

Abstract
The development of chemoresistance and inability in elimination of cancer stem cells are among the key limitations of cancer chemotherapy. Novel molecular therapeutic strategies able to overcome such limitations are urgently needed for future effective management of cancer. In this report, we show that EpCAM-aptamer-guided survivin RNAi effectively downregulated survivin both in colorectal cancer cells in vitro and in a mouse xenograft model for colorectal cancer. When combined with the conventional chemotherapeutic agents, the aptamer-guided survivin RNAi was able to enhance the sensitivity towards 5-FU or oxaliplatin in colorectal cancer stem cells, increase apoptosis, inhibit tumour growth and improve the overall survival of mice bearing xenograft colorectal cancer. Our results indicate that survivin is one of the key players responsible for the innate chemoresistance of colorectal cancer stem cells. Thus, aptamer-mediated targeting of survivin in cancer stem cells in combination with chemotherapeutic drugs constitutes a new avenue to improve treatment outcome in oncologic clinics.
Introduction
Cancer stem cells are regarded as the ‘roots of cancer’ and therefore are an attractive therapeutic target to inhibit tumour growth, expansion and metastasis1, 2. Current approaches in treating cancer patients include surgical resection with chemotherapy or radiotherapy. Although they are effective in eliminating the majority of the cancer cells, their inability to eliminate the cancer stem cell population remains a limitation for current regimen for cancer treatment3–7. Indeed, cancer stem cells have been shown to be resistant to conventional anticancer drugs through a variety of mechanisms including overexpression of ABC transporters, active DNA-repair capacity, overexpression of antiapoptotic proteins, and elevated autophagy8–10.
5-fluorouracil (5-FU) is a chemotherapeutic drug widely used for the treatment of colorectal cancer and is often included in the first line treatment. However, cancer stem cells in colorectal cancer have been shown to be resistant to 5-FU as well as other chemotherapeutic drugs such as oxaliplatin and irinotecan11. Furthermore, the use of these drugs could lead to the enrichment of cancer stem cells with high tumourigenic capacities in a number of solid cancers11–13. Thus, despite the efficacy of these drugs against bulk cancer cells, they remain mostly ineffective against cancer stem cells14, 15. We hypothesized that aptamer-mediated survivin RNAi enables 5-fluorouracil to eliminate colorectal cancer stem cells and improves the survival of the mice bearing xenograft tumour.
Aptamers are single stranded DNA or RNA that fold into defined 3-D structure and bind to their targets specifically. As ‘chemical antibodies’, aptamers offer significant advantages over antibodies in terms of smaller size, lower immunogenicity, increased stability, ease of synthesis and modification16–19. As a targeting module, aptamers allow for improved delivery of drugs to tumour cells while minimizing the side effects often associated with the drugs. Our laboratory has developed the first RNA aptamers that target the cancer stem cell markers EpCAM. Upon binding to target cells, our EpCAM aptamers undergo receptor-mediated endocytosis which allows for controlled release of drugs intracellularly20, 21. Here we describe the use of the EpCAM aptamer in delivering siRNA to target survivin expression in order to eliminate colorectal cancer stem cells.
Survivin, a member of inhibitors of apoptosis proteins family, is an attractive target for anticancer therapies for its role in inhibiting apoptosis and its close association with stem cells22, 23. In addition to its critical role in inhibiting cell death, survivin is expressed at low levels or absent in terminally differentiated healthy cells, but its expression is significantly elevated in human tumours and its overexpression is further enhanced when exposed to traditional anticancer treatments24–26. However, the role of survivin in the development of chemoresistence in solid tumours remains largely unexplored. Here we demonstrate that by silencing survivin expression in colorectal cancer stem cells using an aptamer-siRNA chimera, the innate chemoresistance to traditional chemotherapeutic agents in colorectal cancer stem cells was reversed, transforming an old anticancer drug into a cancer stem cell killer.
Results
Effective downregulation of survivin via EpCAM Aptamer-guided RNAi in colorectal cancer cells
In this study, we targeted survivin expression in colorectal cancer stem cells using a chimeric structure composed of an EpCAM-specific RNA aptamer as the binding moiety and a survivin siRNA sequence (Supp. Fig. 1). The efficacy of EpCAM aptamer-survivin siRNA chimera in the downregulation of survivin was initially tested on EpCAM-positive colorectal cancer cell line HT-29 with the EpCAM-negative cell line HEK-293T and a survivin RNAi-deficient variant chimera as negative controls (Supp. Fig. 1)27. Both cell lines were treated with 20 nM of the chimera or the negative control chimera for 24 hours or 48 hours in the absence of any transfection agent and subjected to qRT-PCR and Western analysis, respectively. As shown in Fig. 1a,d,e, the treatment of chimera resulted to a ~70% knockdown of both survivin mRNA and protein levels in EpCAM-positive HT29 cells but not in the EpCAM-negative control HEK293T cells. Having demonstrated the efficacy and selectivity of EpCAM-directed survivin RNAi in vitro, we proceeded to evaluate the chimera-mediated survivin RNAi in vivo. To this end, a 20-kDa polyethylene glycol (PEG) was attached to the chimera to increase circulatory half-life of the chimera in mice (Supp. Fig. 1)27.
Figure 1.
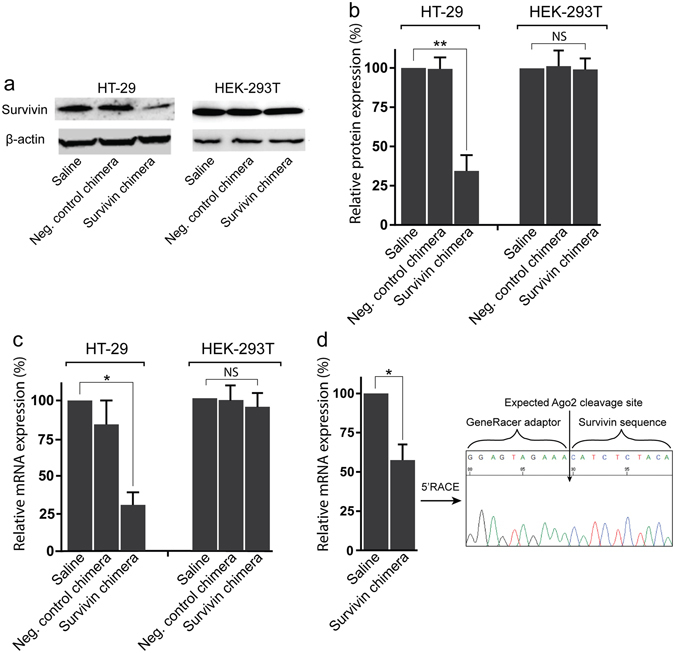
EpCAM aptamer-guided RNAi effectively silenced survivin. (a) Specificity and efficacy of EpCAM-aptamer guided RNAi in knocking down survivin mRNA. Chimera or negative control chimera were incubated with HT-29 or HEK-2913T cells for 24 hours and the total RNA was extracted for qRT-PCR analysis of survivin mRNA levels. GAPDH was used as an internal control. (b,c) HT-29 Tumour-bearing mice were treated with 2 nmol/mouse of PEG-labelled chimera for 48 hours. The tumours were collected for RNA extraction followed by qRT-PCR analysis of survivin mRNA expression (b) and 5′RACE assay (c). (d) Effective downregulation of survivin protein via EpCAM aptamer-guided RNAi. Chimera or negative control chimera were incubated with HT-29 or HEK-2913T cells for 48 hours and the survivin protein levels were analyzed using Western blot analysis. β-actin was used as a loading control. (e) The bar graph shows the survivin protein levels in various treatment groups. Data shown are means ± SEM, n = 3. *p < 0.05, **p < 0.005. NS, no statistically significant difference.
Next, we studied efficacy and mode of action of aptamer-guided survivin RNAi in vivo. The HT29 xenograft tumour-bearing NOD/SCID mice were treated with a single dose of 2 nmol/mouse of the PEG-chimera i.v. Forty-eight hours after the treatment, the tumours were removed and the survivin mRNA content in the treated HT29 xenograft tumour was evaluated. As shown in Fig. 1b, a single dose of 2 nmol/mouse of the chimera resulted in the reduction of survivin mRNA in the xenograft tumour by ~40%. Furthermore, a 5′RACE assay on the RNA extracted from the treated xenograft tumour suggests specific survivin mRNA cleavage at the expected siRNA cut site through the Argonaute2 (Ago2) mechanism (Fig. 1c). Taken together, these data indicate that aptamer-siRNA chimera is able to effectively downregulate survivin in EpCAM-positive colorectal cancer cells both in vitro and in vivo in the absence of transfection reagent.
Survivin silencing combined with 5-FU target colorectal cancer stem cells in vitro
Next, we explored if the downregulation of survivin would lead to the increased sensitivity of colorectal cancer stem cells to standard chemotherapy agents, such as 5-FU, in vitro and in vivo. For the evaluation of the elimination of colorectal cancer stem cells rather than the bulk cancer cells, we used self-renewal and tumour-formation as the key experimental endpoints as such properties reflect the defining functional characteristics of the cancer stem cell population28–30.
For an in vitro assessment of the self-renewal capacity of cancer stem cells, HT-29 cells were treated with the EpCAM aptamer-survivin siRNA chimera or negative control chimera, followed by further incubation in the presence or absence of 2 µM 5-FU for 5 days. A tumoursphere formation assay was used to enumerate the number of tumourspheres formed. As shown in Fig. 2 and Suppl. Fig. 2, a statistically significant 3-fold decrease in self-renewal was observed when HT-29 cells were treated with combined survivin chimera and 5-FU. In contrast, the treatment with either chimera or 5-FU alone, negative control chimera or saline did not lead to significant decrease in self-renewal capacity of the HT-29 cells in vitro. These data indicate that only the combinatorial treatment of survivin knockdown and 5-FU but not the mono-treatment regimen or the negative controls, effectively impaired the self-renewal capacity of HT-29 in vitro. To further verify if the combinatorial treatment leads to the impairment of tumourigenicity of colorectal cancer stem cells in vivo, we performed tumour xenograft experiments under the limiting dilution conditions. Treated HT-29 cells were implanted subcutaneously into the immunodeficient NOD/SCID mice at limiting dilutions with an inoculation cell dose between 1 × 103 to 5 × 104 cells per mouse. As shown in Table S1, there was no significant decrease in tumourigenicity potential in HT-29 cells treated with 5-FU alone or with the negative control chimera. Similarly, targeting survivin expression by mono-treatment with the chimera had little impact on the tumour-forming potential compared to that of saline-treated control (Table S1). In contrast, a significant decrease in HT-29 tumour formation for HT-29 cells treated with the combinatorial regimen was evident, indicating that although survivin knockdown as a mono-treatment is not sufficient to eliminate HT-29 cancer stem cells, the combination with survivin RNAi with classical chemotherapeutic drugs, such as 5-FU, is able to eliminate colorectal cancer stem cells. The downregulation of survivin in empowering traditional chemotherapeutic agents to eliminate colorectal cancer stem cell is not limited to 5-FU as the combined treatment with aptamer-survivin siRNA chimera and oxaliplatin had a similar efficacy in the impairment of self-reviewal capacity of HT-29 colorectal cancer stem cells (Supp. Fig. 3).
Figure 2.
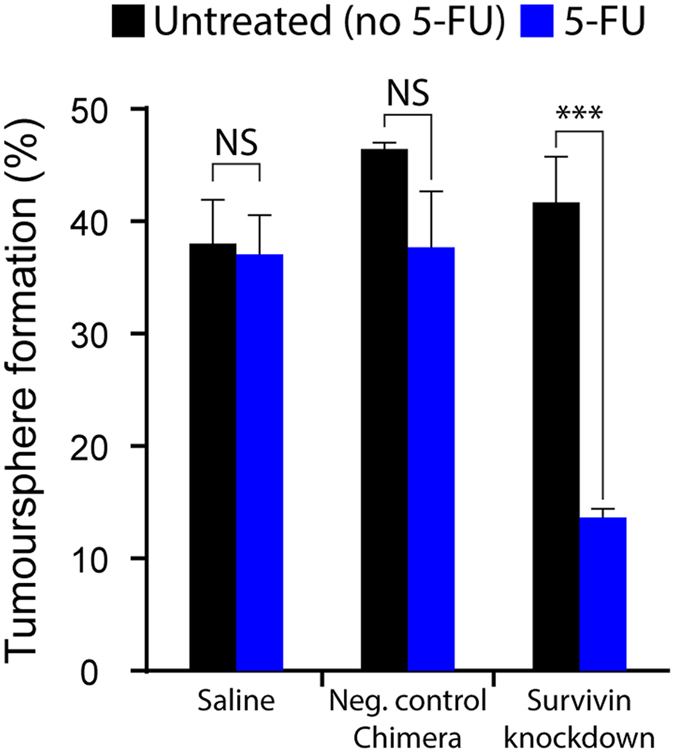
EpCAM aptamer mediated survivin knockdown transforms 5-FU into a colorectal cancer stem cell killer. Colorectal cancer HT-29 cells were first treated with EpCAM aptamer-survivin siRNA chimera for 24 hours followed by treatment with 2 μM 5-FU for 5 days. Tumoursphere formation assay was used to examine the self-renewal capacities of treated cells. The bar graph shows the estimated stem cell frequencies. Data shown are means ± SEM, n = 3. ***p < 0.001. NS, no statistically significant difference.
Combinatorial survivin knockdown and 5-FU treatment in vivo eliminates colorectal cancer stem cells
To evaluate if the combinatorial treatment of chimera and 5-FU leads to the elimination of HT-29 cancer stem cells in vivo, we treated HT-29 xenograft tumour-bearing NOD/SCID mice using PEG-aptamer-siRNA chimera with or without 5-FU. Mice bearing 60 mm3 HT-29 tumours received i.v. injections of aptamer-siRNA chimera (2 nmol/mouse) on days 1, 3, and 5 with or without 30 mg/kg of 5-FU injections on days 3, 5, and 7. Forty-eight hours after the last treatment, tumours were removed (Supp. Fig. 4) and single-cell suspensions were subjected to tumoursphere formation assay to analyze their self-renewal capacities. As shown in Table 1 and Supp. Fig. 5, xenograft tumours from mice that received chimera in the absence of 5-FU showed no significant change in self-renewal when compared with saline-treated mice. Similarly, 5-FU alone or control chimera had no significant impact on self-renewal either. Interestingly, the HT29 xenograft tumours from the mice that received aptamer-survivin siRNA chimera in combination with 5-FU showed significant reduction in self-renewal (Table 1).
Table 1.
Elimination of cancer stem cells via combined in vivo survivin knockdown and 5-FU treatment in vivo.
| Treatment group | Number of cells/well | Estimated stem cell frequency | 95% confidence interval | ||||
|---|---|---|---|---|---|---|---|
| 50 | 20 | 10 | 5 | 1 | |||
| Saline | 4/10 | 3/10 | 1/10 | 1/10 | 0/10 | 1.3% | 0.65–2.5% |
| Saline + 5-FU | 5/10 | 1/10 | 1/10 | 0/10 | 0/10 | 0.99% | 0.47–2.1% |
| Neg. chimera control + 5-FU | 3/9 | 1/7 | 1/10 | 0/10 | 0/10 | 0.76% | 0.32–1.8% |
| Chimera | 3/7 | 1/10 | 1/10 | 0/10 | 0/10 | 0.81% | 0.30–1.9% |
| Chimera + 5-FU | 1/10 | 1/10 | 0/10 | 0/10 | 0/10 | 0.24% | 0.06–0.97% |
As 5-FU is known to kill cancer cells via the induction of apoptosis, we proceeded to investigate the mechanism of action in colorectal cancer stem cells upon combinatorial treatment of chimera and 5-FU in vivo. For this purpose, dissociated tumour cells and tumour sections from mice were fixed and underwent TUNEL staining to enumerate apoptotic cells (Fig. 3 and Supp. Fig. 6). Tumours from mice that received mono-treatments of chimera or 5-FU were found to have 7.23% ± 2.038 and 9.132% ± 0.5787 apoptotic cells, respectively (Fig. 3a,b). Similarly, tumours from mice that received negative control chimera with 5-FU treatment showed 3.975% ± 3.213 of the population to have undergone apoptosis. In contrast, tumours from mice that received combinatorial treatment of chimera and 5-FU displayed enhanced apoptosis as 20.88% ± 3.32 of the tumour cell population had undergone apoptosis. Thus, EpCAM aptamer-guided RNAi of survivin enhanced the 5-FU-mediated cell death in colorectal cancer cells via enhanced apoptosis. Furthermore, tumour sections stained for Ki-67 demonstrated 4-fold decrease of tumour cell proliferation when treated with chimera and 5-FU combinatorial treatment (Supp. Fig. 7).
Figure 3.
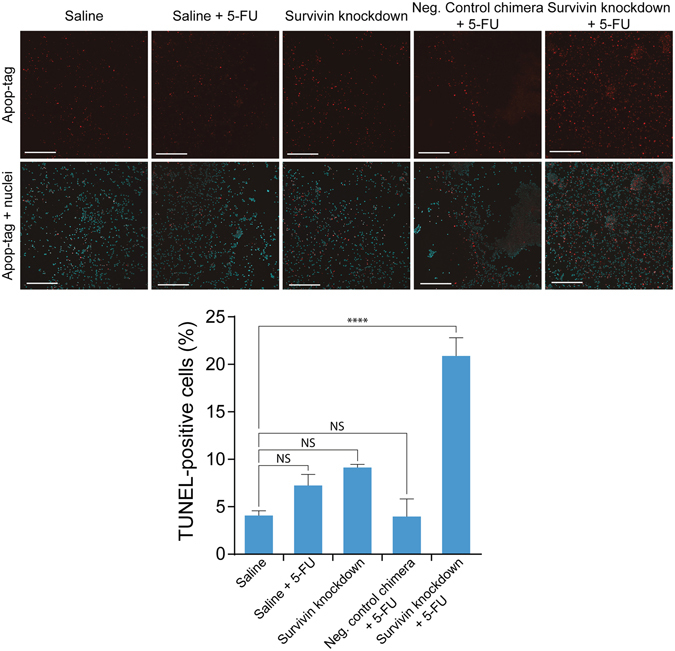
Survivin knockdown in vivo enhances 5-FU-induced apoptosis in HT-29 tumour cells. (a) Representative images of TUNEL apoptosis assay on dissociated HT-29 xenograft tumours after in vivo treatment with chimera and 5-FU. NOD/SCID mice bearing HT-29 tumours (60 mm3) were treated intravenously with 3 injections of 2 nmol/mouse of chimera with or without 3 additional treatment of 30 mg/kg of 5-FU. Two days after the final treatment, tumours were dissociated by collagenase digestion and subjected to TUNEL apoptosis assay. (b) Percentage of apoptotic cells in treated tumours. Data shown are means ± SEM, n = 3. ****p < 0.001. NS, no statistically significant difference.
EpCAM-aptamer guided in vivo survivin RNAi enhanced treatment efficacy of 5-FU
Next, we examined if the suppression of self-renewal and increased apoptosis conferred by the combinatorial treatment of aptamer-guided survivin siRNA and 5-FU in vivo would result in an improved treatment efficacy in mice bearing HT-29 xenograft tumour. NOD/SCID mice bearing HT-29 xenograft colorectal tumours were treated with 2 nmol/mouse of PEG-chimera or negative control chimera on days 1, 3, and 5, with or without 30 mg/kg 5-FU on days-3, 5, 7, 9, and 11. The changes in tumour volume were monitored daily until the animals reached their experimental endpoints. The mice that underwent mono-treatment with the chimera showed no signs of reduced rate of tumour growth (Fig. 4a) nor improvement in survival (Fig. 4b) when compared to the saline control group. The tumour-bearing mice that received 5-FU mono-treatment with or without the negative control chimera showed slower tumour growth compared to the saline group (Fig. 4a). In contrast, the combinatorial treatment of chimera and 5-FU led to the significantly slower rate of tumour growth compared to the mice that received 5-FU treatment with or without negative control chimera (Fig. 4a), and improved overall survival within the first month of treatment (Fig. 4b).
Figure 4.
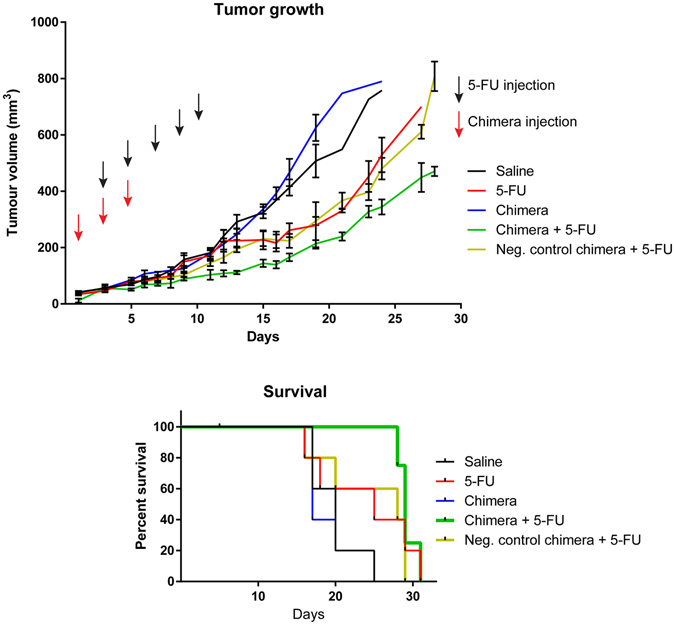
Combined chimera and 5-FU treatments improves therapeutic outcome in HT-29 tumour-bearing mice. (a) The graph represents the tumour volume in mice in response to various treatments as indicated. (b) Survival rate of mice observed over the course of various treatments. Data shown are mean ± SEM, n = 4–5.
Discussion
Survivin is known to be overexpressed after chemotherapy and radiotherapy in many solid cancers including colorectal cancer, and such overexpression is largely associated with drug resistance, tumour aggressiveness and poor prognosis31. Our experience with chemotherapeutic drugs in the past 60 years indicates that despite the limited success in eliminating the bulk cancer cells, these anti-cancer agents, such as 5-FU, remain largely ineffective against cancer stem cells14, 15. Here we describe the use of an oligonucleotide chimera made up of an EpCAM-specific aptamer and a survivin siRNA to downregulate survivin in both the bulk and colorectal cancer stem cells without the aid of transfection agent or other delivery vehicles such as viral vectors or nanodrug carriers. When combined with the co-treatment with conventional chemotherapy drugs, the aptamer-guided survivin RNAi resulted in enhanced sensitivity of the cancer stem cells towards 5-FU or oxaliplatin. The aptamer-siRNA chimera used here was previously established to specifically target EpCAM-expressing cancer cells, undergo receptor-mediated endocytosis upon binding to target cells, and engage cell’s endogenous RNAi machinery27. Notably, EpCAM is overexpressed on both bulk cancer cells and cancer stem cells32, 33. We purposely engineered an EpCAM aptamer with a moderate dissociation constant so that it will preferentially be enriched at the site of tumour as cancer cells express up to 1000-fold higher EpCAM than normal epithelial cells34, 35. Therefore, the EpCAM aptamer-survivin siRNA chimera used in this study can effectively target both the cancer stem cells and non-cancer stem cells in vivo. Such dual targeting capacity is critical for the success of clinical oncology since it has been established that cancer stem cells and non-cancer stem cell may interconvert, and survivin has been shown to be involved in the regulation of such interconversion36, 37. It is thus imperative not to limit the targeted therapies against cancer stem cells but also target the non-cancer stem cell population simultaneously. To our knowledge, this fully synthetic aptamer-siRNA chimera is the shortest of its kind ever developed, providing relative ease for large scale chemical synthesis and modification27.
Depending on the experimental systems and approaches used, inhibition of survivin function alone could lead to decreased tumour cell viability, increased apoptosis, or inhibition of tumour growth38, 39. It could also lead to no detectable changes in cell viability, stemness or tumour growth as found in our studies and by others27, 40. Indeed, clinical trials with survivin inhibition have revealed that the anti-cancer efficacy of the inhibition of survivin is more pronounced when combined with chemotherapy41–44. This is in agreement with our finding that survivin silencing with the chimera was only effective against cancer stem cells when combined with 5-FU, resulting in reduced self-renewal and tumour growth and enhanced apoptosis (Figs 2–4). Therefore, our data suggest that survivin is one of the key players responsible for the innate chemoresistance of colorectal cancer stem cells. Taken together, our results indicate that targeting survivin in cancer stem cells in combination with chemotherapeutic drugs constitutes a new avenue to improve treatment outcome in oncologic clinics.
Chemotherapy is one of the important treatment strategies used for managing patients with cancer. For colorectal cancer, 5-FU and oxaliplatin are the major chemotherapeutic agents used by oncologists for treating colorectal cancer. Such drugs are known to be largely ineffective in killing cancer stem cells. Furthermore, innate chemoresistance is responsible for the drug-resistance frequently encountered in colorectal cancer patients. Our data presented here suggest that downregulation of survivin results in the reversal of the innate chemoresistance to both 5-FU and oxaliplatin in colorectal cancer stem cells (Figs 2, 3 and 4, Table 1). By combining targeted siRNA delivery with the treatment of 5-FU, we have transformed a standard chemotherapeutic drug known to be ineffective against cancer stem cells into an effective colorectal cancer stem cell killer. In addition to in vitro and ex vivo data which showed promising results for the combinatorial treatment of chimera and 5-FU in enhancing apoptosis and eliminating cancer stem cells, in vivo treatment of tumour-bearing mice using PEG-labeled chimera with 5-FU via tail vein injection resulted in enhanced tumour apoptosis and reduced tumour self-renewal (Figs 3 and 4). As shown in Fig. 4, the most prominent effect of the combinatorial treatment of aptamer-guided survivin siRNA and 5-FU occurred within the first 25 days of treatment. The limited number of chimera injections may have been insufficient in maintaining survivin downregulation in the HT-29 tumours throughout the duration of the combinatorial treatment. Additionally, the continuation of 5-FU treatments without the chimera potentially elicited pro-survival responses such as autophagy, contributing to drug resistance and tumour growth45–47. Thus, our results suggest that maintaining survivin downregulation simultaneously with chemotherapy may be required in order to enhance the anti-tumour efficacy and improved outcome in oncologic clinics.
In conclusion, EpCAM aptamer-guided delivery of survivin siRNA is able to target both the bulk and cancer stem cells in a xenograft colorectal cancer model. The aptamer-guided knockdown of survivin reverses the innate chemoresistance of colorectal cancer stem cells. The aptamer-mediated siRNA delivery to cancer stem cells in vivo opens a new avenue to target specific cancer cell populations and to modulate genes that are critical for cancer cell survival and chemoresistance.
Material and Methods
Animals
Female 6-week old immunodeficient NOD/SCID mice were purchased from the Animal Resources Centre (Perth, Australia). All experiments were performed in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and Guidelines to Promote the Wellbeing of Animals Used for Scientific Purposes from Australian Government’s National Health and Medical Research Council. The Deakin University Animal Ethics Committee approved all experimental protocols.
Cell culture
HT-29 (human colorectal adenocarcinoma, ATCC® HTB38™) cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen, Australia) supplemented with 10% fetal bovine serum (FBS, Hyclone, Canada), 50 U/L penicillin and streptomycin (Invitrogen, Australia) and 1× Glutamax (Life Technologies). Cell cultures were kept in a humidified atmosphere with 5% CO2 at 37 °C. For aptamer-siRNA treatment, 20 nM of either chimeras were added to the cell culture for 48 and 24 hours for total protein and RNA extraction, respectively.
Western analysis
Western analysis was performed as described in detail in our previous publication27. The primary antibodies used in the western blot were mouse anti-human β-actin (1:5000, Sigma) and mouse anti-human survivin (1:400, Cat. No: SC-17779, Santa Cruz). The secondary antibodies were HRP-conjugated goat anti-mouse secondary antibody (1:10000, Thermo Fisher) or goat anti-rabbit secondary antibody (1:5000, Pierce). Quantification of ECL signals was conducted using a LAS-4000 Imaging System (GE Healthcare Life Sciences) with β-actin as a loading control.
RNA extraction and qrRT-PCR
Total RNA was extracted using Trizol (Invitrogen) following the manufacturer’s protocol, and cDNA was generated from the extracted RNA using High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Fast SYBR Green Master Mix (Invitrogen) and Stratagene Mx3000 P system (Agilent Technologies) were used to carry out qRT-PCR assays with GAPDH as an internal control. The primers used for qRT-PCR assays were survivin (forward primer: 5′-GAACTGGCCCTTCTTGGAG-3′, reverse primer: 5′-AAGTCTGGCTCGTTCTCAGT-3′) and GAPDH (forward primer: 5′-GAAATCCCATCACCATCTTCCAGG-3′, reverse primer: 5′-GAGCCCCAGCCTTCTCCATG-3′).
In vitro tumoursphere and ex vivo xenograft assays
The tumoursphere assay was performed in accordance with previously reported protocols48. Cells were collected by trypsinization and resuspended as single cells in cancer stem cell media containing serum-free DMEM/F12 (Invitrogen) supplemented with 100 units/mL B27 (Gibco), 10 µg/mL Insulin (Sigma), 20 ng/mL EGF (Sapphire Bioscience), and 20 ng/mL bFGF (Sapphire Bioscience). The cells were plated in a round-bottom 96-well ultra-low attachment plates (Corning) at densities of 1, 5, 20, and 50 cells/well. Sphere formation was recorded 5–7 days after incubation at 37 °C in a humidified atmosphere with 5% CO2. Cancer stem cell frequency was calculated using the limiting dilution software package (ELDA) on the website of Walter and Eliza Hall Institute of Medical Research (http://bioinf.wehi.edu.au/software/elda/index.html)49. Only spheres with a size larger than 50 µm in diameter were counted. For ex vivo xenograft assay, cells were injected subcutaneously into the left flanks of female NOD/SCID mice at three cell densities (5 × 104, 1 × 104, and 1 × 104) in serum-free DMEM with Matrigel at 1:1 ratio. Tumours were detected by palpation.
Mice treatment for in vivo tumoursphere and xenograft assays
Female NOD/SCID mice with HT-29 tumours (60 mm3) in the left flanks received 2 nmol of aptamer-siRNA chimera intravenously via the tail-vein on days 1, 3, and 5, and PBS on day 7, while mice with 5-FU treatment received PBS i.v. injection on day 1 and 30 mg/kg of 5-FU via on days 3, 5, and 7. The tumours were collected 48 hours after the final injection and dissociated into single cell suspensions to be used for in vitro tumoursphere assay (50, 20, 10, 5, 1 cells/well) and xenograft assay (1 × 105, 1 × 104, 1 × 103, 1 × 102 cells/mouse). The cancer stem cells frequency was measured using the limiting dilution software package on the website of Walter and Eliza Hall Institute of Medical Research (http://bioinf.wehi.edu.au/software/elda/index.html)49.
Tumour dissociation
Tumours collected from xenografts were washed thoroughly with Hank’s buffer containing 1% penicillin/streptomycin to remove excess blood and any extraneous material. The tumours were placed in a sterile petri dish and minced into smaller pieces (approximately 2–4 mm3) with a scalpel. The chopped tissues were collected and rinsed with Hank’s buffer, then suspended in dissociation medium containing DMEM, 20% FBS, 2% penicillin/streptomycin, 100 units/mL B27, 10 µg/mL Insulin, 20 ng/mL EGF, 20 ng/mL bFGF, and 50 U/mL collagenase II (Sigma). The dissociation medium was used on the chopped tumours at a ratio of 6 ml per gram of tumour and incubated at 37 °C overnight on a rotating orbit mixer incubator. Cells were collected after incubation and centrifuged at 1,000 g for 5 minutes. The pellets were suspended and washed with PBS twice by centrifugation at 500 g to remove any residual debris.
TUNEL assay
The dissociated tumour cells were washed with PBS and fixed on glass slides with 1% paraformaldehyde. Apoptotic cells were detected using TUNEL Apoptosis Detection Kit (Millipore) following the manufacture’s protocol. Fixed cells were counterstained with mounting medium (Vectashield, Vector Laboratories, Burlingame, CA) containing 1 µg/mL DAPI, and the cells were visualized with Fluoview FV10i laser scanning confocal microscope (Olympus, NSW, Australia).
In vivo treatment for tumour growth rate and survival
Female NOD/SCID mice with HT-29 tumours (60 mm3) in the left flanks received 2 nmol of aptamer-siRNA chimera on days 1, 3, and 5, and PBS on days 7, 9, and 11, while mice with 5-FU treatment received intravenous injection of PBS on day 1 and 30 mg/kg of 5-FU via IV injections on days 3, 5, 7, 9 and 11. The weights and tumour dimensions of mice were monitored daily. The endpoints set for the experiments were defined by rapid weight loss by 20% of the body weight, tumour diameter exceeding 13 mm, and any health deteriorations impacting on the welfare of the animal as set by the Deakin University Animal Ethics Committee. Tumour diameters were measured with a digital caliper and tumour volume was calculated with the formula: Volume = Length × Width2/2.
Electronic supplementary material
Author Contributions
W.D. conceived the study and designed the experiments. H.A., T.W., D.X., W.Y. performed experiments. H.A., T.W., D.X., R.B., collected and analyzed the data. H.A., W.D. wrote the manuscript, P.T., P.Z., Y.L., L.K., K.L., S.Z., S.S., Y.H. provided materials and/or revised the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Hadi AlShamaileh and Tao Wang contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05859-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: An evolving concept. Nat Rev Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- 2.Huang EH, Wicha MS. Colon cancer stem cells: implications for prevention and therapy. Trends Mol Med. 2008;14:503–509. doi: 10.1016/j.molmed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Connor ML, et al. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014;344:180–187. doi: 10.1016/j.canlet.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 5.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Diehn M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–2845. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 9.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: A link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/S0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 10.Ojha R, Bhattacharyya S, Singh SK. Autophagy in Cancer Stem Cells: A Potential Link Between Chemoresistance, Recurrence, and Metastasis. Biores Open Access. 2015;4:97–108. doi: 10.1089/biores.2014.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson BJ. ABCB5 identifies a therapy-refractory tumor cell population in colorectal cancer patients. Cancer Res. 2011;71:5307–5316. doi: 10.1158/0008-5472.CAN-11-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dylla SJ. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todaro M. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Maugeri-Sacca M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17:4942–4947. doi: 10.1158/1078-0432.CCR-10-2538. [DOI] [PubMed] [Google Scholar]
- 15.Dallas NA, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–265. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147–159. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 18.Radom F, Jurek PM, Mazurek MP, Otlewski J, Jeleń F. Aptamers: Molecules of great potential. Biotechnol Adv. 2013;31:1260–1274. doi: 10.1016/j.biotechadv.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Panowksi S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6:34–45. doi: 10.4161/mabs.27022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shigdar S, et al. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102:991–998. doi: 10.1111/j.1349-7006.2011.01897.x. [DOI] [PubMed] [Google Scholar]
- 21.Shigdar S, et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330:84–95. doi: 10.1016/j.canlet.2012.11.032. [DOI] [PubMed] [Google Scholar]
- 22.Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J. 2010;430:199–205. doi: 10.1042/BJ20100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vegran F, et al. Apoptosis gene signature of Survivin and its splice variant expression in breast carcinoma. Endocr Relat Cancer. 2011;18:783–792. doi: 10.1530/ERC-11-0105. [DOI] [PubMed] [Google Scholar]
- 24.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 25.Altieri DC. Targeting survivin in cancer. Cancer Letters. 2013;332:225–228. doi: 10.1016/j.canlet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taubert H, et al. Expression of survivin detected by immunohistochemistry in the cytoplasm and in the nucleus is associated with prognosis of leiomyosarcoma and synovial sarcoma patients. BMC cancer. 2010;10:65. doi: 10.1186/1471-2407-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang T, et al. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics. 2015;5:1456–1472. doi: 10.7150/thno.11692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kreso, A. & O’Brien, C. A. Colon cancer stem cells. Curr Protoc Stem Cell Biol Chapter 3, Unit 3 1 (2008). [DOI] [PubMed]
- 29.Mo J, et al. The in-vitro spheroid culture induces a more highly differentiated but tumorigenic population from melanoma cell lines. Melanoma Res. 2013;23:254–263. doi: 10.1097/CMR.0b013e32836314e3. [DOI] [PubMed] [Google Scholar]
- 30.Korkaya H, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7:e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duffy MJ, O’Donovan N, Brennan DJ, Gallagher WM, Ryan BM. Survivin: a promising tumor biomarker. Cancer Lett. 2007;249:49–60. doi: 10.1016/j.canlet.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 32.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martowicz A, et al. EpCAM overexpression prolongs proliferative capacity of primary human breast epithelial cells and supports hyperplastic growth. Mol Cancer. 2013;12:56. doi: 10.1186/1476-4598-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Went PT, et al. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–128. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 35.Cimino A, et al. Epithelial cell adhesion molecule (EpCAM) is overexpressed in breast cancer metastases. Breast Cancer Res Treat. 2010;123:701–708. doi: 10.1007/s10549-009-0671-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaffer CL, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahan P, et al. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: possible involvement in radioresistance. Cell Death Dis. 2014;5:e1543. doi: 10.1038/cddis.2014.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aliabadi HM, Landry B, Mandipoor P, Uludag H. Induction of Apoptosis by Survivin Silencing through siRNA Delivery in a Human Breast Cancer Cell Line. Mol Pharm. 2011;8:1821–1830. doi: 10.1021/mp200176v. [DOI] [PubMed] [Google Scholar]
- 39.Guvenc H, et al. Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res. 2013;19:631–642. doi: 10.1158/1078-0432.CCR-12-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong SE, et al. S6K1 inhibition enhances tamoxifen-induced cell death in MCF-7 cells through translational inhibition of Mcl-1 and survivin. Cell biology and toxicology. 2013;29:273–282. doi: 10.1007/s10565-013-9253-2. [DOI] [PubMed] [Google Scholar]
- 41.Satoh T, et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3872–3880. doi: 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- 42.Lewis KD, et al. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2011;29:161–166. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- 43.Giaccone G, et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 44.Cheson BD, et al. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012;118:3128–3134. doi: 10.1002/cncr.26510. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Hou N, Faried A, Tsutsumi S, Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer. 2010;46:1900–1909. doi: 10.1016/j.ejca.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 46.Sasaki K, et al. Resistance of colon cancer to 5-fluorouracil may be overcome by combination with chloroquine, an in vivo study. Anticancer drugs. 2012;23:675–682. doi: 10.1097/CAD.0b013e328353f8c7. [DOI] [PubMed] [Google Scholar]
- 47.Tang JC, Feng YL, Liang X, Cai XJ. Autophagy in 5-Fluorouracil Therapy in Gastrointestinal Cancer: Trends and Challenges. Chin Med J (Engl) 2016;129:456–463. doi: 10.4103/0366-6999.176069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw FL, et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia. 2012;17:111–117. doi: 10.1007/s10911-012-9255-3. [DOI] [PubMed] [Google Scholar]
- 49.Hu Y, Smyth GK. J Immunol Methods. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays; pp. 70–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.