

Abstract
Isoprostanoids are a group of non-enzymatic oxygenated metabolites of polyunsaturated fatty acids. It belongs to oxylipins group, which are important lipid mediators in biological processes, such as tissue repair, blood clotting, blood vessel permeability, inflammation and immunity regulation. Recently, isoprostanoids from eicosapentaenoic, docosahexaenoic, adrenic and α-linolenic namely F3-isoprostanes, F4-neuroprostanes, F2-dihomo-isoprostanes and F1-phytoprostanes, respectively have attracted attention because of their putative contribution to health. Since isoprostanoids are derived from different substrate of PUFAs and can have similar or opposing biological consequences, a total isoprostanoids profile is essential to understand the overall effect in the testing model. However, the concentration of most isoprostanoids range from picogram to nanogram, therefore a sensitive method to quantify 20 isoprostanoids simultaneously was formulated and measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The lipid portion from various biological samples was extracted prior to LC-MS/MS evaluation. For all the isoprostanoids LOD and LOQ, and the method was validated on plasma samples for matrix effect, yield of extraction and reproducibility were determined. The methodology was further tested for the isoprostanoids profiles in brain and liver of LDLR(-/-) mice with and without docosahexaenoic acid (DHA) supplementation. Our analysis showed similar levels of total F2-isoprostanes and F4-neuroprostanes in the liver and brain of non-supplemented LDLR(-/-) mice. The distribution of different F2-isoprostane isomers varied between tissues but not for F4-neuroprostanes which were predominated by the 4(RS)-4-F4t-neuroprostane isomer. DHA supplementation to LDLR(-/-) mice concomitantly increased total F4-neuroprostanes levels compared to F2-isoprostanes but this effect was more pronounced in the liver than brain.
Graphical abstract
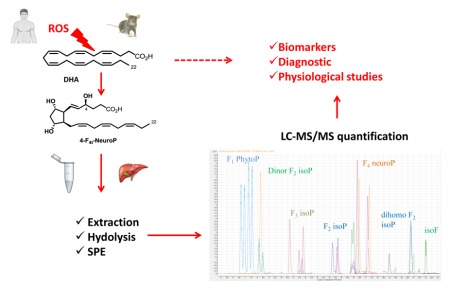
Isoprostanoids are a group of non-enzymatic oxygenated metabolites of polyunsaturated fatty acids. It belongs to oxylipins group, which are important lipid mediators in biological processes, such as tissue repair, blood clotting, blood vessel permeability, inflammation and immunity regulation. Recently, isoprostanoids from eicosapentaenoic, docosahexaenoic, adrenic and α-linolenic namely F3-isoprostanes, F4-neuroprostanes, F2-dihomo-isoprostanes and F1-phytoprostanes, respectively have attracted attention because of their putative contribution to health. Since isoprostanoids are derived from different substrate of PUFAs and can have similar or opposing biological consequences, a total isoprostanoids profile is essential to understand the overall effect in the testing model. However, the concentration of most isoprostanoids range from picogram to nanogram, therefore a sensitive method to quantify 20 isoprostanoids simultaneously was formulated and measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The lipid portion from various biological samples was extracted prior to LC-MS/MS evaluation. For all the isoprostanoids LOD and LOQ, and the method was validated on plasma samples for matrix effect, yield of extraction and reproducibility were determined. The methodology was further tested for the isoprostanoids profiles in brain and liver of LDLR−/− mice with and without docosahexaenoic acid (DHA) supplementation. Our analysis showed similar levels of total F2-isoprostanes and F4-neuroprostanes in the liver and brain of non-supplemented LDLR−/− mice. The distribution of different F2-isoprostane isomers varied between tissues but not for F4-neuroprostanes which were predominated by the 4(RS)-4-F4t-neuroprostane isomer. DHA supplementation to LDLR−/− mice concomitantly increased total F4-neuroprostanes levels compared to F2-isoprostanes but this effect was more pronounced in the liver than brain.
1. Introduction
Excessive free radicals in vivo have been implicated in a number of human diseases such as neurodegenerative, cardiovascular, pulmonary disorder and cancer[1] [2]. The most common free radicals are reactive oxygen species (ROS), which can modify lipids, proteins and nucleic acids. Of the lipids in particular, the polyunsaturated fatty acids (PUFA) form a wide variety of oxygenated metabolites [3] [4]. Among them, the isoprostanes (IsoPs) appears to be a promising group of biomarkers to be assessed for oxidative stress (OS) assessment in vivo for over two decades due to its specificity and sensitivity[5] [6]. These compounds are formed in situ on membrane phospholipids and then released into their free form via phospholipase A2 and platelet activating factor hydrolase for circulation. Elevation of IsoPs, in particular those originated from arachidonic acid (AA, 20:4 n-6) also known as F2-IsoPs in biological fluids (e.g. plasma and urines) are recognized as the reference biomarker for lipid peroxidation and OS in most biological systems. Beyond their capacity of OS as biomarker, IsoPs from n-3 PUFA also demonstrated to be biologically active[7] [8] [9]. Therefore it is crucial to be able to quantify the different isoforms in a large panel of biological samples to integrate this chemical and biological complexity.
Unlike PUFAs, the isoprostanoids are quite complex to assess since the concentration range is very low (from picogram to nanogram) in most biological samples. Moreover, depending on the parent PUFAs, a large diversity of molecule has been discovered as shown in Figure 1. Analysis of these metabolites in biological samples is a challenge and depends on the robustness of the analytical instrumentation. Further, it requires one or several preparation steps, including hydrolysis and extraction from their biological matrix before analysis by radio immunological methods (RIA) or gas chromatography-mass spectrometry (GC-MS) or liquid chromatography-mass spectrometry (LC-MS), which are often coupled to another mass spectrometer (MS/MS) to increase the sensitivity[10]. It is well known to analysts, that RIA is not specific enough to provide efficient quantification of different IsoPs [11] To date, LC-MS/MS is the most common technique to quantify these biomarkers [12], even if the mass spectrometry is not the perfect method to perform absolute quantification compared to GC-MS because of the various ionization efficiency between different molecules. These changes can be very important when comparing compounds with very close structures especially for lipids including the IsoPs. In order to optimize ionization efficacy for each compound, it is essential to have the pure standard to develop a rigorous quantitative method. Although some standards are available commercially, many of the novel ones are unavailable. Through total synthesis, Durand’s group was able to synthesize [13–21] these novel standards, for example dihomo-IsoPs from adrenic acid (C22:4 n-6, AdA) for mass spectrometry analysis.
Figure 1.
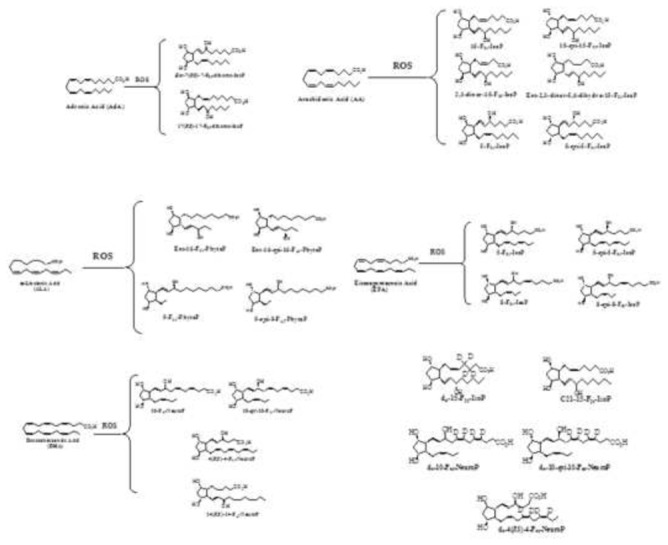
Chemical structure of isoprostanes derived from non-enzymatic oxidation of n-6 PUFA, adrenic acid (AdA) and arachidonic acid (AA), and n-3 PUFA, α-linolenic acid (ALA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) measured in this study.
In this study, we developed a complete quantitative profiling of IsoPs by LC-MS/MS. As IsoPs are present in a very low concentration it was imperative to improve largely the sensitivity of the method therefore we also tried two different derivatization procedures of the carboxylic acid function to improve the ionization of molecules. For both profiles, with and without derivatization, chromatographic separation has been optimized and sensitivity compared. The two methods have been tested on human plasma and the best one was applied in this study. The final methodology was then validated on plasma sample and applied to other biological samples, namely cerebrospinal fluid (CSF), urine, and brain, liver and muscle tissues. Our methodology was finally checked on a mouse model, in which the goal was to determine the isoprostanoids profiling in the brain and liver of LDLR−/− mice and to investigate the effect of docosahexaenoic acid (C22:6 n-3, DHA) supplementation on these profiles.
2. Material and methods
2.1. Chemicals
Commercially available IsoP standards (d4-15-F2t-IsoP and 2,3-dinor-15-F2t-IsoP) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Others standards Ent-16-epi-16-F1t-PhytoP, Ent-16-F1t-PhytoP, 9-F1t-PhytoP, 9-epi-9-F1t-PhytoP, Ent-15(RS)-2,3-dinor-5,6 dihydro-15-F2t-IsoP, 8-F3t-IsoP, 8-epi-8-F3t-IsoP, 5-F3t-IsoP, 5-epi-5-F3t-IsoP, 15-F2t-IsoP, 15-epi-15-F2t-IsoP, 5-F2t-IsoP, 5-epi-5-F2t-IsoP, 10-F4t-NeuroP, 10-epi-10-F4t-NeuroP, 14(RS)-14-F4t-NeuroP, 4(RS)-4-F4t-NeuroP, Ent-7(RS)-7-F2t-dihomo-IsoP, 17(RS)-17-F2t-dihomo-IsoP, C21-15-F2t-IsoP, d4-10-epi-10-F4t-NeuroP, d4-10-F4t-NeuroP, d4-4(RS)-4-F4t-NeuroP were synthesized according to our published procedures13. Hexane, ethanol absolute, acetic acid potassium hydroxide (KOH), methanol (MeOH; HPLC gradient Grade), butylated hydroxytoluene (BHT) and formic acid were purchased from Sigma Aldrich (Saint Quentin Fallavier, France). Acetonitrile (ACN; HPLC grade) was obtained from Acros Organics (Illkirch, France). Ammonia solution 30 % (NH4OH) was purchased from Carlo Erba Reagenti (Cornaredo, Italy). Water used in this study was purified on a milliQ system (Millipore). The 96 well-plate for solid extraction (SPE) (Oasis Max, 60 mg) was purchased from Waters (Saint-Quentin en Yvelines, France).
2.2. Standards preparation for linearity and reproducibility assessment
Standard solutions with or without derivatization were prepared in MeOH at the following concentrations, 0.06, 0.12, 0.24, 0.49, 0.98, 1.95, 3.91, 7.81, 15.63, 31.25, 62.5, 125, 250, and 500 ng mL−1 for all primary standards. The concentration of the deuterated internal standards (IS) used 5 ng taken from 250 ng mL−1 stock solution. Calibration curves were calculated by the area ratio of the analyte and the internal standard. The linearity and the accuracy of the detection were determined and the limit of detection (LOD : lower point with s/n > 5) and limit of quantification (LOQ: lower point with s/n > 10) were defined for the 20 compounds.
2.3. Biological fluid extraction
Healthy human plasma (1 mL) or CSF (600 μL), LDLR−/− mice plasma (≥200 μL) or urine (≥200 μL) were collected, and supplemented with BHT (1% in ethanol), and stored at −80°C. For the extraction, the samples were thawed and spiked with 5 ng of each internal standard. A volume of 985 μL of hydrolysis solution (KOH 1M in MeOH) was added. The resulting mixture was mixed and incubated at 40°C for 30 minutes (excepted for urine). After cooling in room temperature, 2 mL of 40 mM formic acid (pH 4.5 adjusted with 1 M NaOH) was added. Thereafter, the samples including urine were cleaned and extracted by solid-phase extraction (SPE) on 96-well plate OASIS MAX 60 mg (Waters, USA) modified from Lee et al. method [22]. Briefly, the wells were cleaned with 2 mL of MeOH and conditioned with 2 mL of 40 mM formic acid (pH 4.5). After loading the samples, the wells were washed with 2 mL of 2% NH4OH followed by 2 mL of MeOH/20 mM formic acid (20:80 v/v) and 2 mL of hexane. The IsoPs were eluted with 2 ml hexane/ethanol/acetic acid (70:29.4:0.6 v/v/v). After drying under nitrogen gas, the samples were re-dissolved with 20 μL of MeOH. A part of the sample (5 μl) was taken for LC-MS/MS analysis and the remaining samples were derivatized prior to LC-MS/MS measurement.
2.4. Biological tissue preparation
The tissue samples were stored at −80°C before preparation. To a total of 200 mg of thawed tissue (brain, liver or muscle) sample, 1 mL of Folch solution (CHCl3:MeOH, 2:1, v/v) containing 10 μL BHT (1 % in ethanol) was added and spiked with 5 ng of each internal standard. The mixture was homogenized with a Fast Prep instrument (MP Biomedicals) for 30 s at 6.5 m/s. Then the homogenized tissue was further extracted with 1.5 mL ice-cold Folch solution (CHCl3: MeOH, 2:1, v/v) and 0.5 mL of ultrapure water. The mix was shaken for 30 s and centrifuge for 10 min at room temperature to separate the aqueous and organic layers. The lower organic layer was carefully removed and transferred to a pyrex tube and then evaporated under nitrogen gas. The extracted lipid was dissolved in 1 mL of hydrolysis solution (KOH 1M in MeOH) and incubated at 40°C for 30 minutes. After cooling in room temperature, 3 mL of 40 mM formic acid was added. The samples were then cleaned and extracted by SPE as described in section 2.3.
2.5. Derivatization of the extracted samples
To a set of extracted samples, 10 μL of freshly prepared solution of 10 mM triphenylphosphine (TPP), 10 mM 2,2’-dipridyl disulfide (DPDS) and 10 μg 2-picolyamine (PA) prepared separately in acetonitrile were added successively. To another set of the extracted samples, freshly prepared 10 μL of TPP and DPDS, and 10 μL of freshly prepared 10 μg 2-hydrazinopyridine (HP) in acetonitrile were added. The sample mixture was incubated at 60°C for 10 min. The mixture was dried under nitrogen and then reconstituted in MeOH for LC-MS/MS analysis.
2.6. LC-MS/MS analysis
High performance liquid chromatography (HPLC) was performed using an Agilent 1290 Infinity equipped with a thermostated autosampler, a binary pump and a column oven. The analytical column was a Zorbax SB-C18 Rapid Resolution HD (2,1 x 100 mm; 1,8 μm) (Agilent Technologies, USA) and maintained at 25°C. The mobile phases consisted of water: formic acid (99.9:0.1;v/v) (A) and acetonitrile : formic acid (99.9:0.1, v/v) (B). The linear gradient was set as follows for the non-derivatized IsoPs analysis: 20% B at 0 min, 30% B at 15 min, 35% B at 20 min, 100% B at 23 min, 100% B at 26 min, and 20% B at 26.5 min for 1.5 min of equilibration. For the derivatized IsoPs, the gradient was set to 18% B at 0 min, 30% B at 22 min, 35% B at 26 min, 100% B at 28 min, 100% B at 29 min, and 18% B at 29.5 min for 1.5 min of equilibration. The flow rate was set at 0.3 mL/min. The autosampler was fixed at 5 C and the injection volume was 5 μL per analysis. The HPLC system was coupled on-line to an Agilent 6460 triple quadrupole MS (Agilent Technologies, USA) equipped with electrospray ionization (ESI). The ESI was performed in negative ion mode for non-derivatized IsoPs and positive mode for derivatized IsoPs. The MS source parameters were set as follows: source temperature 325ºC, nebulizer gas (nitrogen) flow rate 10 L min−1, sheath gas temperature 350ºC, sheath gas (nitrogen) flow rate 12 L min−1 and the spray voltage adjusted to −3000 V. The dwell time used was 10 ms. The analysis was performed in Selected Reaction Monitoring (SRM) detection mode using nitrogen as the collision gas. The SRM of each compound without (Table 1) or with derivatization (Table 2) were pre-determined by MS/MS analysis. Peak detection, integration and quantitative analysis were performed by Mass Hunter Quantitative analysis software (Agilent Technologies, USA). Concentration of the analytes was calculated by calibration curves obtained in Section 2.2.
Table 1.
Selected reaction monitoring (SRM) of the isoprostanes derived from polyunsaturated fatty acids.
| Compounds | RT (min) | Precursor ion (m/z) | Product ion (m/z) | F (V) | CE (V) |
|---|---|---|---|---|---|
| Adrenic acid | |||||
| Ent-7(RS)-7-F2t-dihomo-IsoP | 20.76 | 381 | 143 | 120 | 18 |
| 17(RS)-F2t-dihomo-IsoP | 20.90 | 381 | 337 | 120 | 12 |
| Arachidonic acid | |||||
| 15-epi-15-F2t-IsoP | 14.24 | 353 | 193 | 120 | 20 |
| 15-F2t-IsoP | 14.69 | 353 | 193 | 120 | 20 |
| 5-F2t-IsoP | 15.85 | 353 | 115 | 120 | 12 |
| 5-epi-5-F2t-IsoP | 16.01 | 353 | 115 | 120 | 12 |
| 2,3-dinor-15-F2t-IsoP | 8.29 | 325 | 237 | 100 | 5 |
| Ent-15(RS)-2,3-dinor-5,6-dihydro-15-F2t-IsoP | 8.30 | 327 | 283 | 120 | 20 |
| d4-15-F2t-IsoP | 14.61 | 357 | 197 | 120 | 20 |
| C21-15-F2t-IsoP | 18.97 | 368 | 193 | 120 | 22 |
| alpha-Linolenic acid | |||||
| Ent-16-epi-16-F1t-PhytoP | 6.66 | 327 | 283 | 120 | 15 |
| Ent-16-F1t-PhytoP | 6.94 | 327 | 283 | 120 | 15 |
| 9-F1t-PhytoP | 7.30 | 327 | 283 | 120 | 15 |
| 9-epi-9-F1t-PhytoP | 7.58 | 327 | 283 | 120 | 15 |
| Eicosapentaenoic acid | |||||
| 8-F3t-IsoP | 10.70 | 351 | 127 | 120 | 18 |
| 8-epi-8-F3t-IsoP | 11.82 | 351 | 127 | 120 | 18 |
| 5-F3t-IsoP | 11.48 | 351 | 115 | 120 | 15 |
| 5-epi-5-F3t-IsoP | 11.72 | 351 | 115 | 120 | 15 |
| Docosahexaenoic acid | |||||
| 10-F4t-NeuroP | 16.43 | 377 | 153 | 120 | 15 |
| 10-epi-10-F4t-NeuroP | 17.31 | 377 | 153 | 120 | 15 |
| 4(RS)-F4t-NeuroP | 19.50 | 377 | 101 | 120 | 15 |
| d4-10-epi-10-F4t-NeuroP | 17.19 | 381 | 157 | 120 | 15 |
| d4-10-F4t-NeuroP | 16.31 | 381 | 157 | 120 | 15 |
| d4-4(RS)-4-F4t-NeuroP | 20.90 | 382 | 239 | 120 | 15 |
The deuterated form of IsoP and NeuroP, and C21-15-F2t isoP were used as internal standards for quantification of samples in this study. IsoP: isoprostane; NeuroP: neuroprostane; F: Fragmentor; CE: collision energy; N : number.
Table 2.
Selected reaction monitoring (SRM) of the derivatized isoprostanes derived from polyunsaturated fatty acids.
| Compound | RT (min) | Precursor ion (m/z) | Product ion (m/z) | F (V) | CE (V) |
|---|---|---|---|---|---|
| Adrenic acid | |||||
| Ent-7(RS)-7-F2t-dihomo-IsoP | 24.17 | 455 | 109 | 130 | 30 |
| 17(RS)-F2t-dihomo-IsoP | 23.41 | 455 | 109 | 130 | 30 |
| Arachidonic acid | |||||
| 15-epi-15-F2t-IsoP | 14.73 | 427 | 109 | 120 | 25 |
| 15-F2t-IsoP | 15.34 | 427 | 109 | 120 | 25 |
| 5-F2t-IsoP | 18.32 | 427 | 109 | 120 | 30 |
| 5-epi-5-F2t-IsoP | 18.50 | 427 | 109 | 120 | 30 |
| Ent-15(RS)-2,3-dinor-5,6-dihydro-15-F2t-IsoP | 8.89 | 401 | 109 | 120 | 30 |
| d4-15-F2t-IsoP | 15.26 | 431 | 109 | 120 | 25 |
| C21-15-F2t-IsoP | 20.83 | 442 | 109 | 120 | 20 |
| alpha-Linolenic acid | |||||
| Ent-16-epi-16-F1t-PhytoP | 5.97 | 401 | 109 | 120 | 30 |
| Ent-16-F1t-PhytoP | 6.21 | 401 | 109 | 120 | 30 |
| 9-F1t-PhytoP | 6.62 | 401 | 109 | 120 | 30 |
| 9-epi-9-F1t-PhytoP | 6.86 | 401 | 109 | 120 | 30 |
| Eicosapentaenoic acid | |||||
| 8-F3t-IsoP | 10.58 | 425 | 109 | 120 | 35 |
| 8-epi-8-F3t-IsoP | 11.91 | 425 | 109 | 120 | 35 |
| 5-F3t-IsoP | 12.77 | 425 | 109 | 130 | 30 |
| 5-epi-5-F3t-IsoP | 13.03 | 425 | 109 | 130 | 30 |
| Docosahexaenoic acid | |||||
| 10-F4t-NeuroP | 16.80 | 451 | 109 | 120 | 20 |
| 10-epi-10-F4t-NeuroP | 18.17 | 451 | 109 | 120 | 20 |
| 14(RS)-14-F4t-NeuroP | 18.53 | 451 | 109 | 130 | 25 |
| 4(RS)-F4t-NeuroP | 23.81 | 451 | 109 | 130 | 25 |
| d4-10-epi-10-F4t-NeuroP | 16.64 | 455 | 109 | 120 | 20 |
| d4-10-F4t-NeuroP | 18.04 | 455 | 109 | 120 | 20 |
| d4-4(RS)-4-F4t-NeuroP | 23.90 | 455 | 109 | 140 | 30 |
The deuterated form of IsoP and NeuroP, and C21-15-F2t isoP were used as internal standards for quantification of samples in this study. IsoP: isoprostane; NeuroP: neuroprostane; isofurane ; F: Fragmentor; CE: collision energy.
2.7. Accuracy and precision
Repeatability and precision were respectively assessed using relative standard deviation (% RSD) and accuracy at 3 concentrations (3.91, 31.25 and 250 ng mL−1) of pure standards in triplicate determination. The concentration was subsequently calculated using the standard curves generated. For inter-day variation, the samples were analyzed on 2 different days, with 15 days in between interval.
2.8. Validation of sample preparation
The preparation of human plasma sample was validated through the yield extraction and the matrix effect. Briefly, three sets in triplicate were prepared : 500 μL of plasma (n = 3) were spiked with 5 ng of IS stock solution and 2 different concentrations of standards (31.25 and 250 ng mL−1) and were extracted as described in Section 2.3: 1) 500 μL of plasma (n = 3) were extracted and then spiked with 5 ng of IS stock solution with 2 different concentrations of standards (31.25 and 250 ng mL−1), and 2) a separate set of pure 5 ng of IS stock solution and standards solutions (31.25 and 250 ng mL−1) in the absence of plasma extract were prepared in MeOH. All sets (of three samples) were analyzed in triplicate using the LC-MS/MS. The yield extraction was determined as the percent difference between peak areas of standards in pre-spiked and post-spiked samples. The matrix effect was determined as the percentage difference between peak areas of standards added to the extracted samples and pure standard. The plasma matrix effect and yield extraction were calculated for each compound measured in the method described.
2.9. Biological samples
With permission, a sub-group of LDLR−/− mice from a previous study [23] was used to determine the effect of DHA supplementation on the profile of the isoprostanoids in the brain and liver. Briefly, from 8 weeks of age and for 20 weeks, the mice received by daily oral gavages (50 μL, 5 days/week) either oleic acid rich sunflower oil (Lesieur, Asnières-sur-Seine, France; Control group) or a mixture of oleic acid rich sunflower oil and DHA rich tuna oil (OMEGAVIE DHA90TG, Polaris Nutritional Lipids, France containing 90% of DHA as TG) providing 2% (or 35.5 mg/d/mouse) of energy as DHA. At the end of the supplementation, the mice were anaesthetized (40 mg pentobarbital/kg body weight) and the tissue samples were rapidly removed and snap-frozen in liquid nitrogen and stored at −80°C until analysis. Quantification of DHA and AA were performed on brain and liver sample. In brief, after an organic extraction in presence of internal standard, the total fatty acid were methylated, analysed and quantified on a gas chromatography-flame ionization detector (GC-FID) system [24].
3. Results and discussion
3.1. LC-MS/MS method development
In order to achieve the necessary selectivity and sensitivity of the method, the mass detection and chromatographic separation of each standard with or without derivatization were individually optimized.
3.1.1. Mass detection
IsoPs analyzed in this study have different structures comparatively. Nevertheless, due to their common carboxylic acid moiety, they were all detected in the negative ion mode as [M–H]– ions. Firstly, the fragmentor voltage was optimized for each compound in product ion scan mode. This parameter promotes the transmission of the ions between the ionization source and the first quadrupole. Low voltages lead to poor transmission efficiency whereas too high voltage values lead to excessive fragmentation. The optimum value corresponded to a maximum transmission of the [M–H]– ions without fragmentation. The second optimized parameter was the collision energy for each MS/MS transition to monitor; the most abundant one was selected. In this study, one specific fragment was selected for each compound and the collision energy was optimized for each SRM transition (Table 1). SRM transitions observed for this study are divided according to the PUFA type, that includes 15-F2 series (m/z 353 to m/z 193), 5-F2 series (m/z 353 to m/z 115), 8-F3 series (m/z 351 to m/z 127), and 5-F3 series (m/z 351 to m/z 115) [25]. For the F1-PhytoP, the carboxylate portion was lost to give SRM m/z 327 to m/z 283, whereas 10-F4-NeuroP seems to fragment in the same way as 8-F3 series (m/z 377 to m/z 153). For the remaining compounds, the fragmentations were not as definite.
Since the concentration of the IsoPs is low in biological samples, analysts may opt for a derivatization procedure to increase the volatility and polarity of the compounds. To enhance the detection responses of carboxylic acids in ESI-MS/MS several chemical derivatization procedure can be applied and measured in the positive mode of the LC-MS/MS[26] [27]. However, the derivatization reagents are not always commercially available and the preparation can be long and time consuming. In our study, we tested two simple reagents 2-hydrazinopyridine (HP) and 2-picolylamine (PA), which can be derivatized in one step under mild conditions[28]. When the reagents react with the acidic function of the IsoPs, the HP and PA form a hydrazide and an amide bond, respectively. The sensitivity obtained was preferably for PA derivatives than HP derivatives (Figure 2), therefore MS parameters were optimized only for PA (Table 2). An important drawback of this derivatization method is that for all species a unique PA fragment of m/z = 109 is obtained, which creates poor specificity of the IsoP tested.
Figure 2.
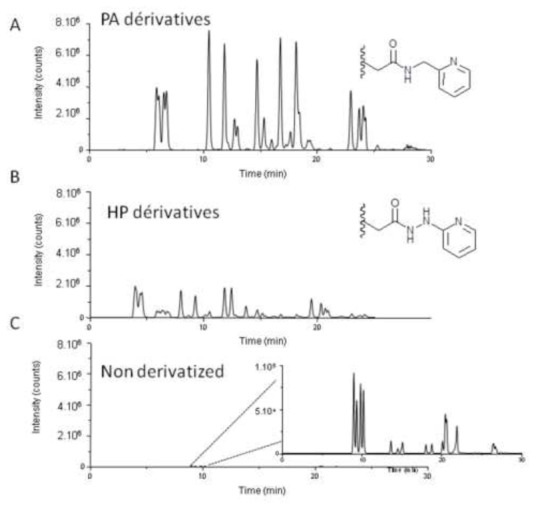
Total ion current chromatogram of IsoP: with PA derivatization (A), with HP derivatization (B), and without derivatization (C) of the same sample mix.
3.1.2. Chromatographic separation
The IsoP determined in this study have similar molecular mass and structure. As a result, the liquid chromatographic separation of each metabolite is a crucial step where each isomer of the IsoPs needs to be optimized to be separated in the chromatogram for detection. In this study, the eluent phase was acidic and a semi-linear gradient elution allows the chromatographic resolution of most compounds on a 10 cm C18 reverse phase column within 30 minutes. The gradient was also designed for the measurement of PA derivatives. As shown in Figure 3, F1t-PhytoPs eluted first, followed by 2,3-dinor-15-F2t-IsoP, F3t-IsoPs, F2t-IsoPs, then F4t-NeuroPs and F2t-dihomo-IsoPs. The isobaric compounds bearing the same transition such as F1t-PhytoPs were successfully separated (Table 1). Furthermore, all the isomers were resolved in the chromatographic separation including 8-F3t- and 8-epi-8-F3t-IsoPs (Figure 4A and B), while for 5-F2t- and 5-epi-5-F2t-IsoPs the separation between the two peaks showed an overlap at the tail of the chromatographic peak (Figure 4C and D). The separation was comparable with PA derivatives indicating derivatization procedure did not improve the separation.
Figure 3.
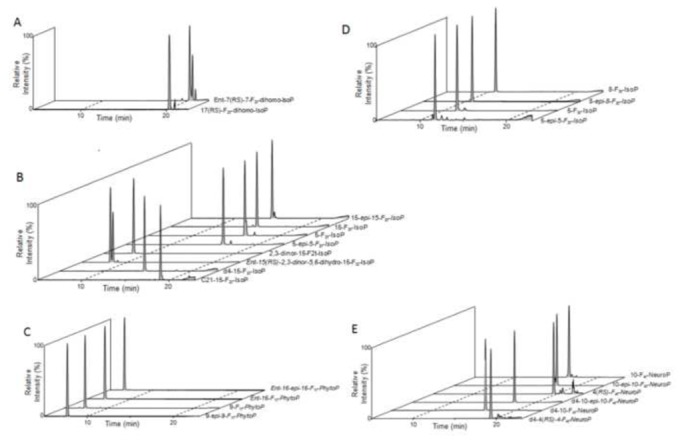
Chromatogram of selected reaction monitoring (SRM) of metabolites from each PUFA A: adrenic acid, B: arachidonic acid, C: α-linolenic acid, D: eicosapentaenoic acid and E: docosahexaenoic acid.
Figure 4.
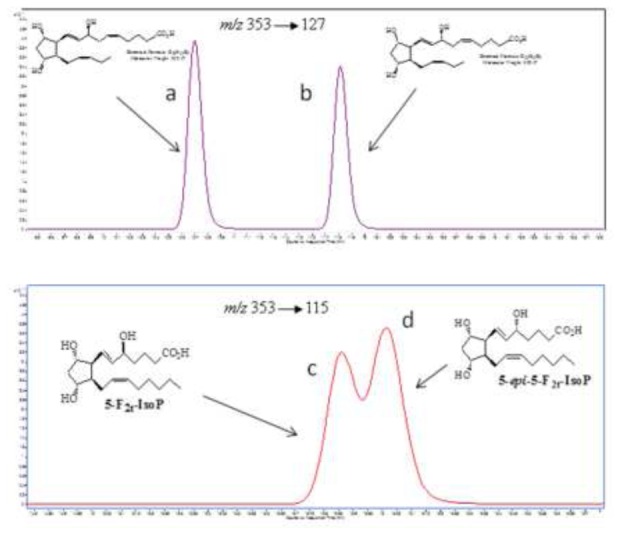
Optimum chromatographic separation of diastereoisomers for 8-F3t-IsoP (a) and 8-epi-8-F3t-IsoP (b). The diastereoisomers of 5-F2t-IsoP (c) and 5-epi-5-F2t-IsoP (d) were unable to resolve as well in the chromatographic analysis.
3.1.3. Sensitivity
To quantify our isoprostanoids we first had to choose the appropriate internal internal standard for each metabolite. NeuroPs and 15-F2t-IsoP were quantified through their deuterated equivalent and for other IsoPs, d4-15-F2t-IsoP and C21-15-F2t-IsoP were used. Based on the internal standard, each calibration curve was obtained with 10 concentrations of the pure IsoPs ranging from 0.9 to 500 ng/mL. The curves were fitted using linear regression model with 1/X factors. The linearity of the method was assessed for each metabolite by evaluating the correlation coefficient (Table 3). The LOD and LOQ of our method were evaluated, and in general LOD corresponding to the lowest concentration had signal to noise ratio above 3 and LOQ corresponding to the lowest concentration had signal to noise ratio above 10. Both of these values however depended on the type of isoprostanoids. For PA derivatives the LOD values ranged from 0.24 ng/mL to 1 ng/mL and the LOQs from 0.12 ng/mL to 2 ng/mL. As shown in Table 3, the LOD values ranged from 0.49 ng/mL to 15.6 ng/mL and the LOQs from 0.98 ng/mL to 31.25 ng/mL. The sensitivity obtained for F2-IsoP, which is the main IsoP in the literature, is in agreement with LOQ previously reported by others [29] [30]. The sensitivity observed was better than the isoprostanoids with derivatization, in particular for PA. The lack of sensitivity and specificity by PA derivatization indicates that it was not suited for the analysis of IsoPs and more so in biological samples, which often have complicated matrix structure that could further affect the precision of the measurement.
Table 3.
Limit of detection (LOD) and limit of quantification (LOQ) of the pure compounds analyzed.
| Compound | Linear regression | R2 | LOD (ng/mL) | LOQ (ng/mL) | IS |
|---|---|---|---|---|---|
| Adrenic acid | |||||
| Ent-7(RS)-7-F2t-dihomo-IsoP | y = 0.0044 x − 0.0339 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| 17(RS)-F2t-dihomo-IsoP | y = 0.0065 x − 0.0639 | 0.998 | 7.81 | 15.63 | d4-15-F2t isoP |
| Arachidonic acid | |||||
| 2,3-dinor-15-F2t-IsoP | y = 0.0079 x − 0.0558 | 0.998 | 0.98 | 1.95 | d4-15-F2t isoP |
| Ent-15(RS)-2,3-dinor-5,6-dihydro-15-F2t-IsoP | y = 0.0079 x − 0.0747 | 0.998 | 3.91 | 7.81 | d4-15-F2t isoP |
| 15-epi-15-F2t-IsoP | y = 0.0024 x − 0.0183 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| 15-F2t-IsoP | y = 0.0027 x − 0.0208 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| 5-F2t-IsoP | y = 0.0032 x − 0.0240 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| 5-epi-5-F2t-IsoP | y = 0.0042 x − 0.0348 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| Alpha-Linolenic acid | |||||
| Ent-16-epi-16-F1t-PhytoP | y = 0.0136 x − 0.1393 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| Ent-16-F1t-PhytoP | y = 0.0133 x − 0.1109 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| 9-F1t-PhytoP | y = 0.0166 x − 0.1316 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| 9-epi-9-F1t-PhytoP | y = 0.0160 x − 0.1293 | 0.998 | 1.95 | 3.91 | d4-15-F2t isoP |
| Eicosapentaenoic acid | |||||
| 8-F3t-IsoP | y = 0.0042 x − 0.0314 | 0.998 | 0.98 | 1.95 | d4-15-F2t isoP |
| 8-epi-8-F3t-IsoP | y = 0.0035 x − 0.0299 | 0.998 | 0.98 | 1.95 | d4-15-F2t isoP |
| 5-F3t-IsoP | y = 0.0018 x − 0.0186 | 0.998 | 3.91 | 7.81 | d4-15-F2t isoP |
| 5-epi-5-F3t-IsoP | y = 0.0012 x − 0.0111 | 0.998 | 3.91 | 7.81 | d4-15-F2t isoP |
| Docosahexaenoic acid | |||||
| 10-F4t-NeuroP | y = 0.0017 x − 0.0117 | 0.998 | 0.49 | 0.98 | d4-10-F4t-NeuroP |
| 10-epi-10-F4t-NeuroP | y = 0.0023 x − 0.0162 | 0.998 | 0.98 | 1.95 | d4-10-epi-10-F4t-NeuroP |
| 14(RS)-14-F4t-NeuroP | y = 6.2450.10−5 x − 6.7300.10−4 | 0.998 | 15.63 | 31.25 | d4-10-epi-10-F4t-NeuroP |
| 4(RS)-F4t-NeuroP | y = 1.8386.10−4 x − 0.0019 | 0.998 | 7.81 | 15.63 | d4-4(RS)-F4t-NeuroP |
The deuterated form of IsoP and NeuroP were used as internal standards to determine the LOD and LOQ of the analytes. IsoP: isoprostane; NeuroP: neuroprostane.
3.2. Plasma sample preparation and analysis
The lipid portion of the tissues or cells was extracted using Folch solution in the presence of antioxidant (0.005% BHT). It is known approximately 70% of the IsoPs are conjugated to phospholipid through ester bond [31], therefore hydrolysis step is required to analyse the total concentration. The lipid extract or fluid (except urine) was treated with 1 M KOH prepared in methanol for 30 minutes at 40 ºC. This step is not required for measurement of non-esterified IsoPs i.e. free form. A purification process is needed to reduce matrix effect and to increase sensitivity of the quantification by LC–MS/MS analysis. Different SPE cartridges were tested in this procedure namely, C18[32], HRX[33] and MAX[22]. It was found MAX cartridge, which is composed of mixed anionic exchange phase provided the cleanest sample and the best LC-MS/MS evaluation. A part of the lipid extracts from SPE were taken for derivatization process. The data obtain from plasma with and without derivatization were compared (data not shown). Despite the peak response and area being bigger for the derivatized samples, the background noise of the chromatogram was much more compared to the non-derivatized samples. Moreover, the derivatization caused the formation of few additional peaks very close to the target ones, making it more difficult to differentiate for peak integration. This observation further support that derivatization procedure is not suitable for IsoP analysis by LC-MS/MS for plasma and likely for other biological samples. Therefore, in order to avoid overestimation in our quantification, we adopted the IsoP measurements without derivatization process in this study.
3.3. Validation of sample processing
3.3.1. Matrix effect and yield of extraction
The efficiency of the sample processing was assessed by measuring the matrix effect and the extraction yield using plasma in triplicate. These two parameters were calculated for two concentrations of IsoPs (250 and 31.25 ng/mL). The peak areas of the chromatogram for each IsoP were compared before and after addition plasma extraction to obtain the matrix effect, as summarized in supporting material Table 1. The matrix effect ranged between 51.4% to 92.7% for low concentration and between 56.7% to 77.5% for high concentration. The values were relatively homogeneous in each group of IsoP. The yield of extraction (supporting material Table 1) was calculated for each IsoP for two concentrations comparing the quantity recovered in presence of plasma to the native one. The yield ranged from 52.0% to 85.3% for low concentration and from 40.5% to 69.2% for high concentration. Surprisingly the yield extraction of F3t-IsoPs was above 100%, and no interference signal was observed for the non-spiked plasma extract; the reason for this observation is unknown.
3.3.2. Performance of the method
Repeatability and precision were then evaluated for intra- and inter-day at 3 concentrations: 3.91, 31.25 and 250 ng/mL (supporting material Table 2) were performed in triplicate. The intra-day accuracy ranged from 81.73% to 114.21% for all IsoP evaluated. The RSD values for 3 injections were ≤ 8%. The inter-day variations were assessed by re-analyzing the samples every 15 days (n=2). The accuracies obtained were between 80.9% and 115.53% with a precision ≤ 15%. These data indicate that the method is highly reproducible for the 20 IsoP compounds analyzed.
3.4. Application on various biological samples
Our extraction and LC-MS/MS methods were applied to quantify non-enzymatic oxidized lipids from PUFAs in different biological samples. The different samples (Table 4) measured include human plasma and CSF, mice plasma, urine, liver, brain and muscle tissues A typical profile for human plasma is displayed in Figure 5. The structural matrix was taken into account for the quantity used for each type of samples and the size is shown in Table 4. Apart from CSF, IsoPs were detected and it appears to vary depending on the type of sample. No IsoPs were found in CSF, and it likely due to small volume used and concentration maybe below our limit of detection.
Table 4.
Types of isoprostanes quantified in human and mice biological samples using the method described in Section 3.
| Biological samples | Sample size | IsoPs detected |
|---|---|---|
| Human plasma | 1 mL | 15-F2t-IsoP, 15-epi-15-F2t-IsoP, 5-F2t-IsoP, 5-epi-5-F2t-IsoP, 10-F4t-NeuroP, 10-epi-10-F4t-NeuroP, 4(RS)-F4t-NeuroP |
| Human CSF | 600 μL | n.d |
|
| ||
| Mouse Plasma | ≥ 200 μL | 15-F2t-IsoP, 15-epi-15-F2t-IsoP, 5-F2t-IsoP, 5-epi-5-F2t-IsoP |
| Mouse Urine | ≥ 200 μL | Ent 15 (RS) 2,3 dinor 5,6 dihydro 15-F2t-isoP, 15-F2t-IsoP, 15-epi-15-F2t-IsoP, 5-F2t-IsoP, 5-epi-5-F2t-IsoP |
| Mouse Liver | 200 mg | 15-F2t-IsoP, 15-epi-15-F2t-IsoP, 5-F2t-IsoP, 5-epi-5-F2t-IsoP, 10-F4t-NeuroP, 10-epi-10-F4t-NeuroP, 4(RS)-F4t-NeuroP |
| Mouse Brain | 200 mg | 15-F2t-IsoP, 15-epi-15-F2t-IsoP, 5-F2t-IsoP, 5-epi-5-F2t-IsoP, 10-F4t-NeuroP, 10-epi-10-F4t-NeuroP, 4(RS)-F4t-NeuroP, Ent-7(RS)-F2t-Dihomo IsoP |
| Mouse Muscle | 200 mg | 5-F2t-IsoP, 5-epi-5-F2t-IsoP |
n.d. not detected; IsoP: isoprostane; NeuroP: neuroprostane
Figure 5.
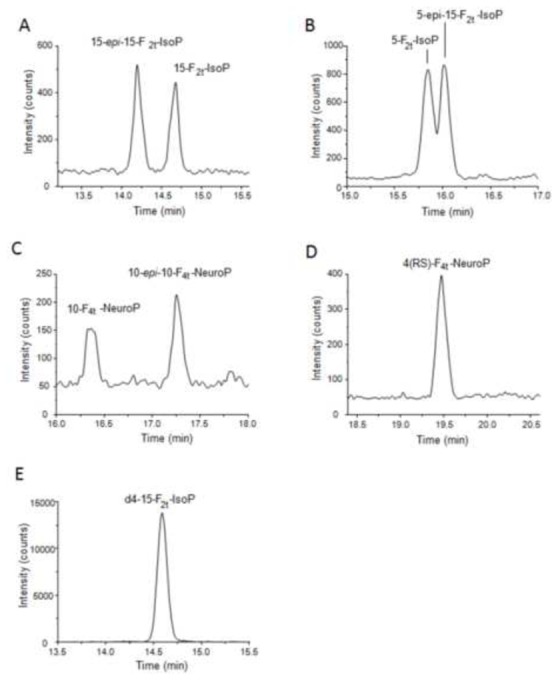
Chromatogram of selected reaction monitoring (SRM) of metabolites detected in human plasma : 15-F2t-IsoP, 15-epi-15-F2t-IsoP (A) ; 5-F2t-IsoP, 5-epi-5-F2t-IsoP (B) ; 10-F4t-NeuroP, 10-epi-10-F4t-NeuroP (C) ; 4(RS)-F4t-NeuroP (D) ; internal standard d4-15-F2t-IsoP (E).
The method developed was also used to perform the isoprostanoids profiles in the liver and brain of LDLR−/− mice and to investigate if DHA supplementation could affect the profiles. The overall levels i.e. total summation of all the related isomers measured for F2-IsoPs (19 pg/mg vs 14 pg/mg) and for F4-NeuroPs (19 pg/mg vs 17 pg/mg) in the liver and brain of control mice (white bars, Figures 6A and 6C) were slightly higher in the liver than the brain (+36% for F2-IsoP and +11% for F4-NeuroP). Furthermore, the total levels of F4-NeuroPs in the control mice liver and brain were similar to the levels of F2-IsoPs even though the concentration of DHA was 6 times and 34 times higher than AA in the liver and brain respectively (data not shown). Our observation suggest that the presence of high DHA concentration may contribute in protecting the liver and brain from ROS attack despite it being more prone to peroxidation due to extra double bonds in the structure compared to AA [8]. When looking at the different isomers of the isoprostanoids (Figure 6B and 6D), it is interesting to note that the distribution of the different F2-IsoP isomers is slightly different in the liver and brain of the control mice with 15-epi-F2t-IsoP and 15-F2t-IsoP being more abundant in the brain; for the liver, the abundance of the different F2-isomers was as follows: 15-F2t<5-Epi-F2t>15-Epi-F2t>5-F2t.
Figure 6.
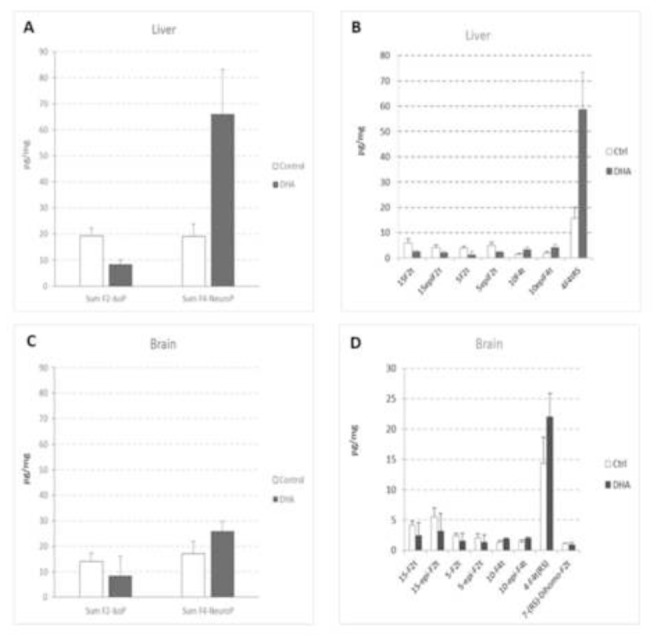
Isoprostanoids levels in the liver and brain of LDLR−/− mice given either oleic acid rich sunflower oil (Ctrl, n=3) or a mixture of oleic acid rich sunflower oil and DHA rich tuna oil providing 2% of energy as DHA (DHA, n=3). The sum of F2-IsoPs and sum of F4-NeuroPs represents the ‘total’ sum of the isomers measured for the respective group.
Among the F4-NeuroPs, the most abundant isomer was 4(RS)-4-F4t-NeuroP regardless of the tissue type. This finding is consistent with previous analysis performed in rat brain and heart tissues[34]. When the mice were supplemented with DHA (Figure 6, dark bars), the total level of F2-IsoPs decreased in the brain (−40% for the sum of F2-IsoP) and the liver (−57% for the sum of F2-IsoP) whereas the total level of F4-NeuroPs increased by 51% in the brain and 247% in the liver. The concomitant decrease of F2-IsoPs and increase of F4-NeuroPs could be attributed at least in part by the replacement of AA by DHA in the membrane phospholipids. It should nevertheless be noted that the modulation of isoprostanoids profiles is much more pronounced in the liver than in the brain emphasizing the high “plasticity” of liver towards DHA supplementation. Consistently, correlations between levels of AA and F2-IsoP as well as DHA and F4-NeuroP were strong in the liver (R2=0.71 and 0.96 respectively, Figure 7A and B) whereas they were much weaker in the brain (R2=0.02 and 0.16 respectively, data not shown).
Figure 7.
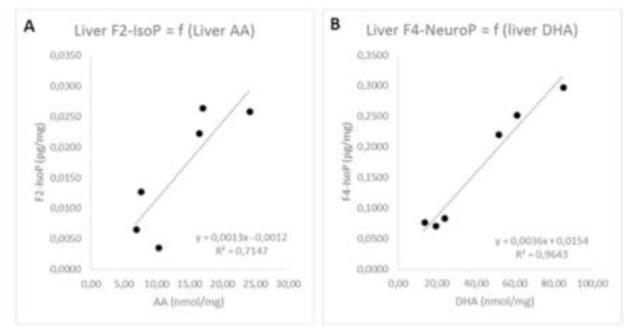
Correlations between the levels of AA or DHA and the corresponding isoprostanoids (i.e. sum of F2-IsoPs and sum of F4-NeuroPs) in the liver (A and B).
It should be noted in this report that we profiled the isoprostanoids in the liver and brain of an atherosclerotic mice and not normal mice. It is also anticipated that the profile of a normal mice may be different from our observation. Regardless, the objective of this study is to understand the differential changes between tissues. Our observation particularly displayed the importance of performing an integrated analysis of isoprostanoids levels since biological interpretation regarding tissue distribution and dietary modulation of lipid peroxidation is complex, and may lead to incorrect interpretation of experimental findings[10]. Moreover, the distribution of different types of isomers depends on the tissue type, which indicates the importance of tissue selection for studies evaluating bioactivity and organ crosstalk.
In summary, we have described a LC-MS/MS methodology allowing simultaneous quantification of several isoprostanes derived from n-3 and n-6 PUFA which are potential biomarkers in biological systems. Using the LC-MS/MS, we first characterized the analytical and quantification parameters of the 20 studied IsoPs and the 4 internal standards including LOD and LOQ concentration range. We optimized the sample preparation and the extraction process of these isoprostanoids which include Folch extraction, basic hydrolysis and SPE purification to obtain low matrix effect and good yield of extraction. The method was validated on human plasma (repeatability and accuracy). The optimized chromatographic separation permits to separate nearly all the isomers in 30 minutes with a good sensitivity. We then applied this method to human samples (plasma and CSF) and mice samples (plasma, urine, liver, brain, muscle) to optimize the quantity required to be able to profile the isoprostanoids. Finally, the method was tested on brain and liver samples of LDLR−/− mice with and without DHA supplementation to observe the change on the isoprostanoids profiles. We found a variation in the distribution of different F2-IsoP isomers between tissues but not for F4-NeuroP. DHA supplementation concomitantly increased F4-NeuroP levels. OS is a key feature in a number of human diseases, since ROS are likely to be involved in all disease stages. Many of these diseases are associated to PUFA therefore it is important to identify and evaluate the IsoP compounds simultaneously, not only as reliable biomarkers but also for its functional roles in the PUFA metabolism; we believe that the information we obtained from such profiling will allow us to understand the interaction of the compounds in diet and disease studies.
4. Conclusion
In this report, we developed a quick and robust method to determine multiple numbers of non-enzymatic oxidized lipid products of PUFAs, namely isoprostanoids in various biological sample in particular human plasma by LC-MS/MS. Unlike other reports, we were able to measure and incorporate new isoprostanoids from AdA and ALA as well as some isomers of AA, DHA and EPA into our method. However, it should be noted that not all 20 of the products determined were found in all biological samples therefore care must be taken when selecting them in metabolism studies.
Highlights.
Isoprostanoids are a group of non-enzymatic oxygenated metabolites of polyunsaturated fatty acids which are key intermediates in a lot of physiological mechanism. An quantitative LC-MS/MS profiling of these biomarkers was developed, validated and applied it on various biological sample. This method will be highly useful to follow biological process dealing with ROS.
Supplementary Material
Supporting material Table 1. Plasma matrix effect and extraction efficiency of healthy human plasma.
Supporting material Table 2 Repeatability and accuracy of the method for isoprostanes evaluation in human plasma.
Acknowledgments
Grant support
This work was supported by the Fondation pour la Recherche Médicale (grant ING20121226373), the Agence Nationale de la Recherche, the Institut National de la Santé et de la Recherche Medical and the French National Infrastructure MetaboHUB ANR-11-INBS-010.
Abbreviations
- AA
Arachidonic acid
- AdA
Adrenic acid
- ALA
α-Linolenic acid
- BHT
Butylated hydroxytoluene
- CSF
Cerebrospinal fluid
- DHA
Docosahexaenoic acid
- DPDS
2,2′-dipyridyl disulfide
- AGTA
Ethylene glycol tetra acetic acid
- EPA
Eicosapentaenoic acid
- ESI
Electrospray ionization
- HP
2-hydrazinopyridine
- HPLC
High-pressure liquid chromatography
- IsoPs
Isoprostanes
- LOD
limit of detection
- LOQ
limit of quantification
- m/z
Mass-to-charge ratio
- MRM
Multiple reaction monitoring
- MS
Mass spectrometry
- NeuroPs
Neuroprostanes
- OS
Oxidative Stress
- PhytoPs
Phytoprostanes
- PA
2-picolylamine
- PUFAs
Polyunsaturated fatty acids
- ROS
Reactive oxygen species
- SPE
Solid-phase extraction
- SRM
Selected-reaction monitoring
- S/N
signal to noise ratio
- TPP
Triphenylphosphine
References
- 1.Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401(1):1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- 2.Frijhoff J, et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid Redox Signal. 2015 doi: 10.1089/ars.2015.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jahn U, Galano JM, Durand T. Beyond prostaglandins--chemistry and biology of cyclic oxygenated metabolites formed by free-radical pathways from polyunsaturated fatty acids. Angew Chem Int Ed Engl. 2008;47(32):5894–955. doi: 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]
- 4.Milne GL, et al. Isoprostane generation and function. Chem Rev. 2011;111(10):5973–96. doi: 10.1021/cr200160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milne R, Brownstein S. Advanced glycation end products and diabetic retinopathy. Amino Acids. 2013;44(6):1397–407. doi: 10.1007/s00726-011-1071-3. [DOI] [PubMed] [Google Scholar]
- 6.Milne GL, Dai Q, Roberts LJ., 2nd The isoprostanes--25 years later. Biochim Biophys Acta. 2015;1851(4):433–45. doi: 10.1016/j.bbalip.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minghetti L, et al. Nonenzymatic oxygenated metabolites of alpha-linolenic acid B1- and L1-phytoprostanes protect immature neurons from oxidant injury and promote differentiation of oligodendrocyte progenitors through PPAR-gamma activation. Free Radic Biol Med. 2014;73:41–50. doi: 10.1016/j.freeradbiomed.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 8.Galano JM, et al. Non-enzymatic cyclic oxygenated metabolites of adrenic, docosahexaenoic, eicosapentaenoic and alpha-linolenic acids; bioactivities and potential use as biomarkers. Biochim Biophys Acta. 2015;1851(4):446–55. doi: 10.1016/j.bbalip.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Gladine C, et al. Neuroprostanes, produced by free-radical mediated peroxidation of DHA, inhibit the inflammatory response of human macrophages. Free Radic Biol Med. 2014;75(Suppl 1):S15. doi: 10.1016/j.freeradbiomed.2014.10.590. [DOI] [PubMed] [Google Scholar]
- 10.Galano JM, et al. Special Issue on "Analytical Methods for Oxidized Biomolecules and Antioxidants" The use of isoprostanoids as biomarkers of oxidative damage, and their role in human dietary intervention studies. Free Radic Res. 2015;49(5):583–98. doi: 10.3109/10715762.2015.1007969. [DOI] [PubMed] [Google Scholar]
- 11.Klawitter J, et al. Quantification of 15-F2t-isoprostane in human plasma and urine: results from enzyme-linked immunoassay and liquid chromatography/tandem mass spectrometry cannot be compared. Rapid Commun Mass Spectrom. 2011;25(4):463–8. doi: 10.1002/rcm.4871. [DOI] [PubMed] [Google Scholar]
- 12.Vigor C, et al. Non-enzymatic lipid oxidation products in biological systems: assessment of the metabolites from polyunsaturated fatty acids. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;964:65–78. doi: 10.1016/j.jchromb.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 13.Durand T, et al. Syntheses and preliminary pharmacological evaluation of the two epimers of the 5-F-2t-isoprostane. Bioorganic & Medicinal Chemistry Letters. 2001;11(18):2495–2498. doi: 10.1016/s0960-894x(01)00473-5. [DOI] [PubMed] [Google Scholar]
- 14.Durand T, et al. Total syntheses of four metabolites of 15-F-2t-isoprostane. European Journal of Organic Chemistry. 2001;(4):809–819. [Google Scholar]
- 15.Durand T, et al. Total synthesis of (15R)- and (15S)-F-2t-isoprostanes by a biomimetic process using the cyclization of acyclic dihydroxylated octa-5,7-dienyl radicals. Journal of Organic Chemistry. 2002;67(11):3615–3624. doi: 10.1021/jo0109624. [DOI] [PubMed] [Google Scholar]
- 16.El Fangour S, et al. Total synthesis of the eight diastereomers of the syn-anti-syn phytoprostanes F1 types I and II. J Org Chem. 2004;69(7):2498–503. doi: 10.1021/jo035638i. [DOI] [PubMed] [Google Scholar]
- 17.Guy A, et al. Total synthesis of ent-15(RS)-2,3-dinor-5,6-dihydro-8-epi-PGF2 alpha. Tetrahedron Letters. 1998;39(34):6181–6184. [Google Scholar]
- 18.Guy A, et al. Oxygenated Metabolites of n-3 Polyunsaturated Fatty Acids as Potential Oxidative Stress Biomarkers: Total Synthesis of 8-F3t-IsoP, 10-F4t-NeuroP and [ D4]-10-F4t-NeuroP. Chemistry-a European Journal. 2014;20(21):6374–6380. doi: 10.1002/chem.201400380. [DOI] [PubMed] [Google Scholar]
- 19.Oger C, et al. Stereocontrolled Access to Isoprostanes via a Bicyclo[3.3.0]octene Framework. Organic Letters. 2008;10(21):5087–5090. doi: 10.1021/ol802104z. [DOI] [PubMed] [Google Scholar]
- 20.Oger C, et al. The Handy Use of Brown's P2-Ni Catalyst for a Skipped Diyne Deuteration: Application to the Synthesis of a [D-4]-Labeled F-4t-Neuroprostane. Chemistry-a European Journal. 2010;16(47):13976–13980. doi: 10.1002/chem.201002304. [DOI] [PubMed] [Google Scholar]
- 21.Oger C, et al. Total Synthesis of Isoprostanes Derived from Adrenic Acid and EPA. European Journal of Organic Chemistry. 2012;(13):2621–2634. [Google Scholar]
- 22.Lee CY, Jenner AM, Halliwell B. Rapid preparation of human urine and plasma samples for analysis of F2-isoprostanes by gas chromatography-mass spectrometry. Biochem Biophys Res Commun. 2004;320(3):696–702. doi: 10.1016/j.bbrc.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 23.Gladine C, et al. Lipid profiling following intake of the omega 3 fatty acid DHA identifies the peroxidized metabolites F4-neuroprostanes as the best predictors of atherosclerosis prevention. PLoS One. 2014;9(2):e89393. doi: 10.1371/journal.pone.0089393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ducheix S, et al. Essential fatty acids deficiency promotes lipogenic gene expression and hepatic steatosis through the liver X receptor. J Hepatol. 2013;58(5):984–92. doi: 10.1016/j.jhep.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Berdeaux O, et al. F-2-isoprostanes: Review of analytical methods. Current Pharmaceutical Analysis. 2006;2(1):69–78. [Google Scholar]
- 26.Cartwright AJ, et al. Derivatisation of carboxylic acid groups in pharmaceuticals for enhanced detection using liquid chromatography with electrospray ionisation tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19(8):1058–62. doi: 10.1002/rcm.1883. [DOI] [PubMed] [Google Scholar]
- 27.Pettinella C, et al. Targeted quantitative analysis of fatty acids in atherosclerotic plaques by high sensitivity liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;850(1–2):168–76. doi: 10.1016/j.jchromb.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 28.Higashi T, et al. Simple and practical derivatization procedure for enhanced detection of carboxylic acids in liquid chromatography-electrospray ionization-tandem mass spectrometry. J Pharm Biomed Anal. 2010;52(5):809–18. doi: 10.1016/j.jpba.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Petrosino T, Serafini M. Matrix effect in F(2)-isoprostanes quantification by HPLC-MS/MS: a validated method for analysis of iPF(2)alpha-III and iPF(2)alpha-VI in human urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;965:100–6. doi: 10.1016/j.jchromb.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Prasain JK, et al. Simultaneous quantification of F2-isoprostanes and prostaglandins in human urine by liquid chromatography tandem-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;913–914:161–8. doi: 10.1016/j.jchromb.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrow JD, et al. Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci U S A. 1992;89(22):10721–5. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milatovic D, Montine TJ, Aschner M. Measurement of isoprostanes as markers of oxidative stress. Methods Mol Biol. 2011;758:195–204. doi: 10.1007/978-1-61779-170-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Faouder P, et al. LC-MS/MS method for rapid and concomitant quantification of pro-inflammatory and pro-resolving polyunsaturated fatty acid metabolites. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;932:123–33. doi: 10.1016/j.jchromb.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 34.de la Torre A, et al. Total syntheses and in vivo quantitation of novel neurofuran and dihomo-isofuran derived from docosahexaenoic acid and adrenic acid. Chemistry. 2015;21(6):2442–6. doi: 10.1002/chem.201405497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting material Table 1. Plasma matrix effect and extraction efficiency of healthy human plasma.
Supporting material Table 2 Repeatability and accuracy of the method for isoprostanes evaluation in human plasma.