

Abstract
The androgen receptor (AR) is widely expressed in breast cancer (BC) and evidence suggests dependence on AR signaling for growth and survival. AR antagonists such as enzalutamide and seviteronel have shown success in pre-clinical models and clinical trials of prostate cancer, and are currently being evaluated in BC. Reciprocal regulation between AR and the HER2/PI3K/mTOR pathway may contribute to resistance to HER2- and mTOR-targeted therapies; thus, dual inhibition of these pathways may synergistically inhibit BC growth. HER2+ and triple-negative BC cell lines were treated with AR antagonist plus anti-HER2 monoclonal antibody trastuzumab or mTOR inhibitor everolimus. Apoptosis, cell proliferation and drug synergy were measured in vitro. Pathway component genes and proteins were measured by qRT-PCR, western blot, and reverse phase protein array. In vivo, HER2+ BC xenografts were treated with enzalutamide, everolimus, trastuzumab, and combinations of these drugs. AR antagonists inhibited proliferation of both HER2+ and TNBC cell lines. Combining AR antagonist and either everolimus or trastuzumab resulted in synergistic inhibition of proliferation. Dihydrotestosterone caused increased phosphorylation of HER2 and/or HER3 that was attenuated by AR inhibition. Everolimus caused an increase in total AR, phosphorylation of HER2 and/or HER3, and these effects were abrogated by enzalutamide. Growth of trastuzumab-resistant HER2+ xenograft tumors was inhibited by enzalutamide, and combining enzalutamide with everolimus decreased tumor viability more than either single agent. AR antagonists synergize with FDA-approved BC therapies such as everolimus and trastuzumab through distinct mechanisms. Treatment combinations are effective in trastuzumab-resistant HER2+ BC cells in vivo.
Keywords: Breast cancer, Androgen receptor, HER2, mTOR, synergy
Introduction
The estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) have been the principal targets of breast cancer (BC) therapies for the last several decades. The androgen receptor (AR) has recently emerged as another promising target in BC. AR is expressed in 77% of all BC and is more widely expressed than estrogen receptor (ER); it is present to varying degrees in all BC subtypes, including luminal A, luminal B, HER2 enriched, and triple negative BCs (TNBC) (1). Recent preclinical studies of AR in BC indicate it is required for tumor cell survival, and may contribute to metastatic progression (2-7).
Although like ER, AR is associated with more indolent disease (8-11), and yet ER is a therapeutic target because the tumors are dependent on estrogens and ER. Some tumors may similarly become growth dependent on AR, as a higher ratio of AR to ER positive cells correlates with poor outcome in breast cancers (4) and anti-androgens reduce tumor viability in preclinical models of ER+ breast cancer (4, 5). In TNBC it is clear that AR drives tumor growth in preclinical models, and tumor growth is inhibited by anti-androgens (3, 4, 7).
Recent clinical trials of anti-androgen therapies in BC have demonstrated significant clinical benefit (12, 13). Notably, administration of the AR antagonist enzalutamide (Xtandi, Medivation, Inc) in patients with advanced TNBC results in better than expected survival (13). Enzalutamide (Enza) is a second-generation AR antagonist, which is a competitive inhibitor that prevents AR nuclear localization. Several other next-generation AR antagonists are currently in pre-clinical and clinical development, including seviteronel (VT-464, Innocrin, Inc), and apalutamide (ARN-509 Janssen Research & Development, LLC). Seviteronel (Sevi) is a selective CYP17A1 inhibitor, and therefore inhibits synthesis of androgens and estrogens (14). Sevi also functions as a direct competitive antagonist of AR (15). Based on the availability of targeted therapies and promising pre-clinical data in BC, AR inhibition has the potential to be widely effective across multiple BC subtypes and disease settings, including those that have become resistant to other therapies.
Anti-HER2 therapies such as the monoclonal antibodies trastuzumab (Tras) and pertuzumab are now standard of care for HER2+ BC, but acquired resistance and progression while on therapy remain major challenges. AR expression is enriched in HER2+ BC, where it has been shown to promote tumor growth via upregulation of HER3 through the Wnt signaling pathway (16). Inhibition of HER2 signaling with the small molecule lapatinib also enhanced the anti-tumor effect of Enza in a model of castrate-resistant prostate cancer in mice (17). Thus, combining an anti-androgen with HER2-taregeted therapy may represent a new option for combating therapeutic resistance in HER2+ BC and warrants pre-clinical testing.
Downstream of HER2, the mammalian target of rapamycin (mTOR) is a recognized driver of BC growth and is a potential therapeutic target in a number of solid tumor types. Therapeutic inhibition of mTOR has shown promise in pre-clinical studies (18) and clinical trials. A recent phase III trial of the second-generation mTOR inhibitor everolimus (Everol) (Afinitor, Novartis Inc;) in BC demonstrated significant clinical benefit in patients with ER+/HER2- disease (19), leading to FDA approval of this agent for patients with advanced ER+ BC. However, results have been disappointing in trials of patients with HER2+ disease (20) and TNBC (21), as we address further in the discussion. Given the heavily integrated nature of mTOR signaling, converging signals from well-established BC drivers such as HER2, PI3K, and ER, therapeutic inhibition of mTOR may have unintended compensatory effects within the cell, leading to resistance and continued BC growth. Indeed, this observation was part of the rationale to design clinical trials to dually inhibit mTOR and HER2; however, two phase III clinical trials testing this hypothesis resulted in only marginal survival improvements (20, 22). Further compounding the issue, molecular determinants of Everol efficacy remain elusive.
In the current study, the interplay between AR and mTOR in the context of HER2+ BC and TNBC was examined, as well as AR and HER2 in HER2+ BC models. We sought to determine whether dual inhibition of these pathways would synergistically inhibit BC growth, thus providing rationale for a new therapeutic combination that could increase the efficacy of HER2-targeted and mTOR-targeted therapies.
Materials and Methods
Cell lines and reagents
BC cell line MDA-MB-453 was purchased from ATCC in 2012. BT474, SKBR3, BT474-HR20, and SKBR3-Pool2 were a gift from Dr. Bolin Liu and received in 2009. The trastuzumab-resistant cell lines BT474-HR20 and SKBR3-Pool2 have been previously described (23, 24). Briefly, these Tras-resistant cell lines were generated by chronically treating cells with increasing concentrations of Tras; upon establishment of resistance, cells were then cultured in 20ug/mL Tras. These cell lines have a better take-rate in vivo compared to their parental counterparts. Cell lines were authenticated at the University of Colorado Tissue Culture Core facility. Cell line molecular subtypes, mutation status, and culture conditions are listed in Supplementary Table 1.
Enza was supplied by Medivation, Inc. (Medivation, Inc. was acquired by Pfizer, Inc. in September 2016). Everol was purchased from Selleckchem, Inc. Tras was obtained from Genentech (Roche). Sevi was supplied by Innocrin, Inc.
Cellular assays
Proliferation was measured either by colony formation assay using crystal violet, or by using an IncuCyte ZOOM Live Cell Imaging System (Essen Biosciences, Inc.) following the manufacturer's protocol. Cells were first stably transfected with a plasmid containing nuclear red-fluorescent protein (RFP) and proliferation was measured either by change in percent confluence or change in fluorescence over time, with individual well images acquired every 4 hours for 6 days. Percent inhibition of growth was calculated by dividing by growth of vehicle-treated cells. Apoptosis was measured on an IncuCyte using a red fluorescent cleaved caspase 3/7 antibody to quantify changes in apoptosis over time. Colony formation assay was performed by plating 500 cells/well in 6-well plates and treating with 10nM Everol, 10uM Enza, or the combination, for two weeks, then removing treatment and allowing cells to re-grow for 1 additional week.
Patient-derived xenograft sponge culture explants
A patient derived xenograft, HCI-009, which is AR+ and TNBC (25), was harvested from a NOD/SCID mouse and cut into 1mm3 sections, as described previously (26). Sections were immediately placed on dental sponges which were grown in culture dishes with RPMI media and 10% FBS, containing vehicle, 20uM Enza, 10nM Everol, or combination. Sponge cultures were grown for 48 hours in the presence of drug, then formalin-fixed and paraffin embedded for immunohistochemistry (IHC).
Immunoblotting
For cell lines, cells were lysed in RIPA buffer (150mM NaCl, 1% IGEPAL, 0.5% Na-Deoxycholate, 0.1%SDS, 50mM Tris, 1mM EDTA) containing protease inhibitor and phosphatase inhibitor. For xenografts, lysates were generated by first homogenizing tumors with a rotor-stator homogenizer, then lysing in RIPA buffer with protease and phosphatase inhibitor. Whole cell protein lysates were separated on SDS-PAGE gels, and transferred to PVDF membranes. Membranes were blocked in 5% BSA in Tris-buffered saline-Tween and incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: AR (PG-21, 1:500 dilution, EMD Millipore); phospho-HER2 (Y1248, 1:1000 dilution, Cell Signaling); total-HER2 (D8F12, 1:1000 dilution, Cell Signaling); phospho-HER3 (Y1197, 1:1000 dilution, Cell Signaling); total-HER3 (1B2E, 1:1000 dilution, Cell Signaling); phospho-S6 (Ser235/236, 1:1000 dilution, Cell Signaling); S6 (5G10, 1:1000 dilution, Cell Signaling); α-tubulin (B-5-1-2; 1:20,000 dilution, Sigma Aldrich); β-actin (8H10D10, 1:10000 dilution, Cell Signaling). Following incubation in secondary antibody, results were detected using an Odyssey CLx Imager (Licor Inc); densitometry quantification was performed using Image Studio Lite Version 4.0 and reported as a ratio normalized with loading control α-tubulin or β-actin.
Quantitative RT-PCR
Total RNA was isolated using the RNeasy Plus Mini Kit (Qiagen) according to manufacturer's instructions. cDNA was synthesized using qScript cDNA SuperMix (Quanta Biosciences) following manufacturer's instructions. qPCR was performed on an ABI 7600 FAST thermal cycler using Absolute Blue qPCR SYBR Green Low ROX Mix (Thermo Scientific). Target gene expression was normalized to 18s rRNA. Experiments were repeated at least twice. qPCR primers were as follows: AR Fwd: 5′-CTCACCAAGCTCCTGGACTC-3′; AR Rev: 5′-CAGGCAGAAGACATCTGAAAG-3′; PIP Fwd: 5′-TCCCAAGTCAGTACGTCCAAA-3′; PIP Rev: 5′-CTGTTGGTGTAAAAGTCCCAG-3′; 18s Fwd: 5′-GTAACCCGTTGAACCCCATT-3′; 18s Rev: 5′-CCATCCAATCGGTAGTAGCG-3′.
Reverse Phase Protein Array (RPPA)
BT474-HR20 cells were rinsed twice with ice-cold phosphate buffered saline. Subsequently, cells pellets were lysed in buffer containing T-PER(Thermo-Fisher Scientific) supplemented with 300 mM NaCl, 1 mM sodium orthovanadate, 2 mM Pefabloc (Roche), 5 μg/mL aprotinin (Sigma), 5 μg/mL leupeptin (Sigma) and 5 μg/mL pepstatin A (Sigma). Individual samples were vortexed for 1min. Samples were incubated on ice for 20 minutes and then centrifuged at 10,000 rpm for 5 minutes at 4°C. Supernatant was transferred to fresh tubes and protein concentrations were measured using BCA assay (Thermo Fisher). Lysates were then diluted to 0.5 μg/μL in extraction buffer containing equal volumes of T-PER (Thermo Fisher Scientific) and 2× Tris-Glycine SDS sample buffer (Invitrogen) supplemented with 2.5% β-mercaptoethanol (Thermo Fisher Scientific) in preparation for array printing. Diluted lysates were boiled for 8 min at 100°C and stored at −80°C prior to printing of arrays.
RPPA printing and analysis was conducted as previously described (27-29). Total protein levels were assessed in each sample by staining with Sypro Ruby Protein Blot Stain (Invitrogen) according to manufacturer's instructions. Antibody staining intensities were quantified using the MicroVigene v5 Software Package (Vigenetech).
Signaling pathway activation was evaluated by staining the arrays with 196 antibodies against key signaling proteins, mainly phosphorylated and cleaved protein products. Before use for RPPA analysis, antibody specificity was confirmed by Western blot and analysis as previously described (27). For heatmap generation, two-way unsupervised hierarchical clustering was performed using the Ward method in JMP v 5.1.2 (SAS; Cary, NC).
Tumor xenografts and in vivo treatments
Xenograft studies were approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC protocol 83614(01)1E). All experiments were conducted in accordance with the NIH Guidelines of Care and Use of Laboratory Animals. Tras-resistant BT474-HR20 BC cells were stably transfected with the NES-TGL vector, which contains GFP-luciferase. A total of 2×106 BT474-HR20 cells were mixed in 100uL growth factor reduced Matrigel (BD Biosciences) and injected bilaterally into the mammary fat pads of female NOD/SCID mice (Taconic). Mice also received estradiol pellets made in-house using silastic tubing and 1.5mg pharmaceutical-grade estradiol; these pellets allow for extended release of estradiol and minimal toxicity in NOD/SCID mice. Tumor growth was measured weekly by caliper and by luciferase signal using an in vivo preclinical imaging system (IVIS). When tumors reached an average of 50mm3, mice were randomized into 6 treatment groups based on caliper measurements and total IVIS signal (Supplementary figure 1). Mice received Enza via their chow (equivalent to 50mg/kg dose). Enza was mixed with ground mouse chow at a concentration of 0.43 mg/g chow (Research Diets, Inc.). Everol was administered intraperitoneally twice weekly at a dose of 2mg/kg. Tras was administered intraperitoneally twice weekly at a dose of 5mg/kg. Mice were euthanized by CO2 asphyxiation and cervical dislocation. Tumors, mammary glands, and colons were harvested for immunohistochemical and gene expression analyses.
Statistical Analyses
Data were analyzed with GraphPad Prism 6, using Student's t tests for comparisons of 2 conditions, or 1-way ANOVA with Bonferroni correction when making multiple comparisons. P-values <0.05 were considered statistically significant. Standard deviations are indicated by error bars, except for in vivo studies, where standard error of the mean is indicated by error bars. Synergy was calculated using CalcuSyn Software (Biosoft Inc), which uses the Median Effect method(30), where a combination index (CI)<0.9 indicates synergy, CI = 0.9-1.1 indicates additivity, and CI>1.1 indicates antagonism. Experiments were performed in biological triplicate, and mean values were imported to CalcuSyn for synergy calculations.
To compare the effect of treatment on tumor growth over time, a repeated measures design was used. Assumptions for different types of repeated measures analyses were tested (i.e., normal distribution, equal variances, balanced data, no missing data or unequal time measurements). Normal distributions were determined by graphing the data to check for a symmetrical data distribution without outliers and by the Shapiro-Wilk test. Data failing this assumption were transformed. A repeated measures ANOVA was used if there were no missing data, there were equal numbers in each treatment group, the measurement time points were equal, and there were no missing data points. If this model failed the assumptions of sphericity (Mauchly's Test), either a p-value correction (Huynh-Feldt) was reported or a multivariate ANOVA was used to determine differences in treatment groups over time. If there were missing data, or unbalanced data, or unequal time points, a repeated measures mixed models approach was used. The appropriate covariance structure for the mixed model was tested and the covariance structure leading to the best model fit (lowest Akaike Information Criterion and Bayesian Information Criterion values) was used. The data were transformed by taking the square root to meet the assumption of normality. Adjusted p-values using Tukeys' method were used to determine differences between individual treatment groups over time. The repeated measures analyses were performed using SAS ver 9.4 (SAS Institute, Cary, NC). Significance was set at p<0.05.
Results
Enzalutamide inhibits phosphorylation of HER2 and HER3, and enhances trastuzumab efficacy in vitro
Treatment of HER2-amplified cell line SKBR3 with Enza, Tras, or the combination resulted in significant inhibition of growth, with the combination treatment inhibiting proliferation more than either single agent (Figure 1A). Additionally, treatment of the Tras-resistant cell line SKBR3-Pool2 with Enza significantly inhibited proliferation, and combining Enza with Tras resulted in more inhibition of growth than either single agent (Figure 1B). Treatment of parental SKBR3 cells with the AR ligand dihydrotestosterone (DHT) for 48 hours resulted in an upregulation of AR, phospho-HER2 and total HER2, but not HER3, and treatment with Enza abrogated these effects (Figure 1C). To determine if Enza inhibited growth or caused cytotoxicity, we analyzed cleavage of caspase 3/7 over time when cells were treated with Enza, Tras, or the combination. In the parental SKBR3 cells, either single agent caused a significant increase in apoptosis over time, and combination treatment promoted more apoptosis than either single agent (Figure 1D). In the Tras-resistant SKBR3-Pool2 cells, Tras alone had no effect on apoptosis (Figure 1E). While Enza and Tras+Enza caused an increase in apoptosis, there was not a significant difference in apoptosis between these two groups.
Figure 1. Enza inhibits proliferation and promotes apoptosis in HER2+ breast cancer cells.
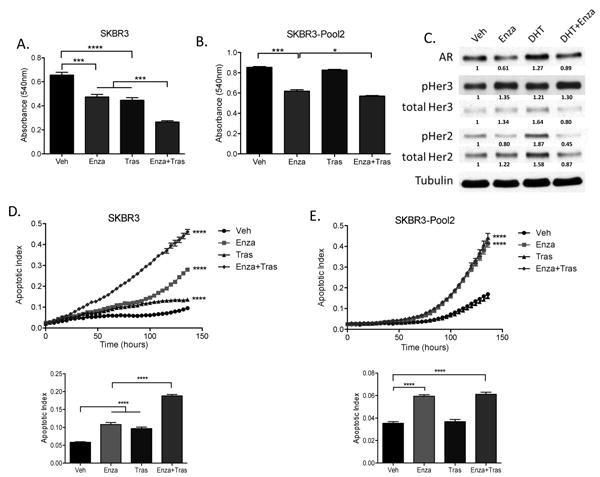
(A) SKBR3 cells and (B) Tras-resistant SKBR3-Pool2 cells were grown in the presence of 10uM Enza and/or 20ug/mL Tras for 5 days, and cell growth was measured by crystal violet staining by measuring absorbance at 540nm. (C) Parental SKBR3 cells were hormone-stripped for 72 hours, then cells were treated with vehicle, 10uM Enza, and/or 10nM DHT for 48 hours, then whole cell lysates were immunoblotted for the indicated proteins. (D) Parental SKBR3 cells and (E) Tras-resistant SKBR3-Pool2 cells were treated with 10uM Enza and/or 20ug/mL Tras, and IncuCyte Caspase-3/7 reagent. Apoptosis was measured on an IncuCyte Imager as a function of changing fluorescence over time. Bar graphs in (D) and (E) indicate apoptotic index at 68 hours. Cell viability and apoptosis experiments were performed in biological triplicate, and repeated at least twice, with one representative experiment shown. For western immunoblot, experiments were repeated twice, with a representative experiment shown. *p<.05, *** p<.001, ****p<.0001 by ANOVA with Bonferroni's multiple comparison test.
mTOR inhibition promotes AR expression and activity
Two BC cell lines (BT474-HR20 and MDAMB453) harboring PIK3CA mutations treated with Everol for 48 hours demonstrated an increase in AR protein (Figure 2A and 2B) and gene expression (Figure 2C and 2D). Additionally, Everol treatment resulted in increased total- and phospho-HER3 protein in the BT474-HR20 cells (Figure 2A), and phospho-HER2 and phospho-HER3 in the MDAMB453 cells (Figure 2B). These Everol-dependent increases were at least partially abrogated by Enza in BT474-HR20 cells. Interestingly, treatment of the PIK3CA-wild type cell line SKBR3 with Everol did not result in upregulation of AR, phospho-HER2, or phospho-HER3 (Supplemental Figure 2). As expected, Everol treatment resulted in decreased phospho-S6, a downstream readout of mTOR activity. Enza treatment also caused a decrease in pS6 expression. In MDA-MB-453 cells and Tras-resistant BT474-HR20 cells, an increase in AR gene expression occurred within 12 hours of Everol treatment that was maintained through 48 hours (Figure 2C, 2D). An AR target gene, PIP (prolactin-induced protein) displayed a similar expression pattern, suggesting that AR transcriptional activity is increased by mTor inhibition with Everol (Figure 2C, 2D).
Figure 2. Everol upregulates AR protein expression and transcriptional activity.

(A) Tras-resistant BT474-HR20 and (B) MDAMB453 cells were grown in either charcoal-stripped serum (DCC) or full serum (FBS) for 48 hours. Cells grown in DCC were then treated with 10nM DHT, 10uM Enza, 10nM Everol, or combinations for 48 hours, as shown. Cells grown in full serum were treated with either 10uM Enza, 10nM Everol, or combination for 48 hours. (C) BT474-HR20 cells and (D) MDAMB453 cells were treated with 10nM Everol for the indicated times and RNA was harvested at timepoints shown. RT-qPCR was performed for AR and an AR target gene, PIP (prolactin-induced protein). GAPDH was used as a housekeeping gene for qPCR. (E) A TNBC PDX was explanted and grown in culture medium containing vehicle, Everol, or Enza + Everol, for 48 hours. Explants were harvested and embedded, then stained for AR protein expression by immunohistochemistry. Experiments were repeated at least twice, with one representative result shown. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To further confirm AR upregulation by Everol, an AR+, TNBC patient derived xenograft (PDX) that contains an activating PIK3CA mutation (E542K) was explanted and cultured ex vivo on dental sponges in full serum for 48 hours in the presence of Enza, Everol, or the combination. AR protein, as measured by immunohistochemical staining, was increased when the PDX pieces were treated with Everol compared to vehicle, and was downregulated when explants were treated with Enza+Everol (Figure 2E).
RPPA based protein pathway activation mapping was performed measuring 196 phospho- and total-proteins was performed to identify potential downstream effects of Everol possibly contributing to AR upregulation. Over a time-course of Everol treatment from 5 minutes through 48 hours, significant dynamic changes in a number of signaling pathways occurred (Figure 3A). p70S6K and S6RP, targets of mTOR activity, were downregulated by Everol at all timepoints measured relative to vehicle-treated cells (Figure 3A, 3B). A number of phosphoproteins were activated within 5 minutes and 30 minutes of Everol treatment, including growth factor receptors (ERBB2, EGFR, FGFR) and STAT family members (STAT4, STAT5, STAT6), and subsequently decreased at later timepoints (Figure 3A). Phospho-AKT increased significantly over time with Everol treatment. Changes in total AR expression due to Everol treatment were dynamic over time, with an early increase, followed by a decrease for several hours, and finally a large increase at 48 hours (Figure 3B). Interestingly, phospho-AR levels at position Ser-650 gradually increased over time with Everol treatment, whereas AR-Ser81 decreased over time. A known AR target, ELK1, followed the expression pattern of AR-Ser-650 activity, with a gradual increase in ELK1 phosphorylation at Ser-383 over time with Everol treatment.
Figure 3. Everol dynamically regulates multiple signaling pathways over time.
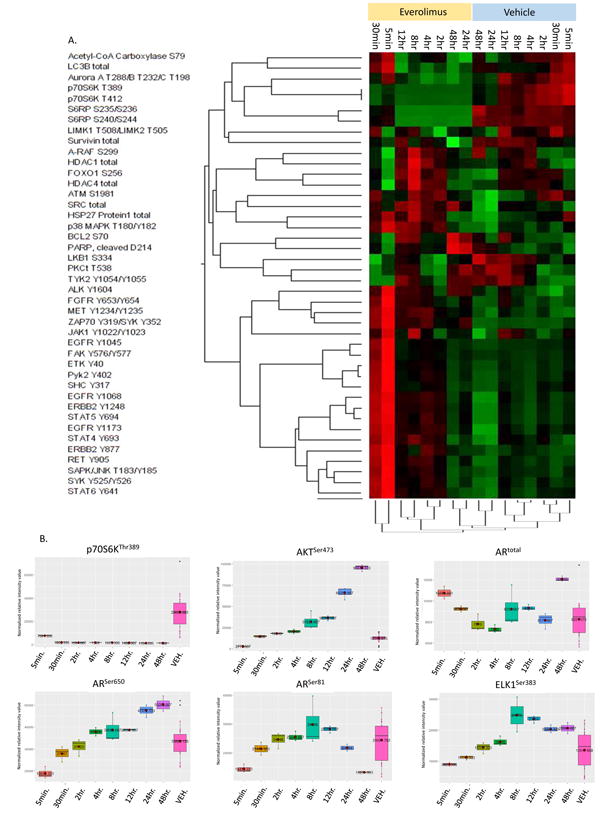
BT474-HR20 cells were treated with 10nM Everolimus for 48 hours in biological triplicate. Cells were harvested at the indicated timepoints and lysates were printed onto reverse phase protein arrays. Protein signaling activation was evaluated by staining arrays with 196 antibodies against key signaling proteins, mainly phosphorylated and cleaved protein products. At each timepoint, the two groups (vehicle vs. Everol) were compared, and a Kruskall-Walis multiavariable analysis was performed. The heatmap indicates proteins that had significantly altered expression when treated with Everol compared to the matched vehicle-treated timepoint. Within the heatmap, red color indicates higher relative intensity values, black are intermediate and green indicate lower relative intensity values. (B) Effects of Everol over time on select target proteins. Each panel represents a phosphoprotein or total protein that was significantly differentially expressed/activated at ≥1 timepoint when compared to all vehicle-treated cells (pink box). The far right box-and-whisker plot (pink) in each panel is the combined value of vehicle-treated cells at all timepoints.
AR and mTOR antagonists synergistically inhibit BC cell proliferation
BC cells were treated with a dose matrix of Everol in combination with either of two AR antagonists, Enza or Sevi, at three clinically relevant doses per drug. Combining Everol plus Enza or Everol plus Sevi synergistically inhibited proliferation in BT474 and MDAMB453 cells (Figure 4A-B). In the BT474 cells in particular, there was a synergistic inhibition at all dose combinations measured when Enza was combined with Everol, whereas in MDA453 cells the synergistic effect was strongest with Sevi + Everol. Additionally, assessment of the Dose Reduction Index (DRI), which measures the fold-change by which a drug may be reduced when administered in combination versus the drug alone, indicated that co-administering an AR antagonist with Everol allowed for a significant reduction in Everol and the same effect on growth inhibition (Figure 4A-B).
Figure 4. AR antagonists synergize with everolimus to inhibit BC proliferation in vitro.
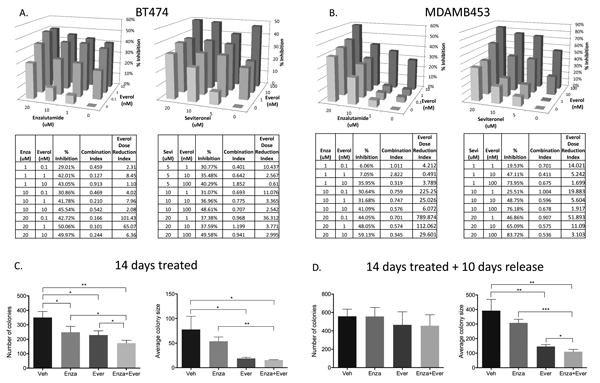
(A) BT474 and (B) MDAMB453 cells were labeled with nuclear red fluorescent protein and treated with Everol and either Enza or Sevi at doses indicated for six days in biological triplicate, and repeated at least twice. Proliferation was measured on an IncuCyte live cell imager using nuclear-RFP and percent confluence. Percent inhibition of growth was used to calculate synergistic interaction between the two drugs, using CalcuSyn software. A combination index (CI) <0.9 indicates synergy at a given dose combination, CI 0.9-1.1 indicates additivity, and CI>1.1 indicates antagonism. Everolimus Dose Reduction Index indicates the fold-change by which everolimus dose could be reduced when given in combination, as compared to Everol alone. (C) 2D colony formation assay of BT474-HR20 cells treated with 10uM Enza, 10nM Everol, or combination for two weeks, then (D) taken off treatment for an additional ten days and allowed to regrow. Colony formation assays were performed in biological triplicate *p<0.05, **p<0.01, ***p<0.001.
In order to test the effects of combination over a longer timecourse, a colony formation assay was performed using BT474-HR20 cells and treating with 10uM Enza, 10nM Everol, or the combination. Combination treatment inhibited colony formation significantly more than either single agent (Figure 4C). In order to more closely model administration of therapy in the clinical setting, after 14 days of treatment cells were taken off treatment and allowed to regrow for another 10 days (Figure 4D). On treatment, colony number was significantly less with the combination of Enza and Everol compared to either treatment alone. The colony size was significantly less with either Enza and Everol, but the combination did not significantly differ from Everol alone. However, when treatments were removed for 10 more days, although there were small colonies reforming, the average size of the colonies remained significantly smaller in the combination group as compared to either treatment alone.
Anti-tumor activity of AR antagonist enzalutamide combined with everolimus or trastuzumab
In order to determine if combining Enza with Everol would inhibit tumor growth more than either single-agent treatment, Tras-resistant BT474-HR20 cells, which are ER-positive, HER2-amplified, and harbor an activating PIK3CA mutation, were injected orthotopically into NOD/SCID mice. Mice were treated with either single agent or the combination for 26 days. Tumor viability over time as measured by IVIS signal, was analyzed using a repeated measures mixed model approach. Tumors in all treatment groups increased in size over time (p<0.0001), and the treatment groups grew at different rates (p<0.0001, Figure 5 A-D). Mice treated with either single-agent Enza or single-agent Everol had significantly reduced tumor viability when compared to mice treated with vehicle. The Enza + Everol treated tumors had significantly less viability as measured by IVIS signal than all other treatment groups (p<0.0001) and grew at a slower rate (p<0.0001). Tumors treated with Everol or Everol+Enza had significantly decreased cell proliferation, as measured by BrdU staining (p=0.008, p=0.011, respectively, Figure 5E). Tumors treated with single-agent Enza showed a trend for decreased BrdU staining relative to vehicle; however this did not reach statistical significance (p=0.14).
Figure 5. Combination treatment with Enza and Everol or Tras inhibits the growth of Tras-resistant tumors significantly more than single agent treatment.
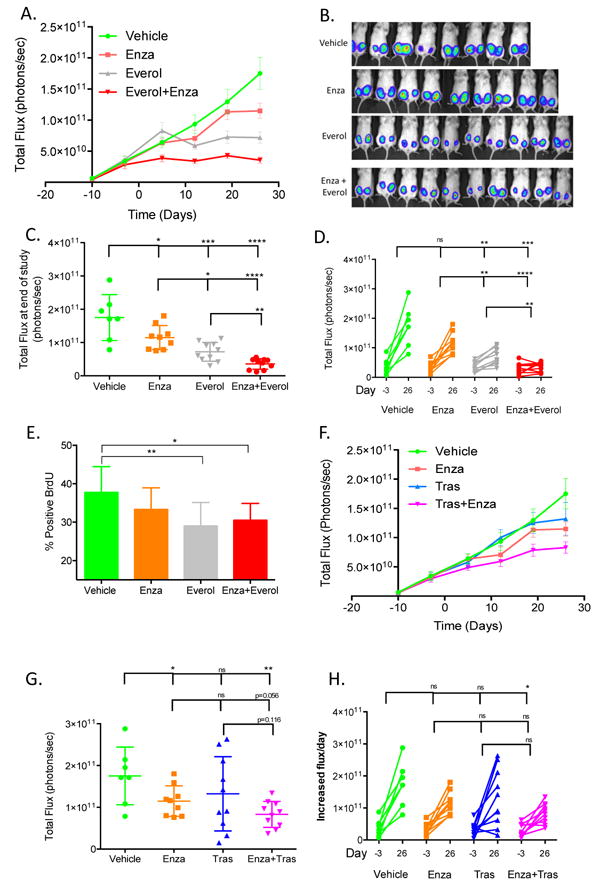
(A) Total flux growth curve of BT474-HR20 xenografts treated with Enza, Everol, or the combination. Mice (n=10/group) were randomized to one of four treatment groups at day -3 and treatment was initiated at day 0. Mice were sacrificed at day 28 and tumors were harvested. (B, C) IVIS signal on last day of study. (D) Change in total flux from day of randomization to end of study on day 26. (E) BrdU immunohistochemical staining from all tumors in each group. (F) Total flux growth curve of BT474-HR20 xenografts treated with Tras, Enza, or the combination (n=10/group). (G, H) IVIS signal on the last day of study for mice treated with Enza, Tras, or the combination *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Treatment of BT474-HR20 cells with a combination of Enza and Tras inhibited viability significantly more than vehicle. There was a trend for decreased viability when combination treatment was compared to Enza alone (p=0.056) or Tras alone (p=0.116) (Figure 5F-H).
Discussion
Everolimus targets the mTOR signaling axis, which is highly integrated with multiple pathways critical for BC progression, including ER and HER2 signaling. The BOLERO-2 phase III clinical trial was designed in part to address this, with ER+ BC patients receiving Everol in combination with the aromatase inhibitor exemestane. Indeed, the results of this trial indicated that dual inhibition of mTOR and ER significantly improved progression-free survival for patients (19). However, two other phase III clinical trials, BOLERO-1 (22), and BOLERO-3 (20), were designed to dually inhibit HER2 and mTOR in Tras-refractory BC, but did not achieve the same clinical benefit. Patients in these trials who received Everol did achieve a statistically significant improvement in PFS when compared to the non-Everol arm; however, this benefit was clinically insignificant, with a survival benefit of only one month in the BOLERO-3 trial. The results of BOLERO-1 and BOLERO-3 suggest that some HER2+ BCs may not respond to combined mTOR/HER2 inhibition due to compensatory activation of another pathway. Studies in prostate cancer indicate there is cross-regulation between the mTOR signaling axis and AR, including upregulation of AR protein expression and activity when mTOR is inhibited (31, 32). Additionally, activating PIK3CA mutations are associated with increased AR expression (33). A pre-clinical study in TNBC showed that the combination of the anti-androgen bicalutamide with PI3K (phosphoinositide 3-kinase) inhibitors slowed tumor growth (34). However, this study demonstrated only an additive, not synergistic, effect when both pathways were inhibited, possibly due to bicalutamide being a less potent inhibitor of AR, and targeting of PI3K instead of mTOR having different effects on downstream signaling cascades.
RPPA based protein pathway activation mapping analysis of the effects of Everol treatment yielded several interesting findings. The transcription factor ELK1 was upregulated with Everol treatment. ELK1 is a member of the ETS transcription factor family. It is upregulated by activated AR in bladder cancer cells and its ability to induce tumorigenicity is dependent on AR (35). The ELK1 S383 phosphorylation site is primarily regulated by MAPK/ERK (36, 37). Additionally, loss of PTEN and subsequent activation of PI3K signaling has been shown to stimulate MAPK signaling through HER2 activation in breast cancer. This raises the possibility that total levels of ELK1 are regulated by AR and activity is stimulated by de-inhibition of a potentially already activated PI3K signaling axis. Upstream of AR, RPPA analysis showed that phospho-Akt was upregulated by Everol treatment. In prostate cancer, Akt upregulates AR activity (38), suggesting a mechanism of everolimus-induced activation of AR. Additionally, AR-S650 is phosphorylated by p38 and JNK (39), both of which were upregulated in the RPPA data presented here. AR-S650 has been previously associated with nuclear export of AR and decreased transcriptional activity (40). However, the increase in AR-S650 we observed by RPPA may be a compensatory effect of upregulation of total AR, whereby saturation of AR in the cell may stimulate a negative feedback response, including increased phosphorylation at the S650 site to keep some AR out of the nucleus in an attempt to decrease its activity.
We used the MDA-MB-453 cell line for in vitro experiments, including synergy and examination of pathway component genes and proteins. This cell line has been previously classified as either TNBC or HER2-amplified (41, 42). Our own analysis of this cell line by fluorescence in situ hybridization and western blotting indicates that it is equivocal for HER2 gene amplification (FISH ratio HER2:Cep17 = 2.1), and not overexpressing by western blot compared to other HER2-amplified cell lines. Therefore, we refer to it in this study as representing the luminal AR subtype (LAR) of TNBC.
The results presented in the current study demonstrate regulation of AR by mTOR in BC. mTOR-mediated downregulation of AR and subsequent synergistic inhibition of proliferation with simultaneous AR and mTOR inhibition was observed in HER2-amplified cell lines as well as TNBC cell lines, but only in those that harbor an activating PIK3CA mutation. This may be due to the mutation-induced hyper-activation of the pathway, where mTOR is downstream of PI3K, and may not depend on HER2 status, or ER and PR status. However, given the limited number of cell lines tested here, larger clinical trials in which an mTOR inhibitor is combined with an AR antagonist should include a biomarker component for testing PIK3CA status to confirm our preliminary findings. The in vivo study presented here involved treatment with Enza, Everol, or combination. Enza is a potent inducer of cytochrome CYP3A, which is one of the primary CYP enzymes that metabolizes Everol (43). Therefore, a combination of Enza plus Everol may necessitate a dose escalation of Everol, which would increase the risk of treatment-associated toxicities. A clinical trial of Enza plus Everol is currently underway in prostate cancer (NCT02125084), and results from these studies will inform clinical trial design in BC. Combining Sevi with Everol would circumvent these pharmacokinetic complications, or alternatively a different mTOR inhibitor that is not metabolized by CYP3A could be utilized in combination with Enza.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Figure 6. Proposed mechanisms of interaction between AR antagonists, HER2 therapeutic antibody, and mTOR inhibitor.
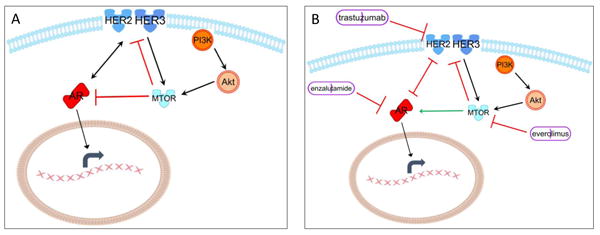
The cross-regulation in breast cancer cells between the HER2/HER3 signaling axis, mTOR integration, and AR is depicted. (A) The feedback and regulatory loops in the normal state are depicted. (B) The effect on these feedback loops when they are therapeutically inhibited is depicted.
Acknowledgments
Enzalutamide was provided by Astellas, Inc and Medivation Inc (Medivation, Inc. was acquired by Pfizer, Inc. in September 2016). Project was funded by DOD BCRP Clinical Translational Award BC120183 W81XWH-13-1-0090 to JKR and AE, R01 CA187733-01A1 to JKR, and the Natural Science Foundation of Zhejiang Province, China partially supported HG (LY17H160058). The authors wish to acknowledge the University of Colorado Shared Resource (CCSG – P30CA046934).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Collins LC, Cole KS, Marotti JD, Hu R, Schnitt SJ, Tamimi RM. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod Pathol. 2011;24:924–31. doi: 10.1038/modpathol.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barton VN, D'Amato NC, Gordon MA, Christenson JL, Elias A, Richer JK. Androgen Receptor Biology in Triple Negative Breast Cancer: a Case for Classification as AR+ or Quadruple Negative Disease. Horm Cancer. 2015;6:206–13. doi: 10.1007/s12672-015-0232-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barton VN, D'Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL, et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015;14:769–78. doi: 10.1158/1535-7163.MCT-14-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D'Amato NC, et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014;16:R7. doi: 10.1186/bcr3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Amato NC, Gordon MA, Babbs B, Spoelstra NS, Carson Butterfield KT, Torkko KC, et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol Cancer Res. 2016 doi: 10.1158/1541-7786.MCR-16-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131–5. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adamczyk A, Niemiec J, Janecka A, Harazin-Lechowska A, Ambicka A, Grela-Wojewoda A, et al. Prognostic value of PIK3CA mutation status, PTEN and androgen receptor expression for metastasis-free survival in HER2-positive breast cancer patients treated with trastuzumab in adjuvant setting. Pol J Pathol. 2015;66:133–41. doi: 10.5114/pjp.2015.53009. [DOI] [PubMed] [Google Scholar]
- 9.Agrawal A, Ziolkowski P, Grzebieniak Z, Jelen M, Bobinski P, Agrawal S. Expression of Androgen Receptor in Estrogen Receptor-positive Breast Cancer. Appl Immunohistochem Mol Morphol. 2015 doi: 10.1097/PAI.0000000000000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9:R6. doi: 10.1186/bcr1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wenger CR, Beardslee S, Owens MA, Pounds G, Oldaker T, Vendely P, et al. DNA ploidy, S-phase, and steroid receptors in more than 127,000 breast cancer patients. Breast Cancer Res Treat. 1993;28:9–20. doi: 10.1007/BF00666351. [DOI] [PubMed] [Google Scholar]
- 12.Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013;19:5505–12. doi: 10.1158/1078-0432.CCR-12-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traina TAM K, Yardley DA, et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC) J Clin Oncol. 2015;33(suppl) abstr 1003. [Google Scholar]
- 14.Eisner JR, Abbott DH, Bird IM, Rafferty SW, Moore WR, Schotzinger RJ. VT-464: A novel, selective inhibitor of P450c17(CYP17)-17,20 lyase for castration-refractory prostate cancer (CRPC) J Clin Oncol. 2012;30:198. [Google Scholar]
- 15.Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES, et al. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol Cancer Ther. 2015;14:59–69. doi: 10.1158/1535-7163.MCT-14-0521. [DOI] [PubMed] [Google Scholar]
- 16.Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011;20:119–31. doi: 10.1016/j.ccr.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiota M, Bishop JL, Takeuchi A, Nip KM, Cordonnier T, Beraldi E, et al. Inhibition of the HER2-YB1-AR axis with Lapatinib synergistically enhances Enzalutamide anti-tumor efficacy in castration resistant prostate cancer. Oncotarget. 2015;6:9086–98. doi: 10.18632/oncotarget.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin LA, Pancholi S, Farmer I, Guest S, Ribas R, Weigel MT, et al. Effectiveness and molecular interactions of the clinically active mTORC1 inhibitor everolimus in combination with tamoxifen or letrozole in vitro and in vivo. Breast Cancer Res. 2012;14:R132. doi: 10.1186/bcr3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–9. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andre F, O'Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15:580–91. doi: 10.1016/S1470-2045(14)70138-X. [DOI] [PubMed] [Google Scholar]
- 21.von Minckwitz G, Loibl S, Untch M, Eidtmann H, Rezai M, Fasching PA, et al. Survival after neoadjuvant chemotherapy with or without bevacizumab or everolimus for HER2-negative primary breast cancer (GBG 44-GeparQuinto)dagger. Ann Oncol. 2014;25:2363–72. doi: 10.1093/annonc/mdu455. [DOI] [PubMed] [Google Scholar]
- 22.Hurvitz SA, Andre F, Jiang Z, Shao Z, Mano MS, Neciosup SP, et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015;16:816–29. doi: 10.1016/S1470-2045(15)00051-0. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, et al. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70:1204–14. doi: 10.1158/0008-5472.CAN-09-3321. [DOI] [PubMed] [Google Scholar]
- 24.Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64:3981–6. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 25.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17:1514–20. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Centenera MM, Raj GV, Knudsen KE, Tilley WD, Butler LM. Ex vivo culture of human prostate tissue and drug development. Nat Rev Urol. 2013;10:483–7. doi: 10.1038/nrurol.2013.126. [DOI] [PubMed] [Google Scholar]
- 27.Sheehan KM, Calvert VS, Kay EW, Lu Y, Fishman D, Espina V, et al. Use of reverse phase protein microarrays and reference standard development for molecular network analysis of metastatic ovarian carcinoma. Mol Cell Proteomics. 2005;4:346–55. doi: 10.1074/mcp.T500003-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Wulfkuhle JD, Berg D, Wolff C, Langer R, Tran K, Illi J, et al. Molecular analysis of HER2 signaling in human breast cancer by functional protein pathway activation mapping. Clin Cancer Res. 2012;18:6426–35. doi: 10.1158/1078-0432.CCR-12-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wulfkuhle JD, Speer R, Pierobon M, Laird J, Espina V, Deng J, et al. Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J Proteome Res. 2008;7:1508–17. doi: 10.1021/pr7008127. [DOI] [PubMed] [Google Scholar]
- 30.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 31.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schayowitz A, Sabnis G, Goloubeva O, Njar VC, Brodie AM. Prolonging hormone sensitivity in prostate cancer xenografts through dual inhibition of AR and mTOR. Br J Cancer. 2010;103:1001–7. doi: 10.1038/sj.bjc.6605882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, Carey M, Agarwal R, Meric-Berstam F, et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin Cancer Res. 2009;15:2472–8. doi: 10.1158/1078-0432.CCR-08-1763. [DOI] [PubMed] [Google Scholar]
- 34.Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC, et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014;16:406. doi: 10.1186/s13058-014-0406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawahara T, Shareef HK, Aljarah AK, Ide H, Li Y, Kashiwagi E, et al. ELK1 is up-regulated by androgen in bladder cancer cells and promotes tumor progression. Oncotarget. 2015;6:29860–76. doi: 10.18632/oncotarget.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruzalegui FH, Cano E, Treisman R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene. 1999;18:7948–57. doi: 10.1038/sj.onc.1203362. [DOI] [PubMed] [Google Scholar]
- 37.Tian J, Karin M. Stimulation of Elk1 transcriptional activity by mitogen-activated protein kinases is negatively regulated by protein phosphatase 2B (calcineurin) J Biol Chem. 1999;274:15173–80. doi: 10.1074/jbc.274.21.15173. [DOI] [PubMed] [Google Scholar]
- 38.Ha S, Ruoff R, Kahoud N, Franke TF, Logan SK. Androgen receptor levels are upregulated by Akt in prostate cancer. Endocr Relat Cancer. 2011;18:245–55. doi: 10.1530/ERC-10-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daniels G, Pei Z, Logan SK, Lee P. Mini-review: androgen receptor phosphorylation in prostate cancer. Am J Clin Exp Urol. 2013;1:25–9. [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S, Kesler CT, Paschal BM, Balk SP. Androgen receptor phosphorylation and activity are regulated by an association with protein phosphatase 1. J Biol Chem. 2009;284:25576–84. doi: 10.1074/jbc.M109.043133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shim JS, Rao R, Beebe K, Neckers L, Han I, Nahta R, et al. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J Natl Cancer Inst. 2012;104:1576–90. doi: 10.1093/jnci/djs396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vranic S, Gatalica Z, Wang ZY. Update on the molecular profile of the MDA-MB-453 cell line as a model for apocrine breast carcinoma studies. Oncol Lett. 2011;2:1131–7. doi: 10.3892/ol.2011.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobsen W, Serkova N, Hausen B, Morris RE, Benet LZ, Christians U. Comparison of the in vitro metabolism of the macrolide immunosuppressants sirolimus and RAD. Transplant Proc. 2001;33:514–5. doi: 10.1016/s0041-1345(00)02116-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.