

We hypothesize that selection during dog domestication targeted CNVs associated with hypersociability.
Abstract
Although considerable progress has been made in understanding the genetic basis of morphologic traits (for example, body size and coat color) in dogs and wolves, the genetic basis of their behavioral divergence is poorly understood. An integrative approach using both behavioral and genetic data is required to understand the molecular underpinnings of the various behavioral characteristics associated with domestication. We analyze a 5-Mb genomic region on chromosome 6 previously found to be under positive selection in domestic dog breeds. Deletion of this region in humans is linked to Williams-Beuren syndrome (WBS), a multisystem congenital disorder characterized by hypersocial behavior. We associate quantitative data on behavioral phenotypes symptomatic of WBS in humans with structural changes in the WBS locus in dogs. We find that hypersociability, a central feature of WBS, is also a core element of domestication that distinguishes dogs from wolves. We provide evidence that structural variants in GTF2I and GTF2IRD1, genes previously implicated in the behavioral phenotype of patients with WBS and contained within the WBS locus, contribute to extreme sociability in dogs. This finding suggests that there are commonalities in the genetic architecture of WBS and canine tameness and that directional selection may have targeted a unique set of linked behavioral genes of large phenotypic effect, allowing for rapid behavioral divergence of dogs and wolves, facilitating coexistence with humans.
INTRODUCTION
Although decades of research have focused on the unique relationship between humans and domestic dogs, the role of genetics in shaping canine behavioral evolution remains poorly understood. Existing hypotheses on the behavioral divergence between dogs and wolves posit that dogs are more adept at social problem solving (1) because of an evolved human-like social cognition (2, 3). However, mounting evidence suggests that human-socialized wolves can match or exceed the performance of domestic dogs across these sociocognitive domains (4). Empirical demonstrations remain robust that dogs display exaggerated gregariousness, referred to as hypersociability, which is a heightened propensity to initiate social contact that is often extended to members of another species, when compared with wolves into adulthood. Hypersociability, one facet of the domestication syndrome (5), is a multifaceted phenotype that includes extended proximity seeking and gaze (6, 7), heightened oxytocin levels (6), and inhibition of independent problem-solving behavior in the presence of humans (8). This behavior is likely driven by behavioral neoteny, which is the extension of juvenile behaviors into adulthood and increasing the ability for dogs to form primary attachments to social companions (4).
Because of strict selective breeding rules, distinct dog breeds conform to a predictable phenotype. This population structure and isolation present the dog as a powerful model to explore the genetic underpinnings of complex traits such as behavior (9). Many dog breeds have been collectively scored using standardized tests for behavioral personality traits central to their domesticated nature (for example, playfulness, sociability, aggression, trainability, curiosity, or boldness) and breed-specific function (for example, herding, pointing, chasing, working) (9–17). Although there has been strong selection for breed conformation, interindividual variation suggests that genetics play a detectable role in shaping canine social behavior (18).
Here, we focus on sociability as informative on the divergence process of dogs from wolves during domestication. This canine behavioral gestalt was previously implicated in phenotype evolution in the dog genome through a genome-wide association scan of more than 48,000–single-nucleotide polymorphism (SNP) genotypes from 701 dogs from 85 breeds and 92 gray wolves with a Holarctic distribution (19). Using divergence, the top ranking outlier site was located within SLC24A4, a gene known to contain polymorphisms linked to eye and hair color variation in humans (19). The second ranking site was located within WBSCR17, a gene implicated in Williams-Beuren syndrome (WBS) in humans. WBS is a neurodevelopmental disorder caused by a 1.5- to 1.8-Mb hemizygous deletion on human chromosome 7q11.23 spanning approximately 28 genes (20). This syndrome is characterized by delayed development, cognitive impairment, behavioral abnormalities, and hypersociability (21–23). A number of other studies have taken a different approach and targeted genes linked to social behavior in other taxa. For example, targeted variation was surveyed in the dopamine receptor D4 and tyrosine hydroxylase, both genes extensively studied for their roles in the primate brain’s reward system (24). The study found an association between longer repeat polymorphisms with lowered activity and impulsivity in a limited survey of breeds. In a similar approach, variation surveyed at a regulatory SNP in the oxytocin receptor gene, also known to influence human pair bonding, was found to be associated with proximity seeking and friendliness in two dog breeds (25). However, behavioral genetic studies are still plagued with the challenge to understand the genetic architecture of nearly every facet of a complex behavior. Our study seeks to overcome this obstacle in the canine model.
We focus on a candidate chromosomal region implicated in canine sociability, a trait arguably more central to the domestication process than increased social cognition, and the adjacent orthologous region that has been mapped to human WBS. We demonstrate that domestic dogs exhibit some of the key behavioral traits quantified in individuals with WBS, most notably hypersociability in the absence of superior social cognition. We integrate targeted resequenced data of the candidate canine WBS region with behavioral measures of sociability and cognition to disentangle the genetic underpinnings of this multifaceted behavioral trait. We find strong evidence that structural variation (SV) in our target region, which is orthologous to the region of the human genome affected by SV in WBS, also contributes to hypersociability in domestic dogs.
RESULTS
Solvable tasks and sociability measures
We evaluated the human-directed sociability of 18 domestic dogs and 10 captive human-socialized gray wolves using standard sociability (7, 26) and problem-solving tasks (2, 8, 27) commonly used to assess human-directed sociability in canines. Three sociability metrics were constructed to assess behaviors indicative of WBS (22): attentional bias to social stimuli (ABS), hypersociability (HYP), and social interest in strangers (SIS) (tables S1 and S2). Solvable task performance was used to assess attentional bias toward social stimuli and independent problem-solving performance (independent physical cognition). Subjects were given up to 2 min to open a solvable puzzle box (8) that contained half of a 2.5-cm-thick piece of summer sausage, both when alone and with a neutral human present. The trial was considered complete after meeting one of the following conditions: The puzzle box lid was completely removed, the food was obtained, or 2 min had elapsed. All trials were video-recorded and coded for whether the puzzle box was solved and the time to solve it. To compare attention toward the puzzle box versus social stimuli under the human-present condition, we recorded the percentage of time spent looking at the puzzle box, touching the puzzle box, and looking at the human (8). We also had an independent researcher, who was blind to the purpose of this study, code 30% of the videos and found that interrater reliability was very strong (weighted Cohen’s kappa, κ = 0.98; 95% confidence interval, 0.97 to 0.99). Consistent with our hypothesis, domestic dogs spent a significantly greater proportion of trial time gazing at the human when compared to wolves when a human was present during the solvable task (median gaze toward human: dog, 21%; wolf, 0%; two-tailed Mann-Whitney, ndog = 18, nwolf = 10, U = 6, P < 0.0001). Dogs also spent a significantly smaller proportion of trial time looking at the puzzle box (median gaze towards box: dog, 10%; wolf, 100%; two-tailed Mann-Whitney, ndog = 18, nwolf = 10, U = 171.5, P = 0.0001) and a significantly smaller proportion of trial time trying to solve the puzzle (median: dog, 6%; wolf, 98%; two-tailed Mann-Whitney, ndog = 18, nwolf = 10, U = 175, P < 0.0001) compared to wolves, a finding that has been equated with social inhibition of problem-solving behavior in both the canine and human WBS literature (19, 22). Significantly more wolves successfully solved the task when compared to dogs under both the human present and alone conditions (human present: 2 of 18 dogs are successful, 8 of 10 wolves are successful; two-tailed Fisher’s exact test, P = 0.0005; alone: 2 of 18 dogs are successful, 9 of 10 wolves are successful; two-tailed Fisher’s exact test, P = 0.0001). Overall, concordant with WBS, dogs displayed greater ABS than wolves did, corresponding to a reduction in independent problem-solving success (fig. S1).
The sociability test measured human-directed proximity-seeking behavior and was assessed by comparing total sociability scores across all sociability conditions. Each phase occurred twice, once with an unfamiliar human and once with a familiar human, totaling four phases run over eight consecutive minutes. In all phases, the experimenter sat on a familiar chair (dogs) or bucket (wolves) inside a marked circle of 1-m circumference denoting proximity. During the passive phase, the experimenter sat quietly on the chair or bucket and ignored the subject by looking down toward the floor. If the animal sought physical contact, then the experimenter touched the subject twice but did not speak or make eye contact with the animal. During the active phase, the experimenter called the animal by name and actively encouraged contact while remaining in their designated location. Consistent with our hypothesis, dogs spent more time in proximity to humans than did wolves (median percent of time spent within 1 m of humans: dogs, 65%; wolves, 35%; two-tailed Mann-Whitney, ndog = 18, nwolf = 9, U = 30, P < 0.005). Dog and wolf sociability toward an unfamiliar human was used to assess SIS. Consistent with our hypothesis, dogs spent more time within 1 m of a stranger when compared to wolves (median: dogs, 53%; wolves, 28%); however, this difference was not statistically significant (two-tailed Mann-Whitney, ndog = 18, nwolf = 9, U = 76, P = 0.51). In summary, dogs were hypersocial compared to wolves, although there was no significant difference in their SIS (fig. S1).
We reduced the dimensionality of six behavioral traits (table S3) to three components that are orthogonal and uncorrelated to each other, whereas ABS, HYP (hypersociability), and SIS are correlated. Principal component 1 (PC1), PC2, and PC3 accounted for 50, 22, and 14% of total behavioral variation, respectively. We have calculated both Kaiser-Meyer-Olkin (KMO) (KMO = 0.62, with values of >0.6 recommended as informative) and Bartlett’s test, which was significant [χ2(15) = 60.42, P = 2.13 × 10−07]. Analysis of the loadings of the constituent behaviors (table S3 and fig. S1) indicated that PC1 represents an autonomous or independent phenotype, as this component is negatively correlated with all behaviors associated with human-directed sociability with the exception of “proximity unfamiliar passive.” PC1 also had positive loadings from “time look object,” a measure indicating a lack of ABS (fig. S2). Loadings of each behavior were roughly equal, with the exception of proximity unfamiliar passive, which had a loading approximately one-third the average magnitude of the others. Loadings of PC2 were heavily biased toward, and positively associated with, the measures of proximity to an unfamiliar person (average loading of 0.64, as compared to an average loading of −0.14 for the other loadings), suggesting that PC2 reflects boldness. The biological meaning of PC3 is more difficult to interpret, but given that it is strongly and positively loaded by the behavior “time look human” (loading of 0.63 compared to an average loading for all other factors of −0.15), it predominantly reflects reliance on humans in the solvable task test. As expected, given the interpretation of PC1 as socially inhibited phenotype, dogs had lower PC1 values than wolves (Mann-Whitney U test, U = 3, P < 0.00005; median: dogs, −1.18; wolves, 2.31). Dogs and wolves did not have significantly different values for PC2 (Mann-Whitney U test, U = 54, P = 0.57; median: dogs, −0.18; wolves, −0.19) or for PC3 (Mann-Whitney U test, U = 48, P = 0.35; median: dogs, −0.069; wolves, 0.011).
De novo annotation of structural variants
In a subset of animals with quantitative behavioral data (ndog = 16, nwolf = 8), we collected paired-end 2x67nt sequence data from 5 Mb spanning the candidate canine WBS locus on canine chromosome 6 [2,031,491 to 7,215,670 base pairs (bp)], which contains 46 annotated genes, 27 of which are in the human WBS locus (tables S4 and S5; see Materials and Methods). The target region had an average of 15.5-fold sequence coverage (dogs, 15.2; wolves, 16.0) (table S5). We obtained genotypes for 26,296 SNPs, which we further filtered to retain 4844 SNPs with nonmissing polymorphic data (average density of 1 SNP for every 14.4 kb). To confirm this region as containing species-specific variation, we first determined whether this region displays signals of positive selection in the dog genome, an effort to independently validate the original (19). We calculated the composite bivariate percentile score and confirmed that the candidate gene, WBS chromosome region 17 (WBSCR17), is under positive selection as a domestication candidate and was significantly depleted of heterozygosity in dogs (mean HO: dog, 0.01; wolf, 0.37; one-tailed t test with unequal variance, P = 7.4 × 10−38) (fig. S3 and table S6).
Because this candidate region shows SV linked to WBS in humans (20) and is known to vary widely in its functional consequences [for example, neurodevelopmental diseases (28) and autism spectrum disorders (29)], we completed in silico SV annotation in the dog and wolf genomes using three programs—SVMerge (30), SoftSearch (31), and inGAP-sv (32), which together use all available SV detection algorithms: read pair (RP), short reads (SR), read depth (RD), and assembly-based (AS). We annotated 38 deletions, 30 insertions, 13 duplications, 6 transpositions, a single inversion, and 1 complex variant relative to the reference dog genome (tables S7 and S8). There was considerable private variation, with 31 annotated SVs found only in dogs, 26 found only in wolves, and a level of heterogeneity observed in wolves that is comparable to that found in human WBS (mean n: wolf, 21; dog, 15; two-tailed t test, P = 0.026) (table S9) (33).
Candidate region association test
Linear mixed models were used to determine the association of SVs with human-directed sociability. Three univariate models were tested for their association with each of the three behavioral indices (ABS, HYP, and SIS) (Fig. 1). In addition, we tested for the association of SVs with the three behavioral indices collectively, referred to as the behavioral index model, and separately with a model that included the first three PCs (PC model) describing human-directed sociable behavior (Fig. 2). Four genic SVs were significantly associated with human-directed social behavior (adjusted P < 2.38 × 10−3): one SV within GTF2I (Cfa6.66), one SV within GTF2IRD1 (Cfa6.72), and two within WBSCR17 associated with ABS (Cfa6.3 and Cfa6.7) (Table 1). In addition, two intergenic SVs were significantly associated with ABS (Cfa6.69, P = 1.56 × 10−4; Cfa6.27, P = 3.31 × 10−4), and Cfa6.27 was also associated with the PCs (P = 1.24 × 10−4). However, we focused our analyses on genic SVs to infer any potential functional impact. Cfa6.66 was associated with multiple sociability metrics (ABS and SIS) and had the strongest two association signals (P = 1.38 × 10−4 and P = 1.95 × 10−4, respectively) (Table 1). GTF2I and GTF2IRD1 are members of the transcription factor II-I (TFII-I) family, a set of paralogous genes that have been repeatedly linked to the expression of HYP in mice (34, 35), and are specifically implicated in the hypersociable phenotype of persons with WBS (36, 37).
Fig. 1. Association of structural variants with indices of human-directed social behavior.
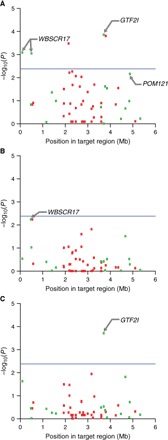
Association with ABS (A), HYP (B), and SIS (C). Manhattan plots show statistical significance of each variant as a function of position in target region. Blue line denotes statistical significance to Bonferroni-corrected level (P = 2.38 × 10−3). Genic and intergenic variants are shown as green and red boxes, respectively.
Fig. 2. Association of structural variants with human-directed social behavior in multivariate regressions.
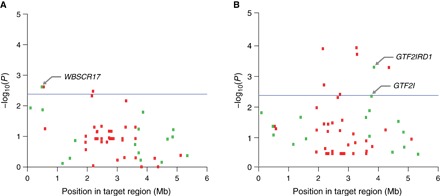
Association in behavioral index model (A) and PC model (B). Manhattan plots show statistical significance of each variant as a function of position in the target region. Blue line denotes significance to Bonferroni-corrected level (P = 2.38 × 10−3); dashed purple line denotes suggestive significance (P = 0.01). Genic and intergenic variants are shown as green and red boxes, respectively.
Table 1. Genic loci associated with indices of human-directed social behavior across dogs and wolves.
NA, not applicable.
| Phenotype | Locus ID | SV type |
Position (Mb)* |
β (SE) |
% Variation explained |
P† |
Candidate gene |
| ABS | Cfa6.66 | Deletion | 5.75 | 0.23 (0.09) | 4.45 | 1.38 × 10−4 | GTF2I |
| Cfa6.3 | Insertion | 2.14 | 0.11 (0.07) | 0.56 | 8.12 × 10−4 | WBSCR17 | |
| Cfa6.7 | Deletion | 2.54 | 0.12 (0.07) | 0.62 | 8.89 × 10−4 | WBSCR17 | |
| SIS | Cfa6.66 | Deletion | 5.75 | −27.0 (24.80) | 11.45 | 1.95 × 10−4 | GTF2I |
| Top three principal components (PC model) |
Cfa6.72 | Deletion | 5.90 | 0.04 (0.052), −0.96 (0.57), 1.11 (0.36) |
NA | 4.98 × 10−4 | GTF2IRD1 |
*See table S7 for locus details.
†P values from likelihood ratio test (adjusted significance threshold P = 2.38 × 10−3).
To disentangle the association of SVs with behavior from an association with species membership, we incorporated species as a covariate (table S10). These analyses were consistent with our initial findings for Cfa6.66, Cfa6.3, and Cfa6.7. Locus Cfa6.66 remained significantly associated with multiple sociability metrics (ABS, P = 2.33 × 10−4; SIS, P = 1.67 × 10−3) and showed the strongest association of any genic SV. Cfa6.3 and Cfa6.7 both retained their associations with ABS (P = 1.06 × 10−3 and P = 9.56 × 10−4, respectively), as did the intergenic SVs Cfa6.69 (P = 1.36 × 10−4) and Cfa6.27 (P = 5.56 × 10−4). Furthermore, the ABS effect size (β) remained stable for the association models with and without species membership as a covariate (ABS β without covariates: Cfa6.3 = 0.11, Cfa6.7 = 0.12, Cfa6.27 = −0.15, Cfa6.66 = 0.23, Cfa6.69 = −0.15; ABS β with covariates: Cfa6.3 = 0.081, Cfa6.7 = 0.10, Cfa6.27 = −0.13, Cfa6.66 = 0.23, Cfa6.69 = −0.14), indicating that the observed effects on sociability are not an artifact of species differences. An association test of each locus with species membership further supports this interpretation as none of the behavior-associated SVs significantly associated with species membership alone (table S11).
Functional impact of annotated structural variants
We next determined whether these behavior-associated SVs were predicted to have a functional impact. We used Ensembl’s Variant Effect Predictor (VEP) v84 (38) with Ensembl transcripts for the CanFam 3.1 reference genome to assign putative functional consequences to all insertions, deletions, and duplications in the filtered set of SVs. Because of a software limitation that VEP is unable to assign consequences for transitions, inversions, and complex SV, we manually inspected seven sites (six TRA, one INV, and one D_I) in the UCSC (University of California, Santa Cruz) genome browser with Ensembl gene models (39). We found three transcription ablations, seven loss-of-start codons, and five transcript amplifications (table S12). All SVs significantly associated with human-directed social behavior were “feature truncations,” except for Cfa6.3, which was a “feature elongation” that is likely due to a lost stop codon or the elongation of an internal sequence feature relative to the reference. Annotation of Cfa6.3, Cfa6.7, Cfa6.66, and Cfa6.72 as modifiers of gene function suggests a direct association between these variants and human-directed social behavior, as quantified by our behavioral measures, mediated by possible interference with WBSCR17, GTF2I, and GTF2IRD1.
PCR validation and analysis of structural variants
The in silico SV detection algorithms applied to the targeted resequencing data can identify the presence or absence of an SV but cannot predict the underlying genotype of an individual for a given SV. To corroborate the in silico findings and investigate the possibility of other genetic models, we used polymerase chain reaction (PCR) amplification and agarose gel electrophoresis to determine the codominant genotypes at the top four loci (Cfa6.6, Cfa6.7, Cfa6.66, and Cfa6.83) (fig. S4). These four SVs overlapped with short interspersed nuclear transposable elements (TEs) with high sequence identity to the reference (182 to 259 bp; 91 to 96% pairwise identity over 193 bp). We further surveyed insertional variation in 298 canids consisting of coyotes, gray wolves (representing populations from Europe, Asia, and North America), American Kennel Club (AKC)–registered breeds, and semidomestic dog populations (see Materials and Methods). We repeated the analysis with the codominant SV genotypes to determine whether there was an association with species membership. Coyotes were excluded from this analysis, and semi-domestic dogs were grouped with domestic dog.
All outlier SVs, now with codominant genotypes, were significantly associated with species membership [Cfa6.6: χ2 = 23.91; P = 1.01 × 10−6; odds ratio (OR), 0.33; Cfa6.7: χ2 = 57.63; P = 3.16 × 10−14; OR, 13.83; Cfa6.66: χ2 = 35.12; P = 3.1 × 10−9; OR, 0.25; Cfa6.83: χ2 = 17.11; P = 3.53 × 10−5; OR, NA), confirming this region’s original identification (19). Similar results were obtained if we only included “modern” breeds, as per the original method that located this region (Cfa6.6: χ2 = 11.9; P = 0.0006; OR, 0.45; Cfa6.7: χ2 = 40.87; P = 1.63 × 10−10; OR, 10.35; Cfa6.66: χ2 = 41.97; P = 9.25 × 10−11; OR, 0.20; Cfa6.83: χ2 = 20.41; P = 6.24 × 10−6; OR, NA) (19), with site-specific patterns (frequency of TE insertion in modern dogs and wolves, respectively: Cfa6.6, 0.52 and 0.32; Cfa6.7, 0.39 and 0.06; Cfa6.66, 0.10 and 0.37; Cfa6.83, 0.17 and 0.00).
We calculated the frequency of insertions per locus by population or species membership. The TEs segregated at low frequencies in coyotes and were variable across wolf populations and dog breeds (fig. S5). Only one coyote carried a single insertion of the TE at locus Cfa6.6, with both Cfa6.6 and Cfa6.7 highly polymorphic across domestic dogs (fig. S5, B and C). Locus Cfa6.66 is found in wolves from China, Europe, and the Middle East and in the WBS study wolves, but only within six dog breeds (boxer, basenji, cairn terrier, golden retriever, Jack Russell terrier, and Saluki), the WBS dogs, two New Guinea singing dogs (NGSDs), and a single pariah dog (fig. S5D). Cfa6.83 appears to be a de novo insertion within domestic dogs because it is lacking entirely within the wild canids (fig. S5E), with a low to moderate frequency within the semidomestic dog populations surveyed (pariah dog, n = 1; village dogs: Africa, n = 1; Puerto Rico, n = 5). Genetic analysis of only WBS dogs and wolves only, coupled with behavioral data, revealed trends per locus as follows: More insertions at Cfa6.6 were correlated with increased ABS and HYP (r = 0.50 and 0.42, respectively), with weaker relationships for SIS (r = 0.11); more insertions at Cfa6.7 correlated with increased ABS and HYP, with an inverse relationship with SIS (r = 0.13, 0.11, and −0.17, respectively); fewer insertions at Cfa6.66 is correlated with higher trait values (r = −0.59, −0.56, and −0.27 for ABS, HYP, and SIS, respectively); more insertions at Cfa6.83 increased all behavioral trait values (r = 0.36, 0.44, and 0.40 for ABS, HYP, and SIS, respectively).
We conducted one-way analysis of variance (ANOVA) using the population or species designation as a predictor of the total number of insertions across four outlier loci. The total number of insertions depends significantly on the population (F23,274 = 19.54, P < 2 × 10−16), with 103 of 276 pairwise population mean comparisons contributing to the ANOVA significance (dog/dog, 46; wolf/dog, 28; coyote/dog, 11; semidomestic/dog, 8; semidomestic/coyote, 3; semidomestic/wolf, 3; wolf/coyote, 2; wolf/wolf, 2; Tukey’s post hoc test, P < 0.05) (fig. S6).
Because the gel-based genotyping method now reveals a codominant genotype compared to the in silico status, we conducted an association scan for each of the four outlier SV loci with the binary phenotype for each AKC breed (40), village dogs, and pariah dogs as “seeks attention” or “avoids attention” using two logistic regression models in R, an additive and dominant model, with sex as a covariate. The use of breed-based stereotypes is supported by the strict genetic isolation and selective breeding efforts that maintain breeds. Hence, many traits strongly determined by genetic variation (including behavioral) can be predicted with high accuracy. The central foundation and advantage of domestication and breed formation are that selection for many traits, including behavior, has been very strong; thus, the number of underlying genes is apt to be small. As proof of principle, Jones et al. (9) successfully mapped a variety of breed-associated traits in a genome-wide association study using dog “stereotypes.” They scored breeds for pointing, herding, boldness, and trainability and identified one locus associated to pointing, three for herding, one for trainability, and, most importantly, five for boldness. These loci contain likely candidate genes, many of which are important in schizophrenia, dopamine receptors, and proteins linked to synaptic junctions. Vaysse et al. (16) also used breed stereotypes to map behaviors, such as boldness, sociability, curiosity, playfulness, chase-proneness, and aggressiveness. They mapped boldness to an intron of HMGA2 and sociability, defined as the “dog’s attitude toward unknown people,” to a gene on the X chromosome after excluding male dogs from the analysis to accurately compare autosomal and sex-chromosome patterns of genetic variation.
We found significant support for an association between three of the four loci and the binary behavioral trait of seeking or avoiding attention (additive model: Cfa6.6, OR, 0.303; P = 2.79 × 10−10; Cfa6.7, OR, 0.398; P = 4.66 × 10−7; Cfa6.83, OR, 2.95; P = 2.83 × 10−4; dominant model: Cfa6.6, OR, 0.184; P = 8.22 × 10−7; Cfa6.7, OR, 0.287; P = 4.31 × 10−5; Cfa6.83, OR, 5.04; P = 6.50 × 10−4; sex was not a significant predictor in any of these models). SV Cfa6.66 was not significant (additive model: OR, 0.852; P = 0.496; dominant model: OR, 0.573; P = 0.124). Further, our logistic regression found that TE copy number could significantly predict the binary breed stereotype behavior of attention seeking or avoidance (OR, 0.676 per insertion; P = 1.13 × 10−5, with no evidence of a sex effect).
Genome-wide SNP survey
To identify additional candidate loci, we collected genome-wide SNP genotypes using the Affymetrix Axiom K9HDSNPA (643,641 loci) and Axiom K9HDSNPB (625,577 loci) arrays. We first conducted a principal components analysis (PCA) on these genome-wide SNP genotypes to ensure the expected spatial clustering pattern of the samples. With a subset of 25,510 uncorrelated and unlinked SNPs, a PCA confirmed the discrete spatial separation of the two species (PC1, 29.9%; PC2, 11.8%) (fig. S7). This finding was further supported by high-average genome-wide differentiation (FST = 0.194), a level comparable to the original finding (19). We next conducted a binary association test on species membership in GEMMA and found support for the candidate locus WBSCR17 as containing species-specific variation (P < 3 × 10−6). Further, we tested each of the quantitative behavioral indices (ABS, HYP, and SIS) in a univariate regression analysis and identified 222 additional SNPs within our 5-Mb target region associated with two behavioral traits (HYP: nSNPs = 84, mean P = 0.002; SIS: nSNPs = 138, mean P = 0.001). Our quantitative association testing identified 77,889 SNPs outside of the resequenced region associated with each behavioral trait (SNPs: ABS, n = 874; HYP, n = 19,373; SIS, n = 57,642; P < 0.005), implicating 221 genes associated with ABS, 3520 genes with HYP, and 3118 genes with SIS. Of these, only a single-gene ontology term associated with ABS (phosphoric ester hydrolase activity), 30 terms with HYP, and 26 with SIS (tables S13 and S14).
DISCUSSION
We present the first study to use behavioral phenotyping and genomic methods to address the underlying genetics of personality and behavioral traits in domestic dogs. We identified and resequenced a candidate locus associated with WBS in humans and known to be under positive selection in the domestic dog genome (19). We found that this region also harbors a large number of highly polymorphic SVs in canines, some of which are private to an individual dog or breed. This finding is concordant with the genetic heterogeneity of WBS in humans, where deletions range from 100 kb to 1.8 Mb in size with variable breakpoints, attributed to chromosomal instability (41–43). Therefore, it is not surprising that the same is true for dogs. Here, we identified SVs found in multiple individuals that were significantly associated with one or more quantified behavioral traits informative on HYP and cognition.
Notably, our study revealed a statistically significant association between SVs in GTF2I and GTF2IRD1, basal transcription factors that regulate vertebrate development (44–48), with measures of human-directed social behavior typical of WBS. Haploinsufficiency of GTF2I and GTF2IRD1 has been repeatedly linked to HYP in knockout mice and WBS patients (34, 35, 37, 48, 49). Tellingly, WBS patients with intact GTF2I and GTF2IRD1 did not exhibit HYP (36, 46). Furthermore, a recent study linked GTF2I polymorphisms to social context–dependent salivary oxytocin levels in humans, suggesting a possible mechanism by which GTF2I may exert its effects on sociability (50). The copy number variation associated with WBS is known to reduce transcription of both genes within and flanking the hemizygous deletions, a molecular signature also found in other human syndromes (for example, Smith-Magenis syndrome and DiGeorge syndrome) (42, 51). The causal SVs have been confirmed in a mouse model to reduce transcription, consistent with changes in gene dosage, and result in HYP, delayed growth rates, and cognitive defects (35).
Our third described gene, WBSCR17, has not been previously associated with sociability. However, this gene is up-regulated in cells treated with N-acetylglucosamine, a glucose derivative, suggesting a role in carbohydrate metabolism (52). SVs in WBSCR17 may represent an adaptation to a starch-rich diet typical of living in human settlements, a speculation concordant with a previous study (53).
Two of the SVs most associated with HYP, a trait uniquely displayed in domestic dogs among the canids, were SINE (short interspersed nuclear element) and LINE (long interspersed nuclear element) TEs, subtypes of retrotransposons that have high rates of insertion [for example, 1 in 108 human births has a de novo L1 insertion (54)]. With large phenotypic consequences due to the amplification of a few loci, these mobile elements have been implicated in the evolution of the canid genome (55, 56), as well as canine disease, syndromes, and morphology (57–62). Because of their recent development and strong selective breeding, a simple genetic architecture controlling many canine traits is expected. This has been well documented for a number of canine complex traits, such as behavior (16, 63, 64), coat color (59, 65), body size (60), and leg length (61).
We surveyed these TEs in an extended sampling of wild and domestic canines and found them to be extremely rare in coyotes, whereas other insertions were derived and found only to segregate within domestic dogs. With a larger sample size and leveraging behavioral phenotypes from breed stereotypes, we found a significant association between TE copy number and behavior. Hence, it is conceivable that selection acting on HYP-associated TEs may have helped shape the evolution of the canid family. We further suggest that canine WBS-linked SVs likely contribute to the developmental delay that facilitates ease of forming interspecies bonds and the juvenile-like HYP exhibited toward these social companions into adulthood. This coupling presents an intriguing parallel to the same processes observed in WBS-affected individuals (20). Together, these findings suggest a major role for the TFII-I family of transcription factors in a defining behavioral phenotype of domestic dogs, thereby mapping canine HYP to the genes associated with HYP in humans with WBS. Our study exemplifies the successful strategy of canine genetic studies to fine-map a heterogeneous region, informed by and relevant to an orthologous complex human trait.
In light of our findings, we propose a unifying hypothesis to explain one aspect of canid domestication, where individuals with hypersocial tendencies were favored under selective breeding, accentuating a behavior likely influenced by SVs in the canine WBS locus. Unlike the “human-like social cognition” hypothesis of domestication (3), which argues that dogs developed advanced forms of social cognition otherwise unique to human beings, the HYP hypothesis presented here posits that adult dogs show exaggerated motivation to seek social contact, which is absent in adult wolves. Our findings provide insight into one genetic mechanism by which the hypersocial response of domestic dogs toward humans compared with human-reared wolves can be acted on and shaped by selection during species domestication. This mechanism is expected to predispose dogs for hypersocial responses toward any bonded companion. This is consistent with the finding that domestic dogs appear to maintain, or even increase, the duration of social engagements with humans and conspecifics as they approach adulthood, with the opposite trend found in wolves (66). In summary, our findings suggest that the same region affected by structural variants in human WBS is associated with the exuberant sociability of domestic dogs. The evidence presented here represents a shift regarding the role of domestication in the evolution of canine behavior, from a vehicle of advanced social cognition to one of HYP.
MATERIALS AND METHODS
Experimental design
Behavioral data
All behaviors are an interaction of genes and individual experience. We have previously argued that some of the behaviors that have been taken to be domestic- or wild species–specific are in fact the consequence of differing individual life experiences, including socialization to human beings during the sensitive period for social development, of difference in developmental stage, or of inequivalent testing environments or conditions (4, 67).
Here, we ensured that both dogs and wolves were in the same developmental stage by only including subjects over 1 year of age, well past the species-specific window for primary socialization. All dogs and wolves were socialized to humans as puppies, received daily contact from human caretakers, and experienced regular free-contact interactions with unfamiliar humans from puppyhood through the time of this study. To ensure that the wolves used in this study had been socialized to accepted standards and were as familiar to their caretakers as possible, we only included wolves that had been hand-reared by humans from before 10 to 14 days of age following the procedures established by Klinghammer and Goodman (68) and those that were still living in the same facility in which they were raised. Wolves experienced 24-hour contact with human caretakers for at least the first 6 weeks of life, followed by contact during daylight hours until 4 months of age and then daily human interaction with caretakers and other humans thereafter. Therefore, in the current study, the lower level of sociability displayed toward familiar individuals by wolves in comparison to pet dogs could not be explained by lack of initial bond formation (socialization) or insufficient familiarity with their caretakers. Wolves did show social interest in their caretakers, approaching them for greetings when they entered during the sociability test in this study. However, they then returned to other activities. This pattern of behavior might be considered a “typical” social greeting for bonded adult animals, whereas the prolonged greeting of pet dogs, sometimes lasting the full 2 min, would be considered exaggerated or hypersocial (7).
To ensure equivalent testing conditions, each species was tested in a controlled setting most constant with their home environment (69); dogs were individually tested at an indoor location in Corvallis, Oregon, USA; wolves were tested in a familiar outdoor enclosure at Wolf Park, Battle Ground, Indiana, USA. Testing procedures were the same for both species. Each subject was assessed using two tests designed to quantitatively probe their human-directed sociability along indices relevant to the clinical presentation of WBS: a solvable task test and a sociability test (7, 8). Data from the solvable task test and sociability test were used to calculate three indices relevant to behaviors that typify WBS in humans: ABS, HYP, and SIS (table S15). Those tests are described in detail in the following sections.
Solvable tasks and sociability measures
The solvable task test was used to measure individual problem-solving performance, attentiveness to humans, and the degree to which a familiar human’s presence interfered with independent problem-solving behavior. Although this problem-solving task is considered challenging, it has previously been validated as physically solvable by wolves, small dogs, and large dogs (8). All subjects were naïve to the problem before testing, and humans were instructed to remain passive and neutral after placing the container on the ground.
The sociability test consisted of a passive and an active phase, each lasting for 2 min. One wolf (ID 2794) was not available for sociability testing; therefore, sociability analysis was conducted on all 18 dogs and 9 wolves. The experimenter spoke to and touched the subject if the animal came close enough to reach while remaining on the bucket or chair. If the animal moved away, then the experimenter called his/her name again to regain the subject’s attention. All trials were recorded on video. For each condition, videos were coded for time spent in proximity to the experimenter and time spent touching the experimenter (7). An independent coder blind to the purpose of this study double-coded 42% of these videos; interrater reliability was determined to be strong using a weighted Cohen’s kappa, κ = 0.75 (95% confidence interval, 0.64 to 0.86) (70).
It should be noted that many of the wolves in the current study have participated and performed as well as or better than pet domestic dogs on tasks related to social cognition (using human cues to solve problems) (26). Here, they quickly approached the humans to initiate a greeting or to receive the puzzle box. The key difference we observed was that adult dogs were more likely to engage in prolonged or exaggerated contact with humans than adult wolves.
Behavioral indices relevant to WBS in humans
Data from the solvable task test and the sociability test were used to quantify canine behavior along indices relevant to the sociable phenotype of WBS, including (i) time spent looking at the puzzle box in the solvable task test (time look box), (ii) time spent looking at the human in the solvable task test (time look human), time spent in proximity to a familiar experimenter in the (iii) active and (iv) passive phases of the sociability test (“proximity familiar active” and proximity familiar passive), and time spent in proximity to an unfamiliar experimenter in the (v) active and (vi) passive phases of the sociability test (“proximity unfamiliar active” and “proximity unfamiliar passive”).
Data from the solvable task test and sociability test were used to calculate three indices relevant to the behavior under selection during dog domestication and analogous to behaviors that typify WBS in humans: ABS, HYP, and SIS. ABS was calculated as the ratio of time spent looking at the experimenter to the sum of the time spent looking at the experimenter and the time spent looking at the puzzle box in the solvable task test and was intended to quantify the proportion of the animal’s attention directed toward the experimenter. HYP was calculated as the sum of the time spent in proximity to the experimenter in each phase of the sociability test and was intended to quantify engagement with humans across social scenarios. SIS was calculated as the sum of the time spent in proximity to the experimenter in the two unfamiliar phases of the sociability test and was intended to quantify engagement with unfamiliar persons (tables S2 and S15).
PCA of behavioral indices
Dog and wolf behavior was also characterized by PCA using data from the solvable task test (8) and sociability test (table S2) (69) with the prcomp function in R (www.r-project.org/). Inclusion of PCs was assessed with the nFactors package in R (71). Most of the component retention analyses indicated inclusion of the top two PCs (Kaiser’s rule, 2; Horn’s parallel analysis, 2; acceleration factor, 2; optimal coordinates, 1). However, we found a relatively low percentage of behavioral variation explained by the first two PCs (cumulatively, 72%) and a lack of an obvious knee in the scree plot (fig. S2). In addition, previous research has shown that inclusion of a greater number of phenotypic PCs significantly increases the power of genome-wide associations (72). Therefore, the top three PCs were selected for use as phenotypes in regression analyses.
Genetic sample collection and genomic enrichment
Following behavioral trials, 2 to 3 ml of blood was collected from each dog and wolf from the cephalic, saphenous, or jugular vein, depending on the individual, temperament, and accessibility of the vein. Blood was deposited into a sterile blood collection tube, labeled, and then immediately placed in a freezer kept below −18°C until shipped overnight on ice for analysis. We chose 24 of 28 samples to sequence (dogs, n =16; wolves, n = 8). Two of the original 18 dogs were removed from sequencing because of their low DNA yield; 2 of the original 10 wolves were excluded from sequencing because of the lack of an opportunity to redraw blood samples from these individuals, either due to our institutional protocols or due to the unavailability of the individual (tables S1 and S5). We prepared genomic DNA from blood samples using QIAamp DNA mini kits (DNeasy Blood and Tissue kit, Qiagen). DNA was quantified using a Qubit 2.0 Fluorometer and checked on a 2% agarose gel for degradation. We followed up on a region under positive selection in the domestic dog genome on chromosome 6 that was identified from a genome-wide scan of 48,036 SNPs (19), through targeted resequencing of a ~5-Mb contiguous block (2,031,491 to 7,215,670 bp) that contained 46 Ensembl-annotated genes (39, 73), 27 of which have been described in WBS (table S4). We used a full-service option offered by MYcroarray for DNA enrichment and genomic library preparation. We designed 80-nucleotide oligomer bait probes to target the region of interest (MYbaits kit design). Genomic DNA was sonicated to approximately 300-bp fragment sizes, 500 ng of which was used to construct Illumina TruSeq sequencing libraries. Each library was dual index–amplified for eight cycles of PCR, yielding between 590 and 1744 ng of the sequencing library. Of this, 500 ng was used for the target enrichment with our custom MYbaits kit. Following enrichment, libraries were amplified for six cycles, yielding between 6.7 and 14.7 ng of the library. Libraries were standardized by pooling 5 ng from each library to a volume of 30 μl at 4 ng/μl for paired-end 2x67nt sequencing in a single lane of Illumina HiSeq 2500. Refer to table S6 for the enrichment summary statistics.
Sequence data processing and bioinformatics
For strict deplexing, we retained sequences with perfect matches between the observed and expected index sequence tags. Reads were trimmed and clipped with cutadapt-1.8.1 (74) to discard reads that were <20 bp in length, exclude sites of low quality (<20), and remove remnant TruSeq adapter sequence. Mean and SDs of library insert sizes were calculated individually for each animal with a custom python script (https://gist.github.com/davidliwei/2323462). All reads were mapped to the unmasked reference dog chromosome 6 (CanFam3.1, Ensembl) generated from a boxer breed individual with BWA-0.7.12 (75). We marked and removed PCR duplicates with picard-tools-1.138 (http://picard.sourceforge.net). BAM files were then indexed and sorted, and VCF files were produced from SAMtools (76), from which we calculated sequencing descriptive statistics. From the sorted BAM files, we next used ANGSD (77) to call SNP genotypes with a minimum depth of 10× sequence coverage, a minimum mapping quality of 30, SNP P < 0.00001 and posterior probability >0.95, and a minimum variant quality of 20. Scores were also adjusted around the insertions/deletions with the --baq flag. Monomorphic sites were excluded.
SNP genotypes were phased with SHAPEIT (78). We scanned the region for signals of positive selection in the dog genome using cross-population extended haplotype homozygosity [XP-EHH (79)] of 4844 SNPs within the resequenced region. Per-SNP FST was calculated with a custom script (19). We normalized both the FST and XP-EHH scores into a z-score to yield a mean of zero and an SD of 1. The product of their z scores represented their composite “bivariate percentile score.” We used the empirical rule to identify outlier loci in the 97.5th percentile or greater (z score, >2). Peaks of selection had to contain at least three outlier loci to be considered.
De novo annotation and genotype calling of structural variants
Briefly, SVMerge is an SV detection platform that implements the RP algorithm BreakDancer (80), RP and SR algorithm Pindel (81), and an algorithm that clusters single-end mapped reads to detect insertions (30). The SVMerge pipeline implements its constituent SV callers, filters, and merges the variant calls and then computationally validates breakpoints by Velvet de novo assembly (31). SoftSearch is an RP and SR algorithm that is also the only available SV detection platform, which has been experimentally validated for high performance with custom resequencing data (32, 82). InGAP-sv is an RD and RP algorithm that uses depth-of-coverage signatures to identify putative SVs and then refines and categorizes the variants based on RP signals (82). By integrating the output of these three programs, we leveraged the strengths of all available SV detection algorithms and incorporated the best available method for custom resequencing data (figs. S8 and S9).
Default parameters were used for each SV calling platform, except where we used a minimum of 25× sequence coverage across all platforms to call an SV and a minimum of five reads to form a single-end cluster (table S16). Because gaps in highly repetitive regions of the reference genome represent the primary source of false positives in SV discovery (83, 84), SV calls from all platforms were filtered with a custom script that removed all variant calls with breakpoints that fell inside gaps, microsatellites, and tandem repeats in the reference genome annotated by the UCSC Table Browser (85). The filtered sets of SV output by each program were merged into a final table and then clustered into a single event if both breakpoints fell within 200 bp of each other (fig. S8) (86). The SV detection platforms used in the pipeline predict the presence or absence of SVs but not whether an animal is homozygous or heterozygous for a given SV. It is more biologically plausible that a given SV is heterozygous due to unequal crossing over that mediates SV in the WBSCR17 in humans, which result in hemizygous changes (20), and that large homozygous deletions are often fatal (49). Thus, SV-positive loci were coded as heterozygous. Genotypes were assigned with a custom script (table S17).
Statistical analysis
Candidate region association test
The univariate linear mixed model implemented in the program GEMMA (87) was used to test for associations between SVs and each of the three behavioral indices. The univariate module of GEMMA fits a set of genotypes and corresponding phenotypes to a univariate linear mixed model that accounts for fixed effects, population stratification, and sample structure. For each variant, the univariate model tested the alternative hypothesis H1 (β ≠ 0) against the null hypothesis H0 (β = 0), using the Wald, likelihood ratio, and score test statistics, where β is the effect size of each variant on the phenotype of interest. Population stratification was accounted for using either a centered or standardized relatedness matrix as a random effect, where the authors recommend a centered matrix for nonhuman organisms. Three univariate models were thus implemented: the first estimating associations between SVs and ABS (ABS model), the second between SVs and HYP (HYP model), and the third between SVs and SIS (SIS model). For each univariate model, the centered relatedness matrix was estimated from SNP genotypes in the target region by GEMMA and was incorporated to account for relatedness and population structure among the samples. SNP genotypes were used in calculating the relatedness matrix in place of SV genotypes because there were more than an order of magnitude more SNP genotypes than SV genotypes (4844 versus 89) on which the estimation was based. Negative values in the relatedness matrix, indicating that there was less relatedness between a given pair of individuals than would be expected between two randomly chosen individuals, were set to zero in the resulting matrix (88, 89). Sex and age were used as covariates. Only SVs with minor allele frequency >0.025 were tested (90). The Bonferroni correction for multiple comparisons was used in conjunction with the simpleM method to account for linkage disequilibrium among variants (91) to establish significance thresholds. With simpleM (http://simplem.sourceforge.net/), we estimated the effective number of independent tests as Meff = 21, corresponding to a significance threshold of P = 2.38 × 10−3 (Bonferroni cutoff of α = 0.05 for 21 independently tested SVs). The likelihood ratio test was used to determine P values. Because the ABS phenotype was calculated as a proportion, the arcsine transformation was applied before all analyses; all other phenotypes were not transformed.
The multivariate linear mixed models of GEMMA estimate the association between a given variant and all phenotypes of interest simultaneously, accounting for the correlation between the phenotypes and generally exhibiting greater statistical power than univariate linear mixed models. Specifically, the multivariate module of GEMMA fits a set of genotypes and corresponding phenotypes to a multivariate linear mixed model that accounts for fixed effects, population stratification, and sample structure. For each variant, GEMMA tests the alternative hypothesis H1 (β ≠ 0) against the null hypothesis H0 (β = 0) using the Wald, likelihood ratio, and score test statistics, where β is the effect size of each variant for all phenotypes. In addition to the univariate models implemented for each phenotype individually, the multivariate linear mixed model of GEMMA was used to estimate associations between SVs and several behavioral phenotypes simultaneously. Two multivariate models were implemented with the same model parameters and data transformation used in the univariate models: one estimating associations between SVs and the indices of human-directed sociability (behavioral index model) and the other estimating associations between SVs and the first three PCs of social behavior (PC model).
To investigate the possibility that SVs are associated with species membership (dog versus wolf), we conducted an association scan of each SV locus with species membership with PLINK (table S11) (92). Variants strongly associated with social behavior, but not with species membership, are particularly robust candidates for mediators of social behavior.
PCR validation and analysis of structural variants
We attempted to design primers flanking all SVs significantly associated with human-directed social behavior (Table 1) as well as two other SVs that were suggestive of an association but did not pass the significance threshold (univariate model: HYP and Cfa6.6, β ± SE = −138.8 ± 33.62, P = 5.75 × 10−3; ABS and Cfa6.83, β ± SE = −0.064 ± 0.09, P = 6.90 × 10−3). Primers were designed on the basis of the dog reference genome (CanFam3.1) with Primer3 (table S18) (93). We were unable to design primers that amplified Cfa6.3 and Cfa6.72, and thus, high-confidence codominant genotypes could only be obtained for Cfa6.6, Cfa6.7, Cfa6.66, and Cfa6.83. We note that Cfa6.3 is ~40 bp downstream of a 300-bp gap in the reference genome. It is possible that this gap caused a false positive during the in silico annotation of this locus, as any sequencing into the gap would not map to the reference and could instead be interpreted as an insertion by SV annotation algorithms.
For the 24 dogs and wolves in the targeted resequencing study, along with a broader sampling of wild canids and dog breeds, we PCR-amplified each SV locus and called genotypes based on banding patterns in agarose gel electrophoresis. PCR conditions were as follows: 0.2 mM deoxynucleotide triphosphates, 2.5 mM MgCl2, bovine serum albumin (0.1 mg/ml), 0.2 μM each primer, 0.75 U of Amplitaq Gold (Thermo Fisher Scientific), 1× Gold buffer, and ~10 ng of genomic DNA. Cycling conditions were 10 m at 95°C, followed by 30 cycles of 30 s at 95°C, 30 s at 60°C (30 s at 55°C for Cfa6.83), and 45 s at 72°C, and a final 10-m extension at 72°C. Ten to 15 μl of PCR product was run on a 1.8% agarose gel and imaged for genotype calling (fig. S4). To confirm that the SVs consist of TEs, PCR products for three individuals per homozygous genotype were Sanger-sequenced, assembled, and aligned to CanFam3.1 in Geneious. Low-complexity regions in the TE at all three loci resulted in poor sequence quality, and locus Cfa6.6 required additional internal primers to sequence across the TE. Alignment to the dog reference genome shows that SV lengths are very similar to the in silico estimates and that, in each case, the TEs are fully contained within the SV. Cfa6.6 is 196 bp (includes 188-bp TE), Cfa6.7 is 229 bp (193-bp TE), Cfa6.66 is 259 bp (187-bp TE), and Cfa6.83 is 216 bp (182-bp TE).
We used PCR to amplify and electrophoresis methods to genotype four SVs in a panel of wild canids (gray wolves: Europe, n = 12; India/Iran, n = 7; China, n = 3; Middle East, n = 14; North America, n = 15; coyotes: n = 13), the 16 domestic dogs from our initial sequencing efforts, and 201 domestic dogs from 13 AKC-registered breeds (dogs: Alaskan malamute, n = 13; Bernese mountain dog, n = 20; border collie, n = 20; boxer, n = 13; basenji, n = 7; cairn terrier, n = 18; golden retriever, n = 16; Great Pyrenees, n = 17; Jack Russell terrier, n = 17; miniature poodle, n = 10; miniature schnauzer, n = 16; pug, n = 19; saluki, n = 15). We also genotyped 17 semidomestic populations represented by NGSDs (n = 3), pariah dogs from Saudi Arabia (n = 4), and village dogs from two locations (Africa, n = 5; Puerto Rico, n = 5). Although an ideal design would include a large sampling of individuals from an experimental dog-wolf cross (for example, F1 hybrids and backcrosses), this is not possible to construct in the United States because it would require generating an animal colony with years of selected breeding. An alternative method would be to explore genome editing with CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9), which has only recently been shown to work in canines (94).
We selected breeds from across multiple breed-type clades, representing the different ancestries and behavioral functions. Each breed was phenotyped according to AKC behavioral stereotypes (40) into a category of either seeking or avoiding attention (seeks attention: Bernese mountain dog, border collie, boxer, golden retriever, Jack Russell terrier, miniature poodle, pug; avoids attention: Alaskan malamute, basenji, cairn terrier, Great Pyrenees, miniature schnauzer, saluki, and all semidomestic dogs). The breeds that were classified as “seeks attention” were those that typically attempted to engage with humans, familiar or unfamiliar (40). We did not require these breeds to be gregarious or hypersocial, in that they actively seek any human attention; rather, they show preference for working with humans, spending time, receiving affection, or offering behaviors to human counterparts. Conversely, the breeds that avoid attention are those that would classically be categorized as “aloof” or “independent.” They were either bred to exist on the periphery of human life or tend to opt for individual pursuits.
Genome-wide SNP survey
We collected genome-wide SNP genotypes using the Affymetrix Axiom K9HDSNPA (643,641 loci) and Axiom K9HDSNPB (625,577 loci) arrays with an average concentration of 26.5 ng/μl for 11 of the 24 individuals with behavioral phenotypes (ndog = 5; nwolf = 6). Samples with a dish quality control value of ≥0.82 and call rate of ≥97% were retained. SNP genotype quality control and processing identified that 794,665 SNPs, 56.3% of K9HDSNPA (250,545 loci) and 87% of K9HDSNPB (544,120 loci), passed filtering metrics. Affymetrix recommended a subset of 544,120 loci (referred to as 544,000 SNPs) to be included for all downstream analyses. We used PLINK to obtain a pruned set of 25,510 uncorrelated and unlinked SNPs with the argument --indep-pairwise 50 5 0.2 and then conducted a PCA with the program flashPCA (fig. S7) (95). We also conducted a binary association test in PLINK on the binary phenotype of species membership. Further, we conducted a quantitative association test using the quantitative behavioral traits and a significance threshold of P < 0.005, testing each of the behaviors (ABS, HYP, and SIS) independently and then jointly. Similar to the regression of the targeted resequencing data described above, we also completed a univariate regression analysis with GEMMA on the 544,000-SNP set and the quantitative behavioral phenotypes of ABS, HYP, and SIS. We incorporated kinship information via a relatedness matrix. We adjusted the likelihood ratio test significance threshold of P < 1st percentile to identify candidate regions. We conducted gene ontology enrichment analysis in WebGestalt (96, 97) using the reference genome as the reference set of genes, the hypergeometric test for evaluating the level of term enrichment, and adjusted the significance threshold due to multiple testing using the method of Benjamini and Hochberg (98). We considered a term significant if the adjusted value was P < 0.05.
Ethics
All subjects were volunteered by their owners/caretakers and remained in their care throughout the study. Experimental procedures were evaluated and approved by Oregon State University’s Institutional Animal Care and Use Committee (IACUC) (protocol #4444). Laboratory methods were conducted under the approved IACUC protocol #2008A-14 of Princeton University. IACUC guidelines for animal subjects were followed.
Supplementary Material
Acknowledgments
We thank G. Rosenlicht, L. Thielke, K. Shreve, C. Kutzler, H. Schoderbeck, and H. Dillman for the assistance with data collection and behavioral coding. We thank B. Gaston, D. Drenzek, and the Wolf Park staff for the assistance with wolf data collection, R. Hylton for the assistance on structural variant amplification and genotype collection, and H. G. Parker and D. L. Dreger for the experimental design advice. We also thank R. K. Wayne for providing the samples from global populations of wild canids to assess an evolutionary perspective of the annotated variants. Funding: The study was partially funded by the Princeton Department of Ecology and Evolutionary Biology, Princeton Office of the Dean of the College, and Princeton Council on Science and Technology, as well as NSF DEB-1245373 support awarded to D.S. and NIH GM086887 and NSF DMS 1264153 awarded to J.S.S. We thank the Oregon State University (OSU) Department of Animal and Rangeland Sciences, OSU Graduate School, and OSU STEM Leaders Program for providing financial support to L.B. and a portion of the behavioral research conducted. C.D.L.W. acknowledges partial support by a grant from the Office of Naval Research (N000141512347). Author contributions: B.M.v. and M.A.R.U. designed the experiment. M.A.R.U., L.B., S.W., and I.J.K. collected the behavioral data. M.A.R.U., E.S., and L.B. analyzed the behavioral data. E.S. and I.J.K. analyzed the sequence data. C.D.L.W. and J.S.S. contributed significantly to the discussion and approach of the analysis. D.S., A.H., and E.A.O. provided additional canid samples for molecular validation. R.Y.K. conducted the molecular validation with the assistance of R.H. and contributed to the associated results. B.M.v., E.S., I.J.K., C.D.L.W., and M.A.R.U. wrote the manuscript. Competing interests: B.M.v., J.S.S., and M.A.R.U. are inventors on a patent application related to this work, filed by B.M.v. with Princeton University (patent/application no. 62527653, filed on 30 June 2017). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Target region FASTQ and sorted BAM files (aligned to CanFam3.1) are available from the National Center for Biotechnology Information Sequence Read Archive (SRP106310). Additional data are archived and available from authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700398/DC1
fig. S1. Differences between dogs and wolves for three behavioral indices used to predict the WBS phenotype.
fig. S2. Scree plot of principal components of human-directed social behavior.
fig. S3. Scan for positive selection using a bivariate percentile score (XP-EHH and FST) to identify outliers (dashed line; bivariate score, >2) indicated as sites in the 97.5th percentile.
fig. S4. Gel electrophoresis banding patterns for three hypersociability-associated SV genotypes.
fig. S5. A dot plot to represent the total number of insertions per population of species for each outlier locus.
fig. S6. Plots from the ANOVA of the total number of SV insertions at four outlier loci.
fig. S7. PCA from 25,510 unlinked genome-wide SNPs from the Affymetrix K9HDSNP array for six wolves and five dogs.
fig. S8. SV discovery pipeline.
fig. S9. Overlap in number of SVs identified by SVMerge, SoftSearch, and inGAP-sv.
table S1. Raw behavioral data.
table S2. Data for indices of human-directed social behavior.
table S3. Loadings of first three PCs of human-directed social behavior.
table S4. Genes in target region on canine chromosome 6.
table S5. Sample information and the total number of raw reads compared to the number of processed reads after using cutadapt to trim/clip paired-end sequences.
table S6. Outlier clusters on chromosome 6 (CanFam3.1) showing signals of positive selection from XP-EHH.
table S7. Summary of de novo annotated structural variants on canine chromosome 6.
table S8. De novo annotated structural variants on canine chromosome 6 (coordinates based on CanFam3.1 assembly).
table S9. Structural variant summary statistics per individual.
table S10. Genic loci associated with indices of human-directed social behavior across dogs and wolves after inclusion of species as a covariate.
table S11. Association to species membership.
table S12. Predicted functional consequences of SVs.
table S13. The significantly enriched (adjusted P < 0.05) gene ontology term from a quantitative association test with each behavioral trait and 544,000 genome-wide SNPs.
table S14. The significantly enriched (adjusted P < 0.05) gene ontology term from the univariate regression analysis conducted in GEMMA with each behavioral trait and 544,000 genome-wide SNPs.
table S15. Behavioral data and description relative to WBS.
table S16. Parameters for in silico annotation of structural variants for the three methods SVMerge, SoftSearch, and InGAP-SV.
table S17. Structural variant genotype per individual.
table S18. Primer sequences used for PCR and gel-based validation of structural variants.
data file S1. Genotypes of the four outlier insertions in wild and domestic canids.
Reference (99)
REFERENCES AND NOTES
- 1.Frank H., Evolution of canine information processing under conditions of natural and artificial selection. Z. Tierpsychol. 53, 389–399 (1980). [DOI] [PubMed] [Google Scholar]
- 2.Miklósi Á., Topál J., Csányi V., Comparative social cognition: What can dogs teach us? Anim. Behav. 67, 995–1004 (2004). [Google Scholar]
- 3.Hare B., Tomasello M., Human-like social skills in dogs? Trends Cogn. Sci. 9, 439–444 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Udell M. A. R., Dorey N. R., Wynne C. D. L., What did domestication do to dogs? A new account of dogs’ sensitivity to human actions. Biol. Rev. Camb. Philos. Soc. 85, 327–345 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Trut L., Oskina I., Kharlamova A., Animal evolution during domestication: The domesticated fox as a model. Bioessays 31, 349–360 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagasawa M., Mitsui S., En S., Ohtani N., Ohta M., Sakuma Y., Onaka T., Mogi K., Kikusui T., Oxytocin-gaze positive loop and the coevolution of human-dog bonds. Science 348, 333–336 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Bentosela M., Wynne C. D. L., D’Orazio M., Elgier A., Udell M. A. R., Sociability and gazing toward humans in dogs and wolves: Simple behaviors with broad implications. J. Exp. Anal. Behav. 105, 68–75 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Udell M. A. R., When dogs look back: Inhibition of independent problem-solving behaviour in domestic dogs (Canis lupus familiaris) compared with wolves (Canis lupus). Biol. Lett. 11, 20150489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones P., Chase K., Martin A., Davern P., Ostrander E. A., Lark K. G., Single-nucleotide-polymorphism-based association mapping of dog stereotypes. Genetics 179, 1033–1044 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker H. G., Kim L. V., Sutter N. B., Carlson S., Lorentzen T. D., Malek T. B., Johnson G. S., DeFrance H. B., Ostrander E. A., Kruglyak L., Genetic structure of the purebred domestic dog. Science 304, 1160–1164 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Serpell J. A., Hsu Y., Effects of breed, sex, and neuter status on trainability in dogs. Anthrozoös 18, 196–207 (2005). [Google Scholar]
- 12.Svartberg K., Breed—typical behavior in dogs—Historical remnants or recent constructs? Appl. Anim. Behav. Sci. 96, 293–313 (2006). [Google Scholar]
- 13.Duffy D. L., Hsu Y., Serpell J. A., Breed differences in canine aggression. Appl. Anim. Behav. Sci. 114, 441–460 (2008). [Google Scholar]
- 14.Ley J. M., Bennett P. M., Coleman G. J., A refinement and validation of the Monash Canine Personality Questionnaire (MCPQ). Appl. Anim. Behav. Sci. 116, 220–227 (2009). [Google Scholar]
- 15.Turcsán B., Kubinyi E., Miklósi Á., Trainability and boldness traits differ between dog breed clusters based on conventional breed categories and genetic relatedness. Appl. Anim. Behav. Sci. 132, 61–70 (2011). [Google Scholar]
- 16.Vaysse A., Ratnakumar A., Derrien T., Axelsson E., Rosengren Pielberg G., Sigurdsson S., Fall T., Seppälä E. H., Hansen M. S. T., Lawley C. T., Karlsson E. K.; The LUPA Consortium, Bannasch D., Vilà C., Lohi H., Galibert F., Fredholm M., Häggström J., Hedhammar Å., André C., Lindblad-Toh K., Hitte C., Webster M. T., Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLOS Genet. 7, e1002316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.J. A. Serpell, D. L. Duffy, Dog breeds and their behavior, in Domestic Dog Cognition and Behavior, A. Horowitz, Ed. (Springer, 2014), pp. 31–57. [Google Scholar]
- 18.Persson M. E., Roth L. S. V., Johnson M., Wright D., Jensen P., Human-directed social behavior in dogs shows significant heritability. Genes Brain Behav. 14, 337–344 (2015). [DOI] [PubMed] [Google Scholar]
- 19.vonHoldt B. M., Pollinger J. P., Lohmueller K. E., Han E., Parker H. G., Quignon P., Degenhardt J. D., Boyko A. R., Earl D. A., Auton A., Reynolds A., Bryc K., Brisbin A., Knowles J. C., Mosher D. S., Spady T. C., Elkahloun A., Geffen E., Pilot M., Jedrzejewski W., Greco C., Randi E., Bannasch D., Wilton A., Shearman J., Musiani M., Cargill M., Jones P. G., Qian Z., Huang W., Ding Z.-L., Zhang Y.-p., Bustamante C. D., Ostrander E. A., Novembre J., Wayne R. K., Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464, 898–902 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schubert C., The genomic basis of the Williams – Beuren syndrome. Cell. Mol. Life Sci. 66, 1178–1197 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer-Lindenberg A., Mervis C. B., Berman K. F., Neural mechanisms in Williams syndrome: A unique window to genetic influences on cognition and behaviour. Nat. Rev. Neurosci. 7, 380–393 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Jones W., Bellugi U., Lai Z., Chiles M., Reilly J., Lincoln A., Adolphs R., Hypersociability in Williams syndrome. J. Cogn. Neurosci. 12, 30–46 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Ewart A. K., Morris C. A., Atkinson D., Jin W., Sternes K., Spallone P., Stock A. D., Leppert M., Keating M. T., Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat. Genet. 5, 11–16 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Wan M., Hejjas K., Ronai Z., Elek Z., Sasvari-Szekely M., Champagne F. A., Miklósi A., Kubinyi E., DrD4 and TH gene polymorphisms are associated with activity, impulsivity and inattention in Siberian Husky dogs. Anim. Genet. 44, 717–727 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Kis A., Bence M., Lakatos G., Pergel E., Turcsán B., Pluijmakers J., Vas J., Elek Z., Brúder I., Földi L., Sasvári-Székely M., Miklósi A., Rónai Z., Kubinyi E., Oxytocin receptor gene polymorphisms are associated with human directed social behavior in dogs (Canis familiaris). PLOS ONE 9, e83993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jakovcevic A., Mustaca A., Bentosela M., Do more sociable dogs gaze longer to the human face than less sociable ones? Behav. Processes 90, 217–222 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Brubaker L., Dasgupta S., Bhattacharjee D., Bhadra A., Udell M. A. R., Differences in problem-solving between canid populations: Do domestication and lifetime experience affect persistence? Anim. Cogn. 20, 717–723 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Walsh T., McClellan J. M., McCarthy S. E., Addington A. M., Pierce S. B., Cooper G. M., Nord A. S., Kusenda M., Malhotra D., Bhandari A., Stray S. M., Rippey C. F., Roccanova P., Makarov V., Lakshmi B., Findling R. L., Sikich L., Stromberg T., Merriman B., Gogtay N., Butler P., Eckstrand K., Noory L., Gochman P., Long R., Chen Z., Davis S., Baker C., Eichler E. E., Meltzer P. S., Nelson S. F., Singleton A. B., Lee M. K., Rapoport J. L., King M. C., Sebat J., Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Cuscó I., Corominas R., Bayés M., Flores R., Rivera-Brugués N., Campuzano V., Pérez-Jurado L. A., Copy number variation at the 7q11. 23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res. 18, 683–694 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong K., Keane T. M., Stalker J., Adams D. J., Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly. Genome Biol. 11, R128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hart S. N., Sarangi V., Moore R., Baheti S., Bhavsar J. D., Couch F. J., Kocher J.-P. A., SoftSearch: Integration of multiple sequence features to identify breakpoints of structural variations. PLOS ONE 8, e83356 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi J., Zhao F., inGAP-sv: A novel scheme to identify and visualize structural variation from paired end mapping data. Nucleic Acids Res. 39, W567–W575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korenberg J. R., Chen X.-N., Hirota H., Lai Z., Bellugi U., Burian D., Roe B., Matsuoka R., VI. Genome structure and cognitive map of Williams Syndrome. J. Cogn. Neuroci. 12, 89–107 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Young E. J., Lipina T., Tam E., Mandel A., Clapcote S. J., Bechard A. R., Chambers J., Mount H. T. J., Fletcher P. J., Roder J. C., Osborne L. R., Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-tagged mice. Genes Brain Behav. 7, 224–234 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H. H., Roy M., Kuscuoglu U., Spencer C. M., Halm B., Harrison K. C., Bayle J. H., Splendore A., Ding F., Meltzer L. A., Wright E., Paylor R., Deisseroth K., Francke U., Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol. Med. 1, 50–65 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle T. F., Bellugi U., Korenberg J. R., Graham J., “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am. J. Med. Genet. A 124, 263–273 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Edelmann L., Prosnitz A., Pardo S., Bhatt J., Cohen N., Lauriat T., Ouchanov L., González P. J., Manghi E. R., Bondy P., Esquivel M., Monge S., Delgado M. F., Splendore A., Francke U., Burton B. K., McInnes L. A., An atypical deletion of the Williams–Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J. Med. Genet. 44, 136–143 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F., Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T., Durbin R., Eyras E., Gilbert J., Hammond M., Huminiecki L., Kasprzyk A., Lehvaslaiho H., Lijnzaad P., Melsopp C., Mongin E., Pettett R., Pocock M., Potter S., Rust A., Schmidt E., Searle S., Slater G., Smith J., Spooner W., Stabenau A., Stalker J., Stupka E., Ureta-Vidal A., Vastrik I., Clamp M., The Ensembl genome database project. Nucleic Acids Res. 30, 38–41 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.American Kennel Club, The New Complete Dog Book: Official Breed Standards and All-New Profiles for 200 Breeds (21st Edition) (Lumina Media, 2014). [Google Scholar]
- 41.Bayés M., Magano L. F., Rivera N., Flores R., Pérez Jurado L. A., Mutational mechanisms of Williams–Beuren syndrome deletions. Am. J. Hum. Genet. 73, 131–151 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reymond A., Henrichsen C. N., Harewood L., Merla G., Side effects of genome structural changes. Curr. Opin. Genet. Dev. 17, 381–386 (2007). [DOI] [PubMed] [Google Scholar]
- 43.G. Merla, L. Micale, C. Fusco, M. N. Loviglio, Molecular Genetics of Williams–Beuren Syndrome (John Wiley & Sons Ltd, 2012). [Google Scholar]
- 44.Bayarsaihan D., Ruddle F. H., Isolation and characterization of BEN, a member of the TFII-I family of DNA-binding proteins containing distinct helix–loop–helix domains. Proc. Natl. Acad. Sci. U.S.A. 97, 7342–7347 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tipney H. J., Hinsley T. A., Brass A., Metcalfe K., Donnai D., Tassabehji M., Isolation and characterisation of GTF2IRD2, a novel fusion gene and member of the TFII-I family of transcription factors, deleted in William–Beuren syndrome. Eur. J. Hum. Genet. 12, 551–560 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Tassabehji M., Hammond P., Karmiloff-Smith A., Thompson P., Thorgeirsson S. S., Durkin M. E., Popescu N. C., Hutton T., Metcalfe K., Rucka A., Stewart H., Read A. P., Maconochie M., Donnai D., GTF2IRD1 in craniofacial development of humans and mice. Science 310, 1184–1187 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Chimge N.-O., Makeyev A. V., Ruddle F. H., Bayarsaihan D., Identification of the TFII-I family target genes in the vertebrate genome. Proc. Natl. Acad. Sci. U.S.A. 105, 9006–9010 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porter M. A., Dobson-Stone C., Kwok J. B. J., Schofield P. R., Beckett W., Tassabehji M., A role for transcription factor GTF2IRD2 in executive function in Williams-Beuren Syndrome. PLOS ONE 7, e47457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sakurai T., Dorr N. P., Takahashi N., McInnes L. A., Elder G. A., Buxbaum J. D., Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 4, 28–39 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Procyshyn T. L., Spence J., Read S., Watson N. V., Crespi B. J., The Williams syndrome prosociality gene GTF2I mediates oxytocin reactivity and social anxiety in a healthy population. Biol. Lett. 13, 20170051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merla G., Howald C., Henrichsen C. N., Lyle R., Wyss C., Zabot M.-T., Antonarakis S. E., Reymond A., Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am. J. Hum. Genet. 79, 332–341 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lau K. S., Khan S., Dennis J. W., Genome-scale identification of UDP-GlcNAc-dependent pathways. Proteomics 8, 3294–3302 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Axelsson E., Ratnakumar A., Arendt M.-L., Maqbool K., Webster M. T., Perloski M., Liberg O., Arnemo J. M., Hedhammar A., Lindblad-Toh K., The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 495, 360–364 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Cowley M., Oakey R. J., Transposable elements re-wire and fine-tune the transcriptome. PLOS Genet. 9, e1003234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang W., Kirkness E. F., Short interspersed elements (SINEs) are a major source of canine genomic diversity. Genome Res. 15, 1798–1808 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Janowitz Koch I., Clark M. M., Thompson M. J., Deere-Machemer K. A., Wang J., Duarte L., Gnanadesikan G. E., McCoy E. L., Rubbi L., Stahler D. R., Pellegrini M., Ostrander E. A., Wayne R. K., Sinsheimer J. S., vonHoldt B. M., The concerted impact of domestication and transposon insertions on methylation patterns between dogs and grey wolves. Mol. Ecol. 25, 1838–1855 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin L., Faraco J., Li R., Kadotani H., Rogers W., Lin X., Qiu X., de Jong P. J., Nishino S., Mignot E., The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98, 365–376 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Pelé M., Tiret L., Kessler J.-L., Blot S., Panthier J.-J., SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum. Mol. Genet. 14, 1417–1427 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Clark L. A., Wahl J. M., Rees C. A., Murphy K. E., Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc. Natl. Acad. Sci. U.S.A. 103, 1376–1381 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sutter N. B., Bustamante C. D., Chase K., Gray M. M., Zhao K., Zhu L., Padhukasahasram B., Karlins E., Davis S., Jones P. G., Quignon P., Johnson G. S., Parker H. G., Fretwell N., Mosher D. S., Lawler D. F., Satyaraj E., Nordborg M., Lark K. G., Wayne R. K., Ostrander E. A., A single IGF1 allele is a major determinant of small size in dogs. Science 316, 112–115 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parker H. G., VonHoldt B. M., Quignon P., Margulies E. H., Shao S., Mosher D. S., Spady T. C., Elkahloun A., Cargill M., Jones P. G., Maslen C. L., Acland G. M., Sutter N. B., Kuroki K., Bustamante C. D., Wayne R. K., Ostrander E. A., An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science 325, 995–998 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gray M. M., Sutter N. B., Ostrander E. A., Wayne R. K., The IGF1 small dog haplotype is derived from Middle Eastern grey wolves. BMC Biol. 8, 16 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karlsson E. K., Lindblad-Toh K., Leader of the pack: Gene mapping in dogs and other model organisms. Nat. Rev. Genet. 9, 713–725 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Boyko A. R., The domestic dog: Man’s best friend in the genomic era. Genome Biol. 12, 216 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anderson T. M., vonHoldt B. M., Candille S. I., Musiani M., Greco C., Stahler D. R., Smith D. W., Padhukasahasram B., Randi E., Leonard J. A., Bustamante C. D., Ostrander E. A., Tang H., Wayne R. K., Barsh G. S., Molecular and evolutionary history of melanism in North American gray wolves. Science 323, 1339–1343 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frank H., Frank M. G., On the effects of domestication on canine social development and behavior. Appl. Anim. Ethol. 8, 507–525 (1982). [Google Scholar]
- 67.Udell M. A. R., Dorey N. R., Wynne C. D. L., The performance of stray dogs (Canis familiaris) living in a shelter on human-guided object-choice tasks. Anim. Behav. 79, 717–725 (2010). [Google Scholar]
- 68.E. Klinghammer, P. Goodman, Socialization and management of wolves in captivity, in Man and Wolf: Advances, Issues, and Problems in Captive Wolf Research, H. Frank, Ed. (Springer, 1987). [Google Scholar]
- 69.Udell M. A. R., Dorey N. R., Wynne C. D. L., Wolves outperform dogs in following human social cues. Anim. Behav. 76, 1767–1773 (2008). [Google Scholar]
- 70.McHugh M. L., Interrater reliability: The kappa statistic. Biochem. Med. 22, 276–282 (2012). [PMC free article] [PubMed] [Google Scholar]
- 71.G. Raiche, nFactors: An R package for parallel analysis and non graphical solutions to the Cattell scree test. R package version 2.3.3 (2010).
- 72.Aschard H., Vilhjálmsson B. J., Greliche N., Morange P.-E., Trégouët D. A., Kraft P., Maximizing the power of principal-component analysis of correlated phenotypes in genome-wide association studies. Am. J. Hum. Genet. 94, 662–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cunningham F., Amode M. R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., Gil L., Girón C. G., Gordon L., Hourlier T., Hunt S. E., Janacek S. H., Johnson N., Juettemann T., Kähäri A. K., Keenan S., Martin F. J., Maurel T., McLaren W., Murphy D. N., Nag R., Overduin B., Parker A., Patricio M., Perry E., Pignatelli M., Riat H. S., Sheppard D., Taylor K., Thormann A., Vullo A., Wilder S. P., Zadissa A., Aken B. L., Birney E., Harrow J., Kinsella R., Muffato M., Ruffier M., Searle S. M. J., Spudich G., Trevanion S. J., Yates A., Zerbino D. R., Flicek P., Ensembl 2015. Nucleic Acids Res. 43, D662–D669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 75.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler Transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Geome Project Data Processing Subgroup, The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Delaneau O., Marchini J., Zagury J.-F., A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Sabeti P. C., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E. H., McCarroll S. A., Gaudet R., Schaffner S. F., Lander E. S.; International HapMap Consortium, Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen K., Wallis J. W., McLellan M. D., Larson D. E., Kalicki J. M., Pohl C. S., McGrath S. D., Wendl M. C., Zhang Q., Locke D. P., Shi X., Fulton R. S., Ley T. J., Wilson R. K., Ding L., Mardis E. R., BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ye K., Schulz M. H., Long Q., Apweiler R., Ning Z., Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tattini L., D’Aurizio R., Magi A., Detection of genomic structural variants from next-generation sequencing data. Front. Bioeng. Biotechnol. 3, 92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.E. J. Hollox, The challenges of studying complex and dynamic regions of the human genome, in Genomic Structural Variants, L. Feuk, Ed. (Springer, 2012), pp. 187–207. [DOI] [PubMed] [Google Scholar]
- 84.Quinlan A. R., Hall I. M., Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 28, 43–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D., The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Decker B., Davis B. W., Rimbault M., Long A. H., Karlins E., Jagannathan V., Reiman R., Parker H. G., Drögemüller C., Corneveaux J. J., Chapman E. S., Trent J. M., Leeb T., Huentelman M. J., Wayne R. K., Karyadi D. M., Ostrander E. A., Comparison against 186 canid whole-genome sequences reveals survival strategies of an ancient clonally transmissible canine tumor. Genome Res. 25, 1646–1655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou X., Stephens M., Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stich B., Möhring J., Piepho H.-P., Heckenberger M., Buckler E. D., Melchinger A. E., Comparison of mixed-model approaches for association mapping. Genetics 178, 1745–1754 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mandel J. R., Nambeesan S., Bowers J. E., Marek L. F., Ebert D., Rieseberg L. H., Knapp S. J., Burke J. M., Association mapping and the genomic consequences of selection in sunflower. PLOS Genet. 9, e1003378 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tabangin M. E., Woo J. G., Martin L. J., The effect of minor allele frequency on the likelihood of obtaining false positives. BMC Proc. 3 (suppl. 7), S41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gao X., Starmer J., Martin E. R., A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 32, 361–369 (2008). [DOI] [PubMed] [Google Scholar]
- 92.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B. C., Remm M., Rozen S. G., Primer3—New capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zou Q., Wang X., Liu Y., Ouyang Z., Long H., Wei S., Xin J., Zhao B., Lai S., Shen J., Ni Q., Yang H., Zhong H., Li L., Hu M., Zhang Q., Zhou Z., He J., Yan Q., Fan N., Zhao Y., Liu Z., Guo L., Huang J., Zhang G., Ying J., Lai L., Gao X., Generation of gene-target dogs using CRISPR/Cas9 system. J. Mol. Cell Biol. 7, 580–583 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Abraham G., Inouye M., Fast principal component analysis of large-scale genome-wide data. PLOS ONE 9, e93766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang B., Kirov S. A., Snoddy J. R., WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741–W748 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang J., Duncan D., Shi Z., Zhang B., WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 41, W77–W83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
- 99.Fusco C., Micale L., Augello B., Teresa Pellico M., Menghini D., Alfieri P., Cristina Digilio M., Mandriani B., Carella M., Palumbo O., Vicari S., Merla G., Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur. J. Hum. Genet. 22, 64–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700398/DC1
fig. S1. Differences between dogs and wolves for three behavioral indices used to predict the WBS phenotype.
fig. S2. Scree plot of principal components of human-directed social behavior.
fig. S3. Scan for positive selection using a bivariate percentile score (XP-EHH and FST) to identify outliers (dashed line; bivariate score, >2) indicated as sites in the 97.5th percentile.
fig. S4. Gel electrophoresis banding patterns for three hypersociability-associated SV genotypes.
fig. S5. A dot plot to represent the total number of insertions per population of species for each outlier locus.
fig. S6. Plots from the ANOVA of the total number of SV insertions at four outlier loci.
fig. S7. PCA from 25,510 unlinked genome-wide SNPs from the Affymetrix K9HDSNP array for six wolves and five dogs.
fig. S8. SV discovery pipeline.
fig. S9. Overlap in number of SVs identified by SVMerge, SoftSearch, and inGAP-sv.
table S1. Raw behavioral data.
table S2. Data for indices of human-directed social behavior.
table S3. Loadings of first three PCs of human-directed social behavior.
table S4. Genes in target region on canine chromosome 6.
table S5. Sample information and the total number of raw reads compared to the number of processed reads after using cutadapt to trim/clip paired-end sequences.
table S6. Outlier clusters on chromosome 6 (CanFam3.1) showing signals of positive selection from XP-EHH.
table S7. Summary of de novo annotated structural variants on canine chromosome 6.
table S8. De novo annotated structural variants on canine chromosome 6 (coordinates based on CanFam3.1 assembly).
table S9. Structural variant summary statistics per individual.
table S10. Genic loci associated with indices of human-directed social behavior across dogs and wolves after inclusion of species as a covariate.
table S11. Association to species membership.
table S12. Predicted functional consequences of SVs.
table S13. The significantly enriched (adjusted P < 0.05) gene ontology term from a quantitative association test with each behavioral trait and 544,000 genome-wide SNPs.
table S14. The significantly enriched (adjusted P < 0.05) gene ontology term from the univariate regression analysis conducted in GEMMA with each behavioral trait and 544,000 genome-wide SNPs.
table S15. Behavioral data and description relative to WBS.
table S16. Parameters for in silico annotation of structural variants for the three methods SVMerge, SoftSearch, and InGAP-SV.
table S17. Structural variant genotype per individual.
table S18. Primer sequences used for PCR and gel-based validation of structural variants.
data file S1. Genotypes of the four outlier insertions in wild and domestic canids.
Reference (99)