

Abstract
Background
Adverse psychosocial exposures in early life, namely experiences such as child maltreatment, caregiver stress or depression, and domestic or community violence, have been associated in epidemiological studies with increased lifetime risk of adverse outcomes, including diabetes, heart disease, cancers, and psychiatric illnesses. Additional work has shed light on the potential molecular mechanisms by which early adversity becomes “biologically embedded” in altered physiology across body systems. This review surveys evidence on such mechanisms and calls on researchers, clinicians, policymakers, and other practitioners to act upon evidence.
Observations
Childhood psychosocial adversity has wide-ranging effects on neural, endocrine, immune, and metabolic physiology. Molecular mechanisms broadly implicate disruption of central neural networks, neuroendocrine stress dysregulation, and chronic inflammation, among other changes. Physiological disruption predisposes individuals to common diseases across the life course.
Conclusions
Reviewed evidence has important implications for clinical practice, biomedical research, and work across other sectors relevant to public health and child wellbeing. Warranted changes include increased clinical screening for exposures among children and adults, scale-up of effective interventions, policy advocacy, and ongoing research to develop new evidence-based response strategies.
Keywords: Adverse childhood experiences, Brain development, Stress, Health promotion, Social disparities, Primary care
Background
Epidemiological studies have demonstrated that adverse childhood experiences, namely exposures such as neglect, abuse, caregiver mental illness, and family or community violence, predict poorer long-term outcomes across health and social domains. Outcomes associated with early adversity include higher risk of type 2 diabetes, obesity, ischemic heart disease, cancers, depression, addictions, and premature mortality, as well as social outcomes including unemployment and lower educational attainment [1–8]. Particularly convincing evidence comes from large birth cohorts and prospective, longitudinal life-course studies exploring predictive relationships [3, 5–9]. Meanwhile, human and animal research has provided insights into candidate molecular mechanisms by which early adversity may become “biologically embedded” in disrupted physiology [10]. Such findings support life-course models of human health describing how early physiological development interacts over time with behavior and ongoing risk environments to shape outcomes holistically [7].
Nevertheless, evidence about the pathogenic effects of childhood psychosocial adversity has not been widely applied in clinical practice or public health initiatives. Such knowledge has the potential to improve screening and intervention strategies aiming to decrease exposure to early adversity (primary prevention), limit resulting pathology (secondary prevention), and help those already suffering effects (tertiary prevention and treatment). Efforts must span the life course, involving pediatric and adult clinicians, researchers, educators, public health practitioners, families, and communities. Awareness of the effects of adversity can furthermore enhance investigations into the roots of human disease.
This review surveys the evidence on biological mechanisms thought to link early childhood adversity to later disease. While prior literature has predominantly described changes in one or a few physiological axes, this review summarizes changes comprehensively across body systems, offering a unified orientation for clinicians and researchers. The specific questions addressed include (1) How can often time-limited early exposures produce durable physiological changes? (2) How do such physiological changes converge to generate disease? (3) What factors underlie “differential susceptibility” to developmental adversity, and how can interventions promote resilience? Finally, we consider how answers to these questions should shape action across social sectors to promote child wellbeing and lifelong health.
Defining early life adversity
In this review, we conceptualize childhood adversity as a negative childhood experience associated with increased lifetime risk of poorer health and social outcomes. The review is limited to postnatal exposures, while separate literature covers important effects of prenatal adversity [11]. We specifically consider psychosocial adversity, namely that involving relationships (to caregivers, family, community, peers) and other social experiences interacting with psychological processes [12]. Examples of psychosocial adversities include childhood maltreatment, violence exposure, caregiver psychopathology, unstable or depriving care environments (e.g., low-quality foster or institutional care), adverse societal exposures such as crime and discrimination, and other causes of psychological stress or trauma. Various childhood adversities are prevalent globally. A recent review found that at least 44% of children in developed countries and 59% in developing countries had been victims of physical, emotional, or sexual violence or had witnessed domestic or community violence in the preceding year [13]. Caregiver poor mental health is also common, with depression currently representing the leading cause of disease-related disability globally [14].
For brevity, we refer to childhood psychosocial adversity as “early life adversity” (ELA), employing an aggregative approach to conceptualize exposures. Such an approach facilitates the synthesis of complex evidence for application, and is supported by observed dose–response effects linking cumulative early adversity to later outcomes [1, 3, 5], and by the “allostatic load” paradigm exploring pathogenic effects of cumulative all-cause stress [15]. Such aggregative approaches require complementary efforts to differentiate effects of exposures varying in nature, timing, and intensity [16]. Here, we do not specifically examine low childhood socioeconomic status (SES) as a psychosocial adversity, as poverty influences health in part via non-psychosocial pathways (e.g., increasing exposure to physical environmental hazards). Meanwhile, some families living in poverty provide safe psychosocial environments despite the challenges posed by socioeconomic disadvantage. Nevertheless, childhood adversities are strongly partitioned by SES, and shaped by inequities intertwined with poverty such as those defined by race, gender, immigration status, class, and other axes of social inequality.
Biological embedding
Biological embedding describes processes by which initially transient, homeostatic responses durably alter physiology [10]. Events early in life may be embedded preferentially due to a preponderance of sensitive periods, or windows of rapid development and heightened plasticity (responsiveness to experience). While traditionally described in neurodevelopment [16], sensitive period effects have been suggested elsewhere, including in the immune [17] and metabolic [18] systems. Epigenetic processes represent a key family of mechanisms driving embedding. Epigenetic change involves stable alteration of gene expression via mechanisms including, among others, attachment of chemical residues (e.g., methyl groups) to DNA or to molecules involved in packaging and transcriptional control (e.g., histones) [19].
Methodological challenges
A key methodological challenge is the difficulty of causal demonstration amidst social complexity. While epidemiological studies statistically explore confounding and mediational pathways, randomized controlled trials – the “gold standard” in causal inference – are often impossible or unethical. This challenge necessitates substantial use of animal models, enabling controlled experimentation and use of targeted molecular manipulations clarifying causal pathways. These models are considered in this review when potentially useful to understand human processes. An additional challenge has been the reliance on retrospective self-reporting of ELA in many studies. Such reports may agree only moderately with prospective measures, and could be more prone to bias, though both types of measures tend to predict similar disease and social outcomes [20]. We therefore focus on the direction (versus size) of effects and on physiological mechanisms, and prioritize studies using prospective, longitudinal designs.
Search strategy
We identified peer-reviewed, academic literature from multiple databases, including PubMed, Medline, and PsycINFO, using search terms specifying timing in early life (e.g., early, child*, infan*) and adverse exposures (e.g., advers*, psychological stress, maltreat*), as well as terms for specific physiological axes as appropriate. Priority was given to more recent studies, major reviews, and prospective human studies. Cross-sectional and animal studies were included where prospective human evidence was unavailable.
Biological embedding by physiological axis
ELA has diverse effects across neural, endocrine, immune, metabolic, and gut microbial axes, as reviewed below. Table 1 summarizes key findings, while Fig. 1 provides a working conceptual model of ELA’s biological embedding.
Table 1.
Selected effects of early life adversity (ELA) on physiological functioning
| Examples of physiological changes observed after ELA | Overall clinical and functional effects | Key reviews |
|---|---|---|
| Brain structure and activity | ||
| Structural variation in gray and white matter | Increased risk of: - Impairments in executive functioning (e.g., working memory, cognitive control) - Impaired emotion regulation and social functioning - Adverse effects on reward processing and stress regulation (e.g., hippocampus, amygdala, PFC) may increase risk of mood and substance use disorders |
Bick & Nelson, 2016 [21] Hart & Rubia, 2012 [24] McEwen, 2013 [50] Nemeroff et al., 2016 [25] |
| 1) Changes in local/global gray matter volumes a) Some evidence for widespread, global gray matter change b) Decreased gray matter volume of PFC and hippocampus c) Complex volumetric changes in amygdala | ||
| 2) Changes in local/global white matter volume and microstructure | ||
| a) Complex white matter volumetric changes in frontal lobes b) Microstructural variation in various white matter tracts that may impair communication between brain regions | ||
| Functional variation in brain activity and functional connectivity | ||
| 3) Aberrant amygdala reactivity to emotional stimuli | ||
| 4) Alterations in amygdala-PFC connectivity | ||
| Altered neurotransmitter metabolism or production | ||
| 5) Potential altered neurotransmitter levels/signaling involving key molecules, e.g., serotonin, dopamine, GABA, glutamate | ||
| Neuroendocrine (HPA) stress response axes | ||
| Hyper-responsiveness | - Both HPA hyper- or hypo- reactivity are characteristic patterns generating excess “allostatic load,” linked to cardiovascular disease, metabolic syndrome, accelerated cellular aging, and various psychopathologies - Downstream effects of aberrant cortisol levels (e.g., neurotoxicity, heightened inflammation, metabolic dysregulation) may drive pathology across other axes |
Doom & Gunnar, 2015 [36] Heim & Binder, 2012 [87] |
| 1) Enhanced ACTH and cortisol response to stress/stimulation | ||
| 2) Evidence of impaired GR-mediated feedback inhibition | ||
| Hypo-responsiveness | ||
| 4) Blunted HPA response (ACTH and cortisol) to stress/stimulation | ||
| 5) Heightened ACTH response with inappropriately blunted cortisol (normal or low) | ||
| Altered basal diurnal rhythms | ||
| 3) Elevated, or suppressed, average cortisol/CRF | ||
| 6) Complex changes to diurnal cortisol rhythms (e.g., lower morning and flatter decline, or higher morning and steeper decline) | ||
| Autonomic functioning | ||
| 1) Complex patterns of sympathetic- or parasympathetic-predominant imbalance of reactivity to acute stress, with alterations in responsiveness and counter-regulatory control | - Both parasympathetic- or sympathetic-predominant autonomic imbalances are linked to diseases of elevated “allostatic load” (discussed above) | Alkon et al., 2012 [55] El-Sheikh et al., 2009 [56] |
| 2) Elevated or decreased sympathetic or parasympathetic basal tone | ||
| Immunity and inflammation | ||
| 1) Systemic immune suppression (e.g., impaired cellular immunity) | - Chronic inflammation linked to increased cardiometabolic and other disease risk - Immunosuppression linked to impaired control of infectious/neoplastic threats |
Slopen et al., 2012 [66] Baumeister et al., 2016 [67] |
| 2) Chronic basal inflammation (e.g., elevated CRP, TNF- α, IL-6) 3) Heightened inflammatory reactivity | ||
| Metabolism | ||
| 1) Impaired peripheral glucose handling with insulin resistance | - Heightened risk of type 2 diabetes, obesity, hyperlipidemia, or other metabolic disease | Maniam et al., 2014 [70] |
| 2) Altered fat metabolism with dyslipidemia | ||
| Microbiome functioning (emergent evidence, animal models only to date) | ||
| 1) Transient microbiome perturbations after stress in infancy linked to aberrant immune development | - May contribute to inflammation, immune-suppression, and/or neurodevelopmental risk | O’Mahony et al., 2015 [74] |
| 2) Possible durable microbiome changes in adults after early stress | ||
PFC prefrontal cortex, ACTH adrenocorticotropic hormone, GR glucocorticoid receptor, CRF corticotropin releasing factor, CRP C-reactive protein, TNF tumor necrosis factor, IL-6 interleukin-6, HPA hypothalamic-pituitary-adrenal
Fig. 1.
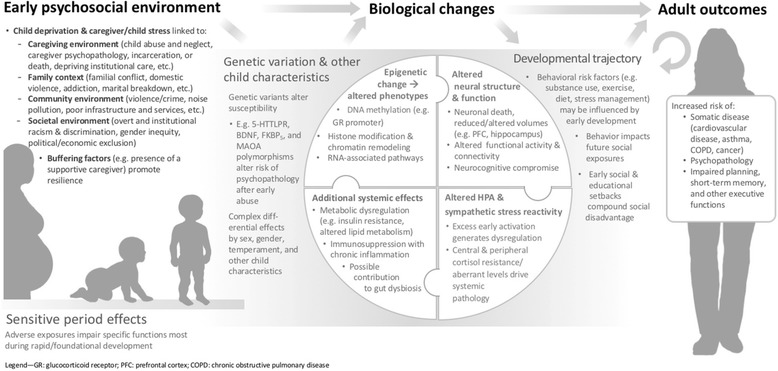
Conceptual model of the biological embedding of early psychosocial adversity. Adapted from [113]
Axis 1: The brain
Human brain maturation is a protracted process beginning in fetal life and continuing into early adulthood [21]. Dramatic growth in gray and white matter occurs in the first 2 years of life, when the brain attains 80–90% of its adult volume before continuing to grow at an attenuated rate [22, 23]. Alongside growth, experience-dependent neural pruning eliminates inactive synapses. Anatomically, the brain matures “from the bottom up,” beginning with primitive brainstem structures and progressing anatomically in anterior-posterior and inferior-superior directions, culminating with the prefrontal cortex (PFC). Functional development similarly progresses from basic sensory and motor capacities to subsequent language and executive functioning (e.g., cognitive control, working memory), and ultimately higher cognition [16]. Normative neurodevelopment thus enables environmental adaptation and progressively complex cognition, but leaves the brain susceptible to negative exposures for an extended period of time.
Extensive literature links ELA to pervasive, quantifiable variation in brain structure and function [15, 21, 24, 25]. Investigation has preferentially examined “stress sensitive” areas dense with glucocorticoid receptors, including limbic structures (e.g., hippocampus and amygdala) key to memory, learning, and emotion regulation, as well as the PFC, critical for higher cognition, executive functioning, and “top-down” control of lower regions [26]. Studies of adolescents and adults provide consistent evidence of smaller PFC gray matter volumes after ELA, paralleling findings from experimental animal models designed to demonstrate causality [21, 24, 25]. Smaller hippocampal volumes have been consistently observed in ELA-exposed adults, though not children, reflecting potential latent effects on a slow-developing structure. Amygdala volumetric effects are complex, including both increases and decreases, likely moderated by exposure timing and type [21, 27].
Considering potential embedding mechanisms, the “neurotoxicity hypothesis” posits that early elevation of stress mediators, particularly glucocorticoids, kills or impedes growth of neurons in stress-sensitive regions via mechanisms including oxidative damage [28]. Stress mediators potentially linked to neurotoxicity in humans include cortisol as well as inflammatory cytokines, excitatory amino acids (e.g., glutamate), and various other molecules (e.g., brain-derived neurotrophic factor (BDNF) and endogenous opioids) [29]. Oxidative stress during early neurodevelopment may also disrupt (delay or extend) neural sensitive periods [30]. Considering epigenetics, experimental animal models show altered expression of genes implicated in basic neurodevelopmental processes (e.g., cell adhesion, sensitive period closure) [31]. Human studies of ELA show genome-wide methylation changes as well as gene-specific effects on neural signaling molecules important to psychological health and neural function, for instance serotonin, glutamate, dopamine, catechol-O-methyl transferase (COMT), and BDNF [19].
Beyond excess stress, environmental deprivation may also play a role in neurodevelopmental compromise, for instance, among children in low-quality institutional care [32]. Broadly, absence of normative psychosocial stimuli (e.g., language exposure or caregiver interaction) during experience-dependent development is proposed to promote excessive synaptic pruning [33]. Indeed, children raised in depriving institutions in infancy show globally decreased cortical thickness [34], a finding possibly paralleled by reduced brain-wide dendritic arborization, spine density, and brain volume in rodent models of early deprivation (e.g., rearing in single-occupancy cages) [35]. Nevertheless, “depriving” exposures (such as caregiver absence) generally evoke potent stress responses [36] while stress mediators regulate synaptic plasticity [37], complicating efforts to discern whether the observed structural changes reflect excess pruning (versus, for instance, glucocorticoid neurotoxicity) and if these mechanisms are, in fact, independent of stress-mediated pathways.
The neurodevelopmental changes described may have far-reaching functional and health implications. Studies suggest that neural-structural changes mediate ELA effects on depression [38], while sensitive period disruption may contribute to schizophrenia and autism pathogenesis [30, 39]. Studies of ELA-associated brain functional changes show deficits in processes including emotion regulation, fear learning, and executive functioning [21]. Functional MRI studies show differences in centrally-driven reward processing that could mediate ELA-related risk of psychopathologies and substance use-related illnesses [40, 41]. Finally, disruption of central stress-regulatory structures may promote neuroendocrine disruption linked to diseases of excess allostatic load [42], as discussed below.
Axis 2: Neuroendocrine stress regulation
ELA broadly impacts stress reactivity as controlled by the hypothalamic-pituitary-adrenal (HPA) and autonomic (sympathetic/parasympathetic) axes. Both axes are under central control by corticolimbic structures, including the PFC, hippocampus, and amygdala [29], and involve common molecular mediators (e.g., corticotropin-releasing factor (CRF), an HPA hormone and autonomic neurotransmitter) [43], suggesting potentially overlapping embedding pathways.
HPA axis
In response to stress, hypothalamic CRF stimulates pituitary adrenocorticotropic hormone (ACTH) release and, in turn, adrenal cortical secretion of glucocorticoids—principally cortisol in humans and corticosterone in many animal species. Glucocorticoids trigger diverse systemic homeostatic responses while exerting negative feedback on the axis. In human studies and animal experimentation, ELA consistently predicts HPA dysregulation generally persisting into adulthood, including patterns of hyper-reactivity, suggesting potential acquired resistance to glucocorticoid negative feedback [29], or hypo-reactivity, suggesting possible attenuated stress sensitivity or exaggerated axis suppression [44]. Differential patterns of dysregulation may reflect variation in factors including timing and type of ELA [45], genotype [46], current age [29], and concurrent psychopathology [47]. Importantly, HPA hyper- and hypo-reactivity both represent prototypical patterns associated with excess allostatic load, and both predict human stress-related chronic illnesses, including cardiovascular, metabolic, and psychiatric diseases linked epidemiologically to ELA [15, 29, 48]. Glucocorticoid dysregulation may also promote oncogenic tumor cell microenvironments (in part via pro-inflammatory effects, as discussed below), fostering growth, migration, invasiveness, and angiogenesis [49], thus potentially contributing to observed links between ELA and cancers [7].
Considering potential mechanisms of HPA changes, animal models of early stress have demonstrated altered expression of the glucocorticoid receptor (GR) (involved preferentially in axis downregulation) and receptors for CRF, ACTH, and other key molecules [50]. In particular, altered serotonin signaling in rats receiving unfavorable maternal care has been shown to induce hypermethylation (silencing) of the GR promoter and related genes [51]. Similar GR hypermethylation was subsequently demonstrated in hippocampal tissue [52] and peripheral lymphocytes [53] of humans maltreated in childhood. Other epigenetic changes shown in animals include genes controlling other key stress-related receptors (e.g., for CRF) and hormones (e.g., CRF, AVP, ACTH, and cortisol), as well as in neurotransmitters/neuropeptides in stress-regulatory brain regions [54].
Autonomic axis
In response to stress, amygdala signaling initiates sympathetic activation via the brainstem, terminating in adrenergic signals to end organs (e.g., liver, heart, digestive tract, and pancreas) and induction of adrenal medullary epinephrine/norepinephrine release producing the prototypical “fight or flight” response. The parasympathetic branch exerts countervailing control, and dynamic sympathetic-parasympathetic balance shapes overall stress physiology [55]. Experimental animal models and observational human studies have consistently linked ELA to autonomic dysregulation, including both hyper- and hypo-responsiveness of sympathetic or parasympathetic pathways. Imbalance in either sympathetic- or parasympathetic-dominant directions again represent manifestations of excess allostatic load and predict stress-related diseases, including heart disease, obesity, type 2 diabetes, cancers, and psychopathologies [55]. Pathology associations may differ by pattern of autonomic imbalance. Several studies, for instance, found that attenuated sympathetic reactivity correlated with antisocial behavior with callous-unemotional traits in ELA-exposed boys, while heightened reactivity correlated with antisocial behavior without callous-unemotional traits [56]. Such findings remain exploratory, and the direction of causal links, if present, is unclear. Among few studies specifically examining mechanisms of autonomic changes, one found that volumetric changes in the amygdala, hippocampus, and PFC statistically mediated autonomic changes as well as risk of psychopathology [57]. Overlapping regulation by corticolimbic structures and core molecular mediators (e.g., CRF) suggests that some HPA-related alterations may also impact autonomic functioning.
Axis 3: Immune functioning
Innate and adaptive immune responses work jointly to control exogenous (e.g., microbial) and endogenous (e.g., necrotic/neoplastic) threats in processes dependent upon inflammatory mediators. When chronically elevated, however, inflammatory mediators contribute to immunosuppression as well as oxidative stress and cytotoxicity [58]. ELA has been linked in human studies and animal experimentation to chronic inflammation [59] and low-level immunosuppression, including impairment of mucosal immunity in children [60] and cellular immunity (e.g., poorer control of latent viral infection) in adolescents [61] and adults [62]. Important work has characterized a "pro-inflammatory phenotype", involving exaggerated cytokine response to bacterial challenge and progressive glucocorticoid receptor desensitization, among ELA-exposed individuals [63]. Considering potential mechanisms, acquired peripheral glucocorticoid resistance may attenuate cortisol’s anti-inflammatory effects [18]. Meanwhile, genome-wide analysis in ELA-exposed individuals has shown increased expression of genes controlling not only cortisol output, but also the activity of key inflammatory mediators like NF-κβ and interleukin-6 (IL-6) [64], with potential antecedents including developmental programming of monocytes for excessive inflammatory responses [18, 65]. Finally, emerging research posits that ELA-related gut dysbiosis may contribute to chronic inflammation, as discussed below.
Health implications of immunosuppression include compromised control of infection and other threats. Meanwhile, inflammatory mediators linked to ELA (e.g., IL-1, IL-6, TNF-alpha, CRP, and fibrinogen) are implicated in risk of cardiovascular and metabolic disease [17, 66, 67]. Inflammation is also a proposed mechanism mediating ELA effects on later depression, age-related diseases [3], neurodevelopmental changes [40], cancers [49], and other systemic effects discussed. Considering cancer risk in particular, immunosuppression impairs control of latent oncogenic viruses [68], while inflammation further promotes oncogenic tumor microenvironments in conjunction with stress mediators, as discussed above [49].
Axis 4: Metabolic health
Interest in metabolic embedding of ELA stems from epidemiological [1, 69] and clinical [70] studies linking ELA to obesity, dyslipidemia, and type 2 diabetes, raising questions about possible causal pathways. While research directly linking ELA to altered development of metabolic physiology remains emergent (versus clear indirect impacts via, e.g., chronic inflammation [3]), potential loci of embedding are multiple. Feeding-related regulation involves, among other networks, dopaminergic reward pathways under top-down control by the PFC, and hypothalamic nuclei integrating nutrient signals to induce hunger or satiety, and systemic shifts between catabolism and anabolism [71]. Peripheral energy homeostasis involves an interplay of anabolic (e.g., insulin) and catabolic (e.g., cortisol, glucagon, epinephrine/norepinephrine) signals promoting increased glycemia and tissue insulin resistance.
Considering mechanisms of potential ELA effects, chronic inflammation, as well as excess catabolic signaling in those with hypercortisolemia, are proposed to drive metabolic dysfunction. Preliminary models also posit that ELA may durably alter hepatic expression of cortisol-activating and -metabolizing enzymes, enhancing tissue-level insulin resistance even in those who later suppress hypercortisolemia [70]. Furthermore, a previous study linked ELA to altered central reward processing promoting excess food intake in some individuals [72]. Additional work is needed to explore the hypothesized pathways.
Axis 5: The microbiome
The gut microbiome represents the collective genome of nearly 100 trillion commensal microorganisms, including over 1000 bacterial species. Dysbiosis, a pathogenic disruption of gut microbial composition or host-microbe interactions, is implicated in diseases including obesity, type 2 diabetes, and depression [73]. While genetically influenced, gut microbial composition responds to factors including stress, diet, infection, drugs, and toxins, making the gut a potential mediator between environment and disease. Various previous studies have suggested profound microbiome effects on neuroendocrine and immune function, such that dysbiosis could compound ELA-related changes including cortisol dysregulation and chronic inflammation [73–77]. Furthermore, growing literature on the “gut-brain axis” describes microbial influence on neural development and functioning [78]. Pathways of influence may include microbial vagus nerve activation, neural signaling by microbial metabolites or molecular patterns, heightened inflammation with downstream neural effects, and induction of epigenetic changes [77, 79, 80]. In animal experimentation and some small human studies, dysbiosis has also been shown to impact relevant brain and behavioral parameters, including cortisol regulation, depressive and anxious symptomatology, and social functioning [77, 79].
Whether ELA itself produces dysbiosis is a question of ongoing interest [74]. A study in rodents found that infant maternal separation durably altered fecal microbiota and increased later inflammatory reactivity [81]. Work in monkeys, meanwhile, found that transient dysbiosis triggered by infant maternal separation predicted durable immune dysfunction, supporting the possibility of early microbiome effects on development in other axes [82]. If human research replicates such findings, the health implications may be considerable.
Interactive effects across axes
The above evidence illustrates how ELA-related physiological changes generate feed-forward synergies; for instance, if glucocorticoid toxicity compromises brain regions tasked with stress regulation [29], or stress-related inflammation further disrupts neural, gut microbial, and metabolic axes to compound HPA dysregulation and further inflammation [83]. Meanwhile, brain functional changes (e.g., altered executive functions and reward processing) may shape health-related behaviors and ongoing social risk exposures [84]. Synergistic effects of ELA thus produce wide-ranging physiological changes marked by aberrant neural function, endocrine activity, chronic inflammation, immunosuppression, insulin resistance and, potentially, dysbiosis. These changes are substantially mediated by altered development of stress-response systems; when acute, activation of these systems generates adaptive changes across body systems (e.g., immune, metabolic, cardiovascular) to address threats. However, chronic or excessive activation contributes to the pathogenic physiological “wear and tear” described within the allostatic load paradigm [15, 29]. In full, ELA-induced changes may mediate epidemiological links to key diseases, including, among others, obesity, dyslipidemia, type 2 diabetes, atherosclerosis, asthma, thromboembolic events (myocardial infarction, stroke), cancer onset and progression, as well as addictions, psychopathology, and adverse social outcomes [1–6, 18].
Differential susceptibility to adversity
Despite described trends, outcomes among ELA-exposed individuals are markedly diverse. A rich literature describes this apparent differential susceptibility to adversity, as selectively reviewed in Table 2 and recommended as further reading [85, 86]. Some observed modifiers of ELA effects include genetics [25, 87–89], child sex and/or gender [19, 90, 91], exposure features (e.g., timing, nature, and intensity) [21, 25], and the presence of other risks or protective factors [36]. Of note, substantial literature suggests that nurturing caregiving is a particularly powerful protective factor mitigating ELA associations with physiological parameters, including elevated allostatic load [92, 93], inflammation [94], cortisol reactivity [95], and cellular aging [96]. Considering neurodevelopment, a prospective study found that caregiving behaviors mediated the association of early childhood socioeconomic stress with hippocampal volumetric change [97]. Such studies suggest that caregiving quality critically shapes psychosocial risk trajectories and developmental effects.
Table 2.
Selected effect modifiers
| Modifier | Examples of findings | Further reading |
|---|---|---|
| Genetic variability | • Genetic polymorphisms found to moderate associations between ELA and various outcomes; Specific examples of outcomes impacted with implicated genes include: | |
| o Emotional and neuroendocrine stress reactivity: 5-HTTLPR | Lester et al., 2006 [86] | |
| o Inflammatory response to stress: 5-HTTLPR | Fredericks et al., 2010 [88] | |
|
o Common forms of psychopathology, including depression, ADHD, and substance addiction: NR3C1, CRHR1, OXTR, 5-HTTLPR, HTR3A, DRD2, MAOA, BDNF, COMT o Atherosclerosis risk: MAOA |
Nemeroff et al., 2016 [25] Heim & Binder, 2012 [87] Zhao et al., 2013 [89] |
|
| Child sex and gender | • Complex sex differences in HPA and autonomic dysregulation after early stress observed in animals and humans | Essex et al., 2013 [19] |
| • Differential effects of maternal vs. paternal stress on boys vs. girls leads some to posit ELA effect moderation by socially embedded gender roles | ||
| • Genetic moderators of the effects of ELA may be sex and/or gender specific o Meta-analysis found stronger effect of MAOA genotype on psychopathology in boys o Different polymorphism on the 5-HTTLPR gene have been linked with increased risk of depression following ELA in males vs. females |
Kim-Cohen et al., 2006 [90] Brummet et al., 2008 [91] |
|
| Other child characteristics | • Pre-existing health conditions, e.g., prematurity, poor physical health status, etc. alter social and physiological consequences of ELA | Doom & Gunnar, 2015 [36] |
| • Child temperament, sensitivity to the environment, and emotion processing are associated with risk for psychopathology and may affect the ways in which children respond to adversity | Lester et al., 2006 [86] | |
| Exposure characteristics | • Characteristics of the exposure, including type (e.g., sexual, physical, emotional abuse, or neglect), chronicity, and intensity, modify associations with physical and mental health outcomes | Nemeroff et al., 2016 [25] |
| • Exposures occurring during early sensitive periods can have heightened impacts on specific developmental domains leading to “timing effects” | Bick & Nelson, 2016 [21] | |
| Social context and caregiving | • Family structure and stability, birth order, caregiver stress and social support, community and societal context may modify effects of specific adversities | Doom & Gunnar, 2015 [36] |
| • Presence of a dependable, supportive caregiver may “buffer” children from effects of otherwise adverse environment | ||
| Cumulative occurrence | • Dose-response relationship between number of adversities and health and social effects are observed in large epidemiological studies | Felitti et al., 1998 [1] Danese et al., 2009 [3] |
ADHD attention deficit hyperactivity disorder, HLA hypothalamic-pituitary-adrenal, ELA early life adversity
Clinical, research, and public health applications
The evidence linking ELA to lifelong health is substantial, with important implications for clinical practice and public health summarized in Table 3. We highlight four recommendations in particular. First, we suggest that screening for ELA should become a routine part of clinical care for children and adults. This aspect of the “developmental history” can provide information about a patient’s risk of major pediatric and adult diseases, facilitating social support, protective intervention, and/or decisions about disease screening and prevention.
Table 3.
Proposed clinical implications of reviewed findings
| Practitioner activity | Recommendations | Recommended resources |
|---|---|---|
| Understanding disease etiology and risk | Consider how ELA contributes to a patient’s risk of common health problems, e.g.: • Mental health disorders: Depression, anxiety, substance use disorders, post-traumatic stress disorder, psychosis • Cardiovascular disease: Ischemic heart disease, hypertension, atherosclerosis • Metabolic pathology: Obesity, type 2 diabetes, dyslipidemia, metabolic syndrome • Neoplasm: Breast, liver, lung cancers |
Results of major epidemiological studies assessing health effects of ELA [1–6] Further reading suggested throughout |
| Screening | • Screen for ELA history • Assess social service and protection needs • Consider ELA history when assessing risk and screening for ELA-related diseases or developmental needs |
Adverse Childhood Experiences Questionnaire [1] WHO Adverse Childhood Experiences International Questionnaire [104] American Academy of Pediatrics Resilience Project Clinical Screening Tools [105] |
| Intervention |
General practice
Provide access to: • Mental healthcare • Early prevention and treatment for other ELA-related diseases • Social services and poverty alleviation • Violence response and prevention interventions Pediatric practice • Family and caregiver support programs • Early development interventions • Services to prevent or respond to ELA exposures, including child protection services |
WHO Preventing Child Maltreatment guide [106] WHO mhGAP Intervention Guide [107] Interventions resources to support healthy child development from Frontiers of Innovation – Center on the Developing Child at Harvard University [108] |
| Transforming care models | Adopt best-practices from “medical home models” to support ELA-exposed patients, including strategies promoting: • Patient- and family-centered wraparound care • Cultural competency • Enhanced access and follow-up |
National Center for Medical Home Implementation Tools & Resources [109] |
| Advocacy | Incorporate evidence on ELA into advocacy relating to: • Access to mental health services • Poverty alleviation, criminal justice reform, and violence prevention • Fair parental leave and high-quality child care • Immigration and refugee policies protecting children and families |
WHO guidance package on Advocacy for Mental Health [110] United Nations Children’s Fund policy advocacy and children's rights tools [111] Children’s Defense Fund policy campaign resources [112] |
ELA early life adversity, WHO World Health Organization
Second, screening for ELA must be matched by investment in scale-up of known effective interventions promoting health by addressing ELA. Considerable evidence suggests that caregiving-focused interventions, for instance, may mitigate the physiological effects of ELA. Some parameters improved by caregiving-focused interventions in longitudinal research include ELA-associated chronic inflammation [98], telomere shortening (accelerated genetic aging) [99], and gray matter volumetric changes [100]. Similarly, cortisol reactivity appears to be sensitive to caregiver-targeted interventions and to psychological support interventions with ELA-exposed individuals [101]. Scale-up investments must include quality monitoring and ongoing assessment of impact at scale. Assessments must disaggregate effects by population subgroups, for instance, as defined by culture, SES, religion, race, or ethnicity, to identify diverse needs [102].
Third, investigators must continue to test new intervention strategies to prevent or reduce the physiological effects of ELA. New approaches should be ever more accurately targeted (e.g., based on genotype-dependent response variation), scalable, effective, and evidence based, making use of the rich literature on biological embedding. In particular, novel approaches are needed to reach the most vulnerable families often least impacted by existing strategies [102]. Efforts should be aided by ongoing development of biomarkers of ELA [103], which can be used to track intervention effects and optimize timing and targeting. Additional research priorities include better characterization of ELA-microbiome links, and consistent use of prospective ELA measures.
Finally, we recommend that practitioners across multiple social sectors recognize ELA as a common soil giving root to various manifestations of poor health over the life course, and better align strategies to advance child welfare and public health. Disease prevention paradigms must move beyond proximal focus on risk behaviors (e.g., diet, substance use) for specific diseases towards life-course models accounting for early influences on lifelong health. Efforts require coordination across health, social services, education, justice, child protection, and other sectors to improve alignment around children’s needs. Among others, relevant priorities might include improving access to mental health services, childcare, and parental leave, expanding family poverty programs, seeking immigration and criminal justice practices that avoid separating children from nurturing caregivers, and addressing racial inequities impacting children.
Conclusions
The findings reviewed here explore various biological mechanisms that may explain links between adverse childhood experiences and disease. These insights can inform efforts to improve health across the life course. As the emergence of novel tools, such as biomarkers of early adversity, drives a new wave of intervention research, strong collaboration is needed between medical and public health practitioners, families, and communities based on a deep appreciation for the effects of early adversity. The understanding of the physiology of biological embedding, as explored here, supports those leading practice-transforming efforts.
Acknowledgements
None.
Funding
Writing of this review was supported by grants MH078829 and MH078829 from the National Institutes of Health (CN), grant OPP1111625 from the Bill and Melinda Gates Foundation (CN, SJ, and AB), and an award from The Sackler Scholar Programme in Psychobiology, an initiative of the Sackler Foundation, to AB.
Availability of data and materials
Not applicable.
Authors’ contributions
AB contributed to conceptualization, drafted the majority of the initial manuscript, and finalized edits. SJ contributed to conceptualization and drafting of the review and offered critical comments and edits. CN oversaw conceptualization of the review, provided scientific guidance, and offered critical comments. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sarah K. G. Jensen, Email: sarah.jensen@childrens.harvard.edu
Charles A. Nelson, III, Email: charles_nelson@harvard.edu.
References
- 1.Felitti VJ, Anda RF, Nordenberg D, Williamson DF, Spitz AM, Edwards V, et al. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults: The Adverse Childhood Experiences (ACE) Study. Am J Prev Med. 1998;14(4):245–58. doi: 10.1016/S0749-3797(98)00017-8. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Croft JB, Chapman DP, Perry GS, Greenlund KJ, Zhao G, Edwards VJ. Relationship between adverse childhood experiences and unemployment among adults from five U.S. states. Soc Psychiatry Psychiatr Epidemiol. 2013;48(3):357–69. doi: 10.1007/s00127-012-0554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danese A, Moffitt TE, Harrington H, Milne BJ, Polanczyk G, Pariante CM, et al. Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers. Arch Pediatr Adolesc Med. 2009;163(12):1135–43. doi: 10.1001/archpediatrics.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giovanelli A, Reynolds AJ, Mondi CF, Ou SR. Adverse childhood experiences and adult well-being in a low-income, urban cohort. Pediatrics. 2016;137(4):e20154016. doi:10.1542/peds.2015-4016. [DOI] [PMC free article] [PubMed]
- 5.Flaherty EG, Thompson R, Dubowitz H, et al. Adverse childhood experiences and child health in early adolescence. JAMA Pediatr. 2013;167(7):622–9. doi: 10.1001/jamapediatrics.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richards M, Wadsworth MEJ. Long term effects of early adversity on cognitive function. Arch Dis Child. 2004;89(10):922–7. doi: 10.1136/adc.2003.032490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly-Irving M, Mabile L, Grosclaude P, Lang T, Delpierre C. The embodiment of adverse childhood experiences and cancer development: potential biological mechanisms and pathways across the life course. Int J Public Health. 2013;58:3–11. doi: 10.1007/s00038-012-0370-0. [DOI] [PubMed] [Google Scholar]
- 8.Kelly-Irving M, Lepage B, Dedieu D, Bartley M, Blane D, Grosclaude P, Lang T, Delpierre C. Adverse childhood experiences and premature all-cause mortality. Eur J Epidemiol. 2013;28:1–14. doi: 10.1007/s10654-013-9832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barboza Solís C, Kelly-Irving M, Fantin R, Darnaudéry M, Torrisani J, Lang T, Delpierre C. Adverse childhood experiences and physiological wear-and-tear in midlife: findings from the 1958 British birth cohort. Proc Natl Acad Sci U S A. 2015;112(7):E738–46. doi: 10.1073/pnas.1417325112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hertzman C. Putting the concept of biological embedding in historical perspective. Proc Natl Acad Sci U S A. 2012;109(Supplement 2):17160–7. doi: 10.1073/pnas.1202203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graignic-Philippe R, Dayan J, Chokron S, Jacquet AY, Tordjman S. Effects of prenatal stress on fetal and child development: a critical literature review. Neurosci Biobehav Rev. 2014;43:137–62. doi: 10.1016/j.neubiorev.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 12.Martikainen P, Bartley M, Lahelma E. Psychosocial determinants of health in social epidemiology. Int J Epidemiol. 2002;31(6):1091–3. doi: 10.1093/ije/31.6.1091. [DOI] [PubMed] [Google Scholar]
- 13.Hillis S, Mercy J, Amobi A, Kress H. Global prevalence of past-year violence against children: a systematic review and minimum estimates. Pediatrics. 2016;137(3):1–13. doi: 10.1542/peds.2015-4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moussavi S, Somnath S, Verdes E, Tandon A, Patel V, Ustun B. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet. 2007;370(9590):851–8. doi: 10.1016/S0140-6736(07)61415-9. [DOI] [PubMed] [Google Scholar]
- 15.McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431–45. doi: 10.1146/annurev-med-052209-100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fox SE, Levitt P, Nelson CA. How the timing and quality of early experiences influence the development of brain architecture. Child Dev. 2010;81:28–40. doi: 10.1111/j.1467-8624.2009.01380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avitsur R, Levy S, Goren N, Grinshpahet R. Early adversity, immunity and infectious disease. Stress. 2015;18(3):289–96. doi: 10.3109/10253890.2015.1017464. [DOI] [PubMed] [Google Scholar]
- 18.Miller GE, Chen E, Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychol Bull. 2011;137:959–97. doi: 10.1037/a0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Essex MJ, Boyce WT, Hertzman C, Lam LL, Armstrong JM, Neumann SM, Kobor MS. Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. 2013;84:58–75. doi: 10.1111/j.1467-8624.2011.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reuben A, Moffitt TE, Caspi A, Belsky DW, Harrington H, Schroeder F, et al. Lest we forget: comparing retrospective and prospective assessments of adverse childhood experiences in the prediction of adult health. J Child Psychol Psychiatry. 2016;57(10):1103–12. doi: 10.1111/jcpp.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bick J, Nelson CA. Early adverse experiences and the developing brain. Neuropsychopharmacology. 2016;41:177–96. doi: 10.1038/npp.2015.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knickmeyer RC, Gouttard S, Kang C, Evans D, Wilber K, Smith JK, et al. A structural MRI study of human brain development from birth to 2 years. J Neurosci. 2008;28(47):12176. doi: 10.1523/JNEUROSCI.3479-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deoni SCL, Dean DC, O'Muircheartaigh J, Dirks H, Jerskey BA. Investigating white matter development in infancy and early childhood using myelin water faction and relaxation time mapping. Neuroimage. 2012;63(3):1038–53. doi: 10.1016/j.neuroimage.2012.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hart H, Rubia K. Neuroimaging of child abuse: a critical review. Front Hum Neurosci. 2012;6:52. doi: 10.3389/fnhum.2012.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nemeroff CB. Paradise lost: the neurobiological and clinical consequences of child abuse and neglect. Neuron. 2016;89:892–909. doi: 10.1016/j.neuron.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 26.McEwen BS, Nasca C, Gray JD. Stress effects on neuronal structure: hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology. 2016;41:3–23. doi: 10.1038/npp.2015.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tottenham N, Sheridan MA. A review of adversity, the amygdala and the hippocampus: a consideration of developmental timing. Front Hum Neurosci. 2010;3:68. doi: 10.3389/neuro.09.068.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uno H, Eisele S, Sakai A, Shelton S, Baker E, DeJesus O, Holden J. Neurotoxicity of glucocorticoids in the primate brain. Horm Behav. 1994;28(4):336–48. doi: 10.1006/hbeh.1994.1030. [DOI] [PubMed] [Google Scholar]
- 29.Danese A, McEwen BS. Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol Behav. 2012;106:29–39. doi: 10.1016/j.physbeh.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Do KQ, Cuenod M, Hensch TK. Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophr Bull. 2015;41(4):835–46. doi: 10.1093/schbul/sbv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarro EC, Sullivan RM, Barr G. Unpredictable neonatal stress enhances adult anxiety and alters amygdala gene expression related to serotonin and GABA. Neurosci. 2014;258:147–61. doi: 10.1016/j.neuroscience.2013.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berens AE, Nelson CA. The science of early adversity: is there a role for large institutions in the care of vulnerable children? Lancet. 2015;386(9991):388–98. doi: 10.1016/S0140-6736(14)61131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsen EI. Sensitive periods in the development of the brain and behavior. J Cogn Neurosci. 2004;16:1412–25. doi: 10.1162/0898929042304796. [DOI] [PubMed] [Google Scholar]
- 34.McLaughlin KA, Sheridan MA, Winter W, Fox NA, Zeanah CH, Nelson CA. Widespread reductions in cortical thickness following severe early-life deprivation: a neurodevelopmental pathway to attention-deficit/hyperactivity disorder. Biol Psychiatry. 2014;76(8):629–38. doi: 10.1016/j.biopsych.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLaughlin KA, Sheridan MA, Lambert HK. Childhood adversity and neural development: Deprivation and threat as distinct dimensions of early experience. Neurosci Biobehav Rev. 2014;47:578–91. doi: 10.1016/j.neubiorev.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doom JR, Gunnar MR. Stress in infancy and early childhood: effects on development. In: Wright JD, Ed. International Encyclopedia of the Social & Behavioral Sciences. Amsterdam: Elsevier Science & Technology; 2015. Vol. 23. pp. 577–82.
- 37.Liston C, Gan WB. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc Natl Acad Sci U S A. 2011;108(38):16074–9. doi: 10.1073/pnas.1110444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jensen SK, Dickie EW, Schwartz DH, et al. Effect of early adversity and childhood internalizing symptoms on brain structure in young men. JAMA Pediatr. 2015;169(10):938–46. doi: 10.1001/jamapediatrics.2015.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hensch TK, Bilimoria PM. Re-opening windows: manipulating critical periods for brain development. Cerebrum. 2012;2012:11. [PMC free article] [PubMed] [Google Scholar]
- 40.Nusslock R, Miller GE. Early-life adversity and physical and emotional health across the lifespan: a neuroimmune network hypothesis. Biol Psychiatry. 2016;80:23–32. doi: 10.1016/j.biopsych.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dillon DG, Holmes AJ, Birk JL, Brooks N, Lyons-Ruth K, Pizzagalli DA. Childhood adversity is associated with left basal ganglia dysfunction during reward anticipation in adulthood. Biol Psychiatry. 2009;66(3):206–13. doi: 10.1016/j.biopsych.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tawakol A, Ishai A, Takx RAP, et al. Relation between resting amygdalar activity and cardiovascular events: a longitudinal and cohort study. Lancet. 2017;389(10071):834–45. doi: 10.1016/S0140-6736(16)31714-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–92. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- 44.Lovallo WR. Early life adversity reduces stress reactivity and enhances impulsive behavior: Implications for health behaviors. Int J Psychophysiol. 2013;90:8–16. doi: 10.1016/j.ijpsycho.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao U, Hammen C, Ortiz LR, Chen L, Poland RE. Effects of early and recent adverse experiences on adrenal response to psychosocial stress in depressed adolescents. Biol Psychiatry. 2008;64:521–6. doi: 10.1016/j.biopsych.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tyrka AR, Price LH, Gelernter J, Schepker C, Anderson GM, Carpenter LL. Interaction of childhood maltreatment with the corticotropin-releasing hormone receptor gene: effects on hypothalamic-pituitary-adrenal axis reactivity. Biol Psyc. 2009;66:681–5. doi: 10.1016/j.biopsych.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA. 2000;284(5):592–7. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- 48.Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554–65. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- 49.Antoni MH, Lutgendorf SK, Cole SW, Dhabhar FS, Sephton SE, McDonald PG, et al. The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nat Rev Cancer. 2006;6(3):240–8. doi: 10.1038/nrc1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McEwen BS. Brain on stress: How the social environment gets under the skin. Proc Natl Acad Sci U S A. 2012;109:17180–5. doi: 10.1073/pnas.1121254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meaney MJ, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci. 2005;28:456–63. doi: 10.1016/j.tins.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 52.McGowan P, Szyf M. The epigenetics of social adversity in early life: Implications for mental health outcomes. Neurobiol Dis. 2010;39:66–72. doi: 10.1016/j.nbd.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 53.Perroud N, Paoloni-Giacobino A, Prada P, Olie E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry. 2011;1(12) doi: 10.1038/tp.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGowan PO, Roth TL. Epigenetic pathways through which experiences become linked with biology. Dev Psychopathol. 2015;27:637–48. doi: 10.1017/S0954579415000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alkon A, Wolff B, Boyce WT. Poverty, Stress, and Autonomic Reactivity. The Oxford Handbook of Poverty and Child Development. New York, NY: Oxford University Press; 2012. [Google Scholar]
- 56.El-Sheikh M, Kouros CD, Erath S, Cummings E, Keller P, Staton L. Marital Conflict and Children's Externalizing Behavior: Interactions between Parasympathetic and Sympathetic Nervous System Activity. Monographs of the Society for Research in Child Development Series. Wiley; 2009. [DOI] [PMC free article] [PubMed]
- 57.Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry. 2009;14:681–95. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- 58.Kanterman J, Sade-Feldman M, Baniyash M. New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol. 2012;22:307–18. doi: 10.1016/j.semcancer.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 59.Fagundes CP, Glaser R, Kiecolt-Glaser JK. Stressful early life experiences and immune dysregulation across the lifespan. Brain Behav Immun. 2013;27:8–12. doi: 10.1016/j.bbi.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shirtcliff EA, Coe CL, Pollak SD. Early childhood stress is associated with elevated antibody levels to herpes simplex virus type 1. Proc Natl Acad Sci U S A. 2009;106(8):2963. doi: 10.1073/pnas.0806660106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Slopen N, McLaughlin KA, Dunn EC, Koenen KC. Childhood adversity and cell-mediated immunity in young adulthood: does type and timing matter? Brain Behav Immun. 2013;28:63–71. doi: 10.1016/j.bbi.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lemieux A, Coe CL, Carnes M. Symptom severity predicts degree of T cell activation in adult women following childhood maltreatment. Brain Behav Immun. 2008;22:994–1003. doi: 10.1016/j.bbi.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller GE, Chen E. Harsh family climate in early life presages the emergence of a proinflammatory phenotype in adolescence. Psychol Sci. 2010;21(6):848–56. doi: 10.1177/0956797610370161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller GE, Chen E, Fok AK, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci U S A. 2009;106(34):14716–21. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Powell ND, Sloan EK, Bailey MT, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via β-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A. 2013;110(41):16574–9. doi: 10.1073/pnas.1310655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Slopen N, Koenen KC, Kubzansky LD. Childhood adversity and immune and inflammatory biomarkers associated with cardiovascular risk in youth: a systematic review. Brain Behav Immun. 2012;26:239–50. doi: 10.1016/j.bbi.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 67.Baumeister D, Akhtar R, Ciufolini S, Pariante C, Mondelli V. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-alpha. Mol Psychiatry. 2016;21(5):642–9. doi: 10.1038/mp.2015.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Godbout JP, Glaser R. Stress-induced immune dysregulation: implications for wound healing, infectious disease and cancer. J Neuroimmune Pharmacol. 2006;1(4):421–7. doi: 10.1007/s11481-006-9036-0. [DOI] [PubMed] [Google Scholar]
- 69.Thomas C, Hypponen E, Power C. Obesity and type 2 diabetes risk in midadult life: the role of childhood adversity. Pediatrics. 2008;121:e1240–9. doi: 10.1542/peds.2007-2403. [DOI] [PubMed] [Google Scholar]
- 70.Maniam J, Antoniadis C, Morris MJ. Early-life stress, HPA axis adaptation, and mechanisms contributing to later health outcomes. Front Endocrinol. 2014;5:73. doi: 10.3389/fendo.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 72.Mason SM, Flint AJ, Field AE, Austin SB, Rich‐Edwards JW. Abuse victimization in childhood or adolescence and risk of food addiction in adult women. Obesity. 2013;21(12):E775–E81. doi: 10.1002/oby.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moloney RD, Desbonnet L, Clarke G, Dinan TG, Cryan JF. The microbiome: stress, health and disease. Mamm Genome. 2014;25:49–74. doi: 10.1007/s00335-013-9488-5. [DOI] [PubMed] [Google Scholar]
- 74.O'Mahony SM, Clarke G, Dinan TG, Cryan JF. Early-life adversity and brain development: Is the microbiome a missing piece of the puzzle? Neuroscience. 2017;342:37–54. doi: 10.1016/j.neuroscience.2015.09.068. [DOI] [PubMed] [Google Scholar]
- 75.Clarke G, O'Mahony SM, Dinan TG, Cryan JF. Priming for health: gut microbiota acquired in early life regulates physiology, brain and behaviour. Acta Paediatr. 2014;103:812–9. doi: 10.1111/apa.12674. [DOI] [PubMed] [Google Scholar]
- 76.Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, et al. Postnatal microbial colonization programs the hypothalamic–pituitary–adrenal system for stress response in mice. J Physiol. 2004;558(1):263–75. doi: 10.1113/jphysiol.2004.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34(46):15490–6. doi: 10.1523/JNEUROSCI.3299-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13:701–12. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- 79.Sampson TR, Mazmanian SK. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 2015;17:565–76. doi: 10.1016/j.chom.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour: Epigenetic regulation of the gut–brain axis. Genes Brain Behav. 2014;13(1):69–86. doi: 10.1111/gbb.12109. [DOI] [PubMed] [Google Scholar]
- 81.O'Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho A, Quiggley EM, et al. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry. 2009;65:263–7. doi: 10.1016/j.biopsych.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 82.Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. 2011;25:397–407. doi: 10.1016/j.bbi.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ganguly P, Brenhouse HC. Broken or maladaptive? Altered trajectories in neuroinflammation and behavior after early life adversity. Dev Cogn Neurosci. 2015;11:18–30. doi: 10.1016/j.dcn.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hertzman C, Boyce T. How experience gets under the skin to create gradients in developmental health. Annu Rev Public Health. 2010;31:329–47. doi: 10.1146/annurev.publhealth.012809.103538. [DOI] [PubMed] [Google Scholar]
- 85.Ellis BJ, Essex MJ, Boyce WT. Biological sensitivity to context: II. Empirical explorations of an evolutionary-developmental theory. Dev Psychopathol. 2005;17:303–28. doi: 10.1017/S0954579405050157. [DOI] [PubMed] [Google Scholar]
- 86.Lester BM, Masten AS, McEwen BS, editors. Resilience in Children. Boston: Blackwell Publications on behalf of the New York Academy of Sciences; 2006. [Google Scholar]
- 87.Heim C, Binder EB. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp Neurol. 2012;233:102–11. doi: 10.1016/j.expneurol.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 88.Fredericks CA, Drabant EM, Edge MD, Tillie JM, Hallmayer J, Ramel W, et al. Healthy young women with serotonin transporter SS polymorphism show a pro-inflammatory bias under resting and stress conditions. Brain Behave Immun. 2010;24(3):350–7. doi: 10.1016/j.bbi.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao J, Bremner JD, Goldberg J, Quyyumi AA, Vaccarino V. MAOA genotype, childhood trauma and subclinical atherosclerosis: a twin study. Psychosom Med. 2013;75(5):471–7. doi: 10.1097/PSY.0b013e318292922a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim-Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, Craig IW, Moffitt TE. MAOA, maltreatment, and gene-environment interaction predicting children's mental health: new evidence and a meta-analysis. Molec Psychiatry. 2006;11(10):903. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- 91.Brummett BH, Boyle SH, Siegler IC, et al. Effects of environmental stress and gender on associations among symptoms of depression and the serotonin transporter gene linked polymorphic region (5-HTTLPR) Behav Genet. 2008;38(1):34–43. doi: 10.1007/s10519-007-9172-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Evans GW, Kim P, Ting AH, Tesher HB, Shannis D. Cumulative risk, maternal responsiveness, and allostatic load among young adolescents. Dev Psychol. 2007;43(2):341–51. doi: 10.1037/0012-1649.43.2.341. [DOI] [PubMed] [Google Scholar]
- 93.Carroll JE, Gruenewald TL, Taylor SE, Janicki-Deverts D, Matthews KA, Seeman TE. Childhood abuse, parental warmth, and adult multisystem biological risk in the Coronary Artery Risk Development in Young Adults study. Proc Natl Acad Sci U S A. 2013;110(42):17149–53. doi: 10.1073/pnas.1315458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen E, Miller GE, Kobor MS, Cole SW. Maternal warmth buffers the effects of low early-life socioeconomic status on pro-inflammatory signaling in adulthood. Mol Psychiatry. 2011;16(7):729–37. doi: 10.1038/mp.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luecken LJ. Parental caring and loss during childhood and adult cortisol responses to stress. Psychol Health. 2000;15(6):841–51. doi: 10.1080/08870440008405586. [DOI] [Google Scholar]
- 96.Brody GH, Yu T, Beach SR. Resilience to adversity and the early origins of disease. Dev Psychopathol. 2016;28:1347–65. doi: 10.1017/S0954579416000894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luby J, Belden A, Botteron K, Marrus N, Harms M, Babb C, et al. The effects of poverty on childhood brain development: the mediating effect of caregiving and stressful life events. JAMA Pediatr. 2013;167(12):1135–42. doi: 10.1001/jamapediatrics.2013.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miller GE, Brody GH, Yu T, Chen E. A family-oriented psychosocial intervention reduces inflammation in low-SES African American youth. Proc Natl Acad Sci U S A. 2014;111(31):11287–92. doi: 10.1073/pnas.1406578111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Drury SS, Theall K, Gleason MM, Smyke AT, De Vivo I, Wong JYY, et al. Telomere length and early severe social deprivation: linking early adversity and cellular aging. Mol Psychiatry. 2012;17:719–27. doi: 10.1038/mp.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brody GH, Gray JC, Yu T, Barton AW, Beach SR, Galván A, et al. Protective prevention effects on the association of poverty with brain development. JAMA Pediatr. 2017;171(1):46–52. doi: 10.1001/jamapediatrics.2016.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Slopen N, McLaughlin KA, Shonkoff JP. Interventions to improve cortisol regulation in children: a systematic review. Pediatrics. 2014;133(2):312–26. doi: 10.1542/peds.2013-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shonkoff JP. Building a new biodevelopmental framework to guide the future of early childhood policy. Child Dev. 2010;81(1):357–67. doi: 10.1111/j.1467-8624.2009.01399.x. [DOI] [PubMed] [Google Scholar]
- 103.Center on the Developing Child at Harvard University. The JPB Research Network on Toxic Stress. 2016. http://developingchild.harvard.edu/science/the-jpb-research-network-on-toxic-stress/. Accessed 1 Nov 2016.
- 104.World Health Organization . Adverse Childhood Experiences International Questionnaire (ACE-IQ) Geneva: WHO; 2014. [Google Scholar]
- 105.American Academy of Pediatrics . The Resilience Project. Elk Grove Village, IL: American Academy of Pediatrics; 2016. [Google Scholar]
- 106.World Health Organization . Preventing Child Maltreatment: A Guide to Taking Action and Generating Evidence. Geneva: WHO; 2006. [Google Scholar]
- 107.World Health Organization . mhGAP Intervention Guide for Mental, Neurological and Substance Use Disorders in Non-specialized Health Settings. Geneva: WHO; 2010. [PubMed] [Google Scholar]
- 108.Center on the Developing Child at Harvard University . Fronteirs of Innovation: Innovation in Action. Cambridge, MA: Harvard University; 2016. [Google Scholar]
- 109.National Center for Medical Home Implementation . Tools & Resources. Elk Grove Village, IL: American Academy of Pediatrics; 2016. [Google Scholar]
- 110.World Health Organization . Advocacy for Mental Health: Mental Health Policy and Service Guidance Package. Geneva: WHO; 2003. [Google Scholar]
- 111.UNICEF . Policy advocacy and partnerships for children's rights. New York, NY: UNICEF; 2016. [Google Scholar]
- 112.Children's Defense Fund . Children's Defense Fund Campaigns. Washington, DC: Children's Defense Fund; 2016. [Google Scholar]
- 113.Bhutta ZA, Guerrant RL. Nelson 3rd CA. Neurodevelopment, nutrition, and inflammation: the evolving global child health landscape. Pediatrics. 2017;139(Suppl 1):S12–S22. doi: 10.1542/peds.2016-2828D. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.