

Abstract
Purpose of review
Peripheral T cell lymphomas (PTCLs) are markedly heterogeneous at the clinical, pathological, and molecular levels. This review will discuss genetic findings in PTCL with special emphasis on how they impact lymphoma classification.
Recent findings
Sequencing studies have identified recurrent genetic alterations in nearly every PTCL subtype. In anaplastic large cell lymphoma, these studies have revealed novel chromosomal rearrangements and mutations that have prognostic significance and may suggest new therapeutic approaches. Angioimmunoblastic T cell lymphoma has been found to have mutations overlapping some cases of PTCL, not otherwise specified with a T follicular helper cell phenotype. Across various subtypes, recurrent mutations and structural alterations affecting genes involved in epigenetic regulation, T cell receptor signaling, and immune response may represent targets for precision therapy approaches.
Summary
New genetic findings are refining the classification of PTCLs and are beginning to be used clinically for diagnosis, risk stratification, and individualized therapy.
Keywords: Peripheral Tcell lymphoma, Molecular genetics, Angioimmunoblastic Tcell lymphoma, Anaplastic large cell lymphoma, Extranodal NK/Tcell lymphoma
Introduction
Peripheral T cell lymphomas (PTCLs) are a group of rare lymphomas originating from mature (i.e., post-thymic or “peripheral”) T lymphocytes and NK cells [1]. PTCLs comprise approximately 10–15% of all non-Hodgkin lymphomas and, with the exception of a few relatively indolent entities, generally are aggressive tumors which carry a poor prognosis [2, 3••]. PTCLs most commonly affect adults and the elderly, and overall are more common in Asia than in the West [4–6]. One likely contributor to this geographic variability is different exposures to viral infectious agents that are associated with specific PTCL entities, including Epstein-Barr virus (EBV) and human T-lymphotropic virus 1 (HTLV-1) [7].
The 2008 World Health Organization (WHO) Classification scheme broadly grouped PTCLs into four categories based on the predominant clinical presentation: leukemic (disseminated), nodal, extranodal, and cutaneous (Table 1) [5]. It is recognized, however, that in practice PTCLs can be difficult to subclassify due to evolving diagnostic criteria, multiple subtypes, overall rarity, and the relative paucity of known, specific genetic abnormalities in comparison to B cell lymphomas. Even within established subtypes, PTCLs can exhibit pronounced clinical, histological, immunophenotypic, cytogenetic, and molecular heterogeneity. However, despite these challenges, significant molecular advances have increased understanding of PTCL pathobiology and impacted current classification. In the 2016 revision of the WHO classification of lymphoid neoplasms, mature T and NK cell lymphomas and lymphoproliferative disorders will encompass 27 distinct entities in addition to PTCL, not otherwise specified (NOS), comprising T cell lymphomas not meeting criteria for one of the more specific categories [3••]. The current review discusses the genetic findings and classification of some of the more common PTCLs.
Table 1.
Classification of mature T and NK cell neoplasms*
| Subtype | Notesa | References |
|---|---|---|
| Predominantly leukemic diseases | ||
| T cell prolymphocytic leukemia | ||
| T cell large granular lymphocytic leukemia | Chronic lymphoproliferative disorder of NK cells remains a distinct, provisional entity | [3••] |
| Aggressive NK cell leukemia | ||
| Adult T cell leukemia/lymphoma | ||
| Predominantly nodal diseases | ||
| Angioimmunoblastic T cell lymphomab | Follicular T cell lymphoma and nodal peripheral T cell lymphoma with T follicular helper cell phenotype included as new provisional entities | [1, 3••, 8] |
| Anaplastic large cell lymphoma, ALK-positiveb | ||
| Anaplastic large cell lymphoma, ALK-negativeb | Cases associated with breast implants represent new provisional entity | [3••, 9, 10] |
| Peripheral T cell lymphoma, not otherwise specifiedb | ||
| Predominantly extranodal diseases | ||
| Extranodal NK/T cell lymphoma, nasal typeb | ||
| Enteropathy-associated T cell lymphoma | Cases previously called “type II,” not associated with celiac disease, now are designated monomorphic epitheliotropic intestinal T cell lymphoma | [3••] |
| Hepatosplenic T cell lymphoma | ||
| Subcutaneous panniculitis-like T cell lymphoma | ||
| Predominantly cutaneous diseases | ||
| Mycosis fungoides/Sézary syndrome | Considered two distinct entities | |
| Primary cutaneous CD30-positive T cell lymphoproliferative disorders | Includes lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma |
Angioimmunoblastic T Cell Lymphoma
Angioimmunoblastic T cell lymphoma (AITL) is one of the most common PTCL subtypes, comprising between 22 and 34% of cases, depending on geographic region [12]. AITL most frequently affects middle-aged adults and the elderly, and is characterized by advanced stage disease, generalized lymphadenopathy, hepatosplenomegaly, skin rash, and polyclonal hypergammaglobulinemia. The clinical course generally is aggressive and often is complicated by infection due to disease-associated immunosuppression. AITL demonstrates a T follicular helper cell (TFH) phenotype and typically is associated with B cells infected by EBV; the neoplastic T cells are EBV-negative. Some patients also develop diffuse large B cell lymphoma, which often is EBV-positive [13].
Recent genetic advances have revealed recurrent abnormalities, including both mutations and structural alterations, in a large proportion of AITLs (Table 2). Somatic mutations in TET2, DNMT3A, RHOA, CD28, and IDH2 have been found in varying frequencies in AITL, offering clues into the pathogenesis of this PTCL subtype [14, 15, 16•, 17•, 18•, 19–21, 46]. RHOAG17V mutations have been identified in both AITL (50–71%) and PTCL, NOS (8–18%), and likely serve a dominant negative function, blocking RHOA GTPase activity [16•, 17•, 18•]. Recently, a novel activating variant, RHOAK18N, has been described [22]. IDH2 mutations are relatively specific for AITL, occurring in 20–45% of cases, and particularly involve IDH2R172 [19, 21]. IDH2R172 mutations lead to accumulation of the (R)-enantiomer of 2-hydroxyglutarate, which inhibits TET family and other enzymes and leads to alterations in DNA and histone methylation. In a study of 190 PTCLs, Lemonnier et al. found TET2 mutations in AITL (47%) and PTCL, NOS (38%), but not in other PTCLs except for 2/10 enteropathy-associated T cell lymphomas. Cases of PTCL, NOS with TET2 mutations were more likely to express TFH markers and/or share common histologic features with AITL. TET2 mutations in both AITL and PTCL, NOS were associated with advanced stage disease, thrombocytopenia, high International Prognostic Index scores, and shorter progression-free survival times [46]. Of note, mutations involving TET2, DNMT3A, RHOA, and IDH2 often co-occur in the same case, including PTCLs in which all four of these genes are mutated [16•, 17•, 18•, 22]. Data suggest that among these mutations, those involving DNMT3A may be particularly early events, and sometimes are present in cells of non-T cell lineage from the same patient [14]. Although the molecular pathogenesis of AITL and other TFH-derived PTCLs remains incompletely understood, the relationship of the aforementioned genes to epigenetic regulation suggests that disruption of gene expression via epigenetic mechanisms may be involved in disease development and/or progression [3••, 19, 47], and might in part explain observed clinical responses of AITL to epigenetic modifying drugs such as histone deacetylase inhibitors [48].
Table 2.
Recurrent genetic alterations in major subtypes of peripheral T cell lymphoma
| Subtype | Genes involved | Notes | References |
|---|---|---|---|
| Angioimmunoblastic T cell lymphoma | |||
| Mutations | TET2, DNMT3A, RHOA, IDH2, CD28 |
|
[14, 15, 16•, 17•, 18•, 19–22] |
| Structural alterations | CTLA4-CD28, CARMA1, MYCBP2 |
|
[23, 24] |
| Anaplastic large cell lymphoma, ALK-positive | |||
| Structural alteration | ALK |
|
[25–27] |
| Anaplastic large cell lymphoma, ALK-negative | |||
| Mutations | JAK1, STAT3, TET2,DNMT3A |
|
[19, 28•] |
| Structural alterations | DUSP22, TP63, ROS1, TYK2, VAV1, ERBB4, TP53, PRDM1 |
|
[28•, 29•, 30–35] |
| Extranodal NK/T cell lymphoma, nasal type | |||
| Mutations | TP53, JAK3, DDX3X, STAT3, STAT5B |
|
[18•, 36–39] |
| Structural alterations | CTLA4-CD28, PRDM1 |
|
[23, 40–43] |
| Peripheral T cell lymphoma, not otherwise specified | |||
| Mutations | TET2, DNMT3A, RHOA, FYN |
|
[14, 16•, 17•, 19] |
| Structural alterations | ITK-SYK, CTLA4-CD28, VAV1, CDKN2A/B |
|
[23, 24, 34, 44, 45] |
Includes entities covered in this review. See also Table 1
Among structural abnormalities, CTLA4-CD28 fusion genes have been identified in AITL and several other PTCL subtypes, though analysis of different PTCL cohorts has uncovered variation in the frequency of this finding [23, 49, 50]. The resultant fusion protein is thought to transform inhibitory T cell signals into activating signals, and may represent a target for anti-CTLA4 immunotherapy [23]. Copy number alterations and loss of heterozygosity also have been demonstrated in AITL and PTCL, NOS; among genes within regions of frequent copy number gains, poor prognosis was associated with overexpression of CARMA1 at 7p22 and MYCBP2 at 13q22 [24].
As noted above, recurrent genetic alterations in AITL also have been found in cases of PTCL, NOS with a TFH phenotype. While the 2008 WHO classification considered these cases within the spectrum of PTCL, NOS, the 2016 revision will introduce two provisional TFH-related entities: follicular T cell lymphoma and nodal PTCL with TFH phenotype [1, 3••, 46].
Anaplastic Large Cell Lymphoma, ALK-Positive
Anaplastic large cell lymphoma (ALCL) is another of the more common PTCL subtypes, accounting for ~12% of all cases [12]. ALCL is a general heading comprising several distinct WHO entities based on clinical presentation and presence or absence of ALK (anaplastic lymphoma kinase) gene rearrangements: ALK-positive ALCL, ALK-negative ALCL (upgraded from provisional to definite entity in 2016 revision), primary cutaneous ALCL, and breast implant-associated ALCL (new provisional entity in 2016 revision) [3••, 25, 51]. Systemic ALCL often presents with advanced stage disease, frequently including B-symptoms such as high fevers. All ALCL subtypes share common pathological features, including the presence of morphologically distinctive cells designated “hallmark” cells (Fig. 1a) and consistent expression of the lymphocyte activation marker, CD30.
Fig. 1.
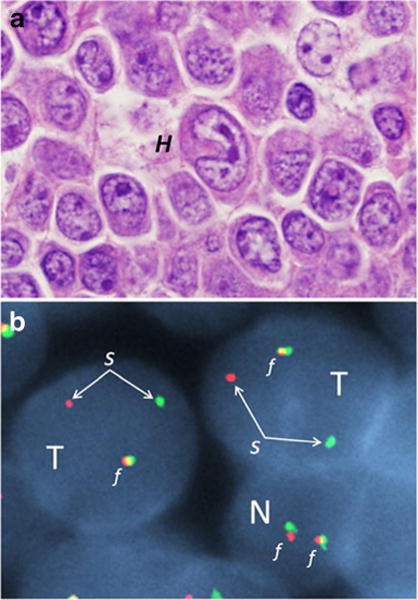
Anaplastic large cell lymphoma, ALK-negative, with DUSP22 rearrangement. a Photomicrograph demonstrating large, pleomorphic tumor cells, including a characteristic “hallmark” cell (H) with a horseshoe-shaped nucleus in the center of the image. b Fluorescence in situ hybridization image of two tumor cells (T) and one smaller, normal cell (N) using a break-apart probe to the DUSP22 gene region. The normal cell has two red-green fusion signals (f), representing two intact alleles. The tumor cells each have one fusion signal and one separated red-green signal (s), indicating a rearrangement of the DUSP22 gene region. This rearrangement has been associated with a favorable prognosis [29•]
ALK-positive ALCL affects a younger patient population than ALK-negative ALCL and other PTCLs, occurring most commonly during the first three decades of life [25]. ALK-positive ALCL was the first PTCL in which a recurrent genetic abnormality was recognized [52]. About half of ALCLs subsequently were found to have ALK rearrangements [25]. The ALK gene was characterized from a t(2;5)(p23;q35) translocation in ALCL, resulting in the fusion of nucleophosmin (NPM1) to ALK and encoding NPM1/ALK fusion transcripts [26]. The t(2;5) translocation is present in approximately 75–85% of ALK-positive ALCLs; more than 20 other ALK partners have been found, accounting for the remaining ALK-positive ALCLs as well as now being recognized in other lymphomas and non-lymphoid cancers [25, 51]. The frequency of the t(2;5) compared to variant ALK rearrangements may be slightly higher in pediatric than in adult ALK-positive ALCLs [53], but identifying the ALK fusion partner is not considered necessary in routine clinical practice.
Both NPM1/ALK and variants lead to expression of ALK fusion proteins with constitutive ALK tyrosine kinase activity [25, 27, 54, 55]. Central to the function of ALK fusion proteins is activation of the downstream oncogenic transcription factor, STAT3 [28•]. ALK-induced STAT3 activation plays a variety of roles in cancer cells, including regulation of availability of hypoxia-inducible factors under hypoxic conditions [56]. ALK tyrosine kinase activity can be targeted by clinically available inhibitors, and pharmacologic ALK inhibition is effective in a variety of clinical scenarios, including relapsed/refractory ALK-positive ALCL [47, 57–59]. Even with conventional cytotoxic chemotherapy, ALK-positive ALCL has a more favorable prognosis than ALK-negative ALCL as a whole (see section below) and most other PTCL subtypes, with overall 5-year survival rates of 70 to 85% [29•, 60, 61]. The ALK partner gene (NPM1 or variant) does not appear to be a significant prognostic factor [62].
Anaplastic Large Cell Lymphoma, ALK-Negative
Systemic PTCLs that are morphologically compatible with ALCL and express CD30, but lack ALK rearrangements, now are considered a separate subtype [3••, 63]. ALK-negative ALCL affects an older patient population than ALK-positive ALCL, and the molecular genetics remain incompletely understood [29•, 51, 63]. Although, as a whole, ALK-negative ALCL generally has been found to have a prognosis inferior to that of ALK-positive ALCL, some studies have found similar overall survival (OS) rates after adjusting for age [60, 64].
Distinguishing ALK-negative ALCL from CD30-positive PTCL, NOS is diagnostically challenging, but is of critical importance due to the potential prognostic and therapeutic implications. Gene expression profiling studies have shown not only that ALK-negative ALCL and ALK-positive ALCL share a common signature, but also that the signature of ALK-negative ALCL is distinct from that of PTCL, NOS [3••, 65•, 66, 67]. A three-gene model (TNFRSF8, BATF3, and TMOD1) has been proposed to help distinguish ALK-negative ALCL from PTCL, NOS in tissue samples, but is not in routine clinical use [68].
Recently, Crescenzo et al. reported that the JAK/STAT3 pathway was constitutively activated via multiple genomic mechanisms in ALK-negative ALCL [28•]. The JAK1 and STAT3 genes each were recurrently mutated in a subset of cases, but were not found in other PTCLs, including ALK-positive ALCL and PTCL, NOS (including 16 CD30-positive cases). In addition, RNA sequencing identified a small subset of ALK-negative ALCLs that lacked these mutations but harbored gene fusions involving non-ALK tyrosine kinase genes, including ROS1 and TYK2, leading to constitutive activation of STAT3. Thus, STAT3 may drive oncogenesis in the majority ALCLs, independent of ALK status.
Recurrent rearrangements of non-tyrosine kinase genes also have been identified in ALK-negative ALCL, notably including rearrangements of the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangements, Fig. 1b) or of TP63 on 3q28 [30, 31]. Parrilla Castellar et al. have shown that these rearrangements have prognostic significance in ALK-negative ALCL [29•]. DUSP22 rearrangements were seen in 30% of ALK-negative ALCLs and were associated with a favorable prognosis (90% 5-year OS), similar to that observed in ALK-positive ALCL (85%). TP63 rearrangements were seen in 8% of ALK-negative ALCLs and were associated with a 5-year OS rate of only 17%; ALCLs lacking rearrangements of ALK, DUSP22, and TP63 (so-called triple-negative cases) had an intermediate 5-year OS rate of 42%. For comparison, PTCL, NOS has been found to have a 5-year OS rate of 32% (and only 19% for cases with CD30 expression in ≥80% of cells) [12].
Scarfo et al. recently identified ERBB4 and COL29A1 as gene expression outliers in 24% of ALK-negative ALCLs [32]. They found that ERBB4 deregulation resulted from expression of two truncated transcripts with intronic transcriptional start sites. Truncated ERBB4 showed in vitro and in vivo oncogenic potential, and an ERBB4-positive patient-derived xenograft model showed sensitivity to pharmacologic ERBB4 inhibition. A genome-wide copy-number study using single nucleotide polymorphism arrays recently reported frequent losses at 6p21 and 17p13, corresponding to the PRDM1 (encoding the tumor suppressor, BLIMP1) and TP53 genes, respectively [33]. PRDM1 was found to be inactivated via multiple mechanisms in 56% of ALK-negative ALCLs and only 6% of ALK-positive ALCLs. Additionally, losses in 6q21 and/or 17p were associated with inferior OS among ALK-negative ALCLs.
It should be noted that systemic ALCL needs to be distinguished from primary cutaneous ALCL (pcALCL), which is an indolent entity with excellent disease-specific survival rates; pcALCL presents in the skin, and although it occasionally involves regional lymph nodes, it rarely disseminates [51, 69, 70]. The reasons that pcALCL is more indolent than systemic ALCL are incompletely understood, but may relate to specific profiles of cytokines and chemokine receptors, partly shared with breast implant-associated ALCLs, that could limit dissemination and suggest distinct initiating events related to chronic antigenic stimulation [71, 72]. Because systemic ALCL can involve the skin secondarily, patients with skin biopsies showing ALCL should undergo staging procedures. Though pcALCL is not specifically covered in this review, it should be noted that it shares some genetic features with systemic ALK-negative ALCL, including DUSP22 rearrangements in 28% [73], TYK2 rearrangements in 13% [74], STAT3 mutations in 5% [28•], and occasional TP63 rearrangements [31]. pcALCL generally is considered an ALK-negative entity, but rare, otherwise similar cases with ALK rearrangements have been reported [75].
Extranodal NK/T Cell Lymphoma, Nasal Type
Extranodal NK/T cell lymphoma, nasal type (NKTCL) is an aggressive lymphoma that most often originates from natural killer (NK) cells, but in some cases is derived from cytotoxic T cells [76, 77]. NKTCL primarily affects adults within Asian and Central and South American populations. The nasal cavity and adjacent structures are the most common sites of presentation, but other extranodal locations may be involved. Patients often present with symptoms of nasal obstruction, epistaxis, or with midfacial destructive lesions (“lethal midline granuloma”). Histologically, NKTCL features an angiocentric and angiodestructive growth pattern. The neoplastic cells are associated with EBV in virtually all cases, suggesting a likely pathogenic role of the virus. The prognosis of NKTCL is generally unfavorable but can vary. Tumors occurring outside the nasal cavity have been found to be highly aggressive and generally do not respond well to chemotherapy. Of note, chemoresistance of NKTCLs is partly attributable to expression of P-glycoprotein, encoded by the multidrug resistance gene, MDR1; drug combinations not affected by P-glycoprotein, particularly L-asparaginase-based regimens, have been associated with improved outcomes [78].
A variety of somatic mutations has been identified in NKTCL, most commonly involving the RNA helicase gene DDX3X (20% of cases) and JAK3 (35% of cases) [36, 37]. Mutations of STAT5B and STAT3 also have been identified in NKTCL and PTCLs derived from γδ-T cells, further emphasizing the role of JAK/STAT signaling in this and related diseases [38]. As discussed above, CTLA4-CD28 fusions may be identified in NKTCLs [23]. Other structural abnormalities include copy number losses at 6q21, including the PRDM1 gene [40].
Peripheral T Cell Lymphoma, Not Otherwise Specified
PTCL, NOS is an intentionally heterogeneous entity, sometimes referred to as a “wastebasket” category, comprising nodal and extranodal PTCLs that do not fulfill criteria for any of the more specific PTCL subtypes [79]. The fact that PTCL, NOS accounts for approximately ~30% of all PTCLs, making it the most common entity, attests to the need for new molecular and other data to increase understanding of these cases. PTCL, NOS most often presents in the lymph nodes; however, any site can be involved. The most frequent extranodal sites of involvement are the gastrointestinal tract and the skin [79]. As noted above, PTCL, NOS has a generally poor prognosis (5-year OS rate, 32%) [12].
The genetic findings in PTCL, NOS are heterogeneous and demonstrate significant overlap with other PTCL subtypes. As discussed above, this overlap may guide development of criteria to place cases in a more specific entity, such as the new provisional categories for non-angioimmunoblastic TFH-derived PTCLs [3••]. Recent gene expression profiling studies have identified at least two major subgroups of PTCL, NOS that are characterized by high expression of either GATA3 or TBX21, with the former associated with inferior prognosis [3••, 65•, 80]. Poor clinical outcomes also were observed when TBX21-associated cases expressed a cytotoxic gene signature.
Various recurrent mutations have been identified in subsets of PTCL, NOS. Palomero et al. have identified novel mutations in the FYN gene, which encodes a tyrosine kinase that plays an important role in T cell activation [16•]. FYN kinase activity was successfully targeted by a multikinase inhibitor in vitro, thereby displaying promise for a therapeutic approach in the small subset of PTCL patients with these mutations. Several additional recurrent genetic mutations in PTCL, NOS were mentioned in the section on AITL, including the epigenetic modulator genes, TET2, DNMT3A, and RHOA [14, 16•, 17,17•, 19]. Similarities between AITL and other TFH-derived PTCLs have been noted at the gene expression profiling level as well, though differences also exist [81, 82]. Interestingly, a recurrent t(5;9)(q33;q22) translocation leading to ITK/SYK fusion and overexpression and activation of spleen tyrosine kinase (SYK) has been identified in a subset of PTCLs with a follicular growth pattern and a TFH phenotype, and rarely in AITL [8, 44, 45, 83]. This fusion appears targetable with SYK inhibitors. Overall, these molecular similarities and differences have contributed to the introduction of the aforementioned provisional TFH-derived PTCL categories, which separate these entities from the broader category of PTCL, NOS but also keep them distinct from AITL [3••].
As discussed above, CTLA4-CD28 fusions occur in a subset of cases of PTCL, NOS [23]. Boddicker et al. recently discovered additional gene fusions in PTCL, NOS by integrating mate-pair DNA sequencing and RNA sequencing [34]. Of particular interest were recurrent VAV1 fusions with varying partners, which were found not only in PTCL, NOS (11%) but also in ALK-negative ALCL (16%) and a single case of ALK-positive ALCL. Cells overexpressing a VAV1-GSS fusion protein showed increased growth and were sensitive to azathioprine, an inhibitor of RAC1 activity. Of note, mutations of VAV1 also have been reported recently in various PTCL subtypes [22, 84].
As mentioned above, Fujiwara et al. have identified various loci with copy number alterations in PTCL, NOS and AITL, some of which were found to have prognostic implications [24]. Similar to AITL, overexpression of CARMA1 was associated with poor prognosis in PTCL, NOS. Additionally, a recurrent loss at 9p21 was associated with reduced expression of CDKN2A, CDKN2B, and MTAP. Reduced levels of CDKN2A were found in 9% of PTCL, NOS samples and were associated with inferior outcomes.
Future Directions and Perspectives
The most recent update to the WHO classification highlights several emergent themes with regard to genetics and high-throughput molecular profiling. First, these approaches, particularly analysis of mutational landscapes and gene expression profiling, have identified areas of commonality between subsets of PTCL, NOS and other PTCL subtypes, as in the case of TFH-derived PTCLs and AITL [3••]. As molecular understanding of PTCLs deepens, this trend is likely to continue, hopefully leading to a decrease in the number of patients given the “wastebasket” diagnosis of PTCL, NOS, albeit likely at the price of an increasing number of overall categories. Second, we are likely to deepen our understanding of the molecular heterogeneity within established entities (e.g., ALK-negative ALCL [29•]), giving rise to new prognostic biomarkers and/or tools for risk-stratifying patients for clinical research studies. Third, we are likely to see the introduction of new molecular tools that help refine the borderlands between related entities and aid clinical diagnosis [65•, 68]; digital gene expression analysis [85] is one technology that may find application here. New “omics” methodologies also may impact classification as they are applied more comprehensively, including epigenomics, proteomics, and metabolomics. Finally, increasing understanding of the scope and targetability of genetic alterations will help determine the role of individualized medicine approaches in clinical practice [86]. These approaches may be particularly applicable to complement the evolving molecular classification of PTCLs (compared to B cell lymphomas, for example), given the overall rarity and molecular heterogeneity of this constellation of disorders.
Conclusions
The recent advances described above have provided insight into the genetic heterogeneity of PTCLs. These discoveries have shed light on the classification, prognosis, and potential targeted therapy of several of the more common PTCL subtypes, but there is still much progress to be made. Treatment outcomes for PTCLs have not yet shown significant improvement despite increased molecular understanding, partly due to a relative paucity of candidate therapeutic targets. As knowledge of the genetic drivers of PTCL increases, cooperative group studies, innovative clinical trial design, and biomarker investigations to guide therapy will be critical to make a tangible impact on patient outcomes.
Acknowledgments
Grant Support This work was supported by award number R01 CA177734 (ALF) from the National Cancer Institute.
Footnotes
This article is part of the Topical Collection on Lymphomas
Conflict of Interest Rosalind F. Sandell, Rebecca L. Boddicker, and Andrew L. Feldman declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Attygalle AD, Cabecadas J, Gaulard P, et al. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward—report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology. 2014;64(2):171–99. doi: 10.1111/his.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armitage JO. The aggressive peripheral T-cell lymphomas: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87(5):511–9. doi: 10.1002/ajh.23144. [DOI] [PubMed] [Google Scholar]
- 3••.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90. doi: 10.1182/blood-2016-01-643569. Recently published summary of the 2016 revision of the WHO classification, including new provisional entities and incorporating genetic data not available when the 2008 WHO classification was published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117(25):6756–67. doi: 10.1182/blood-2010-05-231548. [DOI] [PubMed] [Google Scholar]
- 5.Jaffe ES, Harris L, Stein H, Campo E, Pileri SA, Swerdlow SH. Introduction and overview of the classification of the lymphoid neoplasms. In: Swerdlow SH, Campo E, Harris L, Jaffe ES, Pileri S, Stein H, Thiele J, Vardiman JW, editors. WHO classification of tumors of hematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2008. pp. 158–66. [Google Scholar]
- 6.Perry AM, Diebold J, Nathwani BN, et al. Non-Hodgkin lymphoma in the Far East: review of 730 cases from the international non-Hodgkin lymphoma classification project. Ann Hematol. 2016;95(2):245–51. doi: 10.1007/s00277-015-2543-4. [DOI] [PubMed] [Google Scholar]
- 7.William BM, Armitage JO. International analysis of the frequency and outcomes of NK/T-cell lymphomas. Best Pract Res Clin Haematol. 2013;26(1):23–32. doi: 10.1016/j.beha.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Moreau A, Dupuis J, et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am J Surg Pathol. 2009;33(5):682–90. doi: 10.1097/PAS.0b013e3181971591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miranda RN, Aladily TN, Prince HM, et al. Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol. 2014;32(2):114–20. doi: 10.1200/JCO.2013.52.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455–63. doi: 10.1038/modpathol.3801024. [DOI] [PubMed] [Google Scholar]
- 11••.Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, Thiele J, Vardiman J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4. Lyon: International Agency for Research on Cancer; 2008. Most recent published edition of the WHO classification of lymphoid neoplasms at the time of this writing. [Google Scholar]
- 12.Vose J, Armitage J, Weisenburger D, International TCLP. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–30. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann JC, Chisholm KM, Cherry A, et al. An analysis of MYC and EBV in diffuse large B-cell lymphomas associated with angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma not otherwise specified. Hum Pathol. 2016;48:9–17. doi: 10.1016/j.humpath.2015.09.033. [DOI] [PubMed] [Google Scholar]
- 14.Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366(1):95–6. doi: 10.1056/NEJMc1111708. [DOI] [PubMed] [Google Scholar]
- 15.Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123(9):1293–6. doi: 10.1182/blood-2013-10-531509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16•.Palomero T, Couronne L, Khiabanian H, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral Tcell lymphomas. Nat Genet. 2014;46(2):166–70. doi: 10.1038/ng.2873. Identification of recurrent RHOA mutations in peripheral T-cell lymphomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Sakata-Yanagimoto M, Enami T, Yoshida K, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(2):171–5. doi: 10.1038/ng.2872. Identification of recurrent RHOA mutations in peripheral T-cell lymphomas. [DOI] [PubMed] [Google Scholar]
- 18•.Yoo HY, Sung MK, Lee SH, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(4):371–5. doi: 10.1038/ng.2916. Identification of recurrent RHOA mutations in peripheral T-cell lymphomas. [DOI] [PubMed] [Google Scholar]
- 19.Wang C, McKeithan TW, Gong Q, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. 2015;126(15):1741–52. doi: 10.1182/blood-2015-05-644591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohr J, Guo S, Huo J, Bouska A, Lachel C, Li Y, Simone PD, Zhang W, Gong Q, Wang C, et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia. 2015 doi: 10.1038/leu.2015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cairns RA, Iqbal J, Lemonnier F, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–3. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallois D, Dobay MP, Morin RD, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128(11):1490–502. doi: 10.1182/blood-2016-02-698977. [DOI] [PubMed] [Google Scholar]
- 23.Yoo HY, Kim P, Kim WS, et al. Frequent CTLA4-CD28 gene fusion in diverse types of T-cell lymphoma. Haematologica. 2016;101(6):757–63. doi: 10.3324/haematol.2015.139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujiwara SI, Yamashita Y, Nakamura N, et al. High-resolution analysis of chromosome copy number alterations in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified, with single nucleotide polymorphism-typing microarrays. Leukemia. 2008;22(10):1891–8. doi: 10.1038/leu.2008.191. [DOI] [PubMed] [Google Scholar]
- 25.Delsol G, Falini B, Muller-Hermelink HK, et al. Anaplastic large cell lymphoma, ALK-positive. In: Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, Thiele J, Vardiman J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon: International Agency for Research on Cancer; 2008. pp. 312–6. [Google Scholar]
- 26.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 27.Feldman AL, Vasmatzis G, Asmann YW, et al. Novel TRAF1-ALK fusion identified by deep RNA sequencing of anaplastic large cell lymphoma. Genes Chromosomes Cancer. 2013;52(11):1097–102. doi: 10.1002/gcc.22104. [DOI] [PubMed] [Google Scholar]
- 28•.Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27(4):516–32. doi: 10.1016/j.ccell.2015.03.006. Discovery of varied genetic alterations driving JAK/STAT3 activation in anaplastic large cell lymphomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Parrilla Castellar ER, Jaffe ES, Said JW, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473–80. doi: 10.1182/blood-2014-04-571091. Report on the prognostic significance of DUSP22 and TP63 rearrangements in ALK-negative an-aplastic large cell lymphoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively-parallel genomic sequencing. Blood. 2011;117(3):915–9. doi: 10.1182/blood-2010-08-303305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasmatzis G, Johnson SH, Knudson RA, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012;120:2280–9. doi: 10.1182/blood-2012-03-419937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scarfo I, Pellegrino E, Mereu E, et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood. 2016;127(2):221–32. doi: 10.1182/blood-2014-12-614503. [DOI] [PubMed] [Google Scholar]
- 33.Boi M, Rinaldi A, Kwee I, et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood. 2013;122(15):2683–93. doi: 10.1182/blood-2013-04-497933. [DOI] [PubMed] [Google Scholar]
- 34.Boddicker RL, Razidlo GL, Dasari S, et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood. 2016;128(9):1234–45. doi: 10.1182/blood-2016-03-707141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melard P, Idrissi Y, Andrique L, Poglio S, Prochazkova-Carlotti M, Berhouet S, Boucher C, Laharanne E, Chevret E, Pham-Ledard A, et al. Molecular alterations and tumor suppressive function of the DUSP22 (Dual Specificity Phosphatase 22) gene in peripheral T-cell lymphoma subtypes. Oncotarget. 2016 doi: 10.18632/oncotarget.11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang L, Gu ZH, Yan ZX, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–6. doi: 10.1038/ng.3358. [DOI] [PubMed] [Google Scholar]
- 37.Koo GC, Tan SY, Tang T, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2(7):591–7. doi: 10.1158/2159-8290.CD-12-0028. [DOI] [PubMed] [Google Scholar]
- 38.Kucuk C, Jiang B, Hu X, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-Tor NK cells. Nat Commun. 2015;6:6025. doi: 10.1038/ncomms7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quintanilla-Martinez L, Kremer M, Keller G, et al. p53 Mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159(6):2095–105. doi: 10.1016/S0002-9440(10)63061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kucuk C, Iqbal J, Hu X, et al. PRDM1 is a tumor suppressor gene in natural killer cell malignancies. Proc Natl Acad Sci U S A. 2011;108(50):20119–24. doi: 10.1073/pnas.1115128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iqbal J, Kucuk C, Deleeuw RJ, et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia. 2009;23(6):1139–51. doi: 10.1038/leu.2009.3. [DOI] [PubMed] [Google Scholar]
- 42.Karube K, Nakagawa M, Tsuzuki S, et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood. 2011;118(12):3195–204. doi: 10.1182/blood-2011-04-346890. [DOI] [PubMed] [Google Scholar]
- 43.Huang Y, de Reynies A, de Leval L, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood. 2010;115(6):1226–37. doi: 10.1182/blood-2009-05-221275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia. 2006;20(2):313–8. doi: 10.1038/sj.leu.2404045. [DOI] [PubMed] [Google Scholar]
- 45.Dierks C, Adrian F, Fisch P, et al. The ITK-SYK fusion oncogene induces a T-cell lymphoproliferative disease in mice mimicking human disease. Cancer Res. 2010;70(15):6193–204. doi: 10.1158/0008-5472.CAN-08-3719. [DOI] [PubMed] [Google Scholar]
- 46.Lemonnier F, Couronne L, Parrens M, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7):1466–9. doi: 10.1182/blood-2012-02-408542. [DOI] [PubMed] [Google Scholar]
- 47.Piccaluga PP, Tabanelli V, Pileri SA. Molecular genetics of peripheral T-cell lymphomas. Int J Hematol. 2014;99(3):219–26. doi: 10.1007/s12185-014-1522-1. [DOI] [PubMed] [Google Scholar]
- 48.Pro B, Horwitz SM, Prince HM, Foss FM, Sokol L, Greenwood M, Caballero D, Morschhauser F, Wilhelm M, Iyer SP, et al. Romidepsin induces durable responses in patients with relapsed or refractory angioimmunoblastic T-cell lymphoma. Hematol Oncol. 2016 doi: 10.1002/hon.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoo HY, Kim P, Kim WS, et al. Author reply to Comment on: Frequent CTLA4-CD28 gene fusion in diverse types of T-cell lymphoma, by Yoo et al. Haematologica. 2016;101(6):e271. doi: 10.3324/haematol.2016.148015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong Q, Wang C, Rohr J, Feldman AL, Chan WC, McKeithan T. Comment on: Frequent CTLA4-CD28 gene fusion in diverse types of T-cell lymphoma, by Yoo et al. Haematologica. 2016;101(6):e269–70. doi: 10.3324/haematol.2016.147074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xing X, Feldman AL. Anaplastic large cell lymphomas: ALK positive, ALK negative, and primary cutaneous. Adv Anat Pathol. 2015;22(1):29–49. doi: 10.1097/PAP.0000000000000047. [DOI] [PubMed] [Google Scholar]
- 52.Rimokh R, Magaud JP, Berger F, et al. A translocation involving a specific breakpoint (q35) on chromosome 5 is characteristic of an-aplastic large cell lymphoma (‘Ki-1 lymphoma’) Br J Haematol. 1989;71(1):31–6. doi: 10.1111/j.1365-2141.1989.tb06270.x. [DOI] [PubMed] [Google Scholar]
- 53.Damm-Welk C, Klapper W, Oschlies I, et al. Distribution of NPM1-ALK and X-ALK fusion transcripts in paediatric anaplastic large cell lymphoma: a molecular-histological correlation. Br J Haematol. 2009;146(3):306–9. doi: 10.1111/j.1365-2141.2009.07754.x. [DOI] [PubMed] [Google Scholar]
- 54.Hapgood G, Savage KJ. The biology and management of systemic anaplastic large cell lymphoma. Blood. 2015;126(1):17–25. doi: 10.1182/blood-2014-10-567461. [DOI] [PubMed] [Google Scholar]
- 55.Inghirami G, Pileri SA, European TCLSG Anaplastic large-cell lymphoma. Semin Diagn Pathol. 2011;28(3):190–201. doi: 10.1053/j.semdp.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Martinengo C, Poggio T, Menotti M, et al. ALK-dependent control of hypoxia-inducible factors mediates tumor growth and metastasis. Cancer Res. 2014;74(21):6094–106. doi: 10.1158/0008-5472.CAN-14-0268. [DOI] [PubMed] [Google Scholar]
- 57.Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med. 2011;364(8):775–6. doi: 10.1056/NEJMc1013224. [DOI] [PubMed] [Google Scholar]
- 58.Mosse YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472–80. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gambacorti Passerini C, Farina F, Stasia A, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst. 2014;106(2):djt378. doi: 10.1093/jnci/djt378. [DOI] [PubMed] [Google Scholar]
- 60.Savage KJ, Harris NL, Vose JM, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496–504. doi: 10.1182/blood-2008-01-134270. [DOI] [PubMed] [Google Scholar]
- 61.Gascoyne RD, Aoun P, Wu D, et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999;93(11):3913–21. [PubMed] [Google Scholar]
- 62.Delsol G, Falini B, Muller-Hermelink HK, et al. Anaplastic large cell lymphoma (ALCL), ALK-positive. In: Swerdlow SH, Campo E, Harris L, Jaffe ES, Pileri S, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon: International Agency for Research on Cancer; 2008. pp. 312–6. [Google Scholar]
- 63.Mason DY, Harris L, Delsol G, et al. Anaplastic large cell lymphoma, ALK-negative. In: Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, Thiele J, Vardiman J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon: International Agency for Research on Cancer; 2008. pp. 317–9. [Google Scholar]
- 64.Sibon D, Fournier M, Briere J, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte trials. J Clin Oncol. 2012;30(32):3939–46. doi: 10.1200/JCO.2012.42.2345. [DOI] [PubMed] [Google Scholar]
- 65•.Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915–23. doi: 10.1182/blood-2013-11-536359. Major paper on the role of gene expression profiling in segregating known entities and subgroups within peripheral T-cell lymphomas, not otherwise specified. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Piva R, Agnelli L, Pellegrino E, et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J Clin Oncol. 2010;28(9):1583–90. doi: 10.1200/JCO.2008.20.9759. [DOI] [PubMed] [Google Scholar]
- 67.Piccaluga PP, Fuligni F, De Leo A, et al. Molecular profiling improves classification and prognostication of nodal peripheral T-cell lymphomas: results of a phase III diagnostic accuracy study. J Clin Oncol. 2013;31(24):3019–25. doi: 10.1200/JCO.2012.42.5611. [DOI] [PubMed] [Google Scholar]
- 68.Agnelli L, Mereu E, Pellegrino E, et al. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood. 2012;120(6):1274–81. doi: 10.1182/blood-2012-01-405555. [DOI] [PubMed] [Google Scholar]
- 69.Ralfkiaer E, Willemze R, Paulli M, Kadin ME. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris L, Jaffe ES, Pileri S, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon: International Agency for Research on Cancer; 2008. pp. 300–1. [Google Scholar]
- 70.Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95(12):3653–61. [PubMed] [Google Scholar]
- 71.van Kester MS, Tensen CP, Vermeer MH, et al. Cutaneous anaplas-tic large cell lymphoma and peripheral T-cell lymphoma NOS show distinct chromosomal alterations and differential expression of che-mokine receptors and apoptosis regulators. J Invest Dermatol. 2010;130(2):563–75. doi: 10.1038/jid.2009.270. [DOI] [PubMed] [Google Scholar]
- 72.Kadin ME, Deva A, Xu H, Morgan J, Khare P, MacLeod RA, Van Natta BW, Adams WP, Jr, Brody GS, Epstein AL. Biomarkers provide clues to early events in the pathogenesis of breast implant-associated anaplastic large cell lymphoma. Aesthet Surg J. 2016 doi: 10.1093/asj/sjw023. [DOI] [PubMed] [Google Scholar]
- 73.Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translo-cations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24(4):596–605. doi: 10.1038/modpathol.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Velusamy T, Kiel MJ, Sahasrabuddhe AA, et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood. 2014;124(25):3768–71. doi: 10.1182/blood-2014-07-588434. [DOI] [PubMed] [Google Scholar]
- 75.Oschlies I, Lisfeld J, Lamant L, et al. ALK-positive anaplastic large cell lymphoma limited to the skin: clinical, histopathological and molecular analysis of 6 pediatric cases. A report from the ALCL99 study. Haematologica. 2013;98(1):50–6. doi: 10.3324/haematol.2012.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan JC, Quintanilla-Martinez L, Ferry JA, Peh SC. Extranodal NK/T-cell lymphoma, nasal type. In: Swerdlow SH, Campo E, Harris L, Jaffe ES, Pileri S, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon: International Agency for Research on Cancer; 2008. pp. 285–8. [Google Scholar]
- 77.Pongpruttipan T, Sukpanichnant S, Assanasen T, et al. Extranodal NK/T-cell lymphoma, nasal type, includes cases of natural killer cell and alphabeta, gammadelta, and alphabeta/gammadelta T-cell origin: a comprehensive clinicopathologic and phenotypic study. Am J Surg Pathol. 2012;36(4):481–99. doi: 10.1097/PAS.0b013e31824433d8. [DOI] [PubMed] [Google Scholar]
- 78.Chaudhary RK, Bhatt VR, Vose JM. Management of extranodal natural killer/t-cell lymphoma, nasal type. Clin Lymphoma Myeloma Leuk. 2015;15(5):245–52. doi: 10.1016/j.clml.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 79.Pileri S, Ralfkiaer E, Weisenburger D, et al. Peripheral T-cell lymphoma, not otherwise specified. In: Swerdlow SH, Campo E, Harris L, Jaffe ES, Pileri S, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon: International Agency for Research on Cancer; 2008. pp. 306–8. [Google Scholar]
- 80.Wang T, Feldman AL, Wada DA, et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood. 2014;123(19):3007–15. doi: 10.1182/blood-2013-12-544809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109(11):4952–63. doi: 10.1182/blood-2006-10-055145. [DOI] [PubMed] [Google Scholar]
- 82.Piccaluga PP, Agostinelli C, Califano A, et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res. 2007;67(22):10703–10. doi: 10.1158/0008-5472.CAN-07-1708. [DOI] [PubMed] [Google Scholar]
- 83.Attygalle AD, Feldman AL, Dogan A. ITK/SYK translocation in angioimmunoblastic T-cell lymphoma. Am J Surg Pathol. 2013;37(9):1456–7. doi: 10.1097/PAS.0b013e3182991415. [DOI] [PubMed] [Google Scholar]
- 84.Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304–15. doi: 10.1038/ng.3415. [DOI] [PubMed] [Google Scholar]
- 85.Scott DW, Mottok A, Ennishi D, et al. Prognostic significance of diffuse large B-cell lymphoma cell of origin determined by digital gene expression in formalin-fixed paraffin-embedded tissue biopsies. J Clin Oncol. 2015;33(26):2848–56. doi: 10.1200/JCO.2014.60.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lazaridis KN, McAllister TM, Babovic-Vuksanovic D, et al. Implementing individualized medicine into the medical practice. Am J Med Genet C Semin Med Genet. 2014;166C(1):15–23. doi: 10.1002/ajmg.c.31387. [DOI] [PubMed] [Google Scholar]