyzed Asymmetric Hydrogenation of Ketones. Adv Synth Catal. 2012;354:1105–1113. [Google Scholar]
- 165.Cheng LJ, Xie JH, Chen Y, Wang LX, Zhou QJ. Enantioselective Total Synthesis of (−)-Δ8-THC and (−)-Δ9-THC via Catalytic Asymmetric Hydrogenation and SNAr Cyclization. Org Lett. 2013;15:764–767. doi: 10.1021/ol303351y. [DOI] [PubMed] [Google Scholar]
- 166.Liu C, Xie JH, Li YL, Chen JQ, Zhou QL. Asymmetric Hydrogenation of α,α′-Disubstituted Cycloketones through Dynamic Kinetic Resolution: An Efficient Construction of Chiral Diols with Three Contiguous Stereocenters. Angew Chem, Int Ed. 2013;52:593–596. doi: 10.1002/anie.201207561. [DOI] [PubMed] [Google Scholar]
- 167.Lall MS, Hoge G, Tran TP, Kissel W, Murphy ST, Taylor C, Hutchings K, Samas B, Ellsworth EL, Curran T, et al. Stereoselective Synthesis of (S)-3-(Methylamino)-3-((R)-pyrrolidin-3-yl)propanenitrile. J Org Chem. 2012;77:4732–4739. doi: 10.1021/jo3004716. [DOI] [PubMed] [Google Scholar]
- 168.Chung JYL, Steinhuebel D, Krska SW, Hartner FW, Cai C, Rosen J, Mancheno DE, Pei T, DiMichele L, Ball RG, et al. Asymmetric Synthesis of a Glucagon Receptor Antagonist via Friedel–Crafts Alkylation of Indole with Chiral α-Phenyl Benzyl Cation. Org Process Res Dev. 2012;16:1832–1845. [Google Scholar]
- 169.Rainka MP, Milne JE, Buchwald SL. Dynamic Kinetic Resolution of α-Unsaturated Lactones through Asymmetric Copper-Catalyzed Conjugate Reduction: Application to the Total Synthesis of Eupomatilone-3. Angew Chem, Int Ed. 2005;44:6177–6180. doi: 10.1002/anie.200501890. [DOI] [PubMed] [Google Scholar]
- 170.Liu S, Xie JH, Wang LX, Zhou QL. Dynamic Kinetic Resolution Allows a Highly Enantioselective Synthesis of cis-α-Aminocycloalkanols by Ruthenium-Catalyzed Asymmetric Hydrogenation. Angew Chem, Int Ed. 2007;46:7506–7508. doi: 10.1002/anie.200702491. [DOI] [PubMed] [Google Scholar]
- 171.Magnus NA, Astleford BA, Laird DLT, Maloney TD, McFarland AD, Rizzo JR, Ruble JC, Stephenson GA, Wepsiec JP. Additives Promote Noyori-type Reductions of a β-Keto-γ-lactam: Asymmetric Syntheses of Serotonin Norepinephrine Reuptake Inhibitors. J Org Chem. 2013;78:5768–5774. doi: 10.1021/jo400589j. [DOI] [PubMed] [Google Scholar]
- 172.Prévost S, Ayad T, Phansavath P, Ratovelomanana-Vidal V. Total Synthesis of Symbioramide: A Flexible Approach for the Efficient Preparation of Structural Isomers. Adv Synth Catal. 2011;353:3213–3226. [Google Scholar]
- 173.Chen CY, Weisel M. Concise Asymmetric Synthesis of (+)-Conocarpan and Obtusafuran. Synlett. 2013;24:189–192. [Google Scholar]
- 174.Chen CY, Frey LF, Shultz S, Wallace DJ, Marcantonio K, Payack JF, Vazquez E, Springfield SA, Zhou G, Liu P, et al. Catalytic, Enantioselective Synthesis of Taranabant, a Novel, Acyclic Cannabinoid-1 Receptor Inverse Agonist for the Treatment of Obesity. Org Process Res Dev. 2007;11:616–623. [Google Scholar]
- 175.Rew Y, Sun D, Yan X, Beck HP, Canon J, Chen A, Duquette J, Eksterowicz J, Fox BM, Fu J, et al. Discovery of AM-7209, a Potent and Selective 4-Amidobenzoic Acid Inhibitor of the MDM2–p53 Interaction. J Med Chem. 2014;57:10499–10511. doi: 10.1021/jm501550p. [DOI] [PubMed] [Google Scholar]
- 176.Bai WJ, Xie JH, Li YL, Liu S, Zhou QL. Enantioselective Synthesis of Chiral β-Aryloxy Alcohols by Ruthenium-Catalyzed Ketone Hydrogenation via Dynamic Kinetic Resolution (DKR) Adv Synth Catal. 2010;352:81–84. [Google Scholar]
- 177.Trost BM, Patterson DE, Hembre EJ. Dynamic Kinetic Asymmetric Transformations of Conduritol B Tetracarboxylates: An Asymmetric Synthesis of D-myo-Inositol 1,4,5-Triphosphate. J Am Chem Soc. 1999;121:10834–10835. [Google Scholar]
- 178.Trost BM, Fandrick DR. Palladium-Catalyzed Dynamic Kinetic Asymmetric Allylic Alkylation with the DPPBA Ligands. Aldrichimica Acta. 2007;40:59–72. [Google Scholar]
- 179.Trost BM. Pd- and Mo-Catalyzed Asymmetric Allylic Alkylation. Org Process Res Dev. 2012;16:185–194. doi: 10.1021/op200294r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Trost BM. Metal Catalyzed Allylic Alkylation: Its Development in the Trost Laboratories. Tetrahedron. 2015;71:5708–5733. doi: 10.1016/j.tet.2015.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Butt NA, Zhang W. Transition Metal-Catalyzed Allylic Substitution Reactions with Unactivated Allylic Substrates. Chem Soc Rev. 2015;44:7929–7967. doi: 10.1039/c5cs00144g. [DOI] [PubMed] [Google Scholar]
- 182.Trost BM, Thaisrivongs DA. Palladium-Catalyzed Regio-, Diastereo-, and Enantioselective Benzylic Allylation of 2-Substituted Pyridines. J Am Chem Soc. 2009;131:12056–12057. doi: 10.1021/ja904441a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.You H, Rideau E, Sidera M, Fletcher SP. Non-Stabilized Nucleophiles in Cu-Catalysed Dynamic Kinetic Asymmetric Allylic Alkylation. Nature. 2015;517:351–355. doi: 10.1038/nature14089. [DOI] [PubMed] [Google Scholar]
- 184.Sidera M, Fletcher SP. Cu-Catalyzed Asymmetric Addition of sp2-Hybridized Zirconium Nucleophiles to Racemic Allyl Bromides. Chem Commun. 2015;51:5044–5047. doi: 10.1039/c5cc00421g. [DOI] [PubMed] [Google Scholar]
- 185.Ros A, Estepa B, Ramírez-López P, Álvarez E, Fernández R, Lassaletta JM. Dynamic Kinetic Cross-Coupling Strategy for the Asymmetric Synthesis of Axially Chiral Heterobiaryls. J Am Chem Soc. 2013;135:15730–15733. doi: 10.1021/ja4087819. [DOI] [PubMed] [Google Scholar]
- 186.For recent reviews on atroposelective biaryl synthesis, see refs. 187–191.
- 187.Wencel-Delord J, Panossian A, Leroux FR, Colobert F. Recent Advances and New Concepts for the Synthesis of Axially Stereoenriched Biaryls. Chem Soc Rev. 2015;44:3418–3430. doi: 10.1039/c5cs00012b. [DOI] [PubMed] [Google Scholar]
- 188.Bringmann G, Gulder T, Gulder TAM, Breuning M. Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem Rev. 2011;111:563–639. doi: 10.1021/cr100155e. [DOI] [PubMed] [Google Scholar]
- 189.Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Atroposelective Synthesis of Axially Chiral Biaryl Compounds. Angew Chem, Int Ed. 2005;44:5384–5437. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- 190.Kozlowski MC, Morgan BJ, Linton EC. Total Synthesis of Chiral Biaryl Natural Products by Asymmetric Biaryl Coupling. Chem Soc Rev. 2009;38:3193–3207. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Baudoin O. The Asymmetric Suzuki Coupling Route to Axially Chiral Biaryls. Eur J Org Chem. 2005:4223–4229. [Google Scholar]
- 192.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J Am Chem Soc. 2015;137:4896–4899. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Briones JF, Davies HML. Enantioselective Gold(I)-Catalyzed Vinylogous [3 + 2] Cycloaddition between Vinyldiazoacetates and Enol Ethers. J Am Chem Soc. 2013;135:13314–13317. doi: 10.1021/ja407179c. [DOI] [PubMed] [Google Scholar]
- 194.For recent reviews on donor-acceptor cyclopropanes, see refs. 195–199.
- 195.Grover HK, Emmett MR, Kerr MA. Carbocycles from Donor-Acceptor Cyclopropanes. Org Biomol Chem. 2015;13:655–671. doi: 10.1039/c4ob02117g. [DOI] [PubMed] [Google Scholar]
- 196.De Nanteuil F, De Simone F, Frei R, Benfatti F, Serrano E, Waser J. Cyclization and Annulation Reactions of Nitrogen-Substituted Cyclopropanes and Cyclobutanes. Chem Commun. 2014;50:10912–10928. doi: 10.1039/c4cc03194f. [DOI] [PubMed] [Google Scholar]
- 197.Cavitt MA, Phun LH, France S. Intramolecular Donor-Acceptor Cyclopropane Ring-Opening Cyclizations. Chem Soc Rev. 2014;43:804–818. doi: 10.1039/c3cs60238a. [DOI] [PubMed] [Google Scholar]
- 198.Chen DYK, Pouwer RH, Richard JA. Recent Advances in the Total Synthesis of Cyclopropane-Containing Natural Products. Chem Soc Rev. 2012;41:4631–4642. doi: 10.1039/c2cs35067j. [DOI] [PubMed] [Google Scholar]
- 199.O’Connor NR, Wood JL, Stoltz BM. Synthetic Applications and Methodological Developments of Donor-Acceptor Cyclopropanes and Related Compounds. Isr J Chem. 2016;56:431–444. doi: 10.1002/ijch.201500089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Trost BM, Morris PJ. Palladium-Catalyzed Diastereo- and Enantioselective Synthesis of Substituted Cyclopentanes through a Dynamic Kinetic Asymmetric Formal [3+2]-Cycloaddition of Vinyl Cyclopropanes and Alkylidene Azlactones. Angew Chem, Int Ed. 2011;50:6167–6170. doi: 10.1002/anie.201101684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mei LY, Wei Y, Xu Q, Shi M. Palladium-Catalyzed Asymmetric Formal [3+2] Cycloaddition of Vinyl Cyclopropanes and β,γ-Unsaturated α-Keto Esters: An Effective Route to Highly Functionalized Cyclopentanes. Organometallics. 2012;31:7591–7599. [Google Scholar]
- 202.For an extension of this methodology by the same group see: Mei L-Y, Wei Y, Xu Q, Shi M. Diastereo-and Enantioselective Construction of Oxindole-Fused Spirotetrahydrofuran Scaffolds through Palladium-Catalyzed Asymmetric [3+2] Cycloaddition of Vinyl Cyclopropanes Isatins. Organometallics. 2013;32:3544–3556.
- 203.Xu H, Qu JP, Liao S, Xiong H, Tang Y. Highly Enantioselective [3+2] Annulation of Cyclic Enol Silyl Ethers with Donor–Acceptor Cyclopropanes: Accessing 3a-Hydroxy [n.3.0]Carbobicycles. Angew Chem, Int Ed. 2013;52:4004–4007. doi: 10.1002/anie.201300032. [DOI] [PubMed] [Google Scholar]
- 204.de Nanteuil F, Serrano E, Perrotta D, Jerome Waser J. Dynamic Kinetic Asymmetric [3 + 2] Annulation Reactions of Aminocyclopropanes. J Am Chem Soc. 2014;136:6239–6242. doi: 10.1021/ja5024578. [DOI] [PubMed] [Google Scholar]
- 205.Tran DN, Cramer N. Rhodium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Racemic Allenes by the [3+2] Annulation of Aryl Ketimines. Angew Chem, Int Ed. 2013;52:10630–10634. doi: 10.1002/anie.201304919. [DOI] [PubMed] [Google Scholar]
- 206.Osborne JD, Randell-Sly HE, Currie GS, Cowley AR, Willis MC. Catalytic Enantioselective Intermolecular Hydroacylation: Rhodium-Catalyzed Combination of β-S-Aldehydes and 1,3-Disubstituted Allenes. J Am Chem Soc. 2008;130:17232–17233. doi: 10.1021/ja8069133. [DOI] [PubMed] [Google Scholar]
- 207.Trost BM, Fandrick DR, Dinh DC. Dynamic Kinetic Asymmetric Allylic Alkylations of Allenes. J Am Chem Soc. 2005;127:14186–14187. doi: 10.1021/ja0543705. [DOI] [PubMed] [Google Scholar]
- 208.For another report on the addition of malonates to allenes via DyKAT see: Ogasawara M, Nagano T, Hayashi T. A New Route to Methyl (R, E)-(–)-Tetradeca-2, 4, 5-Trienoate (Pheromone of Acanthoscelides Obtectus) Utilizing a Palladium-Catalyzed Asymmetric Allene Formation Reaction. J Org Chem. 2005;70:5764–5767. doi: 10.1021/jo050684z.
- 209.For recent reviews on hydroaminations, see refs. 210–211.
- 210.Huang L, Arndt M, Gooßen K, Heydt H, Gooßen LJ. Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem Rev. 2015;115:2596–2697. doi: 10.1021/cr300389u. [DOI] [PubMed] [Google Scholar]
- 211.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 212.Zhang Z, Bender CF, Widenhoefer RA. Gold(I)-Catalyzed Dynamic Kinetic Enantioselective Intramolecular Hydroamination of Allenes. J Am Chem Soc. 2007;129:14148–14149. doi: 10.1021/ja0760731. [DOI] [PubMed] [Google Scholar]
- 213.For a related study see: Michon C, Medina F, Abadie M-A, Agbossou-Niedercorn F. Asymmetric Intramolecular Hydroamination of Allenes using Mononuclear Gold Catalysts. Organometallic. 2013;32:5589–5600.
- 214.Zhang Z, Bender CF, Widenhoefer RA. Gold(I)-Catalyzed Enantioselective Hydroamination of N-Allenyl Carbamates. Org Lett. 2007;9:2887–2889. doi: 10.1021/ol071108n. [DOI] [PubMed] [Google Scholar]
- 215.Arnold JS, Nguyen HM. Rhodium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Racemic Tertiary Allylic Trichloroacetimidates with Anilines. J Am Chem Soc. 2012;134:8380–8383. doi: 10.1021/ja302223p. [DOI] [PubMed] [Google Scholar]
- 216.Arnold JS, Stone RF, Nguyen HM. Rhodium-Catalyzed Regioselective Amination of Secondary Allylic Trichloroacetimidates with Unactivated Aromatic Amines. Org Lett. 2010;12:4580–4583. doi: 10.1021/ol1019025. [DOI] [PubMed] [Google Scholar]
- 217.Zhang Q, Stockdale DP, Mixdorf JC, Topczewski JJ, Nguyen HM. Iridium-Catalyzed Enantioselective Fluorination of Racemic, Secondary Allylic Trichloroacetimidates. J Am Chem Soc. 2015;137:11912–11915. doi: 10.1021/jacs.5b07492. [DOI] [PubMed] [Google Scholar]
- 218.Morimoto Y, Shimizu S, Mokuya A, Ototake N, Saito A, Kitagawa O. Enantioselective Synthesis of N–C Axially Chiral Indoles through Chiral Palladium-Catalyzed 5-endo-Hydroaminocyclization. Tetrahedron. 2016;72:5221–5229. [Google Scholar]
- 219.Ototake N, Morimoto Y, Mokuya A, Fukaya H, Shida Y, Kitagawa O. Catalytic Enantioselective Synthesis of Atropisomeric Indoles with an N–C Chiral Axis. Chem–Eur J. 2010;16:6752–6755. doi: 10.1002/chem.201000243. [DOI] [PubMed] [Google Scholar]
- 220.For related studies, see refs. 221–222.
- 221.Kamikawa K, Kinoshita S, Furusyo M, Takemoto S, Matsuzaka H, Uemura M. Stereoselective Synthesis of both Enantiomers of N-Aryl Indoles with Axially Chiral N–C Bonds. J Org Chem. 2007;72:3394–3402. doi: 10.1021/jo0700427. [DOI] [PubMed] [Google Scholar]
- 222.Mori A, Kinoshita S, Furusyo M, Kamikawa K. Induction of Axially Chiral Bonds in N–C Bonds in N-Aryl Acridane and Related Complexes by Chromium Tricarbonyl Migration Reactions. Chem Commun. 2010;46:6846–6848. doi: 10.1039/c0cc00836b. [DOI] [PubMed] [Google Scholar]
- 223.Trost BM, Machacek MR, Tsui HC. Development of Aliphatic Alcohols as Nucleophiles for Palladium-Catalyzed DYKAT Reactions: Total Synthesis of (+)-Hippospongic Acid A. J Am Chem Soc. 2005;127:7014–7024. doi: 10.1021/ja050340q. [DOI] [PubMed] [Google Scholar]
- 224.Miyata K, Kutsuna H, Kawakami S, Kitamura M. A Chiral Bidentate sp2-N Ligand, Naph-diPIM: Application to CpRu-Catalyzed Asymmetric Dehydrative C–, N–, and O–Allylation. Angew Chem, Int Ed. 2011;50:4649–4653. doi: 10.1002/anie.201100772. [DOI] [PubMed] [Google Scholar]
- 225.Khan A, Zheng R, Kan Y, Ye J, Xing J, Zhang YJ. Palladium-Catalyzed Decarboxylative Cycloaddition of Vinylethylene Carbonates with Formaldehyde: Enantioselective Construction of Tertiary Vinylglycols. Angew Chem, Int Ed. 2014;53:6439–6442. doi: 10.1002/anie.201403754. [DOI] [PubMed] [Google Scholar]
- 226.Wang YM, Kuzniewski CN, Rauniyar V, Hoong C, Toste FD. Chiral (Acyclic Diaminocarbene)Gold(I)-Catalyzed Dynamic Kinetic Asymmetric Transformation of Propargyl Esters. J Am Chem Soc. 2011;133:12972–12975. doi: 10.1021/ja205068j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Moncarz JR, Laritcheva NF, Glueck DS. Palladium-Catalyzed Asymmetric Phosphination: Enantioselective Synthesis of a P-Chirogenic Phosphine. J Am Chem Soc. 2002;124:13356–13357. doi: 10.1021/ja0267324. [DOI] [PubMed] [Google Scholar]
- 228.Blank NF, Moncarz JR, Brunker TJ, Scriban C, Anderson BJ, Amir O, Glueck DS, Zakharov LN, Golen JA, Incarvito CD, et al. Palladium-Catalyzed Asymmetric Phosphination. Scope, Mechanism, and Origin of Enantioselectivity. J Am Chem Soc. 2007;129:6847–6858. doi: 10.1021/ja070225a. [DOI] [PubMed] [Google Scholar]
- 229.Kolodiazhni OI. Recent Developments in Asymmetric Synthesis of P-Chiral Phosphorous Compounds. Tetrahedron: Asymmetry. 2012;23:1–46. [Google Scholar]
- 230.Tappe FMJ, Trepohl VT, Oestreich M. Transition-Metal-Catalyzed C–P Cross-Coupling Reactions. Synthesis. 2010;18:3037–3062. [Google Scholar]
- 231.Chan VS, Bergman RG, Toste FD. Pd-Catalyzed Dynamic Kinetic Enantioselective Arylation of Silylphosphines. J Am Chem Soc. 2007;129:15122–15123. doi: 10.1021/ja076457r. [DOI] [PubMed] [Google Scholar]
- 232.Bhat V, Wang S, Stoltz BM, Virgil SC. Asymmetric Synthesis of QUINAP via Dynamic Kinetic Resolution. J Am Chem Soc. 2013;135:16829–16832. doi: 10.1021/ja409383f. [DOI] [PubMed] [Google Scholar]
- 233.Dornan PK, Kou KGM, Houk KN, Dong VM. Dynamic Kinetic Resolution of Allylic Sulfoxides by Rh-Catalyzed Hydrogenation: A Combined Theoretical and Experimental Mechanistic Study. J Am Chem Soc. 2014;136:291–298. doi: 10.1021/ja409824b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kou KGM, Dong VM. Tandem Rhodium Catalysis: Exploiting Sulfoxides for Asymmetric Transition-Metal Catalysis. Org Biomol Chem. 2015;13:5844–5847. doi: 10.1039/c5ob00083a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wu Z, Li F, Wang J. Intermolecular Dynamic Kinetic Resolution Cooperatively Catalyzed by an N-Heterocyclic Carbene and a Lewis Acid. Angew Chem, Int Ed. 2015;54:1629–1633. doi: 10.1002/anie.201410030. [DOI] [PubMed] [Google Scholar]
- 236.Takeda R, Kawamura A, Kawashima A, Sato T, Moriwaki H, Izawa K, Akaji K, Wang S, Liu H, Aceña JL, et al. Chemical Dynamic Kinetic Resolution and S/R Interconversion of Unprotected α-Amino Acids. Angew Chem, Int Ed. 2014;53:12214–12217. doi: 10.1002/anie.201407944. [DOI] [PubMed] [Google Scholar]
- 237.Lin S, Zhao GL, Deiana L, Sun J, Zhang Q, Leijonmarck H, Córdova A. Dynamic Kinetic Asymmetric Domino Oxa-Michael/Carbocyclization by Combination of Transition-Metal and Amine Catalysis: Catalytic Enantioselective Synthesis of Dihydrofurans. Chem–Eur J. 2010;16:13930–13934. doi: 10.1002/chem.201001992. [DOI] [PubMed] [Google Scholar]
- 238.Zhao GL, Ullah F, Deiana L, Lin S, Zhang Q, Sun J, Ibrahem I, Dziedzic P, Córdova A. Dynamic Kinetic Asymmetric Transformation (DYKAT) by Combined Amine- and Transition-Metal-Catalyzed Enantioselective Cycloisomerization. Chem–Eur J. 2010;16:1585–1591. doi: 10.1002/chem.200902818. [DOI] [PubMed] [Google Scholar]
- 239.Afewerki S, Ma G, Ibrahem I, Liu L, Sun J, Córdova A. Highly Enantioselective Control of Dynamic Cascade Transformations by Dual Catalysis: Asymmetric Synthesis of Polysubstituted Spirocyclic Oxindoles. ACS Catal. 2015;5:1266–1272. [Google Scholar]
- 240.Wu W, Liu X, Zhang Y, Ji J, Huang T, Lin L, Feng X. Chiral N,N′-Dioxide–FeCl3 Complex-Catalyzed Asymmetric Intramolecular Cannizzaro Reaction. Chem Commun. 2015;51:11646–11649. doi: 10.1039/c5cc04213e. [DOI] [PubMed] [Google Scholar]
- 241.Russell AE, Miller SP, Morken JP. Efficient Lewis Acid Catalyzed Intramolecular Cannizzaro Reaction. J Org Chem. 2000;65:8381–8383. doi: 10.1021/jo0010734. [DOI] [PubMed] [Google Scholar]
- 242.Ishihara K, Yano T, Fushimi M. Asymmetric Intramolecular Cannizzaro Reaction of Anhydrous Phenylglyoxal. J Fluorine Chem. 2008;129:994–997. [Google Scholar]
- 243.Wang P, Tao WJ, Sun XL, Liao S, Tang Y. A Highly Efficient and Enantioselective Intramolecular Cannizzaro Reaction under TOX/Cu(II) Catalysis. J Am Chem Soc. 2013;135:16849–16852. doi: 10.1021/ja409859x. [DOI] [PubMed] [Google Scholar]
- 244.Chen PH, Sieber J, Senanayake CH, Dong G. Rh-Catalyzed Reagent-Free Ring Expansion of Cyclobutenones and Benzocyclobutenones. Chem Sci. 2015;6:5440–5445. doi: 10.1039/c5sc01875g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ito M, Kitahara S, Ikariya T. Cp*Ru(PN) Complex-Catalyzed Isomerization of Allylic Alcohols and Its Application to the Asymmetric Synthesis of Muscone. J Am Chem Soc. 2005;127:6172–6173. doi: 10.1021/ja050770g. [DOI] [PubMed] [Google Scholar]
- 246.Saitoh F, Nishida H, Mukaihira T, Aikawa K, Mikami; K. Asymmetric Synthesis of Antithrombotic Agents M58163 and M58169: Dynamic Kinetic Resolution in Amide Formation Catalyzed by La-Linked BINOL Complex. Eur J Org Chem. 2006:5454–5457. [Google Scholar]
- 247.Saitoh F, Nishida H, Mukaihira T, Aikawa K, Mikami K. Dynamic Kinetic Resolution for the Catalytic Asymmetric Total Synthesis of Antithrombotic Agents M58163 and M58169. Adv Synth Catal. 2007;349:617–628. [Google Scholar]
- 248.Trost BM, Horne DB, Woltering MJ. Palladium-Catalyzed DYKAT of Butadiene Monoepoxide: Enantioselective Total Synthesis of (+)-DMDP, (–)-Bulgecinine, and (+)-Broussonetine G. Chem–Eur J. 2006;12:6607–6620. doi: 10.1002/chem.200600202. [DOI] [PubMed] [Google Scholar]
- 249.Li H, Belyk KM, Yin J, Chen Q, Hyde A, Ji Y, Oliver S, Tudge MT, Campeau LC, Campos KR. Enantioselective Synthesis of Hemiaminals via Pd-Catalyzed C–N Coupling with Chiral Bisphosphine Mono-oxides. J Am Chem Soc. 2015;137:13728–13731. doi: 10.1021/jacs.5b05934. [DOI] [PubMed] [Google Scholar]
- 250.Yang D, Micalizio GC. Stereochemical Lability of Azatitanacyclopropanes: Dynamic Kinetic Resolution in Reductive Cross-Coupling Reactions with Allylic Alcohols. Chem Commun. 2013;49:8857–8859. doi: 10.1039/c3cc45607b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.For examples of dynamic kinetic systems involving Cp2Zr-imine complexes: see refs. 248–250.
- 252.Tunge JA, Gately DA, Norton JR. Asymmetric Formation of α-Amino Acid Esters through Dynamic Kinetic Resolution: A Cyclic Carbonate as an Optically Active CO2 Synthon. J Am Chem Soc. 1999;121:4520–4521. [Google Scholar]
- 253.Cummings SA, Tunge JA, Norton JR. Direct Measurement of the Rate of Interconversion of Zirconaaziridine Enantiomers. J Am Chem Soc. 2008;130:4669–4679. doi: 10.1021/ja0757935. [DOI] [PubMed] [Google Scholar]
- 254.Li L, Kristian KE, Han A, Norton JR, Sattler W. Synthesis, Structural Characterization, and Reactivity of Cp2- and (CpMe)2-Ligated Titanaaziridines and Titanaoxiranes with Fast Enantiomer Interconversion Rates. Organometallics. 2012;31:8218–8224. [Google Scholar]
- 255.Pearson AJ, Sun H, Wang X. Dynamic Diastereoselectivity during Iron Carbonyl Mediated Spirocyclization Reactions. J Org Chem. 2007;72:2547–2557. doi: 10.1021/jo0625608. [DOI] [PubMed] [Google Scholar]
- 256.Ma YN, Zhang HY, Yang SD. Pd(II)-Catalyzed P(O)R1R2-Directed Asymmetric C–H Activation and Dynamic Kinetic Resolution for the Synthesis of Chiral Biaryl Phosphates. Org Lett. 2015;17:2034–2037. doi: 10.1021/acs.orglett.5b00844. [DOI] [PubMed] [Google Scholar]
- 257.For a related methodology on substrate bearing chiral sulfoxide see: Wesch T, Leroux FR, Colobert F. Atropodiastereoselective C–H Olefination of Biphenyl p-Tolyl Sulfoxides with Acrylates. Adv Synth Catal. 2013;355:2139–2144.
- 258.Hazra CK, Dherbassy Q, Wencel-Delord J, Colobert F. Synthesis of Axially Chiral Biaryls through Sulfoxide-Directed Asymmetric Mild C–H Activation and Dynamic Kinetic Resolution. Angew Chem, Int Ed. 2014;53:13871–13875. doi: 10.1002/anie.201407865. [DOI] [PubMed] [Google Scholar]
- 259.For a related vinylative protocol see: Wesch T, Leroux FR, Colobert F. Atropodiastereoselective C–H Olefination of Biphenyl p-Tolyl Sulfoxides with Acrylates. Adv Synth Catal. 2013;355:2139–2144.
- 260.Achmatowicz O, Jr, Bukowski P, Szechner B, Zwierzchowska Z, Zamojski A. Synthesis of Methyl 2, 3-dideoxy-DL-alk-2-enopyranosides from Furan Compounds: A General Approach to the Total Synthesis of Monosaccharides. Tetrahedron. 1971;27:1973–1996. [Google Scholar]
- 261.Achmatowicz O, Jr, Bukowski P. Total Synthesis of Monosaccharides. Synthesis of Methyl DL-Pentopyranosides with α- andβ-Lyxoβ-Riboα-Xylo and α-Arabino Configurations from Furfuryl Alcohol. Can J Chem. 1975;53:2524–2529. [Google Scholar]
- 262.Wang HY, Yang K, Bennett SR, Guo SR, Tang W. Iridium-Catalyzed Dynamic Kinetic Isomerization: Expedient Synthesis of Carbohydrates from Achmatowicz Rearrangement Products. Angew Chem, Int Ed. 2015;54:8756–8759. doi: 10.1002/anie.201503151. [DOI] [PubMed] [Google Scholar]
- 263.For a report on dynamic kinetic diastereoselective acylation on related substrates see: Wang H-Y, Yang K, Yin D, Liu C, Glazier DA, Tang W. Chiral Catalyst-Directed Dynamic Kinetic Diastereoselective Acylation of Lactols for De Novo Synthesis of Carbohydrates. Org Lett. 2015;17:5272–5275. doi: 10.1021/acs.orglett.5b02641.