

Abstract
Halomonads are moderately halophilic bacteria that are studied as models of prokaryotic osmoadaptation and sources of enzymes and chemicals for biotechnological applications. Despite the progress in understanding the diversity of these organisms, our ability to explain ecological, metabolic, and biochemical traits of halomonads at the genomic sequence level remains limited. This study addresses this gap by presenting draft genomes of Salinicola socius SMB35T, Salinicola sp. MH3R3–1 and Chromohalobacter sp. SMB17, which were isolated from potash mine tailings in the Verkhnekamsk salt deposit area of Russia. The analysis of these genomes confirmed the importance of ectoines and quaternary amines to the capacity of halomonads to tolerate osmotic stress and adapt to hypersaline environments. The study also revealed that Chromohalobacter and Salinicola share 75–90% of the predicted proteome, but also harbor a set of genus-specific genes, which in Salinicola amounted to approximately 0.5 Mbp. These genus-specific genome segments may contribute to the phenotypic diversity of the Halomonadaceae and the ability of these organisms to adapt to changing environmental conditions and colonize new ecological niches.
Keywords: Salinicola, Chromohalobacter, Halomonadaceae, Halophile, Potash mine tailings
Introduction
The family Halomonadaceae encompasses a diverse group of moderately halophilic, aerobic, heterotrophic, Gram-negative bacteria that are ubiquitously present in ocean water, marine sediments, saline lakes, salt pans, salty foods, and saline soils [1]. These microorganisms can grow over a wide range of saline concentrations and received considerable attention as model systems to study the physiological and molecular mechanisms of prokaryotic osmoadaptation [2–4]. Several members of the family have been investigated because of their biotechnological potential, especially as a source of exopolysaccharides, biosurfactants, biodegradable plastics (polyhydroxyalkanoates), salt-tolerant extracellular enzymes, and compatible solutes [5]. Halomonads were used for the development of energy-efficient unsterile fermentation processes that use seawater-based growth medium [5]. Finally, certain strains of Halomonas can mobilize, degrade and neutralize environmental pollutants and may play a vital role in the process of remediation of contaminated saline environments [6].
Members of the Halomonadaceae are heterogeneous, and many currently recognized species were previously assigned to distantly related families. With the advent of molecular typing techniques in the 1990s, the group underwent a major taxonomic revision and now contains 16 validly named genera [7]. Despite the progress in understanding the diversity of these organisms, our ability to explain ecological, metabolic, and biochemical traits of halomonads at the genomic sequence level remains limited. At the time of the manuscript preparation, GenBank listed 54 genomes of bacteria of the Halomonadaceae family. However, two-thirds of these entries originated from strains of a single genus, Halomonas, while other genera within this genomically diverse group remain strongly underrepresented. Here we address this gap by presenting the description, annotation, and analysis of draft genomes of S. socius SMB35 T, Salinicola sp. MH3R3–1 and Chromohalobacter sp. SMB17, which were isolated from soils affected by potash mine tailings in the Verkhnekamsk salt deposit area of Russia. The genome sequences of strains SMB35 T and MH3R3–1 are the first analyzed Salinicola genomes, and one of these genomes represents a type strain of this genus.
Organism information
Classification and features
S. socius SMB35 T (DSM 19940 T), Salinicola sp. MH3R3–1 and Chromohalobacter sp. SMB17 are moderately halophilic Gram-negative, aerobic, catalase-positive and oxidase-negative, non-spore-forming bacteria that belong to the Halomonadaceae family in the class Gammaproteobacteria (Fig. 1, Tables 1, 2, and 3). Strains SMB35 T and SMB17 were isolated from saline soil collected in an area affected by potash mining activity near the town of Solikamsk, Russia. Salinicola sp. MH3R3–1 was isolated from the rhizosphere of salt sand-spurrey (Spergularia salina), a halotolerant plant commonly found around salt mine tailings in the sampled area. The isolation and subsequent maintenance were carried out at 28 °C in RMM medium [8] supplemented with 6% (w/v) NaCl, 0.5% (w/v) tryptone, and 0.25% (w/v) yeast extract. Cells of SMB35 T and SMB17 are straight rods (0.5–0.6 × 1.1–1.4 μm) with a single polar flagellum (Fig. 1). On RMM agar both strains form yellow, circular and convex colonies with smooth margins. Cells of the strain MH3R3–1 are straight motile rods that are 0.5–0.6 × 1.1–1.4 μm in size (Fig. 1). Colonies on spread plates are cream, circular and convex with an entire margin.
Fig. 1.
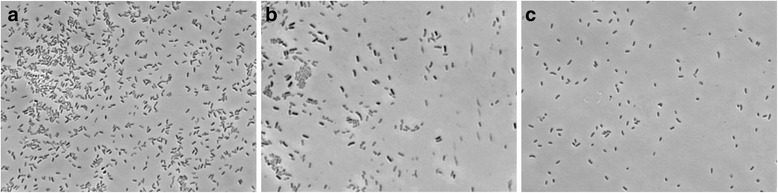
Cellular morphology of S. socius SMB35T(a), Salinicola sp. MH3R3–1 (b), Chromohalobacter sp. SMB17 (c) captured at 100×magnification. Bacteria were cultured in the RMM [8] supplemented with 3% NaCl, fixed and Gram-stained
Table 1.
Classification and general features of S. socius SMB35T
| MIGS ID | Property | Salinicola socius SMB35T | Evidence codea |
|---|---|---|---|
| Classification | Domain: Bacteria | TAS [41] | |
| Phylum: Proteobacteria | TAS [42] | ||
| Class: Gammaproteobacteria | TAS [43] | ||
| Order: Oceanospirillales | TAS [44] | ||
| Family: Halomonadaceae | TAS [1] | ||
| Genus: Salinicola | TAS [1, 9] | ||
| Species: S. socius | TAS [9] | ||
| (Type) strain: SMB35T | TAS [9] | ||
| Gram stain | Negative | TAS [9] | |
| Cell shape | Rod | TAS [9] | |
| Motility | Motile by means of a single polar flagellum | TAS [9] | |
| Sporulation | Non-sporulating | TAS [9] | |
| Temperature range | Mesophilic | TAS [9] | |
| Optimum temperature | 34 °C | TAS [9] | |
| pH range | 6.0–8.0 | TAS [9] | |
| Carbon source | D-glucose, D-galactose, D-xylose, D-maltose, D-trehalose, D-fructose, glycerol, mannitol, sorbitol, acetic acid | TAS [9] | |
| MIGS-6 | Habitat | Saline soil | TAS [9] |
| MIGS-6.3 | Salinity | 0.5–30.0% | TAS [9] |
| MIGS-22 | Oxygen requirement | Aerobic | TAS [9] |
| MIGS-15 | Biotic relationship | Free-living | TAS [9] |
| MIGS-14 | Pathogenicity | Non-pathogen | |
| MIGS-4 | Geographic location | Perm region/Russia | |
| MIGS-5 | Sample collection | July 2002 | |
| MIGS-4.1 | Latitude | N 59.39551 | |
| MIGS-4.2 | Longitude | E 56.76835 | |
| MIGS-4.4 | Altitude | 118 m |
aEvidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project
Table 2.
Classification and general features of Salinicola sp. MH3R3–1
| MIGS ID | Property | Salinicola sp. MH3R3–1 | Evidence codea |
|---|---|---|---|
| Classification | Domain: Bacteria | TAS [41] | |
| Phylum: Proteobacteria | TAS [42] | ||
| Class: Gammaproteobacteria | TAS [43] | ||
| Order: Oceanospirillales | TAS [44] | ||
| Family: Halomonadaceae | TAS [1] | ||
| Genus: Salinicola | TAS [1, 9] | ||
| Species: Salinicola sp. | TAS [45] | ||
| Strain: MH3R3–1 | TAS [45] | ||
| Gram stain | Negative | NAS [45] | |
| Cell shape | Rod | NAS [45] | |
| Motility | Motile | NAS [45] | |
| Sporulation | Non-sporulating | NAS [45] | |
| Temperature range | Mesophilic | TAS [45] | |
| Optimum temperature | Not available | ||
| pH range | Not available | ||
| Carbon source | Not available | ||
| MIGS-6 | Habitat | Rhizosphere of Spergularia salina | TAS [45] |
| MIGS-6.3 | Salinity | 0–25.0% | NAS [45] |
| MIGS-22 | Oxygen requirement | Aerobic | NAS [45] |
| MIGS-15 | Biotic relationship | Free-living | TAS [45] |
| MIGS-14 | Pathogenicity | Non-pathogen | |
| MIGS-4 | Geographic location | Perm region/Russia | |
| MIGS-5 | Sample collection | June 2011 | |
| MIGS-4.1 | Latitude | N 59.56772 | |
| MIGS-4.2 | Longitude | E 56.76833 | |
| MIGS-4.4 | Altitude | 148 m |
aEvidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project
Table 3.
Classification and general features of Chromohalobacter sp. SMB17
| MIGS ID | Property | Chromohalobacter sp. SMB17 | Evidence codea |
|---|---|---|---|
| Classification | Domain: Bacteria | TAS [41] | |
| Phylum: Proteobacteria | TAS [42] | ||
| Class: Gammaproteobacteria | TAS [43] | ||
| Order: Oceanospirillales | TAS [44] | ||
| Family: Halomonadaceae | TAS [1] | ||
| Genus: Chromohalobacter | TAS [1, 7] | ||
| Species: Chromohalobacter sp. | TAS [46] | ||
| Strain: SMB17 | TAS [46] | ||
| Gram stain | Negative | TAS [46] | |
| Cell shape | Rod | TAS [46] | |
| Motility | Motile | TAS [46] | |
| Sporulation | Non-sporulating | TAS [46] | |
| Temperature range | Mesophilic | TAS [46] | |
| Optimum temperature | Not available | ||
| pH range | Not available | ||
| Carbon source | Benzoic acid, p-hydroxybenzoic acid, acetic acid | TAS [47] | |
| MIGS-6 | Habitat | Saline soil | TAS [47] |
| MIGS-6.3 | Salinity | 3–30% | TAS [46] |
| MIGS-22 | Oxygen requirement | Aerobic | NAS [46] |
| MIGS-15 | Biotic relationship | Free-living | TAS [47] |
| MIGS-14 | Pathogenicity | Non-pathogen | |
| MIGS-4 | Geographic location | Perm region/Russia | |
| MIGS-5 | Sample collection | October 2000 | |
| MIGS-4.1 | Latitude | N 59.39564 | |
| MIGS-4.2 | Longitude | E 56.76899 | |
| MIGS-4.4 | Altitude | 115 m |
aEvidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project
S. socius SMB35 T (DSM 19940 T) is the type strain of this species with a well-defined status within the family Halomonadaceae [9, 10], but the taxonomic placement of strains MH3R3–1 and SMB17 was not characterized in details. We compared partial (1333 bp) 16S rDNA sequences of MH3R3–1 and SMB17 to those of type strains (n = 53) representing main phylogenetic groups recognized within the family Halomonadaceae [10, 11]. Results of the analysis revealed that the strain SMB17 belonged to the genus Chromohalobacter and was closely related to C. japonicus, whereas MH3R3–1 was most similar to Salinicola salarius and S. socius (Fig. 2).
Fig. 2.
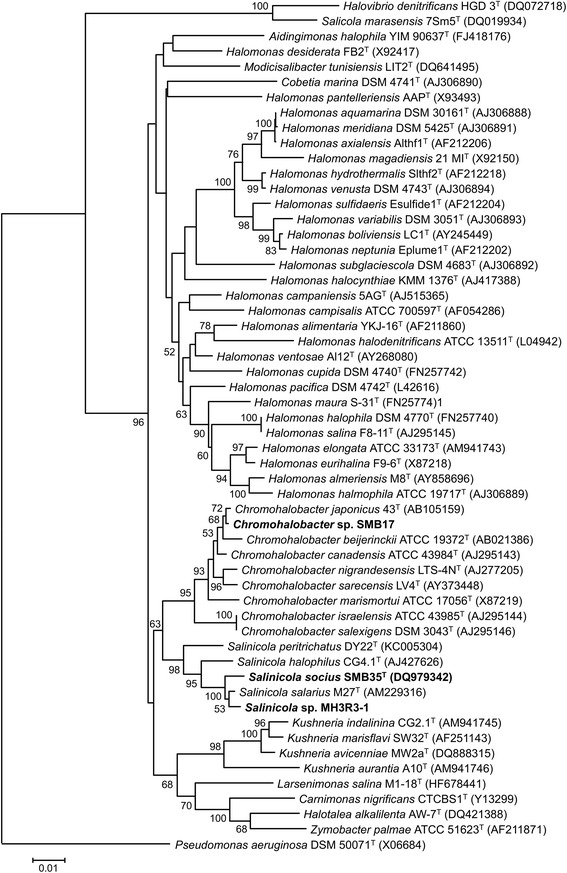
Neighbor-joining phylogeny inferred from data for 16S rRNA sequences of S. socius SMB35T, Salinicola sp. MH3R3–1, Chromohalobacter sp. SMB17 and other species of the Halomonadaceae family. The optimal tree with the sum of branch length = 0.95863591 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method [39] and are in the units of the number of base substitutions per site. The analysis involved 56 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 1241 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 [40]
Genome sequencing information
Genome project history
Strains S. socius SMB35 T, Salinicola sp. MH3R3–1 and Chromohalobacter sp. SMB17 were selected for genome sequencing on the basis of their phylogenetic position and ability to produce compatible solutes ectoine and hydroxyectoine. Genome sequences provide insights into genetic diversity of halophilic bacteria associated with soils affected by potash mining activity in the Perm region of Russia. Genome sequencing was performed by the AgriLife Genomics and Bioinformatics Service at Texas A&M University (College Station, TX). Draft genome sequences of strains SMB35 T, MH3R3–1, and SMB17 are listed in the Genomes Online Database [12] of the JGI under project IDs Gp0136514, Gp0191572, and Gp0191573, respectively. The corresponding WGS projects have been deposited at GenBank under accession numbers MSDO00000000, MSDP00000000, and MSDQ00000000. A summary of the genome sequencing projects and their compliance with MIGS version 2.0 standards [13] is provided in Table 4.
Table 4.
Project information
| MIGS ID | Property | S. socius SMB35T | Salinicola sp. MH3R3–1 | Chromohalobacter sp. SMB17 |
|---|---|---|---|---|
| MIGS 31 | Finishing quality | Draft | Draft | Draft |
| MIGS-28 | Libraries used | Illumina Paired-End library (300 bp insert size) | Illumina Paired-End library (300 bp insert size) | Illumina Paired-End library (300 bp insert size) |
| MIGS 29 | Sequencing platforms | Illumina HiSeq | Illumina HiSeq | Illumina HiSeq |
| MIGS 31.2 | Fold coverage | 302× | 326× | 318× |
| MIGS 30 | Assemblers | ITMO Genome Assembler v0.1.3 | ITMO Genome Assembler v0.1.3 | ITMO Genome Assembler v0.1.3 |
| MIGS 32 | Gene calling method | RAST | RAST | RAST |
| Locus Tag | BTW07 | BTW08 | BTW10 | |
| Genbank ID | MSDO00000000 | MSDP00000000 | MSDQ00000000 | |
| GenBank Date of Release | 01/05/2017 | 01/05/2017 | 01/05/2017 | |
| GOLD ID | Gp0136514 | Gp0191572 | Gp0191573 | |
| BIOPROJECT | PRJNA357614 | PRJNA357619 | PRJNA357624 | |
| MIGS 13 | Source Material Identifier | |||
| Project relevance | Soil remediation | Soil remediation | Soil remediation |
Growth conditions and genomic DNA preparation
Strains SMB35 T, SMB17, and MH3R3–1 were cultured with shaking at 28 °C in liquid RMM medium. The cells were pelleted by centrifugation and the genomic DNA was extracted using the UltraClean Microbial DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA) according to the manufacturer’s recommendations. DNA concentrations were measured fluorometrically by using a DNA Quantitation Kit (Bio-Rad, Hercules, CA, USA). For each strain, ten micrograms of DNA were diluted to concentration of 200 ng/μl and shipped to the AgriLife Genomics and Bioinformatics Service facility for library construction and Illumina sequencing.
Genome sequencing and assembly
Samples of purified DNA from strains SMB35 T, MH3R3–1, and SMB17 were used to construct short-insert paired-end libraries with an average insert size of 320 bp, which were sequenced using a HiSeq 2500 instrument (Illumina, San Diego, CA, USA). Raw reads were filtered using a FastQC toolkit [14] followed by assembly with the ITMO Genome Assembler v. 0.1.3 [15]. For SMB35 T, the sequence run produced 2,530,015 × 2 reads totaling 1,265,007,500 bp of data. A k-mer length of 53 nucleotides was chosen to optimize for the highest N50. The resulting assembly had 164 contigs with a total length of 4.19 Mb (N50, 199,518 bp; Nmax, 370,185 bp; median coverage, 302×). Sequencing of the MH3R3–1 sample yielded 2,690,448 × 2 reads that totaled 1,345,224,000 bp of data. The maximum k-mer value of 63 was chosen for further analysis since it yielded the best combination of N50 and genome length. The final assembly had 159 contigs with a total length of 4,121,883 bp (N50, 139,268 bp; Nmax, 370,744 bp; median coverage, 326×). The final assemblies of SMB35 T and SMB17 genomes were aligned and ordered using progressive Mauve v.2.3.1 [16]. For SMB17, a total of 2,690,448 × 2 reads were obtained that yielded 1,201,941,000 bp of data. The final 3,775,557-bp assembly was based on the k-mer value of 63 and consisted of 118 contigs (N50, 109,330 bp; Nmax, 468,741 bp; median coverage, 318×) that were aligned and ordered against the finished genome of Chromohalobacter salexigens DSM 3043 T [17].
Genome annotation
The genomes were annotated using the RAST v.2.0 pipeline [18]. Additional gene prediction analysis and functional annotation were performed using Geneious v.8.1.8 (Biomatters, Auckland, New Zealand) and tools implemented in PATRIC [19] and JGI Integrated Microbial Genomes [20] databases. Putative membrane and secreted proteins were identified using TMHMM v.2.0 [21] and SignalP 4.1 [22] servers, respectively. For SignalP analysis, the Dmaxcutoff value of 0.57 was used for sequences with transmembrane helices and 0.51 for sequences without. Secondary metabolite production clusters were identified using the antiSMASH program [23]. The analysis of the transporter potential was conducted using the Transporter Automatic Annotation Pipeline [24]. Genomic islands were identified and visualized using the IslandViewer 3 [25]. Circular comparative maps of the genomes of the strains SMB35 T, SMB17, and MH3R3–1 were generated using the BRIG software [26].
Genome properties
The genome of S. socius SMB35 T is comprised of a 4,185,599-bp long chromosome with a 62.2% G + C content. The coding regions accounted for 85.3% of the genome and contained 3,739 protein-coding genes and 89 RNA genes. A total of 2832 genes were assigned a putative function with the remaining annotated as conserved hypothetical or hypothetical. A total of 2037 genes (54.5%) were assigned to COGs. The draft genome of Salinicola sp. MH3R3–1 is 4,121,883-bp long with a 62.0% G + C content. The genome has 3620 protein-coding genes and 130 RNA genes. The coding regions accounted for 86.0% of the whole genome and 2903 genes were assigned a putative function with the remaining annotated as hypothetical proteins. A total of 1870 genes (49.9%) were assigned to COGs. The draft genome of Chromohalobacter sp. SMB17 is 3,775,557-bp long with a 60.5% G + C content. The genome has 3486 protein-coding genes and 123 RNA genes. The coding regions accounted for 87.0% of the whole genome and 2875 genes were assigned a putative function with the remaining annotated as hypothetical proteins. A total of 1947 genes (54.0%) were assigned to COGs. The properties and the statistics of the three sequenced genomes are presented in Table 5. The distribution of genes into COG functional categories is summarized in Table 6.
Table 5.
Genome statistics
| Attribute | S. socius SMB35T | Salinicola sp. MH3R3–1 | Chromohalobacter sp. SMB17 | |||
|---|---|---|---|---|---|---|
| Value | % of Total | Value | % of Total | Value | % of Total | |
| Genome size (bp) | 4,185,599 | 100.0 | 4,121,883 | 100.0 | 3,775,577 | 100.0 |
| DNA coding (bp) | 3,568,310 | 85.3 | 3,547,145 | 86.0 | 3,547,145 | 87.0 |
| DNA G + C (bp) | 2,244,356 | 62.2 | 2,554,139 | 62.0 | 2,284,727 | 60.5 |
| DNA scaffolds | 173 | - | 159 | - | 118 | - |
| Total genes | 3828 | 100.0 | 3750 | 100.0 | 3609 | 100.0 |
| Protein coding genes | 3739 | 97.7 | 3620 | 96.5 | 3486 | 96.6 |
| RNA genes | 89 | 2.3 | 130 | 3.5 | 123 | 3.4 |
| Pseudo genes | - | - | - | - | - | - |
| Genes in internal clusters | - | - | - | - | - | - |
| Genes with function prediction | 2832 | 74.0 | 2903 | 77.4 | 2875 | 79.7 |
| Genes assigned to COGs | 2037 | 54.5 | 1870 | 49.9 | 1947 | 54.0 |
| Genes with Pfam domains | 2652 | 69.3 | 2609 | 69.6 | 2731 | 75.7 |
| Genes with signal peptides | 239 | 6.4 | 238 | 6.6 | 224 | 6.4 |
| Genes with transmembrane helices | 859 | 23.0 | 867 | 24.0 | 815 | 23.4 |
| CRISPR repeats | 0 | - | 0 | - | 0 | - |
Table 6.
Number of genes associated with general COG functional categories
| Code | S. socius SMB35T | Salinicola sp. MH3R3–1 | Chromohalobacter sp. SMB17 | Description | |||
|---|---|---|---|---|---|---|---|
| Value | % | Value | % | Value | % | ||
| J | 188 | 5.03 | 182 | 5.03 | 175 | 5.02 | Translation, ribosomal structure and biogenesis |
| A | 159 | 4.25 | 157 | 4.34 | 164 | 4.70 | RNA processing and modification |
| K | 27 | 0.72 | 27 | 0.75 | 25 | 0.72 | Transcription |
| L | 111 | 2.97 | 107 | 2.96 | 106 | 3.04 | Replication, recombination and repair |
| B | - | - | - | - | - | - | Chromatin structure and dynamics |
| D | 31 | 0.83 | 30 | 0.83 | 30 | 0.86 | Cell cycle control, cell division, chromosome partitioning |
| V | 62 | 1.66 | 73 | 2.02 | 67 | 1.92 | Defense mechanisms |
| T | 101 | 2.70 | 98 | 2.71 | 90 | 2.58 | Signal transduction mechanisms |
| M | 164 | 4.39 | 170 | 4.70 | 157 | 4.50 | Cell wall/membrane biogenesis |
| N | 73 | 1.95 | 72 | 1.99 | 64 | 1.84 | Cell motility |
| U | 210 | 5.62 | 216 | 5.97 | 154 | 4.42 | Intracellular trafficking and secretion |
| O | 82 | 2.19 | 88 | 2.43 | 103 | 2.95 | Posttranslational modification, protein turnover, chaperones |
| C | 100 | 2.67 | 129 | 3.56 | 106 | 3.04 | Energy production and conversion |
| G | 365 | 9.76 | 375 | 10.36 | 421 | 12.08 | Carbohydrate transport and metabolism |
| E | 433 | 11.58 | 442 | 12.21 | 360 | 10.33 | Amino acid transport and metabolism |
| F | 102 | 2.73 | 97 | 2.68 | 106 | 3.04 | Nucleotide transport and metabolism |
| H | 234 | 6.26 | 188 | 5.19 | 253 | 7.26 | Coenzyme transport and metabolism |
| I | 146 | 3.90 | 153 | 4.23 | 156 | 4.48 | Lipid transport and metabolism |
| P | 129 | 3.45 | 104 | 2.87 | 134 | 3.84 | Inorganic ion transport and metabolism |
| Q | 6 | 0.16 | 6 | 0.17 | 4 | 0.11 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 291 | 4.52 | 242 | 6.69 | 262 | 7.52 | General function prediction only |
| S | 122 | 3.26 | 115 | 3.18 | 97 | 2.78 | Function unknown |
| - | 1702 | 45.52 | 1635 | 45.17 | 1539 | 44.15 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Insights from the genome sequence
Phylogenetic analysis revealed that species of Salinicola and Chromohalobacter constitute two closely related yet distinct clades within the Halomonadaceae family (Fig. 2). We compared the newly sequenced Salinicola and Chromohalobacter genomes to each other and the available genome sequences of closely related strains C. japonicus CJ (NZ_CDGZ00000000), C. israelensis 6768 (NZ_JQNW00000000), and C. salexigens DSM 3043 T (CP000285). Results of these comparisons revealed that Salinicola genomes are larger than those of Chromohalobacter by an average of 0.47 Mbp, which amounts to approximately 400 additional protein-coding genes. Our analyses also showed that bacteria of Salinicola–Chromohalobacter group share a core genome containing 2817 protein-coding genes, which represents 75% to 89.8% of the predicted proteome of each strain (Figs. 3 and 4). Genes conserved among all of the genomes encode proteins contributing to many basic housekeeping functions and physiological mechanisms that allow these organisms to cope with deleterious effects of water stress and thrive in hypersaline environments. The mechanisms of osmotolerance in bacteria involve alterations in the permeability or stability of cytoplasmic membrane, regulation of the concentration of intracellular solutes to adjust the cytoplasm osmolality, and production and uptake of inert metabolites collectively known as compatible solutes, osmolytes, or osmoprotectants. Such compounds help to balance the osmotic pressure across the cellular membrane without compromising protein folding or other cellular processes and include certain polyols, sugars, amino acids, amino acid derivatives, and peptides. The analysis of sequenced genomes revealed multiple pathways predicted to function in the de novo synthesis or uptake of osmoprotectants. Most of these determinants have homologs in the well-characterized strain C. salexigens DSM 3043 T [4] and collectively suggest that strains SMB35 T, SMB17, and MH3R3–1 respond to osmotic stress by accumulating the osmoprotectants ectoines, glycine betaine, and trehalose.
Fig. 3.
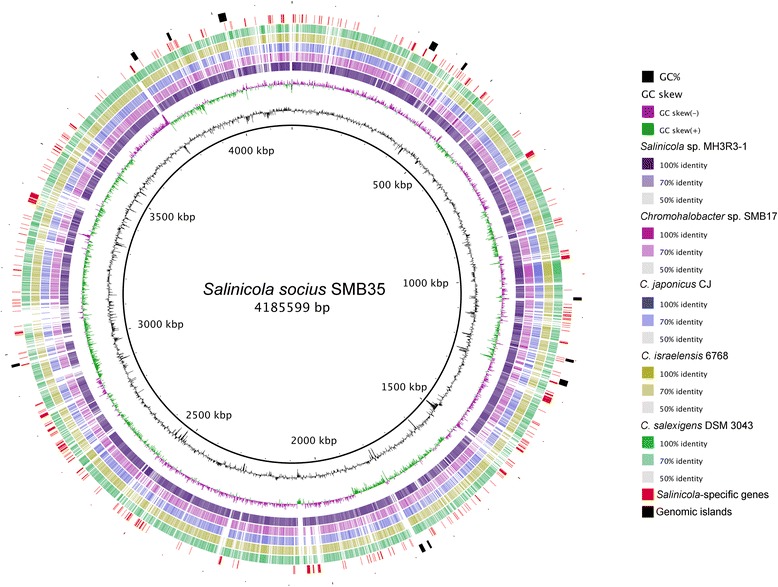
Circular representation of the S. socius SMB35T genome and its comparison to genomes of the Salinicola-Chromohalobacter group. Rings from inside to outside: (1) GC content, (2) GC skew, (3) BLASTn comparison with Salinicola sp. MH3R3–1, (4) BLASTn comparison with Chromohalobacter sp. SMB17, (5) BLASTn comparison with C. japonicus CJ, (6) BLASTn comparison with C. israelensis 6768, (7) BLASTn comparison with C. salexigens DSM 3043T, (8) Genome segments present in both Salinicola genomes, but absent from genomes of all Chromohalobacter spp., (9) Genomic islands predicted in the SMB35T genome. The genome comparisons were visualized using the BRIG software [26]. The inner scales designate the coordinates in kilobase pairs
Fig. 4.
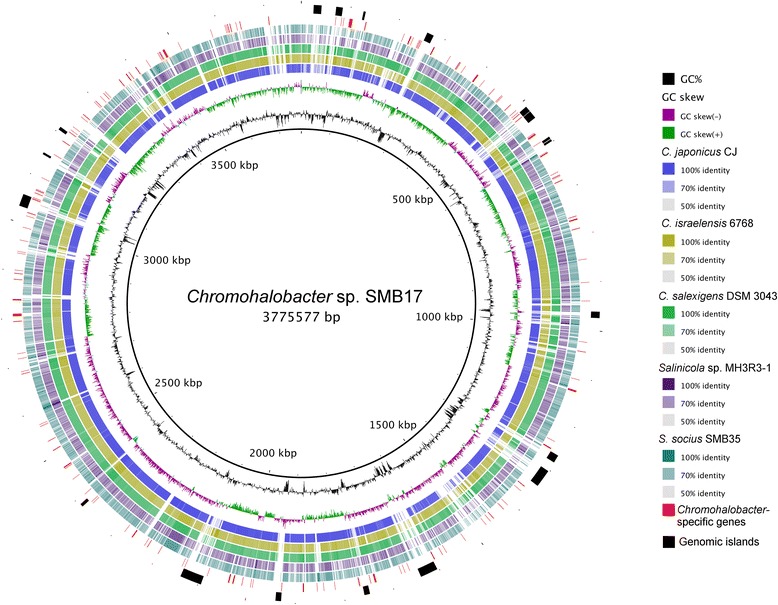
Circular representation of the Chromohalobacter sp. SMB17 genome and its comparison to genomes of the Salinicola-Chromohalobacter group. Rings from inside to outside: (1) GC content, (2) GC skew, (3) BLASTn comparison with C. japonicus CJ, (4) BLASTn comparison with C. israelensis 6768, (5) BLASTn comparison with C. salexigens DSM 3043T, (6) BLASTn comparison with Salinicola sp. MH3R3–1, (7) BLASTn comparison with S. socius SMB35T, (8) Genome segments present in all Chromohalobacter genomes, but absent from genomes of Salinicola spp., (9) Genomic islands predicted in the Chromohalobacter sp. SMB17 genome. The genome comparisons were visualized using the BRIG software [26]. The inner scales designate the coordinates in kilobase pairs
Each of the sequenced strains was predicted to synthesize ectoine from L-aspartate-β-semialdehyde and carried a three-gene operon encoding L-2,4-diaminobutyric acid acetyltransferase 2,4-diaminobutyric acid transaminase and ectoine synthase. Each microorganism also had a gene for the enzyme ectoine hydroxylase, which converts ectoine to 5-hydroxyectoine. However, in contrast to the highly conserved location of the ectABC operon, the ectD gene was present in different parts of the SMB35 T, SMB17, and MH3R3–1 genomes and not associated with ectR, which encodes the transcriptional repressor of the ectoine pathway in C. salexigens DSM 3043 T [2]. In addition to producing ectoines de novo, many bacteria will readily scavenge these metabolites from the environment, and our analyses revealed that all strains featured in this study are capable of taking up ectoines through orthologs of the ectoine-hydoxyectoine ABC-type transport system of Ensifer meliloti [27]. The system consists of an ATPase EhuA, a periplasmic solute-binding protein EhuB, and permease proteins EhuC and EhuD that are encoded in genomes of SMB35 T, SMB17, and MH3R3–1 by the highly conserved ehuABCD operon. It also appears that the studied strains can utilize ectoines as sources of carbon and nitrogen. The three sequenced genomes share a cluster of ectoine catabolism genes doeABXCD that control the degradation of ectoine via hydrolysis to Nα-acetyl-L-2,4-diaminobutyric acid, deacetylation to diaminobutyric acid and ultimate conversion to aspartate.
In addition to ectoines, members of the Halomonadaceae utilize as osmoprotectants glycine betaine and other quaternary ammonium compounds, which are hypothesized to be ubiquitous and relatively abundant in the environment [28]. The three strains share six genes encoding putative BCCT-type transporters, which translocate quaternary ammonium compounds across the membrane using proton/sodium-motive force [29]. The strains also share two gene clusters encoding putative osmoprotectant-specific ATP-binding cassette transporters. The first of these clusters encodes an ortholog of ProVWX, which functions in Escherichia coli as a high-affinity uptake system for GB [30]. At lower affinities, the ProVWX complex also transports choline and many other osmoprotectants, including carnitine, proline, taurine, and ectoine. The second gene cluster encodes an ortholog of the OsmF-YehYXW system, which functions in E. coli as a low-affinity non-osmoregulatory GB-specific transporter [31]. Finally, the sequenced genomes also share at least three genes encoding proteins similar to GB-specific substrate-binding components of ABC transporters. Like most heterotrophic bacteria, SMB35 T, SMB17, and MH3R3–1 lack genes for the de novo synthesis of GB, but are capable of converting choline to GB using choline oxidase BetA and betaine aldehyde dehydrogenase that are encoded by the betIBA operon. The operon also encodes BetI, a choline-sensitive transcriptional regulator of the TetR family of transcriptional repressors. Depending on the environmental conditions, GB can be accumulated as an osmoprotectant or converted to glycine and ultimately catabolized as a source of carbon and nitrogen. The catabolism of GB is a multistep process that starts with the demethylation of GB to dimethylglycine by the GbcAB oxygenase. The resultant dimethylglycine is then converted by a heterodimeric flavin-linked oxidoreductase DgcAB to sarcosine, which is further demethylated to glycine by a heterotetrameric sarcosine oxidase SoxABDG. Genes for all enzymes mentioned above were present in the genomes of SMB35 T, SMB17, and MH3R3–1, along with the AraC-family transcriptional activator GbdR. In other species of bacteria, GbdR senses levels of GB and dimethylglycine and induces transcription of genes involved in the uptake and catabolism of GB, and detoxification of the catabolic byproducts [32].
We further analyzed the sequenced genomes for the presence of genes involved in the synthesis of trehalose, a compatible solute synthesized by many microorganisms in response to stress imposed by high osmolarity, desiccation, heat, freezing, or oxidation [33]. One of the most common ways of synthesizing trehalose by bacteria involves the TPS/TPP pathway, in which the enzyme trehalose-6-phosphate synthase catalyzes the transfer of glucose from UDP-glucose to glucose-6-phosphate, leading to trehalose-6-phosphate [34]. The latter is then dephosphorylated by TPP to release free trehalose. Our analyses revealed that otsAB genes encoding enzymes of the TPS/TPP pathway were present in genomes of strains SMB35 T, SMB17, and MH3R3–1, and other bacteria of the Salinicola–Chromohalobacter lineage. The contribution of the otsAB genes to the stress response was demonstrated in C. salexigens DSM 3043T where trehalose produced via the TPS/TPP pathway was characterized as a secondary solute involved in osmo- and thermoprotection [35].
We also conducted proteome comparisons to identify genes that are specifically associated with Salinicola or Chromohalobacter . Results of these analyses revealed 399 genes that were present in S. socius SMB35 T and Salinicola sp. MH3R3–1, but absent from the genomes of Chromohalobacter spp. (Fig. 3). Almost half of these genes (188 genes, or 47%) encoded conserved hypothetical proteins. Among Salinicola-specific genes with predicted functions were genes for the alternative trehalose synthesis pathway that involves the conversion of the terminal unit of a glucose polymer from a α-(1–4) to an α,α-(1–1) configuration, and then the release of the resulting trehalosyl moiety as free trehalose [36]. The key enzymes of this pathway, maltooligosyl trehalose synthase and maltooligosyltrehalose trehalohydrolase, are encoded by treY and treZ genes, which were identified in Salinicola strains SMB35 T and MH3R3–1 but not in genomes of SMB17 and other Chromohalobacter strains. In contrast to the TPS/TPP pathway, the importance of the treY–treZ pathway for the salinity and temperature tolerance of the Halomonadaceae remains to be determined. The rest of Salinicola-specific genes encoded putative regulatory proteins (transcriptional regulators, a diguanylate cyclase/phosphodiesterase with PAC/PAS sensor domain, and a RpoN-like sigma factor) and different enzymes, some of which constitute pathways for metabolism of phosphonate and the polyamine spermidine. The Salinicola-specific transporter proteins were predicted to function in the transport of sodium, potassium, di- and tricarboxylic acids, and provide resistance to heavy metals such as copper, lead, cadmium, zinc and mercury. Interestingly, SMB35 T also carried the gene for aminocyclopropane-1-carboxylic acid deaminase, which encodes for an enzyme that converts aminocyclopropane carboxylate into ammonia and α-ketobutyrate. In plants, ACC is the immediate precursor of the hormone ethylene that mediates many developmental processes and response to stress. Stressed plants produce excessive amounts of ethylene, which results in smaller roots and delayed aging, abscission, and senescence [37]. Soil bacteria with ACC deaminase lower plant ethylene levels by catabolizing aminocyclopropane carboxylate, which stimulates root growth and improves plant tolerance to biotic and abiotic stress.
We identified a total of 187 genes that were uniquely present in Chromohalobacter spp. and had no counterparts in the sequenced Salinicola genomes (Fig. 4). A significant proportion (37%) of these genes belonged to the conserved hypothetical category. The genes with defined functions encoded regulators (transcriptional factors and components of two-component signal transduction circuits), components of type IV fimbriae, a VapC-like toxin/antitoxin system, and various enzymes, including subunits of a formate dehydrogenase complex and components of a cyanophycin biosynthesis pathway. Other Chromohalobacter-specific proteins were predicted to function in stress response (detoxification of reactive oxygen and resistance to arsenic) or transport phosphate, di-tripeptides, nitrate, branched chain amino acids, and other solutes.
Finally, the three sequenced genomes were screened for the presence of genomic islands using the IslandViewer 3 pipeline [25]. Results of the analysis revealed that Chromohalobacter sp. SMB17 had the highest proportion of DNA assigned to genomic islands (252 kbp, or 6.7%) (Fig. 4), which encoded, among other functions, a betaine-binding subunit of ABC transporter, a tricarboxylate transport system, and several O-antigen biosynthesis enzymes. Other features associated with GIs included multiple transposons, a retron element, and seven integrases, three of which were associated with prophages. Salinicola socius SMB35 T and Salinicola sp. MH3R3–1 had, respectively, 233.8 kbp and 129.0 kbp of DNA assigned to genomic islands, which represents 5.6% and 3.1% of the genome of each strain (Fig. 3). These GIs encoded multiple conserved hypothetical proteins, various enzymes, transport and regulatory proteins. In addition, each strain harbored a unique assortment of intact and decaying mobile genetic elements. For example, S. socius SMB35 T harbored three prophages that collectively encompassed 120.6 kbp of the genome and contained genes for structural bacteriophage proteins, DNA metabolism enzymes, transcriptional regulators, and lytic enzymes. All three SMB35 T prophages contained integrase genes and two of these elements were integrated into tRNA-Gly (CCC) and tRNA-Thr (CGT). The genome of Salinicola sp. MH3R3–1 contained two islands (35.7 and 24.9 kbp) that contained genes for plasmid replication, partitioning, and conjugation and exhibited similarity to integrative and conjugative elements, a class of hybrid genomic islands with elements of both temperate phages and conjugative plasmids [38].
Conclusions
This study provides new insights into the genomic diversity of the family Halomonadaceae by contributing three genome sequences of moderately halophilic heterotrophic bacteria of the Salinicola - Chromohalobacter group. The comparative analysis of genomes of S. socius SMB35 T, Salinicola sp. MH3R3–1, and Chromohalobacter sp. SMB17 underscores the importance of two classes of osmoprotectants, ectoines, and quaternary ammonium compounds, to the capacity of these bacteria to tolerate wide ranges of solute concentrations and adapt to hypersaline environments. The sequenced genomes share highly conserved genes for the synthesis of ectoine (ectABC) and hydroxyectoine (ectD), and conversion of choline to GB (betIBA). The three strains also are capable of achieving osmoprotection by accumulating ectoines and quaternary amines via uptake and share an ectoine-specific ABC-type transporter (EhuABCD), two QA-specific ABC-type transporters (ProVWX and OsmF-YehYXW), and several transporters of the Betaine/Carnitine/Choline-Transport family. The three sequenced strains are further capable of utilizing ectoines and quaternary ammonium compounds as sources of C and N and share a cluster of ectoine catabolism genes (doeABXCD), as well as three operons (gbcAB, dgcAB, and soxABDG) that control the degradation of GB to aspartic acid. SMB35 T, MH3R3–1, and SMB17 also carry otsAB genes that encode enzymes for the synthesis of trehalose, which in this group of bacteria functions as a secondary compatible solute involved in osmotic and thermal protection. Finally, results of this study revealed that Chromohalobacter and Salinicola share a core genome of approximately 2800 genes, which represents 75–90% of the predicted proteome of each strain. However, each genus also harbored a set of unique protein-coding genes, which was particularly significant in Salinicola and amounted to approximately 0.5 Mbp. The genus-specific genome segments encoded enzymes, transcriptional regulators, transporters, conserved hypothetical proteins, and mobile genetic elements. We hypothesize that this flexible gene pool may contribute to the phenotypic diversity of the Halomonadaceae and the ability of these organisms to adapt to changing environmental conditions and colonize new ecological niches.
Acknowledgments
Funding
The genome sequencing project was funded by award #RUB2–7100-PE-13 to DVM and EGP from CRDF Global.
Author’s contributions
DVM, EGP, OVM, ESK, LNA conceived and designed the experiments. DVM, BEO performed the genome sequencing and assembly experiments. DVM, BEO, AAP performed the genome annotation and comparative studies. DVM wrote the manuscript. All authors commented on the manuscript before submission. All authors read and approved the final manuscript.
Abbreviations
- ABC transporters
ATP-binding cassette transporters
- ACC
Aminocyclopropane-1-carboxylic acid
- BCCT
Betaine/Carnitine/Choline-Transport
- BLASTn
Basic local alignment search tool for nucleotide sequences
- BRIG
Blast ring image generator
- COGs
Clusters of orthologous groups of proteins
- DSM
DSMZ-deutsche sammlung von mikroorganismen und zellkulturen
- GB
Glycine betaine
- GI
Genomic island
- ITMO
Saint-Petersburg National Research University of Information Technologies, Mechanics, and Optics
- JGI
Joint Genome Institute
- MIGS
The minimum information about a genome sequence specification
- PAC
Motif C-terminal to PAS motifs
- PAC/PAS sensor domain
PAS - domain named after the Drosophila protein Period (Per), the human arylhydrocarbon receptor nuclear translocator (Arnt), and Drosophila Single-minded (Sim)
- PATRIC
Pathosystems Resource Integration Center
- Pfam
Protein families
- QA
Quaternary amine
- RAST
Rapid annotation using subsystem technology
- RMM
Raymond’s mineral medium
- TPP
Trehalose-6-phosphate phosphatase
- TPS
Trehalose-6-phosphate synthase
- UDP-glucose
Uridine diphosphate glucose
- WGS
Whole genome shotgun
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.de la Haba RR, Arahal DR, Sánchez-Porro C, Ventosa A. The family Halomonadaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes. Gammaproteobacteria. 4th. Berlin Heidelberg: Springer-Verlag; 2014. pp. 325–360. [Google Scholar]
- 2.Vargas C, Argandona M, Reina-Bueno M, Rodriguez-Moya J, Fernandez-Aunion C, Nieto JJ. Unravelling the adaptation responses to osmotic and temperature stress in Chromohalobacter salexigens, a bacterium with broad salinity tolerance. Saline Syst. 2008;4:14. doi: 10.1186/1746-1448-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oren A, Larimer F, Richardson P, Lapidus A, Csonka LN. How to be moderately halophilic with broad salt tolerance: clues from the genome of Chromohalobacter salexigens. Extremophiles. 2005;9:275–279. doi: 10.1007/s00792-005-0442-7. [DOI] [PubMed] [Google Scholar]
- 4.Ates O, Oner ET, Arga KY. Genome-scale reconstruction of metabolic network for a halophilic extremophile, Chromohalobacter salexigens DSM 3043T. BMC Syst Biol. 2011;5:12. [DOI] [PMC free article] [PubMed]
- 5.Yin J, Chen JC, Wu Q, Chen GQ. Halophiles, coming stars for industrial biotechnology. Biotechnol Adv. 2015;33:1433–1442. doi: 10.1016/j.biotechadv.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Maltseva O, McGowan C, Fulthorpe R, Oriel P. Degradation of 2,4-dichlorophenoxyacetic acid by haloalkaliphilic bacteria. Microbiology. 1996;142:1115–1122. doi: 10.1099/13500872-142-5-1115. [DOI] [PubMed] [Google Scholar]
- 7.Parte AC. LPSN—list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014;42(Database issue):D613–D616. doi: 10.1093/nar/gkt1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raymond RL. Microbial oxidation of n-paraffinic hydrocarbons. Develop Ind Microbiol. 1961;2:23–32. [Google Scholar]
- 9.Anan’ina LN, Plotnikova EG, Gavrish EY, Demakov VA, Evtushenko LI. Salinicola socius gen. Nov., sp nov., a moderately halophilic bacterium from a naphthalene-utilizing microbial association. Microbiology. 2007;76:324–330. doi: 10.1134/S0026261707030095. [DOI] [PubMed] [Google Scholar]
- 10.de la Haba RR, Arahal DR, Marquez MC, Ventosa A. Phylogenetic relationships within the family Halomonadaceae based on comparative 23S and 16S rRNA gene sequence analysis. Int J Syst Evol Microbiol. 2010;60:737–748. doi: 10.1099/ijs.0.013979-0. [DOI] [PubMed] [Google Scholar]
- 11.de la Haba RR, Sanchez-Porro C, Marquez MC, Ventosa A. Taxonomic study of the genus Salinicola: transfer of Halomonas salaria and Chromohalobacter salarius to the genus Salinicola as Salinicola salarius comb. nov. and Salinicola halophilus nom. Nov., respectively. Int J Syst Evol Microbiol. 2010;60:963–971. doi: 10.1099/ijs.0.014480-0. [DOI] [PubMed] [Google Scholar]
- 12.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, et al. The genomes OnLine database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40(Database issue):D571–D579. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 20 June 2014.
- 15.Alexandrov A, Kazakov S, Melnikov S, Sergushichev A, Shalyto A, Tsarev F. Combining de Bruijn graph, overlap graph and microassembly for de novo genome assembly. In: Proceedings of Bioinformatics 2012. SocBiN (Society for Bioinformatics in Northern Europe). 2012. http://socbin.org/bioinfo2012/all-abstracts.pdf. Accessed 20 June 2014.
- 16.Darling AE, Treangen TJ, Messeguer X, Perna NT. Analyzing patterns of microbial evolution using the mauve genome alignment system. Methods Mol Biol. 2007;396:135–152. doi: 10.1007/978-1-59745-515-2_10. [DOI] [PubMed] [Google Scholar]
- 17.Copeland A, O'Connor K, Lucas S, Lapidus A, Berry KW, Detter JC, et al. Complete genome sequence of the halophilic and highly halotolerant Chromohalobacter salexigens type strain (1H11T) Stand Genomic Sci. 2011;5:379–388. doi: 10.4056/sigs.2285059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42(Database issue):D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42(Database issue):D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40(Database issue):D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 22.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 23.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren QH, Chen KX, Paulsen IT. TransportDB: a comprehensive database resource for cytoplasmic membrane transport systems and outer membrane channels. Nucl Acids Res. 2007;35(Database issue):D274–D279. doi: 10.1093/nar/gkl925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015;43:W104–W108. doi: 10.1093/nar/gkv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jebbar M, Sohn-Bosser L, Bremer E, Bernard T, Blanco C. Ectoine-induced proteins in Sinorhizobium meliloti include an ectoine ABC-type transporter involved in osmoprotection and ectoine catabolism. J Bacteriol. 2005;187:1293–1304. doi: 10.1128/JB.187.4.1293-1304.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welsh DT. Ecological significance of compatible solute accumulation by microorganisms: from single cells to global climate. FEMS Microbiol Rev. 2000;24:263–290. doi: 10.1111/j.1574-6976.2000.tb00542.x. [DOI] [PubMed] [Google Scholar]
- 29.Ziegler C, Bremer E, Kramer R. The BCCT family of carriers: from physiology to crystal structure. Mol Microbiol. 2010;78:13–34. doi: 10.1111/j.1365-2958.2010.07332.x. [DOI] [PubMed] [Google Scholar]
- 30.Lucht JM, Bremer E. Adaptation of Escherichia coli to high osmolarity environments: osmoregulation of the high-affinity glycine betaine transport system ProU. FEMS Microbiol Rev. 1994;14:3–20. doi: 10.1111/j.1574-6976.1994.tb00067.x. [DOI] [PubMed] [Google Scholar]
- 31.Lang S, Cressatti M, Mendoza KE, Coumoundouros CN, Plater SM, Culham DE, et al. YehZYXW of Escherichia coli is a low-affinity, non-osmoregulatory betaine-specific ABC transporter. Biochemistry. 2015;54:5735–5747. doi: 10.1021/acs.biochem.5b00274. [DOI] [PubMed] [Google Scholar]
- 32.Wargo MJ. Homeostasis and catabolism of choline and glycine betaine: lessons from Pseudomonas aeruginosa. Appl Environ Microbiol. 2013;79:2112–2120. doi: 10.1128/AEM.03565-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arguelles JC. Physiological roles of trehalose in bacteria and yeasts: a comparative analysis. Arch Microbiol. 2000;174:217–224. doi: 10.1007/s002030000192. [DOI] [PubMed] [Google Scholar]
- 34.Strom AR, Kaasen I. Trehalose metabolism in Escherichia coli: stress protection and stress regulation of gene expression. Mol Microbiol. 1993;8:205–210. doi: 10.1111/j.1365-2958.1993.tb01564.x. [DOI] [PubMed] [Google Scholar]
- 35.Reina-Bueno M, Argandona M, Salvador M, Rodriguez-Moya J, Iglesias-Guerra F, Csonka LN, et al. Role of trehalose in salinity and temperature tolerance in the model halophilic bacterium Chromohalobacter salexigens. PLoS One. 2012;7:e33587. doi: 10.1371/journal.pone.0033587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maruta K, Hattori K, Nakada T, Kubota M, Sugimoto T, Kurimoto M. Cloning and sequencing of trehalose biosynthesis genes from Arthrobacter sp. Q36. Biochim Biophys Acta. 1996;1289:10–13. doi: 10.1016/0304-4165(95)00139-5. [DOI] [PubMed] [Google Scholar]
- 37.Glick BR, Todorovic B, Czarny J, Cheng Z, Duan J, McConkey B. Promotion of plant growth by bacterial ACC deaminase. Critical Rev Plant Sci. 2007;26:227–242. doi: 10.1080/07352680701572966. [DOI] [Google Scholar]
- 38.Johnson CM, Grossman AD. Integrative and conjugative elements (ICEs): what they do and how they work. Annu Rev Genet. 2015;49:577–601. doi: 10.1146/annurev-genet-112414-055018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. Nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology, volume 2, the Proteobacteria, part B: the Gammaproteobacteria. 2nd. New York: Springer; 2005. p. 1. [Google Scholar]
- 43.Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class. Nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology, volume 2, the Proteobacteria, part B: the Gammaproteobacteria. 2nd. New York: Springer; 2005. p. 1. [Google Scholar]
- 44.Garrity GM, Bell JA, Lilburn T. Oceanospirillales ord. nov. In: Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology. Volume 2, the Proteobacteria, part B: the Gammaproteobacteria. 2nd. New York: Springer; 2005. pp. 270–323. [Google Scholar]
- 45.Korsakova ES, Anan’ina LN, Nazarov AV, Bachurin BA, Plotnikova EG. Diversity of bacteria of the family Halomonadaceae at the mining area of the Verkhnekamsk salt deposit. Microbiology. 2013;82:249–252. doi: 10.1134/S0026261713020070. [DOI] [Google Scholar]
- 46.Anan’ina LN. A naphthalene-degrading consortium of microorganisms isolated from saline soils. Ph.D. dissertation. Perm: Institute of Ecology and Genetics of Microorganisms, Russian Academy of Sciences; 2007. [Google Scholar]
- 47.Plotnikova EG. The diversity, metabolic properties, and functional genomics of bacteria-destructors of aromatic and halogenated hydrocarbons. D.Sc. Dissertation. Perm: Institute of Ecology and Genetics of Microorganisms, Russian Academy of Sciences; 2010. [Google Scholar]