

Big cat genomes reveal a history of interspecies admixture and adaptive evolution of genes underlying development and sensory perception.
Abstract
The great cats of the genus Panthera comprise a recent radiation whose evolutionary history is poorly understood. Their rapid diversification poses challenges to resolving their phylogeny while offering opportunities to investigate the historical dynamics of adaptive divergence. We report the sequence, de novo assembly, and annotation of the jaguar (Panthera onca) genome, a novel genome sequence for the leopard (Panthera pardus), and comparative analyses encompassing all living Panthera species. Demographic reconstructions indicated that all of these species have experienced variable episodes of population decline during the Pleistocene, ultimately leading to small effective sizes in present-day genomes. We observed pervasive genealogical discordance across Panthera genomes, caused by both incomplete lineage sorting and complex patterns of historical interspecific hybridization. We identified multiple signatures of species-specific positive selection, affecting genes involved in craniofacial and limb development, protein metabolism, hypoxia, reproduction, pigmentation, and sensory perception. There was remarkable concordance in pathways enriched in genomic segments implicated in interspecies introgression and in positive selection, suggesting that these processes were connected. We tested this hypothesis by developing exome capture probes targeting ~19,000 Panthera genes and applying them to 30 wild-caught jaguars. We found at least two genes (DOCK3 and COL4A5, both related to optic nerve development) bearing significant signatures of interspecies introgression and within-species positive selection. These findings indicate that post-speciation admixture has contributed genetic material that facilitated the adaptive evolution of big cat lineages.
INTRODUCTION
Recent evolutionary radiations are powerful systems to investigate the interplay of complex processes, such as the origin of ecological divergence and the effects of secondary admixture (1). Genome-wide analyses provide novel opportunities to address these issues, enabling an in-depth assessment of the selective and demographic forces that have shaped present-day species (2, 3). The genus Panthera is a remarkable group to investigate these issues because it comprises five big cat species (Fig. 1A) that arose from a recent and rapid diversification process (4, 5). Understanding the history of their unique features [for example, the tiger’s stripes, the lion’s mane, and the jaguar’s stocky build, with a massive head and stout forelimbs (6, 7)] depends on resolving the underlying phylogeny of the Panthera clade, a task that has been notoriously difficult to accomplish (4, 5, 8). Recent analyses have indicated that genealogical discordance caused by both incomplete lineage sorting (ILS) and post-speciation admixture has contributed to produce such a complex system (5). Genome-wide comparisons of the five extant species should help illuminate this issue and allow for in-depth investigations of their adaptive divergence.
Fig. 1. Evolutionary history of the great cats.
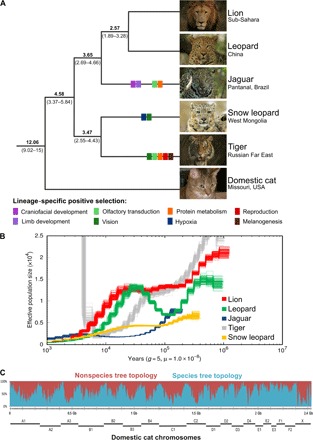
(A) Species tree of the genus Panthera estimated from genome-wide data. All five extant species are represented as follows: lion (Panthera leo), leopard (P. pardus), jaguar (Panthera onca), snow leopard (Panthera uncia), and tiger (Panthera tigris). Numbers above branches indicate the estimated age [in million years ago (Ma)] of the adjacent node, averaged across all genomic windows (100-kb window size, 100-kb steps) that conform to the species tree (95% highest posterior density interval below the respective branch). Colored rectangles on terminal branches indicate phenotypic categories (defined below the tree; see Fig. 3B for more details) affected by species-specific episodes of positive selection. (B) PSMC plot depicting the demographic history of the five Panthera species inferred from genomic data. (C) Genealogical discordance across the genome of Panthera cats, demonstrated by a sliding window analysis (500-kb window size, 100-kb steps) of a full-genome alignment mapped to domestic cat chromosomes (gray lines at the bottom). The y axis indicates the percentage of overlapping windows within a given interval that conform to (blue) or reject (red) the species tree. Photo credits: D. Kantek (jaguar); C. Sperka (others).
RESULTS AND DISCUSSION
To enable genomic comparisons across the Panthera, we sequenced the genome of a male jaguar from the Brazilian Pantanal to ~94x coverage, using three different libraries [180 base pairs (bp) paired-end, 3-kb mate pair, and 8-kb mate pair). We assembled this genome de novo using the ALLPATHS-LG approach (see Materials and Methods), achieving N50 contig and scaffold lengths of 28.6 kb and 1.52 Mb (tables S1 and S2). We annotated the genome with ab initio gene prediction and validated protein-coding genes with transcriptome data from six different tissues. These approaches predicted 25,451 protein-coding genes, of which 24,411 (96%) were supported by our RNA sequencing (RNA-seq) data or other empirical evidence (tables S3 and S4). We also sequenced the genome of a leopard (Panthera pardus) to ~25x coverage, thus obtaining a comparative data set comprising all Panthera species, including previously published tiger, lion, and snow leopard whole-genome sequence (wgs) data (table S5) (9).
We used these data to investigate the historical demography of each species with the pairwise sequentially Markovian coalescent (PSMC) approach. PSMC results (Fig. 1B) revealed population reductions in all five species ca. 100,000 to 300,000 years ago, and a second round of decline (most notably for the lion and leopard) between 10,000 and 20,000 years ago, following the last glacial maximum. In the period between these two rounds of decline, the inferred patterns differ among species, with the leopard showing a rebound ca. 100,000 years ago, whereas the lion and snow leopard remained mostly stable (although at very different effective sizes). The jaguar was the most negatively influenced by the initial decline, stabilizing ca. 30,000 years ago at the lowest effective population size among the extant big cats. Overall, these findings strongly indicate that the population sizes of all Panthera species varied considerably during the Pleistocene, with successive fluctuations likely resulting in cumulative loss of genetic diversity in each of these lineages.
To conclusively resolve the Panthera phylogeny and investigate patterns of genealogical discordance, we aligned the five genomes and those of the domestic cat (as an outgroup). We performed maximum likelihood (ML) phylogenetic analyses using a genomic sliding window approach (with 100- or 500-kb windows) and also extracted all shared protein-coding genes (13,183 loci) to reconstruct individual-gene ML phylogenies. With both window- and gene-based analyses, the same topology (Fig. 1A) was retrieved as the most frequent reconstruction (64% of windows and 23% of genes; see tables S6 and S7). On the basis of estimates of divergence time (see below) and concordance with a Y chromosome–derived phylogeny (8), we conclude that it most likely represents the species tree (a hypothesis that can be further probed with genome-wide analyses of multiple individuals per species). We observed extensive genealogical discordance distributed across all chromosomes and notably prevalent in pericentromeric regions (Figs. 1C and 2). The second and third most frequent topologies varied only in the position of the jaguar with respect to the lion and leopard, with much greater support for lion + jaguar than leopard + jaguar (Fig. 2, A and B). The frequency of these two alternate trees was considerably higher for the X chromosome than for autosomes. The X chromosome was also distinct due to the high frequency of two topologies (12 and 23) in which the leopard is deeply divergent from the remaining species, a striking result most likely caused by ancient admixture between the leopard and an extinct lineage closely related to Panthera. These and other non–species tree topologies were enriched on the X chromosome in blocks corresponding to large recombination deserts (Fig. 2D) (10), indicating that these regions exhibit distinct signatures of historical processes, likely including post-speciation admixture.
Fig. 2. In-depth characterization of phylogenetic discordance in the Panthera.
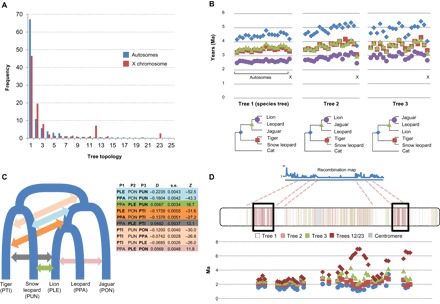
(A) Frequencies of the species tree (Tree 1) and alternative topologies on pooled autosomes (blue) and the X chromosome (red). Trees 12 and 23 show the leopard in a basal position relative to the other species (tables S6 and S7). (B) Chromosome-wide mean divergence times (in Ma), calculated from 100-kb windows that yielded the three most frequent topologies (shown below the graphs; nodes are color-coded). Note the marked drop in divergence time on the X chromosome for non–species tree topologies. (C) Complex patterns of historical admixture among Panthera lineages, inferred from ABBA/BABA tests. Colored arrows indicate the phylogenetic position of significant D statistics (shown in the table) for different species trios, using the domestic cat as the outgroup. Species codes in bold types indicate pairs for which significant evidence of admixture was detected. (D) Patterns of phylogenetic discordance along the X chromosome (estimated in 100-kb windows) correlate with the recombination landscape (10). Regions depicting species tree relationships (Tree 1), shown in white, correspond to higher recombination rates, whereas recombination deserts are enriched for alternative topologies with reduced divergence times (a signature of introgression). Two terminal, lower-recombination regions are enriched for topologies 12 and 23, depicting a basal position for leopard. These regions also harbor tracts of windows with old divergence times between the leopard and the other Panthera.
Mean divergence times were remarkably concordant among autosomes when assessing windows that conformed to the species tree, whereas windows supporting topologies 2 and 3 showed much greater variation (Fig. 2B). Under a scenario of ILS, theory predicts that the average lion + jaguar and leopard + jaguar divergence times should predate species tree (lion + leopard) divergence times (11). Although this prediction was mostly compatible with the autosomal averages, divergence estimates for the X chromosome were significantly younger for topology 2 (less so for topology 3) than for the species tree, indicating that this pattern cannot be explained by ILS alone, and supporting post-speciation admixture as the most likely cause. Using the same rationale, a recent study has reported the opposite pattern for Anopheles mosquitoes, with the X chromosome recording the species tree much more often than the autosomes (2).
To further investigate admixture patterns, we estimated D statistics (also called ABBA/BABA tests), which revealed a complex network of ancestral hybridization among several Panthera lineages (Fig. 2C and table S8). The lion lineage exhibited the most widespread signatures of ancient admixture, likely due to its broad historical range throughout much of the Holarctic region, overlapping with several congeneric species. These results considerably expand the recent evidence for hybridization between the snow leopard and the lion + leopard ancestor [leading to mitonuclear discordance (5)] and reveal a much more complex history of post-speciation admixture in this group than was previously appreciated.
We then examined divergence time outliers (12) using the genome-wide set of 100-kb windows (henceforth referred to as “outlier window test”). We focused specifically on the relationships within the lion-leopard-jaguar clade because these comprised most of the discordant gene trees (Fig. 2A). Following Z transformation of divergence times at each node, we identified 242 windows (0.85% of the total) with dates younger than 2 SDs from the mean (sdm) for both terminal and basal nodes for this trio (that is, a compressed tree). These outliers are most likely caused by post-speciation admixture, leading to a significantly younger age for these particular segments. These windows contained 161 genes (Fig. 3A), which are involved in a variety of cellular processes. When we restricted our analysis to 74 windows with the most extremely reduced basal nodal ages (>3 sdm), we observed that many of them were clustered and showed a precipitous drop in divergence time relative to flanking windows (Fig. 4). Furthermore, 61 (82%) of these windows supported non–species tree relationships. This combined pattern strongly indicates post-speciation admixture, likely followed by selective sweeps that perpetuated the introgressed segments in the recipient species. These “extreme outlier windows” contained 43 genes, including several loci previously implicated in morphology, stature, brain function, and development (DOCK3, BMP4, SHROOM4, PJA1, PPFIA2, and UBE3A), pigmentation (EXOC2), and sensory perception (POU3F4 and COL4A5/6). These results suggested that these phenotypes have undergone adaptive evolution in this group following episodes of interspecies introgression.
Fig. 3. Genomic evidence of natural selection in the Panthera.
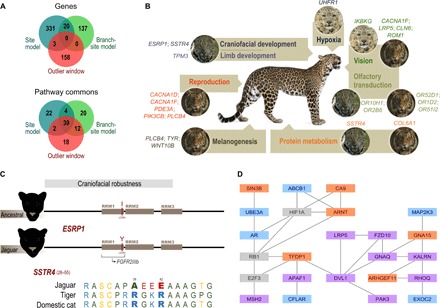
(A) Venn diagrams depicting shared genes (top) and pathways (bottom) among approaches that detect positive selection (site model and branch-site model) and interspecies introgression (Outlier window) (fig. S4 and tables S9 to S13 and S28 to S38). (B) Detailed results of the branch-site test, indicating genes bearing species-specific signatures of positive selection; phenotypic categories (color-coded as in Fig. 1A) were defined on the basis of Kegg and Pathway Commons enrichment results, along with additional literature searches. (C) Two genes with jaguar-specific signatures of positive selection affect craniofacial robustness. Silhouettes on the left depict a jaguar’s robust face relative to the inferred appearance of the Panthera ancestor. The positively selected I298Y substitution in the ESRP1 gene lies in the RRM1 domain, which is known to bind to the FGFR2IIIb gene isoform and affect craniofacial development (17). The two positively selected substitutions (R39A and R42E) in the SSTR4 gene imply substantial physicochemical amino acid changes (green, nonpolar; yellow, polar; blue, basic; red, acid). (D) Schematic representation of the Glypican pathway, showing only genes that were connected with, at most, one intermediate step (in gray) and that exhibited significant signatures of positive selection (purple, site model; orange, tiger branch-site model) or interspecies introgression (blue, outlier window test).
Fig. 4. Genomic evidence of positive selection following interspecific introgression.
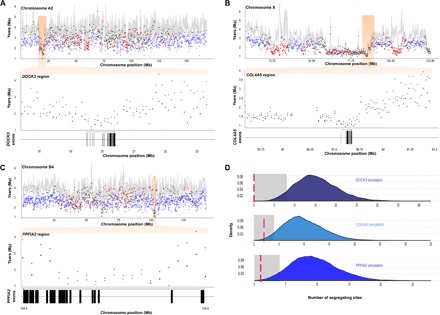
(A to C) Graphs depicting variation in age estimates for the trio jaguar-lion-leopard in 100-kb windows across the length of three chromosomes (top) and specific chromosomal segments containing multiple outlier windows with ages significantly younger than the genome-wide average (bottom), indicating introgression. The gray lines in the top panels (gray circles in the bottom panels) represent the basal age of the trio, whereas colored circles represent the age of the internal node in each window (blue, lion-leopard; red, lion-jaguar; green, leopard-jaguar). Below the bottom panels, the annotated exons of a candidate gene located in each focal region are indicated. (D) Intraspecific signals of positive selection in jaguars affecting the three genes highlighted in (A) to (C). The graphs depict the null distribution of the number of segregating sites per gene based on 10,000 demographic simulations (see text); the gray shading indicates significant departure (P < 0.05) from the null expectation, and the red lines represent the observed value for each gene.
As an independent assessment of adaptive evolution in the Panthera, we surveyed the presence of selection signatures in the 13,183 shared coding genes using site models and branch-site models (both based on dN/dS ratios; see Materials and Methods), applying a novel approach that considers both the species tree and the locus-specific gene tree to account for genealogical discordance (fig. S1). Using a conservative criterion (that is, only keeping genes with significant results for both the species and gene tree analyses), we identified 491 loci with signatures of positive selection (Fig. 3A and tables S9 to S13). Of these, 157 loci exhibited species-specific signatures of selection based on branch-site tests, including genes involved in craniofacial and limb development in the jaguar, hypoxia in the snow leopard, and reproduction and melanogenesis in the tiger, as well as sensory perception (vision and olfaction) and protein metabolism in more than one species (Fig. 3B).
We then inspected the genes bearing species-specific signatures of selection, and for several of them, we observed evidence of relevant functional impact. For example, the jaguar is distinguished among the living Panthera (and the inferred ancestral phenotype) by its distinctively massive head and powerful bite (13) (Fig. 3C). These unique features have been hypothesized to represent jaguar adaptations to a diet largely concentrated on heavily armored reptiles (caiman and freshwater turtles), which may have evolved as a response to the extinction of large mammalian prey at the end of the Pleistocene (14). We identified two genes (ESRP1 and SSTR4) possibly associated with these unique traits because they exhibit jaguar-specific signatures of positive selection and are known to affect craniofacial development (15–17). For both ESRP1 and SSTR4, the particular residues bearing signatures of positive selection in the jaguar [based on the Bayes empirical Bayes (BEB) estimate obtained with the CODEML software; see Materials and Methods] are likely to have important functional roles (see Fig. 3C), laying out a clear hypothesis that can be tested experimentally in model systems.
To test whether signatures of interspecies adaptive divergence (detected by the site and branch-site models) and introgression (detected by the outlier window test) in the Panthera implicate a similar set of phenotypes, we performed functional enrichment analyses of the gene sets retrieved with these different approaches. Although the overlap in genes was very small, there was a remarkable overlap in enriched pathways (Fig. 3A), suggesting that these two evolutionary processes have affected similar phenotypes in this group. Thirty-nine enriched terms were shared among the three data sets, including the IGF-1 (insulin-like growth factor 1) and mTOR (mechanistic target of rapamycin) pathways, known to be involved in phenotypes such as body growth, aging, energy metabolism, and brain development (18, 19). Another term enriched in all three sets was the Glypican pathway, a complex interaction of loci underlying a variety of developmental processes, from bone formation to several neurological phenotypes (20). Remarkably, many of the Glypican pathway loci that we identified were closely connected within this functional network, although their signatures of selection or introgression were detected with different approaches (Fig. 3D). Dissecting the biological processes mediated by these loci is thus a promising avenue for understanding the adaptive evolution of Panthera and likely other mammalian systems.
To investigate whether genes involved in interspecies adaptation and introgression also exhibited intraspecific signatures of positive selection, we designed a custom set of exome capture probes for Panthera, targeting ~19,000 protein-coding genes (~36 Mb), and used it to survey 30 wild-caught jaguars sampled in the Amazon, Pantanal, and Cerrado biomes of South America. We observed extensive variation in nucleotide diversity among genes, with ~10% of the loci (including 12 genes with surveyed coding segments >5 kb) showing zero variation in this set of individuals. We then focused on genes located in extreme outlier windows, that is, with highly significant signatures of interspecies introgression. For each of the 25 captured genes present in these windows, we performed coalescent simulations under the demographic scenarios of stable population size and historical decline based on the PSMC result. To control for a potential effect of the assumed recombination rate, we tested both domestic cat chromosome–specific rates (10) and a conservative scenario with zero recombination. Three genes (DOCK3, COL4A5, and PPFIA2) consistently exhibited the strongest signals of positive selection (P < 0.004) across all four demography/recombination rate scenarios (Fig. 4 and table S14). Two of them (DOCK3 and COL4A5) remained significant (P < 0.05) after a Bonferroni correction, whereas the third was marginally nonsignificant (P = 0.06 to 0.08). To verify whether there was any anomaly (for example, effects of atypical mutation rates and/or background selection) in the genomic regions containing these three genes, we assessed their patterns of genetic divergence in comparisons between each Panthera species and the domestic cat. No anomaly was observed (fig. S2), supporting the interpretation that our results are caused by a combination of interspecies introgression and subsequent positive selection in the jaguar.
All three of these genes are involved in brain development and function. PPFIA2 (also known as Liprin-alpha-2) is a synaptic scaffold protein that plays an important role in synaptic plasticity (21) and has also been recently identified as a strong candidate gene for human high-grade myopia (22). Its signal of interspecies introgression is particularly strong because it includes three extreme outlier windows and six additional outlier windows (Fig. 4), and for all nine 100-kb segments, the species tree is significantly rejected. It is thus a promising candidate gene for analyses of adaptive introgression affecting brain function and visual perception in this group.
An even stronger case for post-introgression adaptation emerges for the two genes bearing significant post-correction signatures of intraspecific positive selection (Fig. 4). Remarkably, both DOCK3 and COL4A5 are involved in axon growth and guidance affecting the optic nerve. DOCK3 is specifically expressed in neurons and has been shown to act directly on axonal outgrowth and optic nerve regeneration (23–25). COL4A5 is implicated in ~85% of cases of Alport syndrome, which affects kidneys, ears, and eyes through defects in the basement membranes of these organs (26, 27). Its effects on ocular function include a critical role in guiding retinal ganglion axons (which form the optic nerve) into the midbrain optic tectum (28, 29). The observation that both genes with significant evidence of adaptive introgression in jaguars affect axon development in the optic nerve is striking and suggests that selection has acted on a vision-related phenotype in this species, taking advantage of previously introgressed genomic segments.
Together, these results illustrate the complex interplay among rapid divergence, post-speciation admixture, and natural selection in the context of a highly successful adaptive radiation. The extent of genome-wide genealogical discordance observed within the Panthera radiation demonstrates that fully resolving such phylogenies is more complex than usually assumed and that post-speciation admixture is a widespread phenomenon that must be accounted for. Finally, the coincidence of low effective population sizes and positive selection following post-speciation admixture suggests that interspecies hybridization may be a recurrent route of evolutionary rescue in this remarkable lineage.
MATERIALS AND METHODS
Jaguar genome sequencing and assembly
The target individual (“Vagalume”) was a healthy, wild-caught male born in the southern Brazilian Pantanal region and currently housed at the Sorocaba Municipal Zoo, Brazil. Certified veterinarians performed all anesthesia and sampling procedures during routine health checkups performed by the zoo staff following Institutional Animal Care and Use Committee guidelines. We collected ca. 100 ml of whole blood in multiple Vacutainer tubes with K2EDTA, stored them at 4°C for less than 48 hours, and performed DNA extraction with the Qiagen Blood and Tissue Kit (Qiagen). Vagalume’s genome was sequenced at ~94x coverage using three libraries and five Illumina HiSeq 2500 lanes [one 180-bp insert paired-end library, sequenced in three lanes, and two mate pair libraries (3- and 8-kb insert size) sequenced in one lane each] (table S1). We applied the standard ALLPATHS-LG (30) de novo assembly pipeline to generate a jaguar whole-genome assembly, with genome size of 2.4 Gb (contig N50 length of 28.6 kb and scaffold N50 length of 1.5 Mb; see table S2).
Jaguar RNA-seq data
To aid in genome annotation, we collected additional blood samples and small biopsies of muscle, testicle, gum, and skin from Vagalume during routine veterinarian procedures conducted at the zoo. These samples were immediately stabilized with RNAlater (Invitrogen) at a 10:1 proportion relative to the sample. Total RNA was extracted with a standard TRIzol RNA extraction protocol (31). We performed RNA quantification and quality assessment using a NanoDrop spectrophotometer (Thermo Fisher Scientific) and Agilent Bioanalyzer. RNA samples with RNA Integrity Number equal or above 8 were used. We constructed a cDNA (complementary DNA) library for each sample using the TruSeq RNA Sample Prep Kit v2 (Illumina) and used it to perform multiplexed/bar-coded RNA-seq with an Illumina HiScan sequencer (table S4).
Leopard genome sequencing
To allow comparisons across all extant Panthera species, we generated wgs data from a male leopard (P. pardus). DNA was extracted from a fibroblast cell line and used to generate a standard 250-bp insert Illumina library, which was sequenced to ~25x coverage using paired-end 125-bp reads using the Illumina HiSeq 2500 platform (table S5).
Annotation of the jaguar genome
The jaguar genome assembly was annotated de novo using a variety of approaches that targeted protein-coding genes, repeats, noncoding RNAs, and nuclear insertions of mitochondrial DNA.
De novo prediction of coding genes
We used MAKER2 (32), a genome annotation and data management tool designed for second-generation genome projects. Ab initio gene predictions were produced by the SNAP (Scalable Nucleotide Alignment Program) version 2013-02-16 (33). To improve annotation quality, we used three different strategies to evaluate gene models in the MAKER2 pipeline. For the first step, we used tiger proteins and RefSeq protein evidence. In the second step, we built the SNAP model (Jaguar.hmm) and used jaguar RNA-seq generated in this study. In the last step, we used Jaguar.hmm to finish the annotation of genes. All predictions were produced in standardized GFF3 format. Evidence-based gene annotations in MAKER2 were produced using default settings. Finally, we aligned the predictions to a TE protein database using BlastP with the E value set to 1 × 10−10. In total, we identified 25,451 genes, whose features are described in table S3.
Functional annotation of coding genes
We used InterProScan5, a tool that combines different protein signature recognition methods into a single resource. This provides an overview of the families that a protein belongs to and the domains and sites it contains, identifying the presence and organization of protein sequence domains as well as critical residues. The genome sequence was submitted in FASTA format. Matches were then calculated against all of the required member database signatures (BlastProDom, FPrintScan, HMMPIR, HMMPfam, HMMSmart, HMMTigr, ProfileScan, HAMAP, PatternScan, SuperFamily, SignalPHMM, TMHMM, HMMPanther, Gene3D, Phobius, and Coils), and the results were output in TSV (a simple tab-delimited file) format. The InterProScan5 approach was able to annotate 22,191 jaguar genes, with only 3260 predicted genes not showing matches with InterPro’s signatures. For the 3260 genes that did not show an InterPro signature, we chose CDD [conserved domain database of the National Center for Biotechnology Information (NCBI)], which provides an online tool to annotate protein domains. In total, protein domains were found in 197 of these jaguar genes.
The predicted genes were characterized through a BLAST search against the UniProt Knowledge Base (E = 1 × 10−10). Genes were assigned to Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology groups using a UniRef Enriched database (UEKO). In total, 21,279 Jaguar genes (83.6%) showed hits to UniProt, and 4020 genes showed matches with KO.
The RNA-seq data set generated in this study from the same jaguar individual whose genome was sequenced was used to validate the annotated coding genes. To do so, the sequence of each gene was treated as a reference and the RNA-seq reads were mapped against them. In total, 16,586 genes (65%) had an RNA-seq coverage of ≥75% of their predicted sequence.
We used OrthoMCL, a tool that provides a scalable method for constructing orthologous groups across multiple eukaryotic taxa, to search for orthologs between the jaguar and tiger genomes. This method uses a Markov Cluster algorithm to group putative orthologs and paralogs. In total, 16,680 genes were clustered using this approach.
To manage and analyze the multiple types of information and integrate all the annotation data, we built a database named Jaguar_SQL, a Structured Query Language (SQL) relational database using MySQL as a database management system. To recover potential genes in this database, the ID of these genes was used in SQL search, and the Final Table was built. For validation of genes, the following criteria were used: (i) genes with sum greater than 1 were considered to have some evidence, and (ii) the gene’s RNA-seq coverage was required to be above 75%.
Annotation of repetitive regions in the jaguar genome
To estimate the overall repeat content of the genome, we used WindowMasker (34). This masked 29.66% of the genome according to the presence of repeated fragments with exact sequences. For the annotation of known repeats, we used the RepeatMasker software (35) and carnivora-specific library from the Repbase Update library version 20140131 (36). RepeatMasker masked 37.48% of genome including 18.92% of long interspersed nuclear elements, 10.40% of short interspersed nuclear elements (SINEs), 5.11% of long terminal repeat elements, and 2.86% of DNA elements (table S15). To mask simple sequence repeats, we used the DUST software (37), and the Jaguar genome contained 11.20%. To detect tandem repeats, we used the Tandem Repeats Finder (TRF) software version 4.07 (38) with mismatch and maximum period parameter values set to 5 and 2000, respectively; TRF output was processed as published previously (39). We found tandem repeats divided into the following groups with the Trevis software: microsatellites, perfect microsatellites, complex tandem repeats, and three groups of large tandem repeats according to array length (table S16). In addition, we computed statistics for 40 of the largest families of microsatellites. Of the two largest families formed by (AG)n and (AC)n microsatellites, 68.72% of (AG)n repeats had imperfect arrays, and 62.1% of (AC)n repeats had perfect microsatellite arrays (table S17).
Annotation of noncoding RNA genes in the jaguar genome
Noncoding RNA genes were annotated according to the Ensembl recommendations (www.ensembl.org/info/genome/genebuild/ncrna.html). All Rfam sequences were aligned against genomic sequences using BLASTN with E = 1 × 10−5. BLAST hits were clustered and used to seed Infernal searches with the corresponding Rfam covariance models (40). The resulting BLAST hits were used as supporting evidence for noncoding RNA genes confirmed by Infernal (41). The noncoding RNA annotation summary is shown in table S18. For the tRNAs, we excluded tRNA predictions from Rfam results and used the results from tRNAscan-SE with default parameters instead (42). Only predictions with a cove score greater than 20 were considered. tRNAscan predicted 169,838 regions with possible tRNA genes. The largest parts are felid-specific SINEs containing SelCys, Arg, Lys, and Gln tRNA fragments [carnivore-specific SINEs (43, 44)]. After removing fragmented Arg, Lys, and Gln tRNAs with stricter parameters (coverage score greater than 60), only 3299 tRNA genes remained, which approaches the 3039 tRNA genes reported in the domestic cat genome (45).
Annotation of NUMTs in the jaguar genome
To annotate nuclear mitochondrial translocations (NUMTs) in the jaguar reference genome, we used a BLAST-based approach. Using the complete jaguar mitochondrial genome (generated in this study) as a query, matches were sought using the following search parameters: (i) a hit with at least 16 bp; (ii) E value threshold of 1 × 10−10; (iii) no DUST filter query; (iv) cost of 0 to open a gap and 2 to extend it; (v) X dropoff value of 40, for preliminary gapped extensions; and (vi) reward for a match and penalty for a mismatch of 1. This search led to the identification of 171 fragments bearing similarity to mitochondrial sequences. These were distributed in 128 scaffolds of the assembled jaguar genome and ranged in length between 86 and 7361 bp (table S19).
In-depth analyses of gene families
Phylome analysis across the Carnivora
We reconstructed the full complement of evolutionary histories of jaguar genes (that is, the jaguar phylome) based on the de novo annotation reported here and 14 additional mammalian genomes: P. pardus (this study), P. tigris (NCBI BioProject PRJNA236771), P. uncia (SRX273036), P. leo (SRX 273034), Felis catus (PRJNA16726), Lynx pardinus (Lynx genome project), Ursus maritimus (www.gigadb.org), Ailuropoda melanoleuca (Ensembl), Odobenus rosmarus (PRJNA167474), Mustela putorius (Ensembl), Canis familiaris (Ensembl), Bos taurus (Ensembl), Mus musculus (Ensembl), and Homo sapiens (Ensembl). This is the most complete phylogenomic analysis of felid genes performed thus far. We reconstructed two versions of this phylome using the PhylomeDB pipeline (46). The first one was based on protein sequences, including all species, and the second one was based on nucleotide sequences and a codon-based evolutionary model, excluding some of the taxa (see below). In this pipeline, for each gene encoded in P. onca, a Smith-Waterman sequence search was performed against the proteomes of the 14 species considered. We used an E value threshold of 1 × 10−5 and required a continuous overlap of 50% over the query sequence. Hits were limited to the closest 150 homologs per gene, which were then aligned in the forward and reverse orientations using three different programs [MUSCLE (47), MAFFT (48), and KALIGN (49)]. The six resulting alignments were then combined with M-COFFEE (50) and trimmed with trimAl v1.3 using gap and consistency thresholds of 0.1 and 0.16667, respectively. These alignments were used to build a protein-based phylome. We then reconstructed ML trees using PhyML v3.0 and the best-fitting evolutionary model among seven different options (JTT, LG, WAG, Blosum62, MtREV, VT, and Dayhoff). The two best-fitting models were determined on the basis of likelihoods of an initial Neighbor Joining tree and using the Akaike information criterion. In all cases, we used four rate categories, inferred the fraction of invariant positions and rate parameters from the data, and computed branch supports by using an approximate likelihood ratio parametric test based on a χ² distribution.
Considering the high levels of similarity across orthologs in the felid species, we decided to back-translate the trimmed protein alignments into their respective codons, using the coding sequences. The low quality of inferred coding DNA sequence (CDS) from the low-coverage genomes (P. uncia, P. leo, and P. pardus) prevented us from using them in the nucleotide-based phylome, so these sequences were removed from the alignments using trimAl. Nevertheless, both phylomes were scanned to predict orthology and paralogy relationships (51) and detect duplication events (52), as described below. We used trimAl to back-translate amino acid residues to their corresponding codons. ML trees were reconstructed on the basis of the codon alignments using codonPhyML v1.0 (PMID 23436912), using GY (Goldman and Yang) as the specific substitution model for codon data and F3X4 as the model for defining the codon frequency from the alignment. In this case, we used a discrete gamma distribution with three rate categories, estimating the gamma parameter from the data. Both phylomes can be browsed and downloaded in PhylomeDB (46) with the PhylomeIDs 583 (protein) and 584 (nucleotide).
Orthology and paralogy relationships were inferred on the basis of phylogenetic evidence from the gene trees contained in the phylome. For this purpose, we used a species-overlap approach as implemented in ETE v2 (53) and used an overlap score of 0. This approach considers a node as speciation-derived when there are no overlapping species between the clades defined by the two daughter branches and a duplication node if otherwise. Orthologs and paralogs are then inferred according to the original orthology definition, that is, orthologous genes are those whose last common ancestor is represented by a speciation event, whereas paralogous genes are those that diverged at duplication events (51). All orthology and paralogy relationships are available through PhylomeDB (46).
The nucleotide-based phylome was analyzed to detect duplication events and establish the lineage in which they occurred. To accomplish this, we used a previously described algorithm (52), as implemented in ETE v2 (79). Using these data, we computed the duplication density (duplications per branch) for all lineages leading to P. onca. HMMER v3.1b2 (PMID 21593126) was used to find domains that contained homology with viral and transposable elements (based on Pfam-A.hmm domains collection). Additional filtration was performed on the basis of Gene Ontology functional terms. A total of 124 proteins were predicted to be transposable elements and were not used in subsequent analyses.
Finally, we constructed a nucleotide data set with the 2151 genes that comprised single-copy orthologs in each of the analyzed carnivore species that has an assembled genome (that is, the low-coverage felid species were not included) and cow and human as successive outgroups. Their trimmed alignments (as constructed in the phylome) were concatenated, and the supermatrix was further trimmed to delete all positions with gaps or missing information, resulting in a final data set containing 1,467,838 nucleotides. We used RAxML 8.0 (54) to reconstruct the ML phylogeny of the included species, incorporating a GTRGAMMA model and applying 1000 bootstrap replicates to verify nodal support.
The agreement between orthology calls between the protein- and nucleotide-based phylomes (for the species that they have in common) was high, with 87% of overlap among predicted orthology relationships. All trees, alignments, and homology relationships for these phylomes, which constitute a valuable resource for felid evolutionary studies, can be accessed at www.phylomedb.org) (46). The phylogenetic tree reconstructed with the 2151 single-copy orthologous genes (below) was consistent with current knowledge of the relationships among carnivoran lineages (55). We used the reconstructed phylogeny to map the number of duplication episodes (and resulting genes) in the three branches leading to the jaguar (fig. S3). This inventory of duplicated genes provides an interesting resource for in-depth investigations of gene family evolutionary dynamics in the Felidae.
Analysis of olfactory receptor genes in the Panthera
Assembled contigs for each species were mined for putative olfactory receptor (OR) genes using the “ORA” BioPerl package (56, 57). ORA uses profile hidden Markov models designed using a data set of mammalian OR genes to scan a target sequence and identify all possible ORs it contains. Tblastx (58) was also used to locate any ORs that ORA may have failed to locate. Sequences with in-frame stop codons or with length less than 650 bp (56) were considered pseudogenes, whereas short OR fragments with an E < 1 × 10−5 were considered as “truncated” nonfunctional OR genes. ORs that contained unresolved regions designated by one or more “N” positions were classified as “unknown,” because their functionality, or lack thereof, could not be determined. Leading or tailing runs of N nucleotides were trimmed while retaining the correct reading frame.
Using BLASTN (58), the OR repertoire from each individual species was compared against the other four. This was performed to count the number of orthologous OR genes showing a conserved functionality or loss of function. BLASTN was also used to identify species-specific gene loss events.
To identify species-specific gene duplications and the subsequent fate of post-duplication ORs, we generated phylogenetic trees for each OR subfamily (13 trees; OR1/3/7, OR2/13, OR4, OR5/8/9, OR6, OR10, OR11, OR12, OR14, OR51, OR52, OR55, and OR5631) using amino acid alignments generated by ClustalO (59) and RAxML (54). The model of protein sequence evolution that best fitted the data was determined using ProtTest (60). Each tree was split into all of its possible subtrees. Subtrees consisting entirely of OR genes from one species were considered to represent gene duplication events.
A total of 5115 putative OR sequences were found across all five species (P. tigris, 1053 ORs; P. leo, 1018 ORs; P. onca, 994 ORs; P. uncia, 1023 ORs; and P. pardus, 1027 ORs). Because of the presence of OR genes with unknown bases in our data set, the total number of functional and nonfunctional could not be fully determined. It is noteworthy that all of the OR gene sequences annotated for P. tigris on GenBank were present in the mined OR data set.
An average of 95 ORs were nonfunctional in each species, with an average of 77 showing conserved stop codons, which indicate loss of function in the most recent common ancestor of Panthera. There were 460 ORs that were functional in all species. Because the loss of function in certain ORs could not be determined, the number of ORs with conserved functionality or loss of function remains conservative.
The total numbers of species-specific loss of function events were as follows: P. tigris, 13 ORs; P. leo, 4 ORs; P. onca, 33 ORs; P. uncia, 18 ORs; and P. pardus, 12 ORs. For OR families 4 and 6, the best model of sequence evolution was determined as JTT + I + G + F; for the remaining families, it was JTT + G + F. Gene trees for each OR family were generated using RAxML, and it was determined that there were three instances of species-specific duplication in P. tigris, three in P. onca, two in P. uncia, and one in P. pardus, with no species-specific duplications observed in P. leo. This yielded a total of nine duplications, giving rise to 18 OR genes, 10 of which have subsequently lost their function. These results lay the basis for in-depth analyses of the functional evolution of ORs in the Panthera.
Analysis of demographic history using genome-wide data
We applied the PMSC analysis (61) to estimate the demographic history of each Panthera species. To call diploid sequences, we generated de novo assemblies for the lion, leopard, and snow leopard using SOAPdenovo2 (62) with k-mer set to 31. All quality-trimmed Illumina sequences of each Panthera species were mapped to their own de novo genome assembly using Burrows-Wheeler Aligner (BWA) (63) with default parameter settings. SAMtools (64) was used to estimate average mapping coverage and to call and filter nucleotide variants. Genome regions with less than half or more than twice the average whole-genome mapping depth were excluded from the final diploid sequences. We applied a mutation rate of 1 × 10−8 and a generation time of 5 years for all five Panthera species. We evaluated the consistency of the PSMC tests by performing 100 bootstrap replicates.
Phylogenomics using the window-based data set
We used TrimGalore (www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to trim the raw Illumina reads and screen for potential adapter sequences in data from all five Panthera species. Filtered reads were mapped to the whole-genome assembly reference of the tiger V1.0 with BWA. We reordered all tiger assembly scaffolds relative to the domestic cat genome assembly (version felCat5) using LAST (65) with default parameter settings. Removal of polymerase chain reaction–induced sequence duplicates and calling and filtering raw single-nucleotide variants (SNVs) were performed using SAMtools. We filtered the SNV data to retain high-quality SNVs (quality > 100). Variants from genomic regions with read-depth variation greater or less than 50% of the genome-wide average (calculated in 10-kb windows) with mapping quality >30 were excluded. We then merged the filtered SNV data into whole-genome alignments conforming to the structure of the domestic cat reference genome assembly. We used this six-species genome alignment (jaguar, tiger, snow leopard, leopard, lion, and domestic cat) to perform sliding window–based phylogenomic analyses. We tested multiple window sizes and step lengths (for example, 500-kb window size and 100-kb step for the analysis shown in Fig. 1C, and 100-kb window/100-kb step for all others shown here) and also performed analyses using approaches that allowed windows to have variable lengths (not shown). All approaches led to consistent results. ML tree searching and bootstrap analysis (200 bootstrap replications) were performed for each window using the software RAxML (54) with a GTR + Γ substitution model. For each window, we used the ML tree and sequence data as input for the program MCMCTree in the software package PAML4 (66) to estimate divergence time variation across the genomes of the five Panthera species. We used two soft constraints: (i) divergence of Pantherinae and Felinae lineages between 9 and 15 Ma and (ii) base of Panthera no earlier than 7 Ma. These constraints are based on the 95% credibility intervals of supermatrix-derived divergence time estimates from previous felid supermatrix analyses (4, 5). Hence, we interpret the ages as relative divergence times rather than absolute estimates, given that these are secondarily derived from fossil calibration–based divergence times. These analyses were performed assuming autocorrelated rates among branches of the tree. To provide a more accurate estimate of the Panthera species divergence time that avoids the confounding effects of post-speciation interspecies introgression, we compiled a submatrix of nonoverlapping whole-genome sliding windows that conformed to the species tree {(tiger, snow leopard),[jaguar,(lion,leopard)]} with strong statistical support [determined using the approximately unbiased (AU) test implemented in CONSEL (67)]. We used this matrix to reestimate the divergence times for all nodes in the genus Panthera using MCMCTree.
Phylogenomics using the gene-based data set
We aligned the genomes of the five Panthera species and the domestic cat (F. catus) using LAST. We used three assembled genomes (jaguar, tiger, and domestic cat) and raw genomic reads for the three species with low-coverage data (lion, leopard, and snow leopard) (table S20). We used the jaguar genome as the reference for mapping and performed the same exercise using the more contiguous tiger genome as the reference. Because the results were consistent, we used the tiger-based alignments in the downstream steps. After mapping, we used SAMtools to compute a species-specific consensus, and its coordinates matched exactly those of the reference (table S21). These alignments could then be used to characterize synteny blocks among jaguar, tiger, and domestic cat genomes as well as to extract coding genes from all species. The latter were inferred to be orthologous among all species based on their genomic position relative to the reference. The set of 13,624 identified orthologous genes (including intron + exon sequences for each locus) was used in the phylogenomic analyses, using ML search with RAxML (GTR + Γ substitution model). We tested whether gene-specific data sets significantly rejected the species tree using the AU test. Finally, to test whether the phylogenetic signal in the gene-based data set could be biased by exon-driven information, we performed a separate set of analyses with introns only (see table S7).
Analysis of interspecies introgression
We applied the ABBA/BABA approach of Green et al. (68) to evaluate the imbalanced frequency of alleles present within alternative tree topologies. All trimmed Illumina sequences were mapped to the repeat-masked genome reference of the domestic cat (felCat5) using BWA. We enforced a minimum mapping quality score of 40. The software package ANGSD (69) was used to calculate D statistics and z scores based on a weighted block jackknife tests (block size of 5 Mb). Statistical significance of the z score was assessed for each replicate by converting the z score into a two-tailed P value. One hundred bootstrap iterations were used to measure the SD of the D statistic.
Detection of signatures of positive selection in the Panthera species
Selection analyses were performed on the basis of dN/dS ratios observed in the CDS retrieved from the gene-based data set. The extracted CDS were aligned and verified (for example, checking for an open reading frame, as well as matching start and stop codon positions, in addition to exon boundaries), and only loci that passed these filtering steps (13,183) were used in the selection screen. We also performed manual, in-depth assessments of comparative gene structure between the jaguar annotation and three other genomes (tiger, domestic cat, and human) in the case of genes identified as bearing particularly relevant signatures of positive selection. Different (nested) models, assuming a neutral model or allowing positive selection, were assessed on the basis of their log likelihoods using the software CODEML within the package PAML4 (66). For every pair of comparable models (M1 versus M2a and M7 versus M8), a likelihood ratio test was used to assess the best-fit scenario for a particular gene. For every comparison, we used the species tree and the locus-specific gene tree (see fig. S1), and only considered genes that exhibited significant results for both. We initially performed a site-model analysis and then specifically assessed every terminal (species-specific) branch of the phylogeny through a branch-site model analysis. In every case, we used a gamma correction for the rate heterogeneity among sites (GTRGAMMA) (tables S9 to S13). We performed a gene enrichment analysis for the genes under selection on each method using WebGestalt (70), with the Human genome as the reference, and searching the following data bases: Gene ontology (71, 72), Pathway Commons (73), KEGG pathway (74, 75), Disease (http://glad4u.zhang-lab.org/index.php), and Phenotypes (76). We used a significance threshold of 0.05 and a multiple testing correction (Benjamini-Hochberg) to control for false discovery. Within genes that significantly rejected neutral models, we identified specific codons with signatures of positive selection based on the BEB estimate produced with CODEML, using a probability threshold of 0.95.
Outlier window test
We estimated divergence times using the genome-wide Panthera-Felis alignments, partitioned into 100-kb windows, as input for the MCMCTree v4.8a software in the PAML4 package. Analyses were run for 100,000 generations with a burn-in of 10,000 generations. Analyses were run twice to check for convergence. We used two soft constraints: (i) the divergence time of Panthera from Felis lineages between 9 and 15 Ma and (ii) the base of Panthera no earlier than 7 Ma. Analyses were performed assuming autocorrelated rates between branches of the tree. We next focused specifically on the distortions on divergence times and relationships within the lion-leopard-jaguar clade, as these constituted most of the discordant gene trees (Fig. 2A). We Z-transformed the complete collection of window-based divergence times at each node and categorized the internal nodes separately (that is, lion + jaguar, lion + leopard, and leopard + jaguar). We selected those windows with divergence time point estimates of 2 or 3 sdm for the basal nodes and >2 sdm for the internal nodes because none were more than 3 sdm. We identified all annotated F. catus genes in which the gene body overlapped or was contained entirely within each window (table S22). For outlier windows, we assessed enrichment of KEGG pathway and disease gene association tests using WebGestalt and gene symbols as input (organism of interest, H. sapiens). Only significant KEGG pathways and disease association categories were reported, using a hypergeometric test and the significance level at 0.05 and the Benjamini-Hochberg multiple test adjustment to control for false discovery (tables S23 to S26).
Analysis of the Glypican pathway
Given the overlapping enrichment results from the selection screens and the outlier window tests, indicating that the Glypican pathway (ID 1459 in the Pathway Commons database) was implicated in both adaptive evolution and interspecies introgression, we performed a specific analysis focusing on the identified genes belonging to this network. Using StringDB (http://string-db.org/), we performed a search for interactions using the coexpression evidence, biochemical experiments data, and curated pathways databases as sources. To expand connections, we allowed up to five unlisted genes to be added to the network. The minimum score to support for these interactions was 0.4.
Jaguar exome capture and sequencing
To assess intraspecific signatures of positive selection, we selected 30 jaguar individuals sampled in three different biomes (Amazon, Pantanal, and Cerrado), using DNA samples obtained from blood or tissue collected during previous field ecology studies and deposited at the LBGM/PUCRS and CENAP/ICMBio collections (table S22). For exome probe design, we used the jaguar transcriptome, both cat and tiger published genomes, available wgs data for the lion and snow leopard (9), and our novel leopard wgs data. We used a custom pipeline (incorporating a reciprocal BLAST search to infer orthology) to annotate the CDS and its flanking region for all genes available for each species, using the domestic cat annotation as a reference. We then used RepeatMasker to remove repeats, especially from the flanking regions, and trimmed those regions that were longer than 500 bp. To maximize sequence reliability and capture efficiency in the Panthera, we submitted the finalized data set for each species to a selection criterion (that is, keeping only one sequence per locus), prioritizing the data source as follows: jaguar (genome and transcriptome data) > tiger (genome) > leopard (mapped reads) = snow leopard (mapped reads) = lion (mapped reads) > domestic cat (genome). We then merged the CDS + flanking regions into a unified fasta file. For the capture experiment, we used a Bioruptor UCD-200 (Diagenode) to sonicate the DNA samples at a low setting. For each sonicated sample, 4.5 μl of product was run on a 1.5% agarose gel at 135 V for ca. 30 min to ensure fragments were appropriately sized (100 to 500 bp; average, 200 to 300 bp). Individual genomic libraries were prepared following the study by Meyer and Kirchner (77), with modifications. Samples were pooled together considering their initial concentration and quality. Sequence quality filtering was performed using a previously published analytical pipeline (78). The parameters involved sequence trimming and removal based on the quality score (phred score, >30) with Trimmomatic (79), removal of sequencing adapters with CutAdapt (80), and preliminary assembly of overlapping paired reads using FLASH (81). This preliminary assembly facilitated further steps, where we mapped the whole exome against our reference. After assessing coverage, we trimmed the data removing individuals and sites with discrepant coverage using SNPcleaner (https://github.com/tplinderoth/ngsQC/tree/master/snpCleaner). On the basis of average depth per individual and per site, we kept up to 70% of individuals with site depth between 2× and 30×. We then calculated the nucleotide diversity per gene (π) for all individuals using VCFtools (82) and used the results to perform gene-based coalescent simulations to test for intraspecific positive selection (table S14).
Assessment of positive selection in jaguar intraspecific data
To test for the occurrence of within-species positive selection affecting genes implicated in interspecies admixture, we identified the loci that were sampled in the exome capture experiment and that were contained in (or overlapped with) extreme outlier windows. For each of the loci, we performed coalescent simulations using MS (83), considering four different demographic/recombination scenarios. We assumed both a stable population size and a past bottleneck based on our PSMC results and two different recombination rates [zero recombination or average chromosome rates based on the domestic cat (10)]. We used the exome-wide average nucleotide diversity per site and the length of each gene (coding sequence only) to simulate the expected number of segregating sites per locus under neutrality. We ran each scenario 10,000 times per gene and used custom-made scripts to generate the null distribution. Genes whose observed diversity was significantly lower (P < 0.05 after Bonferroni correction) than the expected mean were considered to bear a signature of positive selection. To control for the possible effects of anomalous mutation rates or background selection on the genomic regions containing outlier genes, we assessed the divergence (measured as p distances in nonoverlapping 100-kb windows) between each Panthera species and the domestic cat. This was performed across the whole genome, generating a null distribution against which the focal regions were compared (fig. S2).
Supplementary Material
Acknowledgments
Funding: This study was funded by CNPq/Brazil (grants 311327/2011-7 and 487396/2012-0 to E.E. and 309312/2012-4 to G.O.), FAPERGS/Brazil (grant 12/2236-0 to E.E.), FAPEMIG/Brazil (grant RED-00014-14 to G.O.), FAPESP/Brazil, Tetra Pak, Morris Animal Foundation (grants D12FE-019 and D12FE-502 to W.J.M.), and Russian Science Foundation (grant 17-14-01138 to S.J.O.). S.J.O. was supported by the Russian Ministry of Science (mega-grant no. 11.G34.31.0068). Author contributions: E.E., H.V.F., W.J.M., G.L., L.L.C., and G.O. conceived the study. H.V.F., C.S.T., R.A.B., P.M.S.V., and L.L.C. collected the data. E.E., W.J.M., L.S., F.C.C.A., D.K., E.R., A.L.V.N., R.H.F.T., and R.G.M. contributed reagents, biological samples, and resources. H.V.F., G.L., F.J.T., J.A., F.P., G.F., S.H.D.S., G.M.H., A.K., A.A., M.R.R., T.L., K.B., D.L., P.S., T.G., and E.E. performed the data analyses. E.E., W.J.M., H.V.F., G.L., R.N., S.J.O., E.C.T., and T.G. led the interpretation of the results. E.E., H.V.F., W.J.M., and G.L. wrote the manuscript. All authors contributed to the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Jaguar genome data have been deposited in NCBI (BioProject ID PRJNA348348 and BioSample accession no. SAMN05907657). Leopard genome data are available at the NCBI Sequence Read Archive repository. Additional information is available at www.jaguargenome.org.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700299/DC1
fig. S1. Assessment of the effect of genealogical discordance on branch-site tests of positive selection.
fig. S2. Assessment of divergence between domestic cat and Panthera species in windows containing genes with signatures of interspecies introgression and positive selection.
fig. S3. Maximum-likelihood phylogeny constructed with a supermatrix consisting of 2151 concatenated single-copy orthologs for 11 mammals, derived from our phylome analysis.
fig. S4. Enrichment analysis for the site model test.
table S1. DNA sequencing information for the reference jaguar individual.
table S2. Jaguar genome assembly quality metrics.
table S3. Annotated protein-coding genes.
table S4. Summary of the jaguar transcriptome sequencing results.
table S5. DNA sequencing information for the leopard genome.
table S6. Tree topology frequencies in window- and gene-based analyses.
table S7. Tree topology frequencies for 3732 genes and their introns.
table S8. Detailed results of the ABBA/BABA tests.
table S9. Branch-site results for the jaguar.
table S10. Branch-site results for the lion.
table S11. Branch-site results for the snow leopard.
table S12. Branch-site results for the tiger.
table S13. Site model results.
table S14. Results of coalescent simulations based on jaguar exome data.
table S15. Summary of repeat annotation for the jaguar genome.
table S16. Tandem repeat annotation for the jaguar genome.
table S17. Forty largest microsatellite families in the jaguar genome.
table S18. Statistics on noncoding RNA annotation in the jaguar genome.
table S19. Annotation of nuclear insertions of mitochondrial DNA (NUMTS) in the jaguar genome.
table S20. Previously reported genomic resources used in our multispecies analyses.
table S21. Consensus genome information (using the tiger assembly as a reference).
table S22. Outlier window test results.
table S23. Outlier Window KEGG enrichment results.
table S24. Outlier Window Pathway Commons enrichment results.
table S25. Outlier Window Phenotype enrichment results.
table S26. Outlier Window Disease enrichment results.
table S27. Detailed information on the samples used in the exome capture experiment.
table S28. Enrichment results for the jaguar branch-site test.
table S29. Enrichment results for the lion branch-site test.
table S30. Tiger Pathway Commons enrichment results.
table S31. Tiger KEGG enrichment results.
table S32. Tiger Disease enrichment results.
table S33. Site model Pathway Commons enrichment results.
table S34. Site model KEGG enrichment results.
table S35. Site model Disease enrichment results.
table S36. Site model Biological Process enrichment results.
table S37. Site model Molecular Function enrichment results.
REFERENCES AND NOTES
- 1.J. A. Coyne, H. A. Orr, Speciation (Sinauer Associates, 2004). [Google Scholar]
- 2.Fontaine M. C., Pease J. B., Steele A., Waterhouse R. M., Neafsey D. E., Sharakhov I. V., Jiang X., Hall A. B., Catteruccia F., Kakani E., Mitchell S. N., Wu Y.-C., Smith H. A., Love R. R., Lawniczak M. K., Slotman M. A., Emrich S. J., Hahn M. W., Besansky N. J., Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347, 1258524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamichhaney S., Berglund J., Almén M. S., Maqbool K., Grabherr M., Martinez-Barrio A., Promerová M., Rubin C.-J., Wang C., Zamani N., Grant B. R., Grant P. R., Webster M. T., Andersson L., Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518, 371–375 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Johnson W. E., Eizirik E., Pecon-Slattery J., Murphy W. J., Antunes A., Teeling E., O’Brien S. J., The late miocene radiation of modern felidae: A genetic assessment. Science 311, 73–77 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Li G., Davis B. W., Eizirik E., Murphy W. J., Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res. 26, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.M. Sunquist, F. Sunquist, Wild Cats of the World (Univ. Chicago Press, 2002), pp. 287, 306, and 345. [Google Scholar]
- 7.Gonyea W. J., Adaptative differences in the body proportions of large felids. Acta Anat. 96, 81–96 (1976). [DOI] [PubMed] [Google Scholar]
- 8.Davis B. W., Li G., Murphy W. J., Supermatrix and species tree methods resolve phylogenetic relationships within the big cats, Panthera (Carnivora: Felidae). Mol. Phylogenet. Evol. 56, 64–76 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Cho Y. S., Hu L., Hou H., Lee H., Xu J., Kwon S., Oh S., Kim H.-M., Jho S., Kim S., Shin Y.-A., Kim B. C., Kim H., Kim C.-u., Luo S.-J., Johnson W. E., Koepfli K.-P., Schmidt-Küntzel A., Turner J. A., Marker L., Harper C., Miller S. M., Jacobs W., Bertola L. D., Kim T. H., Lee S., Zhou Q., Jung H.-J., Xu X., Gadhvi P., Xu P., Xiong Y., Luo Y., Pan S., Gou C., Chu X., Zhang J., Liu S., He J., Chen Y., Yang L., Yang Y., He J., Liu S., Wang J., Kim C. H., Kwak H., Kim J.-S., Hwang S., Ko J., Kim C.-B., Kim S., Bayarlkhagva D., Paek W. K., Kim S.-J., O’Brien S. J., Wang J., Bhak J., The tiger genome and comparative analysis with lion and snow leopard genomes. Nat. Commun. 4, 2433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li G., Hillier L. D. W., Grahn R. A., Zimin A. V., David V. A., Menotti-Raymond M., Middleton R., Hannah S., Hendrickson S., Makunin A., O’Brien S. J., Minx P., Wilson R. K., Lyons L. A., Warren W. C., Murphy W. J., A high-resolution SNP array-based linkage map anchors a new domestic cat draft genome assembly and provides detailed patterns of recombination. G3. 6, 1607–1616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degnan J. H., Rosenberg N. A., Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Hunter-Zinck H., Clark A. G., Aberrant time to most recent common ancestor as a signature of natural selection. Mol. Biol. Evol. 32, 2784–2797 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meachen-Samuels J., Van Valkenburgh B., Craniodental indicators of prey size preference in the Felidae. Biol. J. Linn. Soc. 96, 784–799 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Emmons L. H., Comparative feeding ecology of felids in a neotropical rainforest. Behav. Ecol. Sociobiol. 20, 271–283 (1987). [Google Scholar]
- 15.Baujat G., Rio M., Rossignol S., Sanlaville D., Lyonnet S., Le Merrer M., Munnich A., Gicquel C., Colleaux L., Cormier-Daire V., Clinical and molecular overlap in overgrowth syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 137C, 4–11 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Bebee T. W., Park J. W., Sheridan K. I., Warzecha C. C., Cieply B. W., Rohacek A. M., Xing Y., Carstens R. P., The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife 4, e08954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warzecha C. C., Sato T. K., Nabet B., Hogenesch J. B., Carstens R. P., ESRP1 and ESRP2 Are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 33, 591–601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Z., p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2, a001057 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crino P. B., The mTOR signalling cascade: Paving new roads to cure neurological disease. Nat. Rev. Neurol. 12, 379–392 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Filmus J., Capurro M., Rast J., Glypicans. Genome Biol. 9, 224 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spangler S. A., Schmitz S. K., Kevenaar J. T., de Graaff E., de Wit H., Demmers J., Toonen R. F., Hoogenraad C. C., Liprin-α2 promotes the presynaptic recruitment and turnover of RIM1/CASK to facilitate synaptic transmission. J. Cell Biol. 201, 915–928 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawthorne F., Feng S., Metlapally R., Li Y.-J., Tran-Viet K.-N., Guggenheim J. A., Malecaze F., Calvas P., Rosenberg T., Mackey D. A., Venturini C., Hysi P. G., Hammond C. J., Young T. L., Association mapping of the high-grade myopia MYP3 locus reveals novel candidates UHRF1BP1L, PTPRR, and PPFIA2. Invest. Ophthalmol. Vis. Sci. 54, 2076–2086 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Namekata K., Harada C., Taya C., Guo X., Kimura H., Parada L. F., Harada T., Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc. Natl. Acad. Sci. U.S.A. 107, 7586–7591 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadea G., Blangy A., Dock-family exchange factors in cell migration and disease. Eur. J. Cell Biol. 93, 466–477 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Laurin M., Côté J. F., Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev. 28, 533–547 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Agtmael T., Bruckner-Tuderman L., Basement membranes and human disease. Cell Tissue Res. 339, 167–188 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Savige J., Sheth S., Leys A., Nicholson A., Mack H. G., Colville D., Ocular features in Alport syndrome: Pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 10, 703–709 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao T., Baier H., Lamina-specific axonal projections in the zebrafish tectum require the type IV collagen Dragnet. Nat. Neurosci. 10, 1529–1537 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Xiao T., Staub W., Robles E., Gosse N. J., Cole G. J., Baier H., Assembly of lamina-specific neuronal connections by slit bound to type IV collagen. Cell 146, 164–176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler J., MacCallum I., Kleber M., Shlyakhter I. A., Belmonte M. K., Lander E. S., Nusbaum C., Jaffe D. B., ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chomczynski P., Sacchi N., Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 (1987). [DOI] [PubMed] [Google Scholar]
- 32.Holt C., Yandell M., MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korf I., Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgulis A., Gertz E. M., Schäffer A. A., Agarwala R., WindowMasker: Window-based masker for sequenced genomes. Bioinformatics 22, 134–141 (2006). [DOI] [PubMed] [Google Scholar]
- 35.A. Smit, R. Hubley, P. Green, RepeatMasker Open-3.0 (1996); www.repeatmasker.org.
- 36.Jurka J., Repbase Update: A database and an electronic journal of repetitive elements. Trends Genet. 16, 418–420 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Morgulis A., Gertz E. M., Schäffer A. A., Agarwala R., A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 13, 1028–1040 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Benson G., Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamazian G., Simonov S., Dobrynin P., Makunin A., Logachev A., Komissarov A., Shevchenko A., Brukhin V., Cherkasov N., Svitin A., Koepfli K. P., Pontius J., Driscoll C. A., Blackistone K., Barr C., Goldman D., Antunes A., Quilez J., Lorente-Galdos B., Alkan C., Marques-Bonet T., Menotti-Raymond M., David V. A., Narfström K., O’Brien S. J., Annotated features of domestic cat—Felis catus genome. Gigascience 3, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardner P. P., Daub J., Tate J. G., Nawrocki E. P., Kolbe D. L., Lindgreen S., Wilkinson A. C., Finn R. D., Griffiths-Jones S., Eddy S. R., Bateman A., Rfam: Updates to the RNA families database. Nucleic Acids Res. 37, D136–D140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nawrocki E. P., Kolbe D. L., Eddy S. R., Infernal 1.0: Inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowe T. M., Eddy S. R., tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walters-Conte K., Johnson D. L. E., Allard M., Pecon-Slattery J., Carnivore-specific SINEs (Can-SINEs): Distribution, evolution, and genomic impact. J. Hered. 102 (suppl. 1), S2–S10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vassetzky N. S., Kramerov D. A., CAN—A pan-carnivore SINE family. Mamm. Genome 13, 50–57 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Chan P. P., Lowe T. M., GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huerta-Cepas J., Capella-Gutiérrez S., Pryszcz L. P., Marcet-Houben M., Gabaldón T., PhylomeDB v4: Zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 42, D897–D902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katoh K., Kuma K.-i., Toh H., Miyata T., MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lassmann T., Sonnhammer E. L. L., Kalign—An accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 6, 298 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallace I. M., O’Sullivan O., Higgins D. G., Notredame C., M-Coffee: Combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34, 1692–1699 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabaldón T., Large-scale assignment of orthology: Back to phylogenetics? Genome Biol. 9, 235 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huerta-Cepas J., Gabaldón T., Assigning duplication events to relative temporal scales in genome-wide studies. Bioinformatics 27, 38–45 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Huerta-Cepas J., Dopazo J., Gabaldón T., ETE: A python Environment for Tree Exploration. BMC Bioinformatics 11, 24 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eizirik E., Murphy W. J., Koepfli K.-P., Johnson W. E., Dragoo J. W., Wayne R. K., O’Brien S. J., Pattern and timing of diversification of the mammalian order Carnivora inferred from multiple nuclear gene sequences. Mol. Phylogenet. Evol. 56, 49–63 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayden S., Bekaert M., Crider T. A., Mariani S., Murphy W. J., Teeling E. C., Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res. 20, 1–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hughes G. M., Gang L., Murphy W. J., Higgins D. G., Teeling E. C., Using Illumina next generation sequencing technologies to sequence multigene families in de novo species. Mol. Ecol. Resour. 13, 510–521 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 59.Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G., Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.D. Darriba, G. L. Taboada, R. Doallo, D. Posada, ProtTest-HPC: Fast Selection of Best-Fit Models of Protein Evolution, in Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics) (Springer, 2011), vol. 6586, pp. 177–184. [Google Scholar]
- 61.Li H., Durbin R., Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y., Tang J., Wu G., Zhang H., Shi Y., Liu Y., Yu C., Wang B., Lu Y., Han C., Cheung D. W., Yiu S.-M., Peng S., Xiaoqian Z., Liu G., Liao X., Li Y., Yang H., Wang J., Lam T.-W., Wang J., SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frith M. C., Hamada M., Horton P., Parameters for accurate genome alignment. BMC Bioinformatics 11, 80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Z., PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Shimodaira H., Hasegawa M., CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Green R. E., Krause J., Briggs A. W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M. H.-Y., Hansen N. F., Durand E. Y., Malaspinas A.-S., Jensen J. D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H. A., Good J. M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E. S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Doronichev V. B., Golovanova L. V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R. W., Johnson P. L. F., Eichler E. E., Falush D., Birney E., Mullikin J. C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S., A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics 15, 356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang J., Duncan D., Shi Z., Zhang B., WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 41, W77–W83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blake J. A., Gene ontology consortium: Going forward. Nucleic Acids Res. 43, D1049–D1056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.The Gene Ontology Consortium; Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G., Gene ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerami E. G., Gross B. E., Demir E., Rodchenkov I., Babur Ö., Anwar N., Schultz N., Bader G. D., Sander C., Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 39, 685–690 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M., KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kanehisa M., Goto S., KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Köhler S., Doelken S. C., Mungall C. J., Bauer S., Firth H. V., Bailleul-Forestier I., Black G. C., Brown D. L., Brudno M., Campbell J., FitzPatrick D. R., Eppig J. T., Jackson A. P., Freson K., Girdea M., Helbig I., Hurst J. A., Jähn J., Jackson L. G., Kelly A. M., Ledbetter D. H., Mansour S., Martin C. L., Moss C., Mumford A., Ouwehand W. H., Park S.-M., Riggs E. R., Scott R. H., Sisodiya S., Van Vooren S., Wapner R. J., Wilkie A. O., Wright C. F., Vulto-van Silfhout A. T., de Leeuw N., de Vries B. B. A., Washingthon N. L., Smith C. L., Westerfield M., Schofield P., Ruef B. J., Gkoutos G. V., Haendel M., Smedley D., Lewis S. E., Robinson P. N., The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 42, D966–D974 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meyer M., Kircher M., Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, prot5448 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Bi K., Vanderpool D., Singhal S., Linderoth T., Moritz C., Good J. M., Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics 13, 403 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
- 81.Magoč T., Salzberg S. L., FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hudson R. R., Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18, 337–338 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700299/DC1
fig. S1. Assessment of the effect of genealogical discordance on branch-site tests of positive selection.
fig. S2. Assessment of divergence between domestic cat and Panthera species in windows containing genes with signatures of interspecies introgression and positive selection.
fig. S3. Maximum-likelihood phylogeny constructed with a supermatrix consisting of 2151 concatenated single-copy orthologs for 11 mammals, derived from our phylome analysis.
fig. S4. Enrichment analysis for the site model test.
table S1. DNA sequencing information for the reference jaguar individual.
table S2. Jaguar genome assembly quality metrics.
table S3. Annotated protein-coding genes.
table S4. Summary of the jaguar transcriptome sequencing results.
table S5. DNA sequencing information for the leopard genome.
table S6. Tree topology frequencies in window- and gene-based analyses.
table S7. Tree topology frequencies for 3732 genes and their introns.
table S8. Detailed results of the ABBA/BABA tests.
table S9. Branch-site results for the jaguar.
table S10. Branch-site results for the lion.
table S11. Branch-site results for the snow leopard.
table S12. Branch-site results for the tiger.
table S13. Site model results.
table S14. Results of coalescent simulations based on jaguar exome data.
table S15. Summary of repeat annotation for the jaguar genome.
table S16. Tandem repeat annotation for the jaguar genome.
table S17. Forty largest microsatellite families in the jaguar genome.
table S18. Statistics on noncoding RNA annotation in the jaguar genome.
table S19. Annotation of nuclear insertions of mitochondrial DNA (NUMTS) in the jaguar genome.
table S20. Previously reported genomic resources used in our multispecies analyses.
table S21. Consensus genome information (using the tiger assembly as a reference).
table S22. Outlier window test results.
table S23. Outlier Window KEGG enrichment results.
table S24. Outlier Window Pathway Commons enrichment results.
table S25. Outlier Window Phenotype enrichment results.
table S26. Outlier Window Disease enrichment results.
table S27. Detailed information on the samples used in the exome capture experiment.
table S28. Enrichment results for the jaguar branch-site test.
table S29. Enrichment results for the lion branch-site test.
table S30. Tiger Pathway Commons enrichment results.
table S31. Tiger KEGG enrichment results.
table S32. Tiger Disease enrichment results.
table S33. Site model Pathway Commons enrichment results.
table S34. Site model KEGG enrichment results.
table S35. Site model Disease enrichment results.
table S36. Site model Biological Process enrichment results.
table S37. Site model Molecular Function enrichment results.