

Abstract
Aim: Vascular calcification (VC) is a risk factor of cardiovascular and all-cause mortality in patients with chronic kidney disease (CKD). CKD–mineral and bone metabolism disorder is an important problem in patients with renal failure. Abnormal levels of serum phosphate and calcium affect CKD–mineral and bone metabolism disorder and contribute to bone disease, VC, and cardiovascular disease. Hypercalcemia is a contributing factor in progression of VC in patients with CKD. However, the mechanisms of how calcium promotes intracellular calcification are still unclear. This study aimed to examine the mechanisms underlying calcium-induced calcification in a rat aortic tissue culture model.
Methods: Aortic segments from 7-week-old male Sprague–Dawley rats were cultured in serum-supplemented medium for 10 days. We added high calcium (HiCa; calcium 3.0 mM) to high phosphate (HPi; phosphate 3.8 mM) medium to accelerate phosphate and calcium-induced VC. We used phosphonoformic acid and the calcimimetic R-568 to determine whether the mechanism of calcification involves Pit-1 or the calcium-sensing receptor.
Results: Medial VC was significantly augmented by HPi+HiCa medium compared with HPi alone (300%, p < 0.05), and was associated with upregulation of Pit-1 protein. Pit-1 protein concentrations in HPi+HiCa medium were greater than those in HPi medium. Phosphonoformic acid completely negated the augmentation of medial VC induced by HPi+HiCa. R-568 had no additive direct effect on medial VC.
Conclusion: These results indicated that exposure to HPi+HiCa accelerates medial VC, and this is mediated through Pit-1, not the calcium-sensing receptor.
Keywords: Calcium, Phosphate, Mönckeberg's arteriosclerosis, Pit-1, Calcium-sensing receptor
Introduction
Vascular calcification (VC) is common in patients with end-stage renal disease and is thought to contribute to increased cardiovascular and all-cause mortality in chronic kidney disease (CKD)1, 2). One of the characteristic types of calcification of uremic artery disease is Mönckeberg's arteriosclerosis. In Mönckeberg's arteriosclerosis, also called arterial medial calcification, calcium deposits are found in the muscular middle layer of arteries. Arterial medial calcification has been observed in young and middle-aged patients without conventional atherosclerotic risk factors3). However, little is known about the adjustment processes of arterial medial calcification.
Hyperphosphatemia is prevalent in patients with CKD and it is linked to an increased risk of cardiovascular mortality in these patients4). Phosphate is arguably the most important biochemical parameter of CKD–mineral and bone metabolism disorder and an important inducer of VC. Phosphate levels above physiological serum levels have been shown to promote VC in experimental models in vivo5) and in vitro6, 7). Phosphate that is transported into cells is primarily mediated by sodium-dependent phosphate cotransporters. Phosphate uptake through Pit-1, a type III sodium-dependent phosphate cotransporter, is essential for calcification of vascular smooth muscle cells (VSMCs)8, 9). We previously reported that phosphate uptake through Pit-1 leads to induction of apoptosis and subsequent calcification of VSMCs in a rat aortic tissue culture model6, 7). Serum phosphate control is an important component in patients with CKD.
In the clinical setting, serum calcium and phosphate are carefully controlled in patients on hemodialysis. Epidemiological studies have shown a direct correlation between serum phosphate and the calcium–phosphate product (Ca × Pi) and VC in patients with end-stage renal disease10, 11). Furthermore, a previous study showed that serum calcium is a risk factor associated with all-cause mortality5). Serum calcium levels are regulated by the action of parathyroid hormone (PTH). Major drivers of PTH hypersecretion and parathyroid cell proliferation are hypocalcemia and hyperphosphatemia. Hypocalcemia and hyperphosphatemia develop in patients with CKD and secondary hyperparathyroidism as a result of low calcitriol levels and decreased kidney function. Management of patients receiving dialysis requires control of secondary hyperparathyroidism and associated extraosseous calcification.
Arterial medial calcification is associated with increased pulse pressure and arterial stiffness12). In the clinical setting, the coronary artery calcium score, the cardio-ankle vascular index, and pulse wave velocity are screening tools to detect coronary artery calcification and arterial sclerosis13, 14). Clinical factors associated with arterial stenosis (AS) are similar to risk factors for VC15). The Ca × Pi product is correlated with the severity of AS16). On the basis of these factors, treatment of hypercalcemia has a beneficial effect on VC and AS. However, the mechanisms underlying calcium-induced calcification are unclear.
Recently, the use of calcimimetics has become a major treatment of secondary hyperthyroidism in patients on dialysis. Calcimimetics increase the sensitivity of the calcium-sensing receptor (CaSR), thereby controlling PTH oversecretion without inducing hypercalcemia. This observation led to the hypothesis that the beneficial effect of calcimimetics on serum biochemistry contributes to slowing the progression of VC. However, whether calcimimetics also modify the progression of atherosclerosis, and whether they act only indirectly via a decrease in serum PTH, calcium, and phosphate levels, or also by a direct effect on vascular CaSRs, remain unclear.
Aim
This study aimed to examine the mechanisms underlying calcium-induced calcification in a rat aortic tissue culture model. We used phosphonoformic acid (PFA) and the calcimimetic R-568 to determine whether the mechanism of calcification involves Pit-1 or CaSR.
Methods
Experimental Animals
We purchased 7-week-old male Sprague–Dawley rats from Kiwa Laboratory Animals Co. (Wakayama, Japan), and maintained them under specific pathogenfree conditions with a 12-h light/dark cycle. After several days (2–7 days) of acclimatization, all of the rats were sacrificed to obtain aortic tissue to culture as described below. All experimental procedures were approved by the Animal Care and Use Committee of Wakayama Medical University and conformed to NIH guidelines for the care and use of laboratory animals.
Aortic Tissue Culture
Cell cultures have many problems, such as cell transformation and lack of an extracellular matrix. Therefore, we used rat aortic tissue culture, which has the structure and matrix of a vessel, and is closer to the in vivo situation than cell cultures. Thoracic aortas below the arch to the diaphragm were removed from 7–8-week-old male Sprague–Dawley rats. After carefully removing the connective tissue, the aortas were cut into several 3–5-mm rings and placed in Dulbecco's modified Eagle's medium (DMEM, Thermo Scientific, Waltham, MA), supplemented with 10% fetal bovine serum, 1% streptomycin, and penicillin. Aortic segments were maintained at 37°C in a 5% CO2 incubator, and the medium was changed every other day. DMEM contained 1.8 mM Ca2+, 0.9 mM PO43-, with a pH of 7.2. NaH2PO4, Na2HPO4, and CaCl2 were added to the serum-supplemented DMEM to create various phosphate and calcium concentrations. Aortic rings were cultured in DMEM with normal phosphate concentrations (CON; phosphate 0.9 mM) or high phosphate concentrations (HPi; phosphate 3.8 mM). Aortic rings in CON and HPi conditions were also cultured with normal calcium concentrations (NCa; calcium 1.8 mM) or high calcium concentrations (HiCa; calcium 3.0 mM) for 10 days, and were visualized by von Kossa stain. We added 1.0 mM PFA (Sigma-Aldrich, Japan, Tokyo, Japan) to the medium to determine whether calcium induced medial calcification with mediation through Pit-1. PFA is a specific inhibitor of sodium-dependent phosphate cotransporters. R-568 was provided by Kyowa-Hakko-Kirin Inc., Tokyo, Japan. R-568 (calcimimetic) concentrations in the medium were adjusted to 0, 10, 100, and 1,000 ng/ml. R-568 (N-(3-[2-chlorophenyl]propyl)-(R)-α-methyl-3-methoxybenzylamine) potentiates the effects of extracellular calcium on parathyroid calcium receptors and inhibits PTH secretion. Aortic segments were cultured at different time intervals according to our study requirements. After completing the culture procedure, the aortic segments were washed three times in Hank's Balanced Salt Solution (–) and were divided for quantification and tissue analysis.
Quantification of Calcification
Aortic segments were weighed and decalcified with 10% formic acid for 24 h at room temperature. The calcium content of the supernatant was determined by the methylxylenol blue method using the Wako Calcium E-Test (Wako Pure Chemical Industries, Osaka, Japan). Results were corrected by wet tissue weight and expressed as mg/g wet weight of tissue.
Western Blot Analysis
After protein extraction, protein concentrations were quantified using a commercial reagent (BCA; Pierce, Biotechnology, Inc., Rockford, IL), which was normalized to 0.5 or 1 µg/µL for all samples. A total of 10 µg protein was applied to each lane of a 15% polyacrylamide gel. The gel was transferred to a polyvinylidene difluoride membrane using the Ezblot (AE-1460; Atto, Tokyo, Japan) reagent and a semi-dry blotting unit (WSE-4110 Powered BLOT One; Atto) according to the instruction manual. After transfer, the blots were blocked with a commercial blocking reagent (PVDF Blocking Reagent for Can Get Signal®; Toyobo, Osaka, Japan) for 1 h at room temperature. After being washed in Tris-buffered saline (50-mM Tris, 150-mM NaCl, pH 7.6) with 0.1% Tween 20 (TBST), the blots were cultured with primary antibodies diluted in immunoreaction enhancer solution (Can Get Signal Solution 1, Toyobo) overnight at 4°C. Primary antibodies used in this study were rabbit polyclonal anti-GAPDH (diluted 1:1500, sc-25778; Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit polyclonal anti-Pit-1 (diluted 1:1000, sc-98814; Santa Cruz Biotechnology). The membrane was washed in TBST and then reacted with horseradish peroxidase-conjugated donkey anti-rabbit IgG (A50-201p; Bethyl Laboratories, Inc., Montgomery, TX), which was diluted in immunoreaction enhancer solution (1:10000, Can Get Signal Solution 2; Toyobo) for 1 h at room temperature. The membrane was then washed again in TBST and processed with an enhanced chemiluminescence procedure (ECL Prime Western Blotting Detection Reagent; GE Healthcare, Japan, Tokyo, Japan). The enhanced chemiluminescence signals on the immunoblots were detected using the WSE-6100 LuminoGraph and quantified using the CS Analyzer version 1.0.2 (Atto).
Statistical Analysis
Data are shown as mean ± standard error (SE). The data were primarily analyzed with Bartlett's test (JMP version 9.0; SAS Institute Inc., Cary, NC) for equality of variance. When homoscedasticity was not confirmed by Levene's test, the Kruskal–Wallis test was applied to the data. On the basis of results of the Kruskal–Wallis test, Steel–Dwass multiple comparisons were further performed to compare the differences among the four conditions. A p value < 0.05 was considered to be statistically significant.
Results
Time Course
Histological images showed severe medial VC in aortic rings that were exposed to HPi+HiCa compared with those cultured in CON and HPi. Histology showed moderate calcification in culture with HPi and diffuse severe calcification in culture with HPi+HiCa (Fig. 1A). The time course of calcium accumulation during 10 days of ex vivo culture was quantified as aortic calcium content (Fig. 1B, C). There was a time course effect on calcium accumulation in the HiCa and NCa conditions. Calcium content was not increased in aortic rings that were cultured in medium with CON, regardless of HiCa during 10 days (Fig. 1B). In medium with HPi, calcium content was gradually increased in a time-dependent manner (Fig. 1C). HiCa caused higher calcification than NCa at each time point (p < 0.05).
Fig. 1.
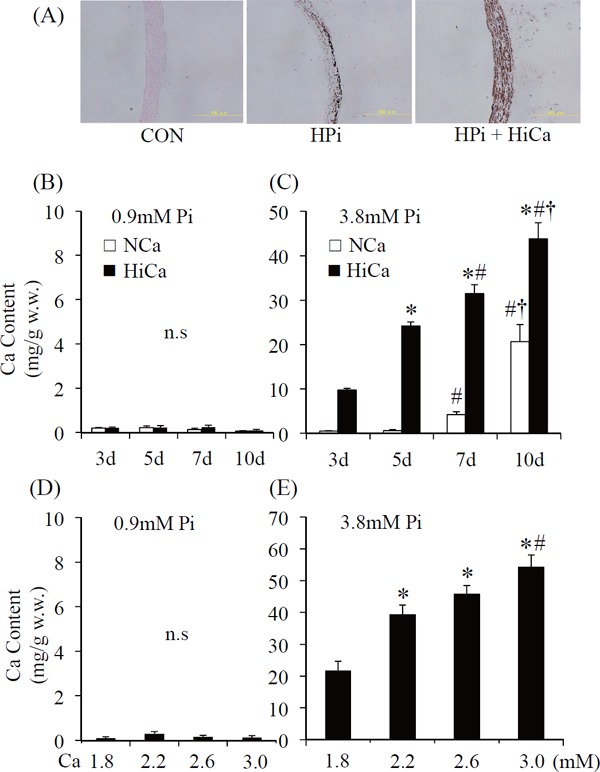
von Kossa staining of aortic rings. Sections show no staining when cultured in a medium with normal concentrations of Pi (0.9 mM, left panel). Sections show moderate calcification in HPi (3.8 mM, center panel) and diffuse severe calcification in HPi+HiCa (Pi 3.8 mM+Ca 3.0 mM; right panel) (A). Calcium content of aortic rings cultured with (B) normal concentrations of inorganic phosphate (CON; 0.9 mM) or (C) high Pi (HPi; 3.8 mM). Time course of calcium accumulation during 10 days of ex vivo incubation with normal calcium medium (NCa; 1.8 mM) and with high Ca medium (HiCa; 3.0 mM). Results are the average of at least 10 rings and represent an independent experiment (n = 15–20 rings obtained from 10 rats). *p < 0.0001 compared with day 3; #p < 0.001 compared with day 5; †p < 0.05 compared with day 7. Dose-response relationship between aortic calcium content and calcium concentration of the medium after 10 days of ex vivo culture with (D) normal concentrations of inorganic phosphate (CON; 0.9 mM) or (E) high Pi (HPi; 3.8 mM). Results are the average of at least 10 rings and represent an independent experiment (n = 15–20 rings obtained from 10 rats). *p < 0.01 compared with 1.8 mM; #p < 0.05 compared with 2.2 mM.
Dose-Dependent Effect of Calcium Concentrations
After 10 days of ex vivo culture, calcification was quantified as aortic calcium content (Fig. 1D, E). Calcium content was not increased in aortic rings that were cultured in medium with CON (Fig. 1D). In medium with HPi, calcium content gradually increased in a dose-dependent manner (Fig. 1E).
Changes in Pit-1 Protein Concentrations
The amount of Pit-1 protein in aortic rings that were cultured in Pi 3.8 mM+Ca 1.8 mM medium was approximately two-fold greater than in those that were cultured in CON (p < 0.05). In addition, Pit-1 protein concentrations in Pi 3.8 mM+Ca 3.0 mM medium were approximately three-fold greater than those in Pi 3.8 mM+Ca 1.8 mM (p < 0.05, Fig. 2). The amount of Pit-1 protein had increased to calcium concentration of medium dependency in Pi 3.8 mM medium conditions.
Fig. 2.
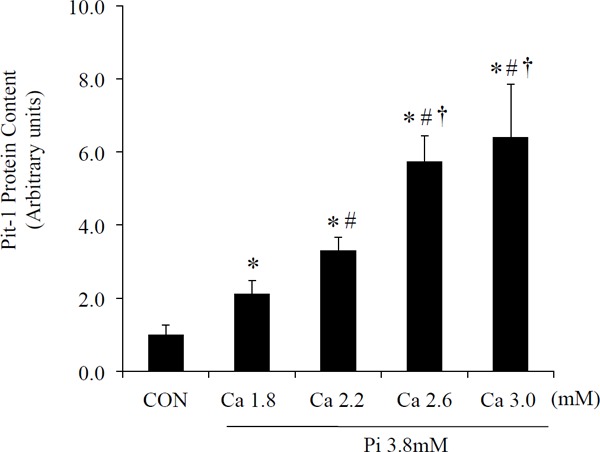
Protein expression of Pit-1 in aortic rings cultured in CON and HPi with some Ca dose (Ca 1.8 mM, 2.2 mM, 2.6 mM, 3.0 mM) medium for 10 days. Pit-1 protein levels were analyzed using western blotting. Pit-1 protein levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. Results are the average of at least 12 rings and represent an independent experiment (n = 12 rings obtained from six rats). *p < 0.05 compared with CON; #p < 0.05 compared with Ca 1.8 mM; †p < 0.05 compared with Ca 2.2 mM. CON, normal concentrations of inorganic phosphate (0.9 mM).
Role of Pit-1 in Medial VC
We used PFA to examine if Pit-1 is involved in the mechanism of calcification induced by HiCa and/or HPi. Tissue was visualized by von Kossa stain. Histology showed medial VC with exposure to HPi and HPi+HiCa. PFA treatment markedly reduced medial VC in the HPi and HPi+HiCa groups (Fig. 3A). PFA treatment markedly reduced aortic calcium content in the HPi and HPi+HiCa groups (p < 0.01) compared with culture without PFA in the same conditions after 10 days of culture (Fig. 3B).
Fig. 3.
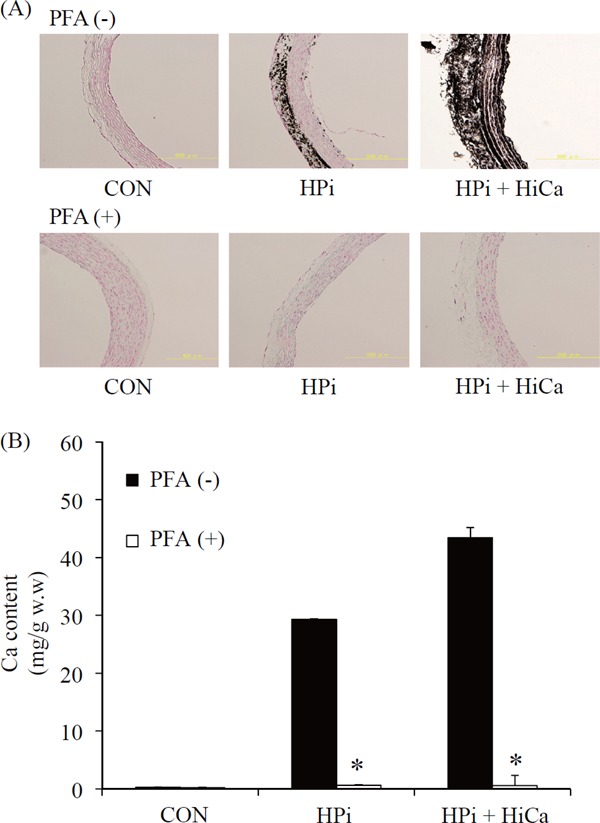
von Kossa staining of aortic rings in various types of medium with/without PFA for 10 days. Culture without PFA is shown in the upper panels and culture with PFA is shown in the lower panels. Sections without PFA show no staining when cultured in a medium with normal concentrations of Pi (0.9 mM, left panel). However, moderate calcification was observed in HPi (3.8 mM, center panel) and diffuse severe calcification was observed in HPi+HiCa (Pi 3.8-mM+Ca 3.0 mM; right panel). Sections with PFA show no staining when cultured in all types of medium (A). Calcium content of cultured aortic rings in various types of medium with/without PFA for 10 days. *p < 0.01, calcium content cultured with PFA was significantly lower than that without PFA in the same condition. Results are the averages of at least 15 rings and represent an independent experiment (n = 15 rings obtained from eight rats) (B). PFA, phosphonoformic acid.
Effect of Calcimimetics in Medial VC
To examine the direct effect of calcimimetics on calcification of aortic rings, we added calcimimetics to the medium, and visualized tissue by von Kossa stain. Histology showed medial VC with exposure to Ca 1.8 mM and Ca 3.0 mM a under HPi conditions. R-568 had no additive effect on medial calcification (Fig. 4A, B).
Fig. 4.
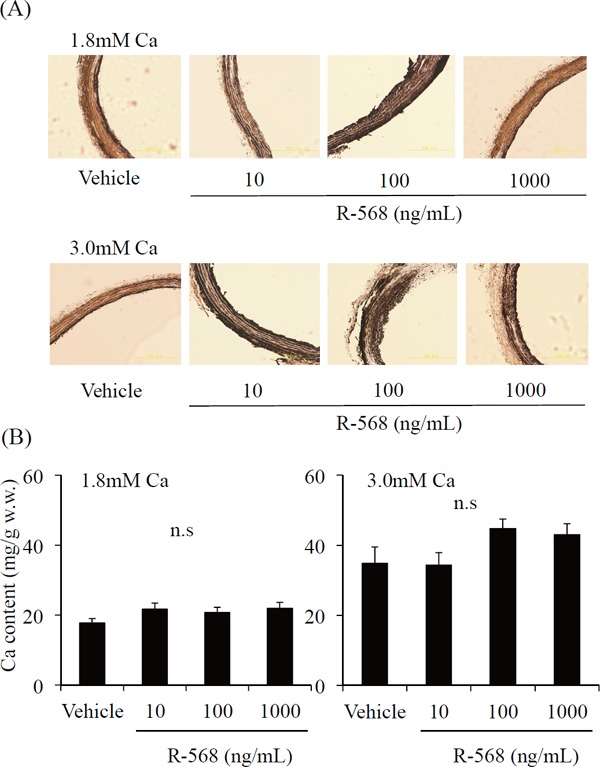
von Kossa staining of aortic rings cultured in normal Ca (1.8 mM) or high Ca (3.0 mM) under HPi (Pi 3.8 mM) with or without R-568. Culture in normal Ca (1.8 mM) under HPi (Pi 3.8 mM) is shown in the upper panels. Culture in high Ca (3.0 mM) under HPi (Pi 3.8 mM) is shown in the lower panels. Sections show diffuse severe calcification when cultured in all types of medium (A). Calcium content of aortic rings cultured in normal Ca (1.8 mM) or high Ca (3.0 mM) under HPi (Pi 3.8 mM) with or without R-568. Results are the average of at least 14 rings and represent an independent experiment (n = 14 rings obtained from eight rats) (B).
Discussion
Once believed to be a passive process, the mechanism of VC is a complex of Ca × Pi that is deposited in the vascular wall. However, currently, VC is recognized as not merely being generated by a passive process of deposition17, 18). Numerous studies have shown that VC is an active, highly regulated, cellular process, mainly mediated by VSMCs19, 20). Among the major events involved in promotion of VC, an increase in extracellular calcium and phosphate levels leads to the transition of VSMCs from a contractile to an osteochondrogenic phenotype, with formation of a procalcifying matrix and vesicles able to nucleate hydroxyapatite21). VC is currently recognized as a cell-regulated process caused by loss of calcification inhibitors and involving osteoblast-like changes in vascular-cell gene expression patterns and matrix development22). In a clinical study, we previously reported that serum calcium concentrations were significantly higher in the aortic calcification area index high group than in the aortic calcification area index low group in patients on hemodialysis2). In the present study, we showed promotion of arterial medial calcification with addition of calcium under HPi by tissue culture. Calcification of aortic rings was enhanced not only by HiCa but also with HPi+HiCa. In a cell culture model of VC, calcium-induced calcification was inhibited by PFA, and thus depended on the activity of Pit-1 or induced expression of Pit-1, as shown by measuring Pit-1 mRNA levels23). In our study, extracellular calcium-induced aortic tissue calcification in the presence of HPi. We also showed that Pit-1 expression increased in association with calcium concentrations under HPi by measuring expression of Pit-1 protein. This study is the first report to show increased expression of Pit-1 protein in tissue culture, indicating that calcium is able to promote calcification with HPi. In HPi+HiCa, calcification was markedly reduced using PFA. This result suggests that Pit-1 is a major factor of VC with calcium-induced calcification.
Hypercalcemia is significantly related to an increased risk of mortality24). Phosphate is thought to be greatly involved in ectopic calcification and mortality. Moreover, calcium should also be considered in this process. Arterial medial calcification is closely associated with the duration of hemodialysis and calcium-containing phosphate binders, including oral doses of elemental calcium prescribed as a phosphate binder5). Calcium-based phosphate binders are inexpensive, effective, and the most widely used for control of secondary hyperparathyroidism; however, there is increasing concern about their association with hypercalcemia. Disturbances in mineral metabolism, such as hyperphosphatemia and hypercalcemia, contribute to progression of calcification. Compared to sevelamer, a non-calcium-containing phosphate binder, calcium-containing phosphate binders are more likely to cause hypercalcemia and progressive coronary and aortic calcification in patients on hemodialysis25). Treatment with calcium-containing phosphate binders significantly decreases the survival rate compared with the use of sevelamer26). Treatment with phosphate binders is independently associated with improved survival in incident hemodialysis patients, but there is not good efficacy in patients with serum calcium concentrations greater than 2.25 mmol/L27). These reports suggest the importance of calcium levels and calcium component medicines in lowering phosphate levels. Serum calcium concentrations need to be kept as low as possible, even if maintained within the target value, to improve the risk of mortality in patients with CKD.
Calcimimetics are allosteric activators of CaSRs and improve upregulation of CaSRs to extracellular calcium in parathyroid cells. In our study, we added R568 to the medium and examined calcification in a rat aortic tissue culture model. VC was not reduced with exposure to R-568. Calcium-induced calcification might not be caused through CaSR signaling. Henaut et al. reported that calcimimetics reduced calcification in human VSMCs28), which is in contrast to our finding of no reduction in calcification with R-568. Smajilovic et al. reported that CaSR is present in vascular intima, but not in vascular media, using a rat aortic tissue culture model29). Moreover, Klein et al. reported that CaSR is present in vascular intima and adventitia, but not in the media of the sheep aorta30). Therefore, this difference in finding between Henaut et al.'s study28) and our study might be because of the differences between cell and tissue culture.
There are some limitations in this study. First, we used a rat aortic tissue culture model, which is more similar to the structure and matrix in vivo. We found that cellular uptake of phosphate and calcium through Pit-1 involved medial VC. Calcification in this model is histologically similar to that observed in uremic human and rat vessels, but is not exactly the same condition as in in vivo.
Second, we found that expression of Pit-1 protein was increased in aortic tissue calcification, but we did not measure phosphate uptake in this model. Our results might not be sufficient to determine whether HiCa under HPi conditions is caused by an increase in intracellular phosphate in response to higher Pit-1 protein levels. We might be able to detect phosphate uptake in this model or other culture conditions. Therefore, further studies on this issue are required.
Conclusion
Our study shows that the amount of Pit-1 protein in aortic rings that are cultured with HPi+HiCa is greater than that in those cultured in only HPi. Our results indicate that exposure to HPi+HiCa accelerates aortic medial calcification through Pit-1, but not CaSR.
Acknowledgments
R-568 was provided by Kyowa-Hakko-Kirin Inc., Tokyo, Japan.
Sources of Funding
This work was supported by JSPS KAKENHI Grant Number 25461251.
Disclosure
Takashi Shigematsu received a lecture fee. However, all results and discussion of this article were not affected by receiving this fee. None of the other authors has a conflict of interest.
References
- 1). Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM: Arterial calcification, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension, 2001; 38: 938-942 [DOI] [PubMed] [Google Scholar]
- 2). Ohya M, Otani H, Kimura K, Saika Y, Fujii R, Yukawa S, Shigematsu T: Vascular calcification estimated by aortic calcification area index is significant predictive parameter of cardiovascular mortality in hemodialysis patients. Clin Exp Nephrol, 2011; 15: 877-883 [DOI] [PubMed] [Google Scholar]
- 3). Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, Elashoff RM, Salusky IB: Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N. Engl. J. Med, 2000; 342: 1478-1483 [DOI] [PubMed] [Google Scholar]
- 4). Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM: Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol, 2004; 15: 2208-2218 [DOI] [PubMed] [Google Scholar]
- 5). London GM, Guérin AP, Marchais SJ, Métivier F, Pannier B, Adda H: Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant, 2003; 18: 1731-1740 [DOI] [PubMed] [Google Scholar]
- 6). Mune S, Shibata M, Hatamura I, Saji F, Okada T, Maeda Y, Sakaguchi T, Negi S, Shigematsu T: Mechanism of phosphate-induced calcification in rat aortic tissue culture: possible involvement of Pit-1 and apoptosis. Clin Exp Nephrol, 2009; 13: 571-577 [DOI] [PubMed] [Google Scholar]
- 7). Sonou T, Ohya M, Yashiro M, Masumoto A, Nakashima Y, Ito T, Mima T, Negi S, Kimura-Suda H, Shigematsu T: Mineral Composition of Phosphate-Induced Calcification in a Rat Aortic Tissue Culture Model. J Atheroscler Thromb, 2015; 22: 1197-1206 [DOI] [PubMed] [Google Scholar]
- 8). Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM: Phosphate regulation of vascular smooth muscle cell calcification. Circ Res, 2000; 87: e10-e17 [DOI] [PubMed] [Google Scholar]
- 9). Li X, Yang HY, Giachelli CM: Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res, 2006; 98: 905-912 [DOI] [PubMed] [Google Scholar]
- 10). Block GA, Hulbert-Shearon TE, Levin NW, Port FK: Association of serum phosphorus and x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am J Kidney Dis, 1998; 31: 607-617 [DOI] [PubMed] [Google Scholar]
- 11). Shigematsu T, Kono T, Satoh K, Yokoyama K, Yoshida T, Hosoya T, Shirai K:Phosphate overload accelerates vascular calcium deposition in end-stage renal disease patients. Nephrol Dial Transplant, 2003; 18: iii86-iii89 [DOI] [PubMed] [Google Scholar]
- 12). Gauthier-Bastien A, Ung RV, Larivière R, Mac-Way F, Lebel M, Agharazii M: Vascular remodeling and media calcification increases arterial stiffness in chronic kidney disease. Clin Exp Hypertens, 2014; 36: 173-180 [DOI] [PubMed] [Google Scholar]
- 13). Chung SL, Yang CC, Chen CC, Hsu YC, Lei MH: Coronary Artery Calcium Score Compared with Cardio-Ankle Vascular Index in the Prediction of Cardiovascular Events in Asymptomatic Patients with Type 2 Diabetes. J Atheroscler Thromb, 2015; 22: 1255-1265 [DOI] [PubMed] [Google Scholar]
- 14). Torii S, Arima H, Ohkubo T, Fujiyoshi A, Kadota A, Takashima N, Kadowaki S, Hisamatsu T, Saito Y, Miyagawa N, Zaid M, Murakami Y, Abbott RD, Horie M, Miura K, Ueshima H, SESSA Research Group : Association between Pulse Wave Velocity and Coronary Artery Calcification in Japanese men. J Atheroscler Thromb, 2015; 22: 1266-1277 [DOI] [PubMed] [Google Scholar]
- 15). Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM: Clinical factors associated with calcific aortic valve disease. J Am Coll Cardiol, 1997; 29: 630-634 [DOI] [PubMed] [Google Scholar]
- 16). Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T: Human aortic valve calcification is associated with an osteoblast phenotype. Circulation, 2003; 107: 2181-2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17). Jono S, Shioi A, Ikari Y, Nishizawa Y: Vascular calcification in chronic kidney disease. J Bone Miner Metab, 2006; 24: 176-181 [DOI] [PubMed] [Google Scholar]
- 18). Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM: Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res, 2011; 109: 697-711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM: Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol, 2004; 15: 2857-2867 [DOI] [PubMed] [Google Scholar]
- 20). Moe SM, Chen NX: Mechanisms of vascular calcification in chronic kidney disease. J Am Soc Nephrol, 2008; 19: 213-216 [DOI] [PubMed] [Google Scholar]
- 21). Giachelli CM: Vascular calcification: in vitro evidence for the role of inorganic phosphate. J Am Soc Nephrol, 2003; 14: S300-304 [DOI] [PubMed] [Google Scholar]
- 22). Speer Mei Y, Giachelli Cecilia M. Regulation of cardiovascular calcification. Cardiovascular Pathology, 2004; 13: 63-70 [DOI] [PubMed] [Google Scholar]
- 23). Yang H, Curinga G, Giachelli CM: Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int, 2004; 66: 2293-2299 [DOI] [PubMed] [Google Scholar]
- 24). Noordzij M, Cranenburg EM, Engelsman LF, Hermans MM, Boeschoten EW, Brandenburg VM, Bos WJ, Kooman JP, Dekker FW, Ketteler M, Schurgers LJ, Krediet RT, Korevaar JC, NECOSAD Study Group : Progression of aortic calcification is associated with disorders of mineral metabolism and mortality in chronic dialysis patients. Nephrol Dial Transplant, 2011; 26: 1662-1669 [DOI] [PubMed] [Google Scholar]
- 25). Chertow GM, Burke SK, Raggi P, Treat to Goal Working Group : Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int, 2002; 62: 245-252 [DOI] [PubMed] [Google Scholar]
- 26). GA Block, P Raggi, A Bellasi, Kooienga L, Spiegel DM: Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int, 2007; 71: 438-441 [DOI] [PubMed] [Google Scholar]
- 27). Isakova T, Gutiérrez OM, Chang Y, Shah A, Tamez H, Smith K, Thadhani R, Wolf M: Phosphorus binders and survival on hemodialysis. J Am Soc Nephrol, 2009; 20: 388-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Hénaut L, Boudot C, Massy ZA, Lopez-Fernandez I, Dupont S, Mary A, Drüeke TB, Kamel S, Brazier M, Mentaverri R: Calcimimetics increase CaSR expression and reduce mineralization in vascular smooth muscle cells: mechanisms of action. Cardiovasc Res, 2014; 101: 256-265 [DOI] [PubMed] [Google Scholar]
- 29). Smajilovic S, Sheykhzade M, Holmegard HN, Haunso S, Tfelt-Hansen J: Calcimimetic, AMG 073, induces relaxation on isolated rat aorta. Vascul Pharmacol, 2007; 47: 222-228 [DOI] [PubMed] [Google Scholar]
- 30). Klein GL, Enkhbaatar P, Traber DL, Buja LM, Jonkam CC, Poindexter BJ, Bick RJ: Cardiovascular distribution of the calcium sensing receptor before and after burns. Burns, 2008; 34: 370-375 [DOI] [PubMed] [Google Scholar]