

Abstract
The roles and characteristics of postzygotic single‐nucleotide mosaicisms (pSNMs) in autism spectrum disorders (ASDs) remain unclear. In this study of the whole exomes of 2,361 families in the Simons Simplex Collection, we identified 1,248 putative pSNMs in children and 285 de novo SNPs in children with detectable parental mosaicism. Ultra‐deep amplicon resequencing suggested a validation rate of 51%. Analyses of validated pSNMs revealed that missense/loss‐of‐function (LoF) pSNMs with a high mutant allele fraction (MAF≥ 0.2) contributed to ASD diagnoses (P = 0.022, odds ratio [OR] = 5.25), whereas missense/LoF pSNMs with a low MAF (MAF<0.2) contributed to autistic traits in male non‐ASD siblings (P = 0.033). LoF pSNMs in parents were less likely to be transmitted to offspring than neutral pSNMs (P = 0.037), and missense/LoF pSNMs in parents with a low MAF were transmitted more to probands than to siblings (P = 0.016, OR = 1.45). We estimated that pSNMs in probands or de novo mutations inherited from parental pSNMs increased the risk of ASD by approximately 6%. Adding pSNMs into the transmission and de novo association test model revealed 13 new ASD risk genes. These results expand the existing repertoire of genes involved in ASD and shed new light on the contribution of genomic mosaicisms to ASD diagnoses and autistic traits.
Keywords: autism spectrum disorder, autistic traits, parental mosaicism, single‐nucleotide mosaicism
1. INTRODUCTION
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impairments in social communication and interaction and restricted repetitive behavior or interests (American Psychiatric Association, 2013). Severe subclinical cognitive or behavioral problems have been reported in relatives of patients with ASD (Davidson et al., 2014; Losh, Childress, Lam, & Piven, 2008). Broadly speaking, both genetic and environmental factors contribute to the etiology of ASD (Gaugler et al., 2014; Hallmayer et al., 2011). However, known ASD risk factors explain the cause of the condition for only a fraction of ASD patients, and much of the genetic etiology of ASD remains unknown (Devlin & Scherer, 2012; Ronemus, Iossifov, Levy, & Wigler, 2014; Sanders et al., 2012). Over the last decade, the key role of de novo germline mutations in the etiology of ASD was established, which led to the discovery of many ASD risk genes (De Rubeis et al., 2014; Dong et al., 2014; Iossifov et al., 2014; Kong et al., 2012; Neale et al., 2012; Sanders et al., 2015; Sanders et al., 2012). The Simons Simplex Collection (SSC), a cohort of simplex ASD families designed to facilitate studies of de novo variations (Fischbach & Lord, 2010), remains a valuable resource for genetic studies of ASD.
Genomic mosaicism describes a phenomenon where an individual developed from a single fertilized egg harbors two or more cell populations with distinct genomes resulting from postzygotic mutations (Strachan & Read, 2004). It has long been known that cancer is predominantly caused by postzygotic mutations (Watson, Takahashi, Futreal, & Chin, 2013; Yates & Campbell, 2012). Recently, mosaicism was shown to play a critical role in many noncancer disorders (Biesecker & Spinner, 2013), and sporadic cases of mosaicism have been reported in clinically unremarkable individuals (Huang et al., 2014; Youssoufian & Pyeritz, 2002). Although the genomic characteristics of postzygotic mosaicism have been thoroughly explored in cancer studies (Alexandrov et al., 2013), its characteristics in individuals not afflicted with cancer require further investigation. Evidence suggesting that some heterozygous lethal mutations could be nonlethal in mosaic form and result in noncancer disorders (Bryant & Zornetzer, 1973; Weinstein et al., 2016) supports a distinct etiological contribution of postzygotic mutations from germline mutations to genetic disorders. Mosaic mutations in causal genes of genetic disorders have also been linked to mild phenotype traits of genetic disorders, but not full diagnosis (Parrini, Mei, Wright, Dorn, & Guerrini, 2004; Xu et al., 2015). Finally, many studies have shown that parental mosaicisms could contribute to recurrent risk of genetic disorders in their children, which were previously under‐recognized (Campbell et al., 2014; Parrini et al., 2004; Xiong et al., 2016; Xu et al., 2015).
In recent years, sporadic case reports (Castermans et al., 2008; Fontenelle, Mendlowicz, Bezerra de Menezes, dos Santos Martins, & Versiani, 2004; Meyer, Axelsen, Sheffield, Patil, & Wassink, 2012; Papanikolaou et al., 2006; Sauter et al., 2003) and one larger study (Yurov et al., 2007) linked mosaicism of large‐scale events such as structural variation and copy‐number variation (CNV) to ASD. More recently, a study explored the contribution of mosaic single‐nucleotide variations (SNVs) to ASD using the whole exomes of 2,388 families in SSC (Freed & Pevsner, 2016). They found a strong bias for mosaic mutations in ASD probands relative to their unaffected siblings. However, their detection strategy, which began with a list of previously identified de novo germline mutations, limited the sensitivity of their study for detecting mosaic mutations with a relatively low mutant allele fraction (MAF). They only validated a small subset of their putative mosaic sites by Sanger sequencing or pyrosequencing, and thus had a limited capacity to distinguish mosaic SNVs from heterozygous SNVs (Contini et al., 2015; Huang et al., 2014; Yamaguchi et al., 2015); therefore, de novo germline mutations could not be convincingly ruled out. Their study focused on ASD probands and their siblings, but they did not investigate the roles of mosaicisms from the parents of their subjects.
The aims of our study were to accurately identify and rigorously validate postzygotic single‐nucleotide mosaicisms (pSNMs) in ASD families, explore the genomic characteristics of pSNMs, investigate the etiological contributions of child pSNMs and de novo SNPs with detectable parental mosaicism to ASD, determine the contributions of pSNMs to autistic traits, and discover new ASD risk genes.
2. MATERIALS AND METHODS
2.1. Subjects
pSNMs in children (“child pSNMs”) and pSNMs in parents that were transmitted to their offspring as heterozygous mutations (“de novo SNPs with detectable parental mosaicism”) were collected from the WES data in the SSC, a large collection of simplex ASD pedigrees (Fischbach & Lord, 2010). Complete WES data were obtained for 2,374 families, including 1,776 quartet pedigrees, 598 trio pedigrees, 1,156 from CSHL, 763 from Yale, and 455 from the Eichler dataset (Supp. Table S1). All Yale whole‐blood samples available for validation are listed in Supp. Table S1.
2.2. Detection of child pSNMs and de novo SNPs with detectable parental mosaicism
The bioinformatics pipeline used to detect child pSNMs and de novo SNPs with detectable parental mosaicism is summarized in Supp. Figure S1. The detection scheme followed a MosaicHunter‐based pipeline as described previously (Huang et al., 2014; Huang et al., 2017). Sequence data from three centers were aligned against the GRCh37.p13 human reference genome by BWA (Li & Durbin, 2009), after which Picard (http://broadinstitute.github.io/picard/) and GATK (McKenna et al., 2010) were used to remove PCR duplicates and perform indel realignment and base quality recalibration. Candidate indels were identified by GATK's UnifiedGenotyper, after which the involved regions were masked for subsequent analyses. Finally, the processed reads were utilized as the input of MosaicHunter.
For child pSNMs, all putative sites were first detected using the trio mode of MosaicHunter. Children with dramatically more putative mosaic mutations (>50 putative pSNMs/subject) than the average number were excluded to remove potential noise from technical artifacts. After exclusion of these individuals, 2,361 families remained for the subsequent analyses, including 1,739 quartet pedigrees and 622 trio pedigrees (Supp. Table S1). We observed significantly more single‐stranded G>T mutations than C>A mutations in the Yale dataset only (P<2.2e‐16, binomial test; Supp. Fig. S2A). Among the single‐stranded G>T mutations, approximately 99% had fewer than eight T alleles, whereas more than 90% had a MAF of less than 10% in the WES data. We randomly selected 80 putative pSNMs for validation using PGM amplicon resequencing, which revealed that 97% (62/64) of the validated single‐stranded G>T mutations were false‐positive sites (Supp. Table S2; Supp. Fig. S2B). The excessive single‐stranded G>T mutations might have been due to oxidative DNA damage during the exome library preparation process (Schmitt et al., 2012), so we removed them by applying an additional filter, which removed Yale putative G>T pSNMs with fewer than eight T alleles. Putative pSNMs existing in more than one individual tended to cluster together and occur in genome regions enriched with CNVs or tandem repeats (Supp. Fig. S3); these putative pSNMs were also filtered from the set used for subsequent analyses. Moreover, child pSNMs having mutant alleles in the WES data from either parent were also filtered. All putative child pSNMs are listed in Supp. Table S3.
For de novo SNPs with detectable parental mosaicism, all genomic sites that were heterozygous in the child and mosaic in at least one parent (posterior probability of parental mosaic genotype ≥ 0.05) were first detected by using the trio mode of MosaicHunter. Next, the single mode of MosaicHunter was applied to the WES data of the corresponding mosaic parent to confirm the mosaic genotype. The pSNMs with mutant alleles that were common in the NCBI Single Nucleotide Polymorphism database (dbSNP) (population allele frequency >0.05) or multiple unrelated ASD pedigrees were also filtered. All putative de novo SNPs with detectable parental mosaicism are listed in Supp. Table S3. The variants were submitted to Database of Single Nucleotide Polymorphisms (dbSNP). Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine. dbSNP accession: {ss2137510534, ss2729283267‐ss2729284734}. Available from: http://www.ncbi.nlm.nih.gov/SNP/.
2.3. Validation of pSNMs
Ultra‐deep amplicon‐based resequencing methods are widely used to validate mosaic variants and are highly accurate (Contini et al., 2015; Nota et al., 2013; Sala Frigerio et al., 2015; Yamaguchi et al., 2015). We employed PGM amplicon sequencing of mosaicism (PASM) (Xu et al., 2015), a PGM ultra‐deep amplicon sequencing method, followed by a Bayesian model to validate putative pSNMs identified in Yale's SSC samples. The average depth‐of‐coverage was approximately 10,000X (Supp. Table S4). All primers used for pSNM validation are listed in Supp. Table S5.
In addition to the experimental validation described above, phasing of the putative pSNMs relative to nearby inherited heterozygous SNVs (informative SNPs), as well as SSC whole‐genome sequencing (WGS) data, were also used as auxiliary in silico validation methods for putative pSNMs (Supp. Table S6). Putative pSNMs were phased to nearby high‐quality heterozygous variants (P>0.05, binomial test, mapping quality ≥ 20, base quality ≥ 20) on reads covering putative pSNMs or read pairs to validate their mosaic status and determine their parental haplotype. For WGS data validation, only those variants that were significantly different from the binomial expectation were classified as mosaic sites (P≤ 0.05, binomial test), whereas variants without mutant alleles and variants following the binomial expectation with a probability of 0.5 were classified as homozygous and heterozygous sites, respectively.
2.4. Extending TADA to include pSNMs
The estimation of the Bayes factor for each gene was performed using the strategy described in the original TADA model (He et al., 2013). We combined evidence from PASM‐validated child pSNMs in Yale's SSC samples, as well as all SSC de novo SNPs with detectable parental mosaicism, with de novo SNVs reported by the Autism Sequencing Consortium and Autism Genome Project (Iossifov et al., 2014). We assumed that the data from each type of mutation were independent. In the first step, we estimated the prior parameters of each type of mutation using the strategy described in the original TADA model (He et al., 2013). Next, we computed the Bayes factor of each gene as the product of the Bayes factor from each type of mutation. In the case of combining de novo mutations, child pSNMs and de novo SNPs with detectable parental mosaicism, the Bayes factor was calculated as follows:
where B denovo is the Bayes factor of each gene from de novo loss‐of‐function (LoF) mutations and de novo missense mutations with deleterious effects, which have been described and calculated previously (He et al., 2013; Sanders et al., 2015); B pSNM is the Bayes factor from functional pSNMs with a high MAF; and is the Bayes factor from de novo functional SNPs with detectable parental mosaicism.
We considered the exonic length, base pair composition, and observed mosaic mutation rate of each gene in non‐ASD siblings (Supp. Table S7) to calculate the expected per‐gene mosaic mutation rate, following the strategy of a previous study of de novo mutations in ASD (Sanders et al., 2012). The mean prior relative risk (γ) of child pSNMs and de novo SNPs with detectable parental mosaicism was estimated using their relative burdens in the SSC. Burden (λ) is defined as the ratio of the number of mutations (child pSNMs or de novo SNPs with detectable parental mosaicism) per ASD proband to the number of mutations per unaffected sibling (Table 1). Based on a previous estimation that 1,000 out of 18,665 Refseq genes contribute to ASD (He et al., 2013), 5.4% (π) of human genes would be randomly considered as ASD risk genes under the null hypothesis. As a result, the mean prior relative risks for missense/LoF child pSNMs with a high MAF and de novo missense/LoF mutations with a low MAF and detectable parental mosaicism were 36.35 and 22.22, respectively, according to the following equation (see details in the Supp. Methods of He et al. [2013]):
Table 1.
Increased burden of pSNMs in ASD probands versus unaffected siblings
| Total number of pSNMs | ||||||
|---|---|---|---|---|---|---|
| Proband | Sibling | |||||
| Category | N = 732/2,321 | N = 571/1,779 | OR (95% CI) | P value | ||
| Yale PGM‐validated child pSNMs | Child pSNMs with high MAF | All | 25 | 18 | NA | 0.447 |
| Missense/LoF | 15 | 4 | 5.25 (3.88, 6.62) | 0.022 | ||
| Other | 10 | 14 | NA | 0.926 | ||
| Child pSNMs with low MAF | All | 85 | 71 | NA | 0.606 | |
| Missense/LoF | 39 | 38 | 0.74 (0.10, 1.37) | 0.801 | ||
| Other | 46 | 33 | NA | 0.295 | ||
| SSC de novo SNPs with detectable parental mosaicism (only present in one child) | Parental pSNMs with high MAF | All | 25 | 17 | NA | 0.351 |
| Missense/LoF | 8 | 8 | 0.53 (0, 1.80) | 0.704 | ||
| Other | 17 | 9 | NA | 0.183 | ||
| Parental pSNMs with low MAF | All | 59 | 26 | NA | 0.012 | |
| Missense/LoF | 28 | 10 | 1.45 (0.50, 2.39) | 0.016 | ||
| Other | 31 | 16 | NA | 0.143 | ||
Thee expression profiles of new ASD risk genes in different brain regions and developmental stages were obtained from the GTEx (GTEx Consortium, 2013) Portal on April 20, 2017 and two previous studies (Johnson et al., 2009; Kang et al., 2011).
2.5. Estimating the contributions of pSNMs to ASD in probands from the SSC
Iossifov et al. (2014) previously described a model for de novo variations in which siblings have a baseline de novo mutation rate, whereas probands have the same baseline rate and an additional burden of mutations. They used the “ascertainment differential” to estimate the contribution of de novo mutations to ASD (Iossifov et al., 2014). The same strategy was applied in our study to estimate the contribution of child and de novo SNPs with detectable parental mosaicism to ASD diagnoses in the SSC.
The occurrence rate of validated missense/LoF pSNMs with a high MAF was about 0.02 in affected probands and about 0.007 in unaffected siblings (P = 0.022, Table 1), so the “ascertainment differential” was about 0.013 (0.02‐0.007 = 0.013). Therefore, we estimated that approximately 65.8% of missense/LoF pSNMs with a high MAF in probands contribute to ASD diagnosis. Given that the occurrence rate of pSNMs in probands and the sensitivity of MosaicHunter for detecting child pSNMs with a high MAF in 80X WES data were approximately 40%, we roughly estimated that child pSNMs with a high MAF increased the risk of ASD by approximately 3.4% in SSC probands. Similarly, since the rate of missense/LoF pSNMs among parental pSNMs with a low MAF (only transmitted to one child) was about 0.012 in affected probands and 0.0056 in unaffected siblings (P = 0.016, Table 1), the ascertainment differential was about 0.0064. Therefore, we estimated that approximately 53.4% of parental missense/LoF pSNMs with a low MAF that were only transmitted to probands contribute to ASD diagnoses. Given the occurrence rate of de novo SNPs with detectable parental mosaicism in probands and the sensitivity of MosaicHunter for detecting de novo SNPs with detectable parental mosaicism with low MAF in 80X WES data (approximately 25%), we roughly estimated that de novo SNPs with detectable parental mosaicism increased the risk of ASD by approximately 2.6% in SSC probands. In total, we estimated that pSNMs might contribute to approximately 6% of the ASD diagnoses in the SSC.
2.6. Protein–protein interaction network analysis
Protein–protein interaction networks were analyzed with DAPPLE (Reich et al., 2006; Rossin et al., 2011) and plotted with Cytoscape (Shannon et al., 2003). The input for the analysis was the combination of the ASD risk genes from Sanders et al. (2015), which were extracted using the original TADA‐Denovo model, and new ASD risk genes identified in this study. All genes within each subnetwork were input into DAVID (Huang da, Sherman, & Lempicki, 2009) to analyze network enrichment in gene ontology terms. We also used the STITCH (Kuhn, von Mering, Campillos, Jensen, & Bork, 2008) and STRING (Szklarczyk et al., 2015) tools to analyze the protein–protein interaction network.
3. RESULTS
3.1. Identification of child pSNMs and de novo SNPs with detectable parental mosaicism
We began our study by detecting pSNMs in children (“child pSNMs”) and pSNMs in parents that were transmitted to offspring as heterozygous mutations (“de novo SNPs with detectable parental mosaicism”) from SSC whole‐exome sequencing (WES) data using a Bayesian genotype named MosaicHunter (Huang et al., 2014; Huang et al., 2017) that we had previously developed. We identified 1,248 putative child pSNMs and 285 putative de novo SNPs with detectable parental mosaicism from the WES data of 2,361 SSC families (Supp. Table S3). We were able to obtain whole‐blood DNA samples from 753 of the SSC families from Yale University (“Yale's SSC families”) and performed experimental validation on their 376 putative child pSNMs and 71 putative de novo SNPs with detectable parental mosaicism using ultra‐deep amplicon resequencing by PGM, a method (named PASM) that we had previously reported (Xu et al., 2015). The study was approved by the Internal Review Boards at Peking University and Yale University. 53% (199/376) of the putative child pSNMs and 44% (31/71) of the putative de novo SNPs with detectable parental mosaicism were validated as true (Fig. 1A and B; Supp. Table S4). As shown in Figure 1C, the mosaic allele fractions estimated by PASM had overall good concordance with the allele fractions estimated from WES data, but mostly due to the much higher depth‐of‐coverage, MAFs estimated by PASM had much narrower 95% credible intervals and higher accuracy. Because we could not obtain the rest of the SSC samples, we performed in silico validations of all putative SSC pSNMs using SSC WGS data and phasing information from informative SNPs (see Materials and Methods), and found similar validation rates (Supp. Table S8).
Figure 1.
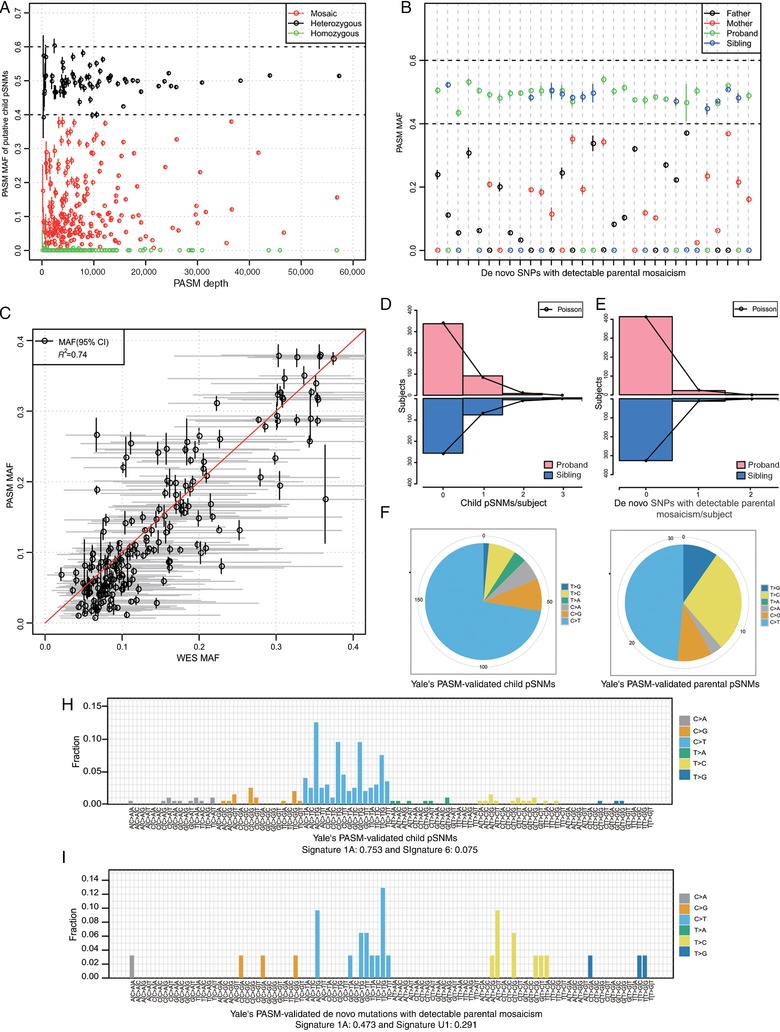
Validation results and characteristics of pSNMs. A and B: PASM validation results of putative child pSNMs (A) and PASM‐validated de novo SNPs with detectable parental mosaicism in all family members (B). C: The mutant allele fractions of the validated pSNMs estimated by PASM had good concordance with the fractions from the WES data. D and E: The per subject distribution of child pSNMs (D) and de novo SNPs with detectable parental mosaicism (E) followed the Poisson distribution. F and G: Decreased ratio of C:G>T:A to T:A>C:G transitions from child pSNMs (F) to de novo SNPs with detectable parental mosaicism (G). H and I: The mutation signatures of child pSNMs (H) and de novo SNPs with detectable parental mosaicism (I) were consistent with the previously reported Signature 1A of cancer mutations (Alexandrov et al., 2013)
3.2. Characteristics of validated child pSNMs and de novo SNPs with detectable parental mosaicism
We studied the characteristics of the experimentally validated pSNMs. The prevalence of the validated pSNMs per subject was 0.152 for child pSNMs and 0.030 for de novo SNPs with detectable parental mosaicism; the pSNM distributions followed the expected Poisson distributions (Fig. 1D and E). We observed no biased parent‐of‐origin for child pSNMs or de novo SNPs with detectable parental mosaicism (Supp. Table S9). We also observed that the ratio of C:G>T:A to T:A>C:G transitions was much lower among de novo SNPs with detectable parental mosaicism compared with that among child pSNMs (Fig. 1F and G), likely due to the biased gene conversion, a meiotic repair bias that favors G/C over A/T alleles (Duret & Galtier, 2009). As shown in Figure 1H and I, the mutational signatures of both child and de novo SNPs with detectable parental mosaicism shared certain similarities with the previously reported mutational signature 1A in human cancers (Alexandrov et al., 2013; Rosenthal, McGranahan, Herrero, Taylor, & Swanton, 2016), with similarity weights of 0.753 and 0.473, respectively. This shared pattern between cancer and noncancer samples could be partly explained by the elevated rate of spontaneous deamination of 5‐methyl‐cytosine at NpCpG trinucleotides (Pfeifer, 2006).
3.3. Contribution of pSNMs to ASD diagnoses
Based on the 199 PASM‐validated pSNMs in 732 probands and 571 siblings from the Yale dataset, the MAFs of probands were significantly higher than those of siblings for missense/LoF pSNMs, but not for other neutral pSNMs (Fig. 2A and B). As demonstrated in Figure 2C, missense/LoF pSNMs with higher MAFs have a higher odds ratio (OR) when probands and siblings are compared, and it reached the significance threshold (P<0.05) when the cut‐off between high MAF and low MAF was 0.2 or higher (Fig. 2C; Table 1). As demonstrated in Figure 2C, we used a sliding‐window strategy to study whether changing the cut‐off value between high and low MAF pSNMs changed our conclusions. Our results suggest that the enrichment of missense/LoF pSNMs with higher MAFs when probands and siblings were compared reached the significance threshold (P<0.05) when the cutoff was 0.2, and the OR became even higher with greater cut‐off thresholds. To further confirm this finding, we applied the Wilcoxon rank sum test to test the burden of functional pSNMs among many possible cutoffs (MAF cutoff = 0, 0.05, 0.10, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, 0.25), and observed a significant burden among many possible cutoffs from 0.15 to 0.25. These results suggested that the enrichment was real and not likely to be explained by chance effects. Interestingly, the significantly increased burden in probands versus siblings was only observed for missense/LoF pSNMs with a high MAF (P = 0.022, one‐sided Wilcoxon rank sum test), but not for missense/LoF pSNMs with low MAF and other pSNMs with neutral functional effects (Fig. 2D; Table 1). We further confirmed our findings using all putative pSNMs identified in 2,321 probands and 1,779 siblings, which revealed a higher burden of pSNMs with high MAFs in probands relative to that of siblings for LoF mutations (P = 0.15, two‐tailed Wilcoxon rank sum test, OR = 2.5) and missense mutations with deleterious effects (Adzhubei et al., 2010) (Mis3 mutations, P = 0.009, two‐tailed Wilcoxon rank sum test, OR = 2.2) (Supp. Table S10).
Figure 2.
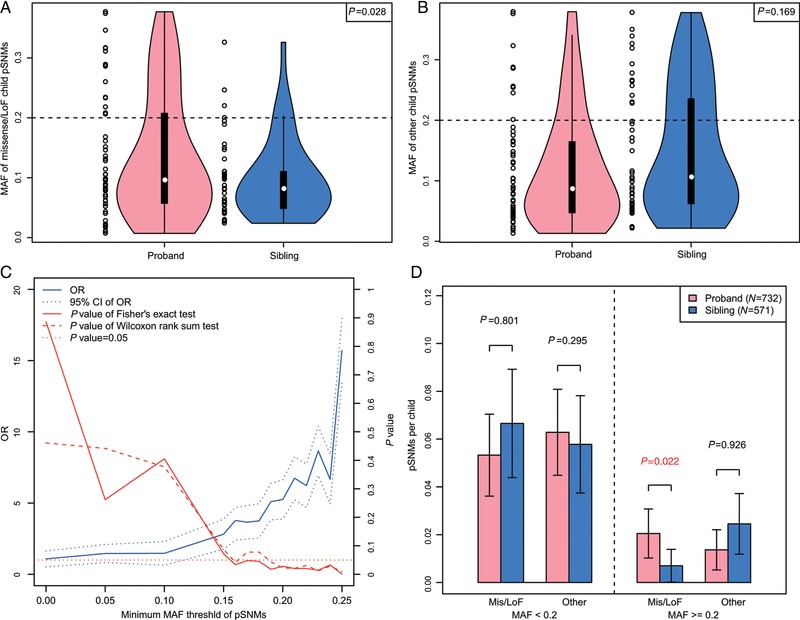
Contribution of missense/LoF child pSNMs with high MAFs to ASD diagnoses. A and B: Higher MAFs of validated pSNMs in ASD probands compared with unaffected siblings were observed for missense/LoF pSNMs (A), but not for other pSNMs with neutral functional effects (B). C: The increased burden of missense/LoF pSNMs with high MAFs between probands and siblings varied across different MAF cutoffs. The increased burden became significant when the MAF cutoff was 0.2 or larger. D: The enrichment of pSNMs in probands versus siblings was observed only for missense/LoF pSNMs with high MAFs
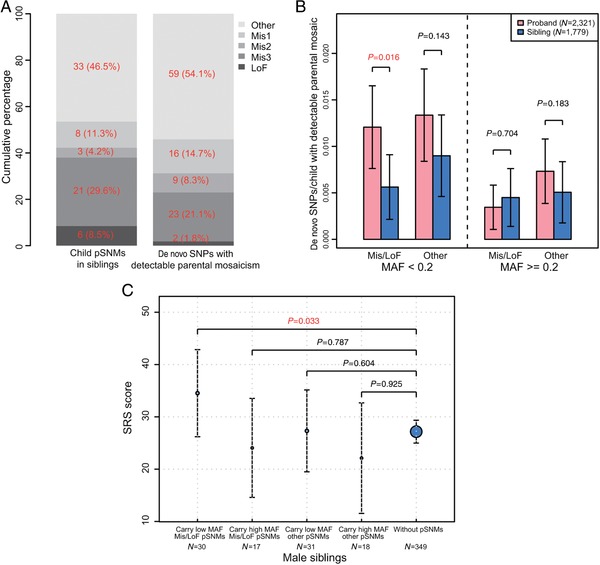
Unlike missense/LoF pSNMs with high MAFs that contribute to ASD diagnoses directly, LoF pSNMs with low MAFs in parents were less likely to be transmitted to offspring than other pSNMs in parents (P = 0.037, one‐sided Fisher's exact test, OR = 5.36; Fig. 3A), and a similar trend was also observed for Mis3 missense pSNMs (OR = 1.63; Figure 3A). Since a large proportion of naturally arising missense/LoF pSNMs were predicted to have deleterious functional effects, they were more likely to be heterozygous lethal and thus could not be transmitted to offspring. Next, we analyzed the transmission bias of missense/LoF parental pSNMs with low MAFs in SSC families, and found a significant twofold enrichment in probands versus siblings for those pSNMs not transmitted to multiple children (P = 0.016, one‐sided Wilcoxon rank sum test; Fig. 3B; Table 1).
Figure 3.
pSNMs with low MAFs contribute to autistic traits, whereas de novo SNPs with detectable parental mosaicism with low MAFs contribute to ASD diagnoses in offspring. A: LoF pSNMs in parents were less likely than other parental pSNMs to be transmitted to offspring. All pSNMs in siblings were validated using PASM, and the validation rate for putative de novo SNPs with detectable parental mosaicism with low MAFs was approximately 70%. B: Missense/LoF parental pSNMs with low MAFs that were transmitted to offspring (each pSNM here was transmitted to one child) showed approximately twofold enrichment in ASD probands versus unaffected siblings. C: Male siblings carrying missense/LoF pSNMs with low MAFs had teacher SRS scores significantly higher than those of male siblings without pSNMs, after all subjects carrying de novo LoF germline mutations (Iossifov et al., 2014) or de novo small deletions were excluded (Sanders et al., 2015)
We next estimated the contribution of pSNMs to ASD diagnoses in the SSC following the strategy of Iossifov et al. (2014) (see Materials and Methods). We estimated that approximately 65.8% of missense/LoF pSNMs with high MAFs in probands and approximately 53.4% of parental missense/LoF pSNMs with low MAFs only transmitted to probands contribute to ASD diagnoses. Taking into account the sensitivity of MosaicHunter for detecting pSNMs, we roughly estimated that child pSNMs with high MAFs increased the risk of ASD by approximately 3.4% in SSC probands, and missense/LoF parental pSNMs with low MAFs (only transmitted to one child) increased the risk of ASD by approximately 2.6% in SSC probands. Therefore, we estimate that pSNMs contribute to approximately 6% of ASD diagnoses in the SSC.
3.4. Autistic traits in siblings carrying pSNMs
We assessed the potential contribution of pSNMs to broader autistic phenotypes in unaffected siblings. After excluding subjects who harbored reported de novo LoF germline mutations (Iossifov et al., 2014) or de novo small deletions (Sanders et al., 2015), we observed that male siblings carrying missense/LoF pSNMs with low MAFs had teacher SRS scores significantly higher than those of male siblings without pSNMs (P = 0.033, one‐sided Wilcoxon rank sum test; Fig. 3C). This increasing trend was not observed in male siblings carrying neutral pSNMs or missense/LoF pSNMs with high MAFs (Fig. 3C). The increasing trend was not observed in female siblings carrying missense/LoF pSNMs (Supp. Fig. S4A), which was consistent with the previously reported female protective effect for ASD (Constantino, 2016; Gockley et al., 2015).
3.5. Autistic traits in parents carrying pSNMs
We also assessed the potential contribution of pSNMs to broader autistic phenotypes in parents carrying pSNMs. After exclusion of parents whose children harbored reported de novo LoF germline mutations (Iossifov et al., 2014) or de novo small deletions, we observed that fathers transmitting missense/LoF pSNMs to probands also tend to have higher social responsiveness scale for adults (SRS‐A) scores (P = 0.173, one‐sided Wilcoxon rank sum test), although this effect did not reach statistical significance, probably due to the small sample size of the analysis (Supp. Fig. S4B). In contrast, this increasing trend was not observed in mothers carrying pSNMs (P = 0.519, one‐sided Wilcoxon rank sum test, Supp. Fig. S4C), which was also consistent with the previously reported female protective effect for ASD (Constantino, 2016; Gockley et al., 2015).
3.6. New ASD risk genes revealed by integrating pSNMs into TADA
The increased burdens of child pSNMs and de novo SNPs with detectable parental mosaicism in ASD pedigrees suggested the hypothesis that, in addition to well‐documented genetic factors such as de novo germline mutations, pSNMs could provide new insight into the identities of ASD risk genes.
To test this hypothesis, we first analyzed the FDR scores of the genes identified by the original TADA model, which considered only de novo germline mutations (TADA‐Denovo). We used a permutation test to estimate the expected distribution of TADA FDR scores. For each observed pSNM, a gene was selected randomly based on the mutation rates of all Refseq genes, which were calculated using base contents and gene lengths. The corresponding TADA FDR q‐value of the randomly selected gene was obtained. The process was repeated 100 times, and the median of the highest FDR scores from 100 iterations was used as the expected value to compare with the highest observed FDR score. This procedure was repeated with the second highest FDR score, third highest FDR score, and so on to account for all observed genes carrying pSNMs. Our analysis was limited to only functional pSNMs with high MAFs in children and de novo functional SNPs in children with detectable mosaicism and low MAFs in parents. As shown in Figure 4A and B, a list of genes carrying pSNMs showed significantly lower FDR scores than expected, which suggested an overlap of the genetic basis of pSNMs and de novo germline mutations contributing to ASD. On the basis of these observations, we decided to integrate child pSNMs and de novo SNPs with detectable parental mosaicism into the TADA‐Denovo model as factors. By applying our extended model, 58 ASD risk genes were identified with FDR≤0.1, including 13 new ASD risk genes that were not identified by the original TADA‐Denovo model (Supp. Table S7). All 13 newly identified genes (ASXL3, CACNA2D3, MED13L, MEGF11, MYH10, MYO9B, PRKAR1B, PYHIN1, RIMS1, TANC2, TBL1XR1, TMEM39B, TSPAN4) had some evidences associated with ASD (Basu, Kollu, & Banerjee‐Basu, 2009; Xu et al., 2012). Of particular interest, we found five genes (MFRP, MYO9B, PTK7, TANC2, MEGF11) carrying both Mis3/LoF de novo mutations (Dong et al., 2014; Iossifov et al., 2014; Sanders et al., 2012) and functional pSNMs in probands, and one additional gene (C3orf35) carrying both a small de novo deletion (Sanders et al., 2015) and a functional pSNM in probands.
Figure 4.
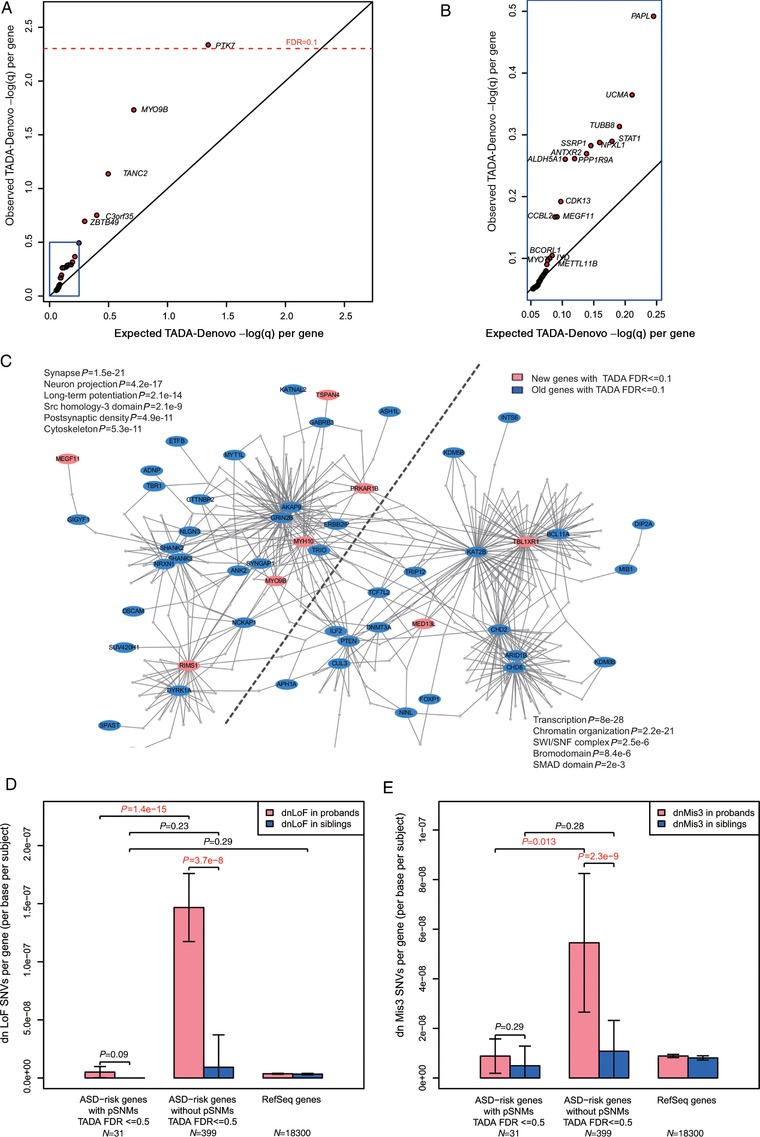
Extending the TADA‐Denovo model with pSNMs revealed new ASD risk genes and protein–protein interaction network analysis. A and B: Genes carrying pSNMs showed significantly higher FDR scores in the original TADA‐Denovo model. The left‐bottom part of (A) is enlarged in (B). C: The new ASD risk genes (labeled in red) shown are highly connected via protein–protein interactions with previously reported ASD risk genes (labeled in blue). The network was divided into two subnetworks, with one subnetwork related to the functions of neurons, the synapse and the cytoskeleton, whereas the other was related to transcription and chromatin organization. D and E: ASD risk genes with pSNMs have more de novo germline LoF/Mis3 SNVs in ASD probands than in unaffected siblings, but these genes carrying pSNMs tend to have fewer de novo LoF/Mis3 germline mutations per base per subject
We next considered the functions of these newly identified ASD risk genes by analyzing protein–protein interactions using DAPPLE (Rossin et al., 2011). Eight of the nine newly identified ASD risk genes with annotations in the DAPPLE database were joined into a single network of previously reported ASD risk genes, with the overall network topology remaining unchanged (Fig. 4C). Consistent with previous findings (Sanders et al., 2015), the network consisted of two functionally related subnetworks, with one subnetwork enriched in genes functioning in neurons, the synapse or the cytoskeleton, whereas the other was enriched in genes related to transcription or chromatin organization (Fig. 4C). Network analysis using two additional tools, STITCH (Kuhn et al., 2008) and STRING (Szklarczyk et al., 2015), confirmed our results. As shown in Supp. Figure S5, STITCH and STRING created network topologies similar to that created by DAPPLE, and most of the new ASD risk genes were robustly included in the network generated by all three tools.
We observed that missense/LoF pSNMs with high MAFs in probands and missense/LoF de novo SNPs with detectable parental pSNMs in probands tended to be enriched in ExAC LoF‐constraint exons (Lek et al., 2016; Samocha et al., 2014) (Supp. Table S11), whereas ASD risk genes with pSNMs tended to have fewer LoF/Mis3 de novo germline mutations per base per subject (Fig. 4D and 4E). Considering that some LoF‐constraint genes and exons carrying pSNMs might not be able to bear heterozygous lethal de novo germline mutations, and thus the contribution of pSNMs to ASD might be underestimated using the TADA‐Denovo model, we relaxed the FDR criterion for genes carrying child pSNMs with high MAFs to 0.5 and reanalyzed the protein–protein interaction networks using DAPPLE. In this way, we found several other new, highly connected hub genes, including SSRP1, SREBF2, and STAT1, and connected the previously isolated network formed by WAC and TNRC6B to the large interaction network through SSRP1 (Supp. Fig. S6).
We further analyzed the expression profiles of the new ASD risk genes in different regions and developmental stages of the brain. As shown in Supp. Figure S7, the 25 new ASD risk genes carrying pSNMs in SSC probands tend to have higher expression in the cerebellar hemisphere (P = 0.057, two‐tailed t‐test) and cerebellum (P = 0.090, two‐tailed t‐test). In addition, most of the highly connected hub genes within the network showed elevated brain expression during the embryonic and fetal periods (Supp. Fig. S8). These results suggest that the newly identified ASD risk genes might play a role in the early development of the brain.
4. DISCUSSION
In this study, we detected and validated child pSNMs and de novo SNPs with detectable parental mosaicism in the SSC dataset, the largest dataset of confident pSNMs in ASD cohorts. We found that pSNMs with varied MAFs have different risk effect sizes for ASD. While missense/LoF pSNMs with high MAFs contribute to ASD diagnoses directly, missense/LoF pSNMs with low MAFs contribute to broad autism phenotypes. In addition, we found that parents carrying missense/LoF pSNMs with low MAFs can transmit their mutant alleles to offspring and thus contribute to sporadic ASD cases, which highlights the importance of identifying pSNMs for genetic counseling. Elevated autistic traits were observed in non‐ASD males, but not in females, confirming the female protective effect for ASD. In addition, we estimated that pSNMs contribute to the etiology of ASD in approximately 6% of ASD patients in the SSC dataset, revealed 13 new ASD risk genes, and included pSNMs in the TADA‐Denovo model.
Missense/LoF pSNMs with higher MAFs had a higher OR when probands and siblings were compared, and it reached the significance threshold (P<0.05) when the cutoff between high MAF and low MAF was 0.2 or higher (Fig. 2C). However, we could not completely rule out the possibility of a chance effect, because there is a lack of publicly available data demonstrating depletion of functional pSNMs with high MAFs in the general population.
In comparison with the most recent work exploring the association of mosaics and ASD by Freed and Pevsner (2016), our study has much better precision (∼53%) for pSNMs detection and much higher sensitivity for detection of pSNMs with relatively low MAFs (among 854 pSNMs we detected with a MAF <20%, they only found 54; among 156 PASM‐validated pSNMs with a MAF <20%, they only found 17). For child mosaicisms, we identified approximately six times (1,248 vs. 221) as many putative pSNMs and experimentally validated approximately 17 times (376 vs. 22) as many pSNMs in comparison with Freed and Pevsner (2016). Moreover, the mosaic sites we identified had a much broader range of MAFs (1%–40% vs. 15%–34%), which enabled us to study the different contributions of pSNMs with varied MAFs. In addition to child mosaicisms, we also systematically identified and validated pSNMs in the parents of ASD cohorts. Among the SNPs in Yale's dataset that were identified in our study, only 32/285 putative de novo SNPs with detectable parental mosaicism and 0/31 SNPs with validated parental mosaicism were present in a previous list of de novo mutations by Iossifov et al. (2014). In addition, we demonstrated that both child and parental mosaicism contribute to ASD and autistic traits. Interestingly, we estimated that approximately 65.8% of missense pSNMs with high MAFs contribute to ASD diagnoses, which was much higher than the proportion (13%) for pathogenic missense de novo heterozygous SNVs (Iossifov et al., 2014). We speculated that the extremely high ASD risk effect sizes of missense child pSNMs might be due to the lack of germline cell competition and natural selection. Our results also implied that mutations that occur at early cell divisions affect more tissues and are more likely to be transferred to the next generation; thus, they might play important roles in human evolution.
Our results highlight the fact that mosaicisms resulting from postzygotic mutations could explain a proportion of the unrecognized genetic etiology of ASD. Given the moderate validation rate of child pSNMs, we only considered child pSNMs that were validated by PASM in Yale's dataset when extending the TADA‐Denovo model. With the development of next‐generation sequencing technologies, identification and validation of pSNMs in larger ASD cohorts will provide a more precise estimation of the contribution of pSNMs to ASD and aid researchers in finding more ASD risk genes.
Supporting information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
ACKNOWLEDGMENTS
The authors thank Drs. Matthew State and Stephan Sanders for sharing the Yale samples. We are grateful to all of the families at the participating Simons Simplex Collection sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin‐Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, and E. Wijsman).
Disclosure statement
The authors declare no conflict of interest.
Dou Y, Yang X, Li Z, et al. Postzygotic single‐nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Human Mutation. 2017;38:1002–1013. https://doi.org/10.1002/humu.23255
Contract grant sponsors: National Natural Science Foundation of China (31530092); Ministry of Science and Technology 863 Grant (2015AA020108); Peking University Clinical Cooperation “985 Project” (PKU‐2013‐1‐06 and PKU‐2014‐1‐1).
Communicated by Bruce R. Gottlieb
Contributor Information
August Yue Huang, Email: huangy@mail.cbi.pku.edu.cn.
Liping Wei, Email: weilp@mail.cbi.pku.edu.cn.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov, L. B. , Nik‐Zainal, S. , Wedge, D. C. , Aparicio, S. A. , Behjati, S. , Biankin, A. V. , … Stratton M. R. (2013). Signatures of mutational processes in human cancer. Nature, 500(7463), 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association (Ed.). (2013). Diagnostic and Statistical Manual of Mental Disorders (DSM 5) (5th ed). Washington, D.C: American Psychiatric Association. [Google Scholar]
- Basu, S. N. , Kollu, R. , & Banerjee‐Basu, S. (2009). AutDB: A gene reference resource for autism research. Nucleic Acids Research, 37(Database issue), D832–D836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker, L. G. , & Spinner, N. B. (2013). A genomic view of mosaicism and human disease. Nature Review Genetics, 14(5), 307–320. [DOI] [PubMed] [Google Scholar]
- Bryant, P. J. , & Zornetzer, M. (1973). Mosaic analysis of lethal mutations in Drosophila. Genetics, 75(4), 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, I. M. , Yuan, B. , Robberecht, C. , Pfundt, R. , Szafranski, P. , McEntagart, M. E. , … Stankiewicz, P. (2014). Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. The American Journal of Human Genetics, 95(2), 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castermans, D. , Thienpont, B. , Volders, K. , Crepel, A. , Vermeesch, J. R. , Schrander‐Stumpel, C. T. , … Devriendt, K. (2008). Position effect leading to haploinsufficiency in a mosaic ring chromosome 14 in a boy with autism. European Journal of Human Genetics, 16(10), 1187–1192. [DOI] [PubMed] [Google Scholar]
- Constantino, J. N. (2016). Data from the Baby Siblings Research Consortium confirm and specify the nature of the female protective effect in autism: A commentary on Messinger et al. Molecular Autism, 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contini, E. , Paganini, I. , Sestini, R. , Candita, L. , Capone, G. L. , Barbetti, L. , … Papi, L. (2015). A systematic assessment of accuracy in detecting somatic mosaic variants by deep amplicon sequencing: Application to NF2 gene. PLoS One, 10(6), e0129099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, J. , Goin‐Kochel, R. P. , Green‐Snyder, L. A. , Hundley, R. J. , Warren, Z. , & Peters, S. U. (2014). Expression of the broad autism phenotype in simplex autism families from the Simons Simplex Collection. Journal of Autism and Developmental Disorders, 44(10), 2392–2399. [DOI] [PubMed] [Google Scholar]
- De Rubeis, S. , He, X. , Goldberg, A. P. , Poultney, C. S. , Samocha, K. , Cicek, A. E. , … Buxbaum, J. D. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515(7526), 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin, B. , & Scherer, S. W. (2012). Genetic architecture in autism spectrum disorder. Current Opinion in Genetics & Development, 22(3), 229–237. [DOI] [PubMed] [Google Scholar]
- Dong, S. , Walker, M. F. , Carriero, N. J. , DiCola, M. , Willsey, A. J. , Ye, A. Y. , … Sanders, S. J. (2014). De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Reports, 9(1), 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret, L. , & Galtier, N. (2009). Biased gene conversion and the evolution of mammalian genomic landscapes. Annual Review of Genomics and Human Genetics, 10, 285–311. [DOI] [PubMed] [Google Scholar]
- Fischbach, G. D. , & Lord, C. (2010). The Simons Simplex Collection: A resource for identification of autism genetic risk factors. Neuron, 68(2), 192–195. [DOI] [PubMed] [Google Scholar]
- Fontenelle, L. F. , Mendlowicz, M. V. , Bezerra de Menezes, G. , dos Santos Martins, R. R. , & Versiani, M. (2004). Asperger syndrome, obsessive‐compulsive disorder, and major depression in a patient with 45,X/46,XY mosaicism. Psychopathology, 37(3), 105–109. [DOI] [PubMed] [Google Scholar]
- Freed, D. , & Pevsner, J. (2016). The contribution of mosaic variants to autism spectrum disorder. PLoS Genetics, 12(9), e1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler, T. , Klei, L. , Sanders, S. J. , Bodea, C. A. , Goldberg, A. P. , Lee, A. B. , … Buxbaum, J. D. (2014). Most genetic risk for autism resides with common variation. Nature Genetics, 46(8), 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gockley, J. , Willsey, A. J. , Dong, S. , Dougherty, J. D. , Constantino, J. N. , & Sanders, S. J. (2015). The female protective effect in autism spectrum disorder is not mediated by a single genetic locus. Molecular Autism, 6, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium . (2013). The Genotype‐Tissue Expression (GTEx) project. Nature Genetics, 45(6), 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer, J. , Cleveland, S. , Torres, A. , Phillips, J. , Cohen, B. , Torigoe, T. , … Risch, N. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Archives of General Psychiatry, 68(11), 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X. , Sanders, S. J. , Liu, L. , De Rubeis, S. , Lim, E. T. , Sutcliffe, J. S. , … Roeder, K. (2013). Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genetics, 9(8), e1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A. Y. , Xu, X. , Ye, A. Y. , Wu, Q. , Yan, L. , Zhao, B. , … Wei, L. (2014). Postzygotic single‐nucleotide mosaicisms in whole‐genome sequences of clinically unremarkable individuals. Cell Research, 24(11), 1311–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A. Y. , Zhang, Z. , Ye, A. Y. , Dou, Y. , Yan, L. , Yang, X. , … Wei, L. (2017). MosaicHunter: Accurate detection of postzygotic single‐nucleotide mosaicism through next‐generation sequencing of unpaired, trio, and paired samples. Nucleic Acids Research. https://doi.org/10.1093/nar/gkx024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da, W., Sherman, B. T. , & Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4(1), 44–57. [DOI] [PubMed] [Google Scholar]
- Iossifov, I. , O'Roak, B. J. , Sanders, S. J. , Ronemus, M. , Krumm, N. , Levy, D. , … Wigler, M. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515(7526), 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. B. , Kawasawa, Y. I. , Mason, C. E. , Krsnik, Z. , Coppola, G. , Bogdanovic, D. , … Sestan, N. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron, 62(4), 494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H. J. , Kawasawa, Y. I. , Cheng, F. , Zhu, Y. , Xu, X. , Li, M. , … Sestan, N. (2011). Spatio‐temporal transcriptome of the human brain. Nature, 478(7370), 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, A. , Frigge, M. L. , Masson, G. , Besenbacher, S. , Sulem, P. , Magnusson, G. , … Stefansson, K. (2012). Rate of de novo mutations and the importance of father's age to disease risk. Nature, 488(7412), 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, M. , von Mering, C. , Campillos, M. , Jensen, L. J. , & Bork, P. (2008). STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Research, 36(Database issue), D684–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … Exome Aggregation Consortium . (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losh, M. , Childress, D. , Lam, K. , & Piven, J. (2008). Defining key features of the broad autism phenotype: A comparison across parents of multiple‐ and single‐incidence autism families. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 147B(4), 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, K. J. , Axelsen, M. S. , Sheffield, V. C. , Patil, S. R. , & Wassink, T. H. (2012). Germline mosaic transmission of a novel duplication of PXDN and MYT1L to two male half‐siblings with autism. Psychiatric Genetics, 22(3), 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, B. M. , Kou, Y. , Liu, L. , Ma'ayan, A. , Samocha, K. E. , Sabo, A. , … Daly M. J. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485(7397), 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nota, B. , Hamilton, E. M. , Sie, D. , Ozturk, S. , van Dooren, S. J. , Ojeda, M. R. , … Salomons, G. S. (2013). Novel cases of D‐2‐hydroxyglutaric aciduria with IDH1 or IDH2 mosaic mutations identified by amplicon deep sequencing. Journal of Medical Genetics, 50(11), 754–759. [DOI] [PubMed] [Google Scholar]
- Papanikolaou, K. , Paliokosta, E. , Gyftodimou, J. , Kolaitis, G. , Vgenopoulou, S. , Sarri, C. , & Tsiantis, J. (2006). A case of partial trisomy of chromosome 8p associated with autism. Journal of Autism and Developmental Disorders, 36(5), 705–709. [DOI] [PubMed] [Google Scholar]
- Parrini, E. , Mei, D. , Wright, M. , Dorn, T. , & Guerrini, R. (2004). Mosaic mutations of the FLN1 gene cause a mild phenotype in patients with periventricular heterotopia. Neurogenetics, 5(3), 191–196. [DOI] [PubMed] [Google Scholar]
- Pfeifer, G. P. (2006). Mutagenesis at methylated CpG sequences. Current Topics in Microbiology and Immunology, 301, 259–281. [DOI] [PubMed] [Google Scholar]
- Reich, M. , Liefeld, T. , Gould, J. , Lerner, J. , Tamayo, P. , & Mesirov, J. P. (2006). GenePattern 2.0. Nature Genetics, 38(5), 500–501. [DOI] [PubMed] [Google Scholar]
- Ronemus, M. , Iossifov, I. , Levy, D. , & Wigler, M. (2014). The role of de novo mutations in the genetics of autism spectrum disorders. Nature Review Genetics, 15(2), 133–141. [DOI] [PubMed] [Google Scholar]
- Rosenthal, R. , McGranahan, N. , Herrero, J. , Taylor, B. S. , & Swanton, C. (2016). DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biology, 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin, E. J. , Lage, K. , Raychaudhuri, S. , Xavier, R. J. , Tatar, D. , Benita, Y. , … Daly, M. J. (2011). Proteins encoded in genomic regions associated with immune‐mediated disease physically interact and suggest underlying biology. PLoS Genetics, 7(1), e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio, C. , Lau, P. , Troakes, C. , Deramecourt, V. , Gele, P. , Van Loo, P. , … De Strooper, B. (2015). On the identification of low allele frequency mosaic mutations in the brains of Alzheimer's disease patients. Alzheimer's Dementia, 11(11), 1265–1276. [DOI] [PubMed] [Google Scholar]
- Samocha, K. E. , Robinson, E. B. , Sanders, S. J. , Stevens, C. , Sabo, A. , McGrath, L. M. , … Daly, M. J. (2014). A framework for the interpretation of de novo mutation in human disease. Nature Genetics, 46(9), 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, S. J. , He, X. , Willsey, A. J. , Ercan‐Sencicek, A. G. , Samocha, K. E. , Cicek, A. E. , … State M. W. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron, 87(6), 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, S. J. , Murtha, M. T. , Gupta, A. R. , Murdoch, J. D. , Raubeson, M. J. , Willsey, A. J. , … State, M. W. (2012). De novo mutations revealed by whole‐exome sequencing are strongly associated with autism. Nature, 485(7397), 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter, S. , von Beust, G. , Burfeind, P. , Weise, A. , Starke, H. , Liehr, T. , & Zoll, B. (2003). Autistic disorder and chromosomal mosaicism 46,XY[123]/46,XY,del(20)(pter –>p12.2)[10]. American Journal of Medical Genetics Part A, 120A(4), 533–536. [DOI] [PubMed] [Google Scholar]
- Schmitt, M. W. , Kennedy, S. R. , Salk, J. J. , Fox, E. J. , Hiatt, J. B. , & Loeb, L. A. (2012). Detection of ultra‐rare mutations by next‐generation sequencing. Proceedings of the National Academy of Sciences of the United States of America, 109(36), 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , … Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan, T. , & Read, A. (2004). An overview of mutation, polymorphism, and DNA repair. In Human molecular genetics 3. New York; London: Garland Science. [Google Scholar]
- Szklarczyk, D. , Franceschini, A. , Wyder, S. , Forslund, K. , Heller, D. , Huerta‐Cepas, J. , … von Mering, C. (2015). STRING v10: Protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Research, 43(Database issue), D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, I. R. , Takahashi, K. , Futreal, P. A. , & Chin, L. (2013). Emerging patterns of somatic mutations in cancer. Nature Review Genetics, 14(10), 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein, M. M. , Kang, T. , Lachman, R. S. , Bamshad, M. , Nickerson, D. A. , Krakow, D. , & Cohn, D. H. (2016). Somatic mosaicism for a lethal TRPV4 mutation results in non‐lethal metatropic dysplasia. American Journal of Medical Genetics Part A, 170, 3298–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, Z. , Yi, L. , Cao, D. , He, W. , Chen, J. , Gao, S. , & Sun, X. (2016). Dravet syndrome with autism inherited from a paternal mosaic heterozygous mutation on SCN1A. Journal of the Neurological Sciences, 369, 53–56. [DOI] [PubMed] [Google Scholar]
- Xu, L. M. , Li, J. R. , Huang, Y. , Zhao, M. , Tang, X. , & Wei, L. (2012). AutismKB: An evidence‐based knowledgebase of autism genetics. Nucleic Acids Research, 40(Database issue), D1016–D1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Yang, X. , Wu, Q. , Liu, A. , Yang, X. , Ye, A. Y. , … Zhang, Y. (2015). Amplicon resequencing identified parental mosaicism for approximately 10% of “de novo” SCN1A mutations in children with Dravet syndrome. Human Mutation, 36(9), 861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, K. , Komura, M. , Yamaguchi, R. , Imoto, S. , Shimizu, E. , Kasuya, S. , … Furukawa, Y. (2015). Detection of APC mosaicism by next‐generation sequencing in an FAP patient. Journal of Human Genetics, 60(5), 227–231. [DOI] [PubMed] [Google Scholar]
- Yates, L. R. , & Campbell, P. J. (2012). Evolution of the cancer genome. Nature Review Genetics, 13(11), 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssoufian, H. , & Pyeritz, R. E. (2002). Mechanisms and consequences of somatic mosaicism in humans. Nature Review Genetics, 3(10), 748–758. [DOI] [PubMed] [Google Scholar]
- Yurov, Y. B. , Vorsanova, S. G. , Iourov, I. Y. , Demidova, I. A. , Beresheva, A. K. , Kravetz, V. S. , … Gorbachevskaya, N. L. (2007). Unexplained autism is frequently associated with low‐level mosaic aneuploidy. Journal of Medical Genetics, 44(8), 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information
Supplymentry Information