

Abstract
Background
IL-1β is a cytokine involved in mediating epithelial barrier dysfunction in the gut. It is known that IL-1β mediates activation of non-muscle myosin light chain kinase in epithelial cells, but the precise mechanism by which epithelial barrier dysfunction is induced by IL-1β is not understood.
Methods and Results
Using a Caco2 cell model, we show that the expression of the tight junction protein, claudin-3, is transcriptionally downregulated by IL-1β treatment. In addition, after assessing protein and mRNA expression, and protein localization, we show that inhibition of nmMLCK rescues IL-1β mediated decrease in claudin-3 expression as well as junction protein redistribution. Using chromatin immunoprecipitation assays, we also show that β-catenin targeting of the claudin-3 promoter occurs as a consequence of IL-1β mediated epithelial barrier dysfunction, and inhibition of nmMLCK interferes with this interaction.
Conclusions
Taken together, this data represent the first line of evidence demonstrating nmMLCK regulation of claudin-3 expression in response to IL-1β treated epithelial cells.
Keywords: claudin-3, interleukin-1β, β-catenin, Caco2, non-muscle myosin light chain kinase
INTRODUCTION
Gut barrier dysfunction is a significant factor in the development of several disease conditions including Crohn’s disease (CD), ulcerative colitis, inflammatory bowel disease, and trauma induced hyperpermeability of the gut. Since gut barrier function is largely maintained by epithelial tight junctions (TJ) lining the intestinal wall, an intense area of study has focused on identifying mechanisms surrounding homeostasis of the gut epithelial barrier (1, 19–21). Specifically, it has been reported that intestinal inflammation in a mouse model for inflammatory bowel disease was preceded by hyperpermeability of the gut epithelium (14), and that restoring epithelial tight junctions attenuated the development of enterocolitis (3). This suggested that a breach in tight junction integrity not only represents a cause of inflammation, but it is also etiologically important.
A major regulatory event leading to increased paracellular permeability involves the phosphorylation of myosin light chain (MLC) by non-muscle myosin light chain kinase (nmMLCK) (29). nmMLCK is a calcium calmodulin-dependent serine/threonine kinase that regulates actin/myosin reorganization and cell contraction in response to various stimuli that mediate short-term contractile events (thrombin, histamine, etc.) as well as mediators of chronic alterations in intestinal permeability (TNFα, IL-1β). Three MLCK isoforms are known: a non-muscle isoform (210 kDa), a short smooth-muscle form (108 kDa), and the non-enzymatic protein telokin (21 kDa). nmMLCK is primarily expressed in endothelial cells, epithelial cells, and neutrophils (6, 29, 30, 32), and is involved in the regulatory mechanisms surrounding inflammation mediated permeability of the gut. For example, MLCK activation plays a significant role in microvascular hyperpermeability induced by neutrophil stimulation or thermal injury (10, 25, 32, 33). In addition, nmMLCK is essential to sodium-nutrient co-transport and TNFα-induced intestinal epithelial barrier loss and mucosal damage (13). Transgenic mice with the nmMLCK isoform deleted showed both endothelial and epithelial barrier property enhancement in the gut, as well as attenuation of burn-induced vascular leakage (25).
IL-1β has been shown to play a significant role in intestinal inflammation resulting from inflammatory bowel disease (IBD) (24). Patients with CD often present elevated IL-1β due to insufficient production of an endogenous IL-1R antagonist (18). Although it has been demonstrated that IL-1β not only plays a role in intestinal inflammation but also contributes to increased permeability across the epithelial barrier in the gut, the precise mechanism of tight junction disruption is unclear. Tight junctions are composed of several types of proteins including ZO-1, occludin, JAM, and the claudin family of proteins. Claudin-3 plays a particularly essential role in intestinal epithelial barrier function (15, 17, 22). It has been shown that claudin-3 expression is positively correlated with epithelial tightness (23), and that disruption of claudin-3 expression negatively affects epithelial barrier function (9). To the best of our knowledge, the role of IL-1β signaling on claudin-3 expression in intestinal epithelial cells has not been reported.
Claudin-3 expression is regulated in a number of ways, one of which is through transcriptional modulation by the dual function protein, β catenin (12). Upon stimulation with inflammatory factors, β-catenin phosphorylation occurs and may translocate to the nucleus where it associates with a number of transcription factors to regulate several genes, including claudin-3 (12). However, the role of β-catenin mediated transcriptional repression of tight junction proteins in epithelial cells resulting from IL-1β treatment has not been investigated.
It has been recently established that IL-1β signaling activates nmMLCK in endothelial and epithelial cells (2). Inhibition of nmMLCK in endothelial and epithelial cells has restored barrier function in response to several inflammatory factors including IL-1β (2). Furthermore, these experiments also showed that nmMLCK was required for IL-1β induced β-catenin nuclear accumulation and FoxO1 mediated claudin downregulation in brain endothelial cells (4). Given the critical role of claudin-3 and nmMLCK in epithelial barrier function, and that IL-1β is known to disrupt epithelial tight junction integrity, here we sought to determine whether nmMLCK plays a role in claudin-3 downregulation by promoting β-catenin association with the claudin-3 promoter in IL-1β treated intestinal epithelial cells.
METHODS
Cell culture
Caco2 cells were obtained from the American Type Culture Collection (ATCC) and cultured per ATCC guidelines. Prior to treatment with IL-1β, cells were serum starved overnight to promote G0 synchronization. Cell treatments (ML-7, IL-1β, Sigma) were dissolved in PBS + 0.1% BSA then added to cell culture media at indicated concentrations. Vehicle control was diluent alone at the same volume as treatment. Cells were assayed for mRNA levels, protein localization, permeability coefficient, and monolayer resistance using the methods described below. Protein expression was assayed using standard Western blotting techniques.
Quantitative Real Time PCR
Total RNA from Caco2 monolayers was harvested with RNAzol (Molecular Research Center) according to the manufacturers protocol. RNA was reverse-transcribed using iScript cDNA Synthesis Kit (Bio-Rad, CA, USA) using 500 ng total RNA per 20 uL reaction. Wet-lab validated primers (PrimePCR Assays; Bio-Rad, CA, USA) for the genes and reference gene (GAPDH) quantified in these studies were used. For real-time PCR reactions, SsoAdvanced SYBR Green Supermix was used, RT-PCR was ran and analyzed on Bio-Rad CFX Connect (Bio-Rad, CA, USA) per manufacturer’s instructions.
Chromatin immunoprecipitation assay (ChIP)
Chromatin immunoprecipitation (ChIP) assays were performed with chromatin isolated from Caco2 monolayers using an agarose ChIP kit (Pierce). Cells were treated with vehicle control (PBS + 0.1% BSA), IL-1β (100 ng/mL, Sigma), or ML-7 (10 μM, Sigma) + IL-1β for 3 hours. DNA was precipitated per standard procedures and amplified in standard qualitative PCR and qRT-PCR reactions using oligonucleotides flanking the putative Cldn3 promoter region. Non-precipitated/Input samples were used as positive controls. ΔCq was calculated by normalizing with Input.
Transepithelial resistance (TER) measurements
Caco2 barrier function was determined as previously described (26) by measuring cell-cell adhesive resistance to electric current using an electric cell-substrate impedance sensing (ECIS) system (Applied Biophysics, Troy, NY). ECIS tracings expressed as transepithelial electric resistance (TER) are normalized to baseline, and comparisons were made between IL-1β ([10 or 100 ng/mL]) and vehicle control ([0.1% BSA in PBS]) treated Caco2 monolayers. TER changes were recorded every 90 seconds and statistical analyses were performed at peak drop in IL-1β TER (n = 3; for 3 independent experiments).
Immunocytochemistry
Caco2 monolayers were grown in 2-chamber slides (Nunc) for immunocytochemistry analysis. Since translocation of transcriptional factors temporally precedes gene regulation, detection of nuclear β-catenin detection was performed 3 hours post treatment (Figure 3), but the detection of claudin-3 and E-cadherin (Figure 4) was performed after IL-1β induced permeability changes were detected (24 hours). In both sets of experiments, cells were treated with vehicle control (PBS + 0.1% BSA), IL-1β (100 ng/mL, Sigma), or ML-7 (10 μM, Sigma) + IL-1β. Fixation and immunolabeling were performed using standard protocols. Briefly, cells were washed twice [PBS with 2 mM CaCl2, 2 mM MgCl2] followed by incubation in 4% paraformaldehyde for 10 minutes. Slides were rinsed twice in PBS, permeabilized [PBS, 0.1% Triton X-100], then blocked [PBS, 3% BSA, 0.01% Tween-20] for 2 hours at room temperature. Cells were incubated with primary antibodies (β-catenin, Cell Signaling; E-cadherin, Millipore; Claudin-3, Life Technologies) overnight at 4°C diluted 1:50 [PBS, 0.01% Tween-20, 3% BSA], washed 3 times for 10 minutes each [PBS, 0.1% Tween-20], incubated with appropriate secondary antibody diluted 1:200 [PBS, 0.01% Tween-20, 2% BSA] (AlexaFluor-conjugated; Life Technologies). Slides were mounted with anti-fade media containing DAPI (Life Technologies). Immunofluorescence confocal microscopy was performed with Olympus FV1000 MPE multiphoton laser scanning microscope (Olympus). Images were performed from 3 independent experiments and representative confocal micrograph images are presented.
Fig. 3.
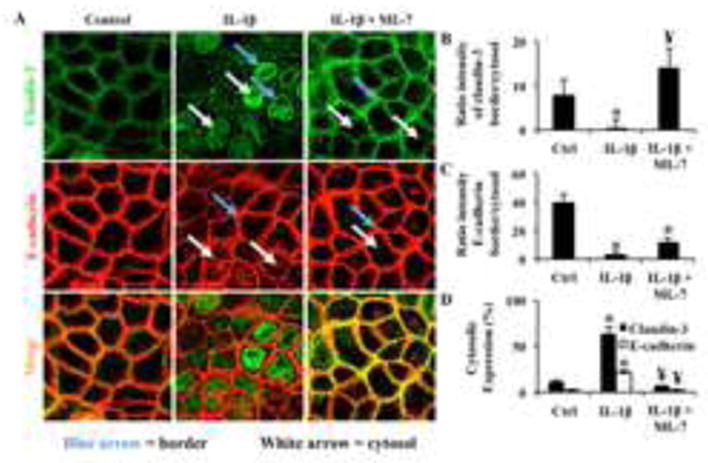
Inhibition of nmMLCK results in attenuated accumulation of cytosolic claudin-3. (A) Caco2 cells were treated with vehicle control or ML-7 (10 μM) followed by IL-1β treatment and claudin-3 protein localization was assessed by immunofluorescent confocal microscopy. Quantitation represents (B) the ratio of intensity of claudin-3 (green) detection at the cell-cell border (blue arrows) vs. cytosolic expression (white arrows), (C) the ratio of E-cadherin intensity at cell-cell borders (blue arrows) vs. cytosolic expression (white arrows), and (D) the percentage of cytosolic claudin-3 or E-cadherin from total intensity for each treatment. Results are representative of 3 experiments. * p<0.05 relative to vehicle control, ¥ p<0.05 relative to IL-1β alone.
Fig. 4.
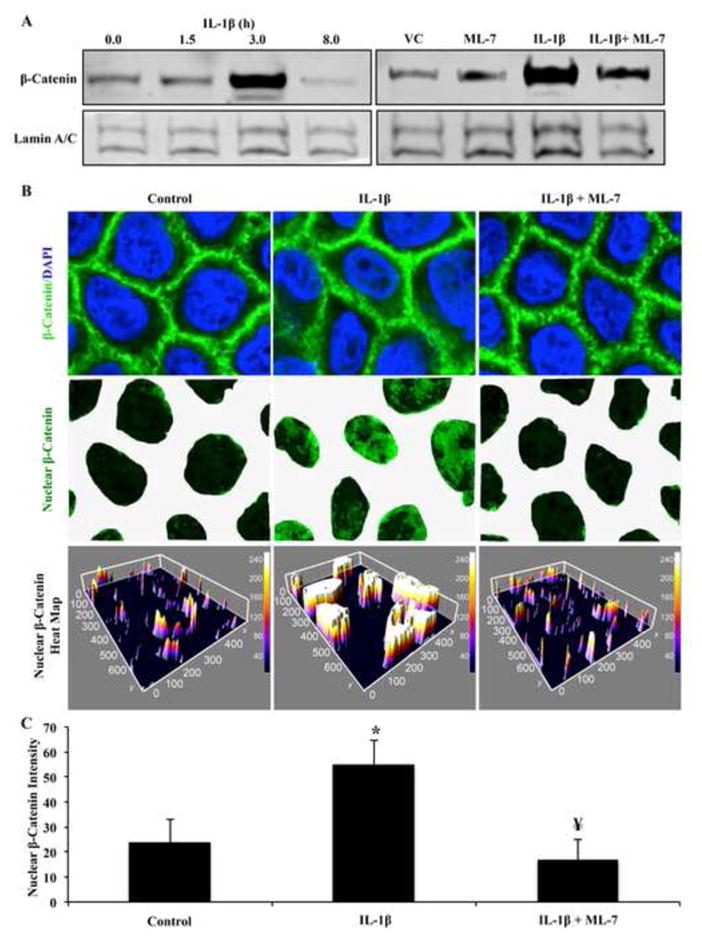
Inhibition of nmMLCK prevents β-catenin translocation to the nucleus. (A) Caco2 cells were treated with IL-1β for 0, 1.5, 3, or 8 hours followed by nuclear fractionation and β-catenin detection by Western blot. Lamin A/C was probed as loading control. (B) Caco2 cells were treated with vehicle control, ML-7, IL-1β, or ML-7 and IL-1β for 3 hours followed by β-catenin detection in the nuclear fraction by Western blot. Results represent results from 4 separate experiments. (C) Confluent Caco2 cells were treated with vehicle control (PBS + 0.1%BSA), IL-1β (100 ng/mL), or IL-1β and ML-7 (10 μM) for 3 hours then subject to nuclear β-catenin (green) detection by immunocytochemistry analysis. Quantitation represents pixel intensity of β-catenin signal detected in nuclear regions. Results are representative of 3 individual experiments. * p<0.05 relative to vehicle control, ¥ p<0.05 relative to IL-1β alone.
Image analysis
Confocal micrographs were analyzed using Image J software. For cytosolic vs. junction detection comparisons, signal intensity was measured at indicated area from raw images, averaged, and plotted. Considering that the border staining for β-catenin strongly diminished nuclear detection, images were edited to highlight the areas of interest. For nuclear β-catenin presentation, nuclear areas were selected from DAPI image then applied to β-catenin staining followed by removal of unselected area. A threshold was set to the resulting images to exclude background and Z-plotted for added clarity. Quantitative assessment of nuclear β-catenin detection was achieved by calculating the mean signal intensity with error bars representing standard error. Results are representative of 3 experiments.
Permeability Assay
Sodium fluorescein-flux assays were conducted as previously described (28). Caco2 cells were plated on collagen I coated polycarbonate 12-well transwell inserts (0.4 μm pore size, Corning) and allowed to reach confluence. The luminal (upper) compartment was treated with sodium fluorescein (2.5 mg/mL) for 15 minutes, then 100 μL aliquots were obtained from each abluminal (lower) compartment and assayed for fluorescence intensity. Permeability coefficients (Ps) were calculated using the formula Ps=[Ab]/t× 1/A×V/[Lu], where [Ab] is the abluminal concentration of sodium fluorescein, t is time in seconds for sodium fluorescein incubation, A is area of membrane in cm2, V is the volume of the abluminal chamber, and [Lu] is the luminal concentration of sodium fluorescein. Results are representative of 4 experiments *p<0.05.
Statistics
One-way ANOVA was used for data involving 3 or more groups with one variable followed by the Student’s t-test to determine significant difference between non-treated controls and indicated treatment group. Significance was noted at p<0.05 and noted with (*) to show significance relative to control or (¥) to indicate significance relative to IL-1β treatment in absence of MLCK inhibitor (ML-7).
RESULTS
nmMLCK inhibition attenuated IL-1β induced barrier dysfunction of Caco2 monolayers
Since nmMLCK inhibition had been shown previously to attenuate IL-1β mediated barrier dysfunction, we aimed to determine whether this finding was also true for intestinal epithelial cells. Therefore, Caco2 barrier function was assayed via ECIS and permeability assays in presence of vehicle control, IL-1β alone, or IL-1β with nmMLCK inhibitor (ML-7, 10 μM). As shown in Figure 1A, IL-1β induced a decrease in TER (~15%) after treatment and did not return to baseline levels, however the addition of ML-7 prevented the IL-1β drop in TER (Figure 1A and Figure 1B). These findings are consistent with previous reports (2). Furthermore, permeability assays showed that sodium fluorescein (~0.38 kDa) accumulation resulting from IL-1β treatment was reduced by greater than 3-fold when co-treated with ML-7 (Figure 1C). These results suggest that nmMLCK plays a prominent role in IL-1β induced barrier dysfunction of intestinal epithelial cells.
Fig. 1.
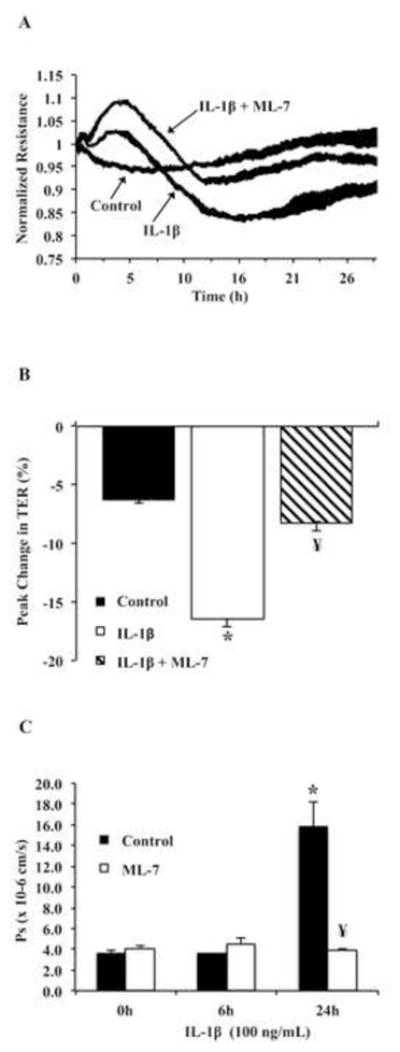
Inhibition of nmMLCK improves IL-1β mediated decrease in TER. (A) Tracing representing resistance measurements of Caco2 monolayers treated with vehicle control (PBS), IL-1β (100 ng/mL), or IL-1β in presence of nmMLCK inhibitor ML-7 (10 μM) (replicates of 4, normalized to timepoint 0). (B) Statistical analysis of nmMLCK inhibition (ML-7, 10 μM) effect on peak change in TER due to IL-1β treatment of Caco2. (* p <0.05 relative to vehicle control, ¥ p<0.05 relative to IL-1β alone). Results are representative of 3 experiments. (C) Caco2 cells were grown on 0.4μM transwell inserts followed by the indicated treatments for 24 hours. Flux to sodium fluorescein was measured as described in methods. * p<0.05 relative to vehicle control, ¥ p<0.05 relative to IL-1β alone.
nmMLCK inhibition diminishes IL-1β mediated decrease in claudin-3 expression
Since claudin-3 is a critical tight junction protein known to be downregulated in diseases involving increased permeability in intestinal epithelium, we hypothesized that claudin-3 expression may be downregulated in response to IL-1β treatment. Therefore, we treated Caco2 cells with IL-1β at different time points (0, 6, and 24 hours) as well as different concentrations of IL-1β (10 or 100 ng/mL, 24 hours) and assessed claudin-3 expression by Western blot. Results showed that IL-1β (10 ng/mL) significantly reduced claudin-3 expression at 24 hours (Figure 2A). Similarly, as shown in Figure 2B, claudin-3 expression was reduced by approximately 40% after 24 hours of IL-1β treatment (10 ng/mL). However, 100 ng/mL IL-1β did not reduce claudin-3 expression significantly relative to the lower dose (Figure 2B). Next, considering nmMLCK inhibition attenuated IL-1β driven claudin-5 downregulation (4), we sought to determine whether nmMLCK inhibition rescued IL-1β induced decrease in claudin-3 expression. Figure 2C shows that IL-1β alone reduced total claudin-3 expression by approximately 50%, yet co-treatment of IL-1β with ML-7 resulted in unperturbed claudin-3 expression despite presence of IL-1β. Similar results were obtained when nmMLCK was targeted with siRNA (data not shown).
Fig. 2.
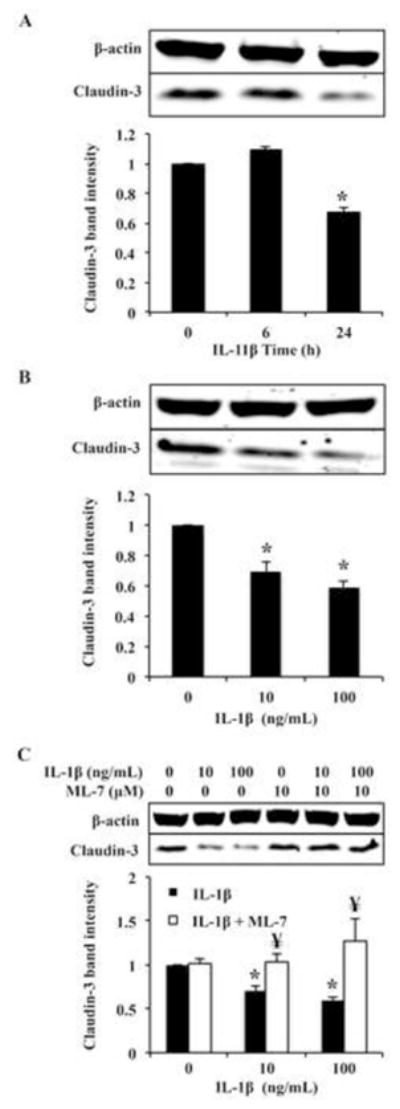
nmMLCK mediates IL-1β induced claudin-3 downregulation in Caco2 cells. (A) Caco2 cells were treated with 100ng/nl IL-1β for 0, 6, or 24 hours, or (B) with 0, 10, or 100 ng/mL for 24 hours. Cell lysates were analyzed by Western blot and degree of claudin-3 expression was assessed by densitometry. β-actin was probed as loading control. (C) Caco2 cells were treated with vehicle control or ML-7 (10 μM) followed by IL-1β treatment (10 ng/mL or 100 ng/mL) and claudin-3 protein expression was assessed by Western blot. Results are representative of 3 experiments.
Inhibition of nmMLCK restores claudin-3 expression at cell-cell junctions
In subsequent experiments, we aimed to identify whether ML-7 improved claudin-3 expression specifically at cell-cell borders when co-treated with IL-1β. Therefore, Caco2 cells were grown to confluence on coverslips, treated with either vehicle control, IL-1β or IL-1β + ML-7, and assayed for claudin-3 detection at cell-cell borders via immunofluorescent microscopy. E-cadherin was stained to show cell-cell border orientation. Results showed that ML-7 co-treatment with IL-1β indeed improved claudin-3 detection at cell-cell borders by over 10-fold (Figure 3B) compared to IL-1β alone. Interestingly, E-cadherin detection at cell junctions was also improved with ML-7 treatment (Figure 3C), although not as robustly as claudin-3. Similarly, cytosolic detection of claudin-3 was approximately 6-fold and E-cadherin cytosolic detection increased approximately 4-fold upon treatment of IL-1β, yet inhibition of nmMLCK in presence of IL-1β attenuated cytosolic detection down to control levels (Figure 3D). These results suggest that nmMLCK plays a role in not only IL-1β induced downregulation of claudin-3, but also in its redistribution.
Inhibition of nmMLCK prevents nuclear accumulation of β-catenin
Since it has been previously established that nmMLCK was required for IL-1β induced nuclear translocation of β-catenin and subsequent repression of the claudin-5 promoter in endothelial cells (4), we hypothesized that nmMLCK was also required for IL-1β induced nuclear translocation of β-catenin in Caco2 cells. Therefore, Caco2 cells were grown to confluence and treated with IL-1β for 0, 1.5, 3, and 8 hours, followed by nuclear fractionation and β-catenin detection by Western blot. As shown in the left panel of Figure 4A, nuclear accumulation of β-catenin was robustly augmented after 3 hours of IL-1β treatment relative to earlier timepoints. Subsequently, Caco2 cells were grown to confluence and treated with IL-1β (100 ng/mL) for 3 hours either in presence or absence of ML-7. These results showed that inhibition of nmMLCK greatly reduced IL-1β mediated nuclear translocation of β-catenin (Figure 4A, right panel), which was confirmed in confocal image analysis (Figure 4B–C). As shown in Figure 4C, nuclear detection of β-catenin increased by approximately 3-fold after 3 hours of IL-1β treatment, but was diminished to control levels when co-treated with ML-7. These data suggest that nmMLCK signaling is involved in the nuclear translocation of β-catenin resulting from IL-1β in Caco2 cells.
nmMLCK inhibition prevents IL-1β induced β-catenin targeting to the claudin-3 promoter
We next determined whether the nmMLCK mediated downregulation of claudin-3 was due to transcriptional regulation by assaying claudin-3 mRNA after IL-1β treatment with or without nmMLCK inhibition with ML-7. As shown in Figure 5A, IL-1β significantly decreased claudin-3 mRNA by roughly 80%, and inhibition of nmMLCK attenuated transcriptional claudin-3 downregulation to control level of expression despite IL-1β treatment. Furthermore, ChIP experiments showed that IL-1β induced β-catenin association with the claudin-3 promoter was approximately 2-fold less when co-treated with ML-7 (Figure 5B, C). These results suggest that nmMLCK plays a role in regulating claudin-3 expression at the level of transcription, and that inhibition of nmMLCK prevents β-catenin targeting to the claudin-3 promoter.
Fig. 5.
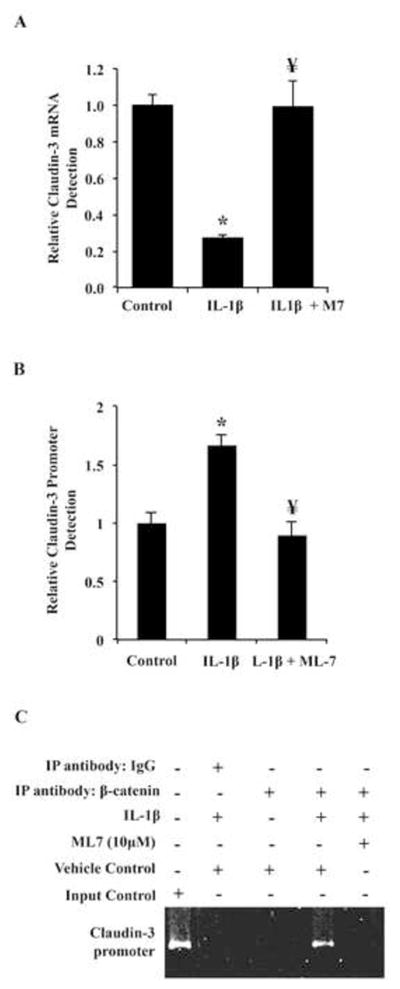
Inhibition of nmMLCK prevents β-catenin targeting to claudin-3 promoter and improves claudin-3 expression. (A) Caco2 cells were treated with vehicle control, ML-7, or ML-7 and IL-1β for 3 hours followed by claudin-3 detection by qPCR. (B,C) ChIP with anti- β-catenin or non-specific IgG antibody was used to precipitate DNA from Caco2 cells treated was subjected to qualitative analysis with PCR primers directed against the claudin-3 promoter. First lane represents non-precipitated DNA as positive control, second lane shows precipitation with non-specific IgG as negative control. Results are representative of 3 experiments. * p<0.05 relative to vehicle control, ¥ p<0.05 relative to IL-1β alone.
DISCUSSION
In this study, we used Caco2 cells to further characterize the signaling pathway for IL-1β mediated epithelial permeability. The novel findings in this report are that 1) IL-1β induced claudin-3 downregulation in Caco2 cells, 2) IL-1β induced β-catenin targeting of the claudin-3 promoter in a manner involving nmMLCK, and 3) the IL-1β directed β-catenin targeting of the claudin-3 promoter was associated with repression of claudin-3. Although nmMLCK had been previously identified as being regulated by IL-1β (2, 13, 31), we show that nmMLCK is involved in IL-1β induced gene regulatory events that lead to downregulation of critical tight junction components in Caco2 cells.
IL-1β mediated epithelial permeability is characterized by several molecular pathways that occur in concert. The initial formation of stress-fibers allows cellular contraction to ultimately form intercellular spaces (7). Interestingly, the initial formation of central stress-fibers contributes a temporary increase in resistance observed on ECIS due to actin polymerization and cell spreading, and was indeed what was seen in our ECIS experiments in the initial 5 hours following IL-1β treatment. Concomitantly, several post-translational events lead to tyrosine phosphorylation, cellular contraction, and cytosolic translocation of junction proteins that possess dual function as transcription regulators (β-catenin, ZO-1, etc.). Indeed, our results indicated that IL-1β promoted β-catenin translocation to the nucleus at 3 hours post IL-1β treatment. We then sought to determine key junction proteins that demonstrated decreased expression in response to IL-1β that may be repressed by nuclear β-catenin. Since we previously demonstrated that β-catenin targeted a claudin promoter in a manner that repressed its expression in IL-1β treated tight junction expressing cells, and nmMLCK regulates permeability in response to IL-1β, we hypothesized that a claudin protein may participate in a similar pathway in IL-1β treated Caco2 cells.
Therefore, we aimed to identify tight junction proteins that are significantly downregulated in response to IL-1β. Results showed IL-1β mediated significant downregulation of claudin-3, but not E-cadherin, ZO-1, occludin, claudin-1, claudin-4, or claudin-15 (data not shown), as well as cytosolic accumulation of E-cadherin and claudin-3. Although E-cadherin is a critical component of the epithelial barrier function, it has been shown that E-cadherin is not required for the maintenance of tight junctions, and its role in dynamic tight junction regulation is largely due to its β-catenin sequestering capacity (5, 27). As Capaldo and others have shown, interruption of E-cadherin function in mature epithelial cells had little to no impact on barrier function, although impaired expression of E-cadherin did delay the development of new tight junctions as well as β-catenin detection at cell borders (5). These factors suggest that cytosolic accumulation of E-cadherin in a mature epithelial monolayer may not directly contribute to IL-1β driven epithelial permeability. Instead, since the role of claudin-3 in intestinal permeability has been clearly shown in vitro and clinically (8, 16, 34), we interpret claudin-3 downregulation and translocation from the tight junction as being a significant factor contributing to the barrier dysfunction seen in IL-1β treated Caco2 cells.
It has been previously established that nmMLCK plays a role in IL-1β induced epithelial barrier dysfunction (2, 13, 31). In the study conducted by Ma, et al, IL-1β induced a dose and time dependent increase in nmMLCK protein expression at the level of mRNA and protein. Furthermore, inhibiting nmMLCK with siRNA improved TER changes induced by IL-1β, which is consistent with our findings. In addition, nmMLCK has been shown to have a direct role in modulating nuclear β-catenin accumulation in a manner that affects tight junction protein expression (4). Specifically, primary cells isolated from nmMLCK knockout mice demonstrated attenuated nuclear accumulation of β-catenin and increased claudin-5 expression relative to IL-1β treated wild-type cells. Consistent with this report, we show here that nmMLCK also plays a role in regulating claudin-3 expression in response to IL-1β in a manner that involves β-catenin targeting of the claudin-3 promoter. Importantly, these results suggest that nmMLCK modulates transcriptional activity in addition to regulating cytoskeletal contraction in Caco2 cells. Therefore, it appears that the involvement of nmMLCK in regulating β-catenin impacts the signaling and transcriptional functions of β catenin.
Another significant finding in this study was that IL-1β induced a direct association of β-catenin to the claudin-3 promoter in an nmMLCK dependent manner. Although other reports have shown that Wnt signaling (and subsequent β-catenin nuclear accumulation) induced Claudin-3 downregulation, the mechanism described was not through direct association of β-catenin with the claudin-3 promoter, but rather by the upregulation of the claudin-3 transcriptional repressor Snail (11). In other reports, β-catenin mediated downregulation of claudin-5 required β-catenin direct association and recruitment of FoxO1 with the claudin-5 promoter in order for claudin-5 downregulation to occur (4). Since we observed direct association of β-catenin to the claudin-3 promoter, it is tempting to speculate that this mechanism of non-Wnt β-catenin signaling may also be driving IL-1β mediated downregulation of claudin-3 in epithelial cells.
In summary, this report identifies a signaling role of nmMLCK in modulating epithelial barrier function (see conclusion Figure 6) in addition to its known role in regulating cytoskeletal contractility. These results support the idea that nmMLCK may serve as a suitable drug target in restoring barrier function in conditions involving inflammation driven epithelial permeability in the gut.
Fig 6.
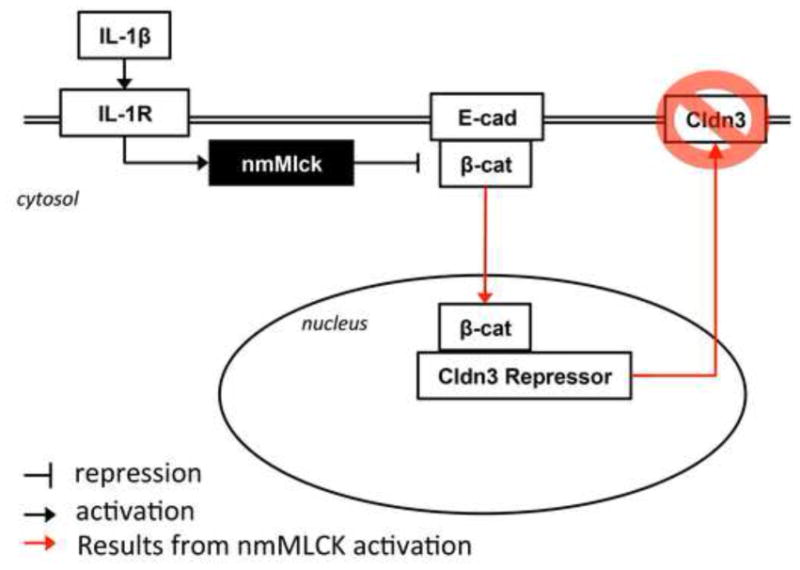
Schematic demonstrating nmMLCK regulation of claudin-3 expression resulting from IL-1β treatment involves β-catenin translocation to the nucleus and transcriptional regulation of claudin-3.
Footnotes
Disclosures: The authors have no conflict of interest to declare. The contents of this publication do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- 1.Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, Houdeau E, Fioramonti J, Bueno L, Theodorou V. Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology. 2012;37:1885–1895. doi: 10.1016/j.psyneuen.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 2.Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1β-Induced Increase in Intestinal Epithelial Tight Junction Permeability. The Journal of Immunology. 2008;180:5653–5661. doi: 10.4049/jimmunol.180.8.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beard RS, Haines RJ, Wu KY, Reynolds JJ, Davis SM, Elliott JE, Malinin NL, Chatterjee V, Cha BJ, Wu MH, Yuan SY. Non-muscle Mlck is required for beta-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1 beta-mediated barrier dysfunction in brain endothelial cells. J Cell Sci. 2014;127:1840–1853. doi: 10.1242/jcs.144550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capaldo CT, Macara IG. Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Molecular biology of the cell. 2007;18:189–200. doi: 10.1091/mbc.E06-05-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clayburgh DR, Rosen S, Witkowski ED, Wang F, Blair S, Dudek S, Garcia JG, Alverdy JC, Turner JR. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. The Journal of biological chemistry. 2004;279:55506–55513. doi: 10.1074/jbc.M408822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia JG, Schaphorst KL, Verin AD, Vepa S, Patterson CE, Natarajan V. Diperoxovanadate alters endothelial cell focal contacts and barrier function: role of tyrosine phosphorylation. Journal of applied physiology. 2000;89:2333–2343. doi: 10.1152/jappl.2000.89.6.2333. [DOI] [PubMed] [Google Scholar]
- 8.Guttman JA, Li Y, Wickham ME, Deng W, Vogl AW, Finlay BB. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cellular microbiology. 2006;8:634–645. doi: 10.1111/j.1462-5822.2005.00656.x. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto K, Oshima T, Tomita T, Kim Y, Matsumoto T, Joh T, Miwa H. Oxidative stress induces gastric epithelial permeability through claudin-3. Biochemical and Biophysical Research Communications. 2008;376:154–157. doi: 10.1016/j.bbrc.2008.08.140. [DOI] [PubMed] [Google Scholar]
- 10.Huang Q, Xu W, Ustinova E, Wu M, Childs E, Hunter F, Yuan S. Myosin light chain kinase-dependent microvascular hyperpermeability in thermal injury. Shock. 2003;20:363–368. doi: 10.1097/01.shk.0000079425.0000.db. [DOI] [PubMed] [Google Scholar]
- 11.Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. Journal of cell science. 2003;116:1959–1967. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- 12.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, Melchner Hv, Plate KH, Gerhardt H, Dejana E. Wnt/β-catenin signaling controls development of the blood–brain barrier. The Journal of Cell Biology. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. American journal of physiology Gastrointestinal and liver physiology. 2005;288:G422–430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 14.Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflammatory bowel diseases. 1999;5:262–270. doi: 10.1097/00054725-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 15.McLaughlin J, Padfield PJ, Burt JP, O’Neill CA. Ochratoxin A increases permeability through tight junctions by removal of specific claudin isoforms. American journal of physiology Cell physiology. 2004;287:C1412–1417. doi: 10.1152/ajpcell.00007.2004. [DOI] [PubMed] [Google Scholar]
- 16.Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N, Bruewer M. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. American journal of physiology Gastrointestinal and liver physiology. 2009;296:G1140–1149. doi: 10.1152/ajpgi.90534.2008. [DOI] [PubMed] [Google Scholar]
- 17.Merilainen S, Makela J, Koivukangas V, Jensen HA, Rimpilainen E, Yannopoulos F, Makela T, Alestalo K, Vakkala M, Koskenkari J, Ohtonen P, Koskela M, Lehenkari P, Karttunen T, Juvonen T. Intestinal bacterial translocation and tight junction structure in acute porcine pancreatitis. Hepato-gastroenterology. 2012;59:599–606. doi: 10.5754/hge11157. [DOI] [PubMed] [Google Scholar]
- 18.Mittal RD, Bid HK, Ghoshal UC. IL-1 receptor antagonist (IL-1Ra) gene polymorphism in patients with inflammatory bowel disease in India. Scandinavian journal of gastroenterology. 2005;40:827–831. doi: 10.1080/00365520510015629. [DOI] [PubMed] [Google Scholar]
- 19.Nalle SC, Turner JR. Intestinal barrier loss as a critical pathogenic link between inflammatory bowel disease and graft-versus-host disease. Mucosal immunology. 2015;8:720–730. doi: 10.1038/mi.2015.40. [DOI] [PubMed] [Google Scholar]
- 20.Neunlist M, Van Landeghem L, Mahe MM, Derkinderen P, des Varannes SB, Rolli-Derkinderen M. The digestive neuronal-glial-epithelial unit: a new actor in gut health and disease. Nature reviews Gastroenterology & hepatology. 2013;10:90–100. doi: 10.1038/nrgastro.2012.221. [DOI] [PubMed] [Google Scholar]
- 21.Nighot PK, Hu CA, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. The Journal of biological chemistry. 2015;290:7234–7246. doi: 10.1074/jbc.M114.597492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinton P, Nougayrede JP, Del Rio JC, Moreno C, Marin DE, Ferrier L, Bracarense AP, Kolf-Clauw M, Oswald IP. The food contaminant deoxynivalenol, decreases intestinal barrier permeability and reduces claudin expression. Toxicology and applied pharmacology. 2009;237:41–48. doi: 10.1016/j.taap.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, Collins JE. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Laboratory Investigation. 2005;85:1139–1162. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 24.Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, Raedler A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clinical and experimental immunology. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynoso R, Perrin RM, Breslin JW, Daines DA, Watson KD, Watterson DM, Wu MH, Yuan S. A role for long chain myosin light chain kinase (MLCK-210) in microvascular hyperpermeability during severe burns. Shock. 2007;28:589–595. doi: 10.1097/SHK.0b013e31804d415f. [DOI] [PubMed] [Google Scholar]
- 26.Rigor RR, Shen Q, Pivetti CD, Wu MH, Yuan SY. Myosin Light Chain Kinase Signaling in Endothelial Barrier Dysfunction. Med Res Rev. 2013;33:911–933. doi: 10.1002/med.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solanas G, Porta-de-la-Riva M, Agusti C, Casagolda D, Sanchez-Aguilera F, Larriba MJ, Pons F, Peiro S, Escriva M, Munoz A, Dunach M, de Herreros AG, Baulida J. E-cadherin controls beta-catenin and NF-kappaB transcriptional activity in mesenchymal gene expression. J Cell Sci. 2008;121:2224–2234. doi: 10.1242/jcs.021667. [DOI] [PubMed] [Google Scholar]
- 28.Sun C, Wu MH, Guo M, Day ML, Lee ES, Yuan SY. ADAM15 regulates endothelial permeability and neutrophil migration via Src/ERK1/2 signalling. Cardiovasc Res. 2010;87:348–355. doi: 10.1093/cvr/cvq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner JR. Intestinal mucosal barrier function in health and disease. Nature reviews Immunology. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 30.Verin AD, Lazar V, Torry RJ, Labarrere CA, Patterson CE, Garcia JG. Expression of a novel high molecular-weight myosin light chain kinase in endothelium. American journal of respiratory cell and molecular biology. 1998;19:758–766. doi: 10.1165/ajrcmb.19.5.3125. [DOI] [PubMed] [Google Scholar]
- 31.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. The American journal of pathology. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Gao XP, Ramchandran R, Zhao YY, Vogel SM, Malik AB. Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nature immunology. 2008;9:880–886. doi: 10.1038/ni.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan SY, Wu MH, Ustinova EE, Guo M, Tinsley JH, De Lanerolle P, Xu W. Myosin light chain phosphorylation in neutrophil-stimulated coronary microvascular leakage. Circulation research. 2002;90:1214–1221. doi: 10.1161/01.res.0000020402.73609.f1. [DOI] [PubMed] [Google Scholar]
- 34.Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]