

Abstract
The electrogenic sodium bicarbonate cotransporter NBCe1 (SLC4A4) expressed in astrocytes regulates intracellular and extracellular pH. Here, we introduce transforming growth factor beta (TGF‐β) as a novel regulator of NBCe1 transcription and functional expression. Using hippocampal slices and primary hippocampal and cortical astrocyte cultures, we investigated regulation of NBCe1 and elucidated the underlying signaling pathways by RT‐PCR, immunoblotting, immunofluorescence, intracellular H(+) recording using the H(+) ‐sensitive dye 2′,7′‐bis‐(carboxyethyl)‐5‐(and‐6)‐carboxyfluorescein, mink lung epithelial cell (MLEC) assay, and chromatin immunoprecipitation. Activation of TGF‐β signaling significantly upregulated transcript, protein, and surface expression of NBCe1. These effects were TGF‐β receptor‐mediated and suppressed following inhibition of JNK and Smad signaling. Moreover, 4‐aminopyridine (4AP)‐dependent NBCe1 regulation requires TGF‐β. TGF‐β increased the rate and amplitude of intracellular H+ changes upon challenging NBCe1 in wild‐type astrocytes but not in cortical astrocytes from Slc4a4‐deficient mice. A Smad4 binding sequence was identified in the NBCe1 promoter and Smad4 binding increased after activation of TGF‐β signaling. The data show for the first time that NBCe1 is a direct target of TGF‐β/Smad4 signaling. Through activation of the canonical pathway TGF‐β acts directly on NBCe1 by binding of Smad4 to the NBCe1 promoter and regulating its transcription, followed by increased protein expression and transport activity.
Keywords: astroglia, epilepsy, growth factor, pH
1. INTRODUCTION
The key physiological roles for central nervous system (CNS) astrocytes include contribution to CNS development, homeostatic function, metabolic support, as well as modulation of neuronal signaling and synaptic transmission. Moreover, astroglia is meanwhile considered as a possible target for medical intervention. Under pathophysiological conditions, astrocytes undergo key changes and morphological alterations, and changes on the expression of ion channels, transporters and receptors, have been established as astrocytic responses to insults.
The electrogenic Na+/ cotransporter (NBCe1) (Majumdar & Bevensee, 2010; Rickmann, Orlowski, Heupel, & Roussa, 2007; Theparambil, Ruminot, Schneider, Shull, & Deitmer, 2014) is crucial for regulating astrocytic intracellular pH and additionally for maintaining ionic homeostasis between intracellular and extracellular space. In turn, ionic composition of the extracellular space affects several cell functions in neurons and astrocytes. In particular, changes on extracellular or intracellular pH maybe associated with several pathophysiological conditions and considerably influence brain function. Glial NBCe1 operates with a stoichiometry of 1Na+: and can transport Na+ and bicarbonate in both directions across the glial cell membrane (Brookes & Turner, 1994; Brune, Fetzer, Backus, & Deitmer, 1994; Deitmer, 1991). Whereas neuronal firing leads to depolarization‐induced alkalinisation in astrocytes via activation of NBCe1, NBCe1 reverses during intracellular alkali load, thereby acting as an acid loader (Theparambil & Deitmer, 2015). NBCe1 plays also a crucial role in neuron‐glial metabolic coupling (Ruminot et al., 2011; Theparambil, Weber, Schmälzle, Ruminot, & Deitmer, 2016) and might also represent a molecular component of neuron‐astrocyte communication (Salameh, Hubner, & Boron, 2017). Changes in expression and function of NBCe1 have been observed in different in vitro (Yao et al., 2016) and in vivo models (Jung, Choi, & Kwon, 2007; Sohn et al., 2011) of ischemia and in seizures (Kang et al., 2002). We have previously shown that the K+ channel blocker 4‐aminopyridine (4AP) regulates NBCe1 transcript, protein, and functional expression in primary astrocyte cultures via c‐jun N‐terminal kinase (JNK), and proto‐oncogene tyrosine‐protein kinase Src signaling in a depolarization‐independent manner (Schrödl‐Häußel, Theparambil, Deitmer, & Roussa, 2015).
The TGF‐β superfamily of cytokines consists of more than 40 members, including TGF‐βs, activins, inhibins, Nodal, bone morphogenetic proteins (BMPs), and growth differentiation factors (GDFs) (reviewed in Derynck & Miyazono, 2008). The three mammalian TGF‐β isoforms (TGF‐β1, TGF‐β2, and TGF‐β3) build the TGF‐β family, are highly conserved between species, and can regulate developmental processes, tissue homeostasis, and tissue repair (Krieglstein, Zheng, Unsicker, & Alzheimer, 2011; Ten Dijke & Arthur, 2007). Collectively, TGF‐βs exert their actions in a cell type‐ and context‐dependent manner. TGF‐β signaling is mediated by a heteromeric complex of two types of transmembrane serine/threonine kinase receptors. Binding of ligand to the receptor complex leads to the phosphorylation of the type II receptor kinase and activation of type I receptor kinase that leads to the propagation of signaling. The canonical, Smad‐mediated, TGF‐β signaling pathway implies phosphorylation of Smad2 and Smad3 proteins, formation of a complex with Smad4, and translocation into the nucleus, where transcription of target genes is activated (Derynck & Miyazono, 2008; Miyazawa, Shinozaki, Hara, Furuya, & Miyazono, 2002; Moustakas & Heldin, 2009, reviewed in Akhurst & Hata, 2012). However, TGF‐β may also signal through other signaling pathways, such as ERK, p38, JNK, and MAPK pathways (Yang et al., 2003) that may act independently or in co‐operation with the canonical pathway.
The role of TGF‐β1 in the context of epilepsy has been studied in several in vitro and in vivo approaches (reviewed in Heinemann, Kaufer, & Friedman, 2012). According to the current notion, disruption of the blood‐brain‐barrier (BBB) causes extravasation of albumin in the extracellular space which in turn interacts with astrocytic TGF‐β receptors and activates TGF‐β signaling cascades, resulting in secretion of TGF‐β1 and increased hyperexcitability. TGF‐β‐dependent activation of astrocytes is accompanied by downregulation of K+ inward‐rectifying channels (Perillan, Chen, Potts, & Simard, 2002), gap junctional proteins, and the glial excitatory amino acid transporters (EAAT1 and EAAT2), together with altered trafficking and surface expression of aquaporin 4 (AQP4). In epithelial cells, TGF‐β1 has been shown to regulate trafficking and expression of transporters that are functionally coupled to NBCe1, among them CFTR (Roux et al., 2010; Yi, Pierucci‐Alves, & Schultz, 2013).
In the present study, we have investigated whether TGF‐β may regulate NBCe1 in astrocytes and analysed the underlying molecular mechanisms.
2. MATERIALS AND METHODS
2.1. Antibodies and reagents/chemicals
Following antibodies were used as primary antibodies: anti‐SLC4A4 rabbit polyclonal from Alomone labs, (Jerusalem, Israel) for western blots; from Atlas Antibodies (Bromma, Sweden) for immunocytochemistry. Anti‐GAPDH (Abcam; Cambridge, UK), anti‐ GFAP, anti‐H3 and anti‐IgG (Merck Millipore, Darmstadt, Germany), anti‐Smad2/3 and anti‐phosphorylated Smad2/3 (Cell Signaling), anti‐phospho Smad4 (ThermoFisher Scientific, IL) anti Na+/K+‐ATPase and anti‐ Aldh1 (Merck Millipore, Darmstadt, Germany), anti‐Actin (Developmental Studies, Hybridoma Bank, IA) were used as primary antibodies. Goat anti‐rabbit or anti‐mouse IgG coupled to AlexaFluor488 or AlexaFluor568, (Jackson Immunoresearch, Newmarket, UK) and coupled to horseradish peroxidase (GE Healthcare, Buckinghamshire, UK) were used as secondary antibodies for immunofluorescence and western blot, respectively. Human recombinant TGF‐β1 and the neutralizing monoclonal mouse anti‐TGF‐β antibody (anti‐TGF‐β) recognizing all three isoforms, was obtained from R&D Systems. Activity of this antibody was determined using the mink lung epithelial cell system (MLEC) (Abe et al.,1994; Krieglstein et al., 2000). 4‐aminopyridine (4AP), Bicuculline methiodide, SP600125, and SB431542 were purchased from Tocris Bioscience (Bristol, UK). SIS3 was purchased from Millipore, and Leptomycin B from Sigma.
2.2. Animals
All protocols were carried out in accordance with German ethical guidelines for laboratory animals and approved by the Institutional Animal Care and Use Committee of the University of Freiburg (authorizations: X‐14/16H and X‐15/06H). Adult C57BL/6N mice of either sex were maintained on a 12‐hr dark/light cycle with food and water ad libitum. Mice were sacrificed by cervical dislocation, and all efforts were made to minimize suffering. Slc4a4 and Tgf‐β2 deficient mice have been described earlier (Gawenis et al., 2007; Sanford et al., 1997).
2.3. Acute hippocampal slices
Acute hippocampal slices were generated from 4‐ to 6‐week old mice (Lein, Barnhart, & Pessah, 2011). Thick slices of 200 µm were put into freshly prepared artificial cerebrospinal fluid (ACSF), containing (mM) NaCl 126, KCl 2.5, NaH2PO4 1.2, Glucose 10, NaHCO3 18, and MgCl2 1.2, and treated either for 20 min with 4AP (100 µM) or for 10 min with 20 µM Bicuculline methiodide (in 0 Mg2+) in the presence or absence of anti‐TGF‐β1,2,3, a function blocking antibody (20 µg/mL), potent to inhibit all endogenous expressed TGF‐βs. Subsequently, slices were processed for immunoblotting.
2.4. Cell culture
2.4.1. Primary hippocampal and cortical astrocyte cultures
For primary culture of mixed glia, mouse pups aged P2/P3 were used, according to McCarthy & de Vellis, 1980, as previously described (Schrödl‐Häußel et al., 2015). For obtaining cultures from Tgf‐β2 deficient mice, embryos at embryonic day 18 (E18) were used. Experiments were carried out after 18–23 days in vitro, 12–18 hr before treatment, serum was deprived from the culture medium. Treatment conditions were 4AP (100 µM) for 20 min, human recombinant TGF‐β (2 ng/mL) for 60 min, and preincubation with anti‐TGF‐β1,2,3 (10 µg/mL), SP600125, a JNK inhibitor (10 µM), SIS3, a pSmad3 blocker (3 µM) and SB431542, an inhibitor of Alk4, Alk5, and Alk7 (10 µM) for 30 min.
2.5. Immunocytochemistry
Immunofluorescence of cultures has been performed as described (Brandes et al., 2007). Primary antibodies (NBCe1 1:200, GFAP 1:1000, Aldh1 1:3000, pSmad2(Ser465/467)/pSmad3(Ser423/425) 1:200, pSmad4(Thr277) 1:50) were diluted in BSA. Cells were incubated with goat anti‐rabbit IgG coupled to Alexa 568 (1:400) and goat anti‐mouse IgG coupled to Alexa 488 (1:400) and goat anti‐rabbit IgG coupled to Alexa 488 (1:400) for 1 hr at RT and viewed with a Leica TCS SP8 confocal microscope (Wetzlar, Germany).
2.6. Immunoblotting
Protein isolation from hippocampal slices or primary astrocytes, electrophoresis, and blotting procedures were performed as described (Schrödl‐Häußel et al., 2015). Protein concentration was determined by Thermo Scientific NanoDrop 2000 spectrophotometer. Primary antibodies were diluted: NBCe1 1:10,000, GAPDH 1:10,000, pSmad2/pSmad3 1:1,000, Smad2/Smad3 1:1,000, Na+/K+ ATPase 1:10,000, TGF‐β 1:500. Blots were developed in enhanced chemiluminescence reagents, and signals were visualized on X‐ray films. Films were scanned and the signal ratio protein of interest:housekeeping gene, was quantified densitometrically. Differences in signal ratio were tested for significance using two tailed unpaired Student's t‐test. For multiple comparisons one way ANOVA and Bonferroni post hoc test has been used. Results with levels of *p < .05, **p < .01, and ***p < .001 were considered significant.
2.7. Cell surface biotinylation
Primary cortical astrocytes were subjected to control or experimental conditions and then kept on ice. Isolation of cell surface proteins was performed using the Pierce® cell surface protein isolation kit following the manufacturer's instructions. Proteins were then processed for immunoblotting with antibodies against NBCe1, Na+/K+‐ATPase, and GAPDH as described above.
2.8. Quantitative real‐time PCR
Total RNA was isolated from cortical astrocytes as described earlier (Oehlke, Schlosshardt, Feuerstein, & Roussa, 2012) and subsequently 1.0 µg was reverse‐transcribed. Primers for NBCe1 were (corresponding to nucleotides 62–114; Genbank accession number AF210250; Giffard et al., 2000): 5′‐ TGGAGGATGAAGCTGTCC ‐3′ as forward primer and 5′‐ ACACACATGTTTAAGGAAGGAA ‐3′ as reverse primer. For GAPDH (Genebank accession number NM_001289726.1), Forward: 5′‐ TGACGTGCCGCCTGGAGAAA‐3′ (nt820–839), Reverse: 5′‐AGTGTAGCCCAAGATGCCCTTCAG‐3′ (nt917–894). Real‐time PCR was performed as described (Rickmann et al., 2007). Cycle conditions: denaturation at 95°C for 10 min, and 40 cycles of PCR amplification at 95°C for 30 s and 58°C for 30 s and elongation at 72°C for 1 min. All PCRs were performed in triplicate on a CFX Connect Realtime PCR (Biorad). The mean ± SEM of the Ct values for NBCe1 and GAPDH were determined and analysed for statistical significance using the Student's t‐test. *p < .05 was considered as statistically significant.
2.9. Intracellular H+ imaging
To measure the intracellular H+ concentration ([H+]i) changes in cultured cortical astrocytes, we used Visitron imaging system and acetoxymethyl ester of a proton‐sensitive dye, 2′,7′‐bis‐(carboxyethyl)‐5‐(and‐6)‐carboxyfluorescein (BCECF‐AM), as described previously (Theparambil et al., 2014). Cells were incubated with 3 µM BCECF‐AM in bicarbonated‐buffered saline solution for 15 min at room temperature. Cells were then mounted on a chamber of the Nikon ECLIPSE TE200 microscope and perfused continuously at room temperature with CO2/ ‐buffered saline solution (in mM): NaCl 114, KCl 5, NaH2PO4 0.5, α‐D‐glucose 2, NaHCO3 26, MgCl2 1, and CaCl2 2, pH 7.4. BCECF was excited consecutively at 488 nm (proton‐sensitive wavelength) and 440 nm (close to isosbestic point), and the changes in fluorescence emission were monitored at >505 nm (using ET540/40 m filter). Images were obtained every 10 s with a 20x water immersion objective. The fluorescence emission intensity of 488 nm excitation changes inversely with a change in [H+]i, whereas the fluorescence emission intensity of 440 nm excitation is largely pH insensitive. The changes in [H+]i were monitored using the ratio F(440)/F(488). The ratio was converted into pH and absolute intracellular proton concentrations ([H+]i) by using the nigericin‐based (4 µM) calibration technique (Theparambil et al., 2014). Cells were perfused with calibration solution (in mM), containing, KCl 145, NaH2PO4 0.4, Na2HPO4 1.6, Glucose 5, MgCl2 6H2O 1, Ca‐DiGluconat H2O 1.3, adjusted at pH 6.5, 7.0, 7.5, and 8.0.
2.10. MLEC assay
Primary cortical astrocytes were treated with or without 4AP for 20 min. Conditioned medium was collected for the mink lung epithelial cell (MLEC) assay. The principle of the assay is that the MLECs, containing a luciferase reporter under the control of a TGF‐β‐responsive truncated plasminogen activator inhibitor (PAI)‐promoter, are able to generate luciferase in a TGF‐ βs dose‐dependent manner. The MLECs only respond to the activated TGF‐ βs. To detect the latent part of TGF‐ βs, the conditioned medium has to be acidified first to convert latent TGF‐ βs into activated ones. To evaluate the levels of activated TGF‐ βs in extracellular environment after 4AP treatment, the MLEC assay was performed as described by Abe et al. (1994). MLECs were placed into 96‐well plates at the density of 2 × 104 cells per well and treated with collected conditioned medium either with or without acidification with 1 M HCL and pH adjustment with 1 M NaOH (to activate latent TGF‐ βs) for 18 hr. Cells were washed with PBS and total proteins were extracted using lysis buffer (Promega). The luciferase activity was analysed in duplicates using a luminometer (GloMax 96 Microplate Luminometer, Promega).
2.11. Chromatin immunoprecipitation assay
Chromatin immunoprecipation (ChIP) assays and PCR were performed on control and TGF‐β treated cortical astrocytes using EZ ChIP kit (Millipore), following the manufacturer's instructions. Briefly, DNA was crosslinked to protein with 1% paraformaldehyde. Cell lysates were obtained by scraping followed by sonication (Diagenode, Bioruptor Sonication Device) to shear cellular DNA. Overnight immunoprecipitations were performed with Histone 3 (H3), IgG, and an anti‐Smad4 antibody. On the next day the crosslinks were reversed, and bound DNA was purified. Subsequently, PCR was performed using primers specific for Smad4 promoter sequences in mouse NBCe1. Primer sequences (with their starting positions in the promoter sequence) are: Smad4 forward: 5′‐AGCCACCAATGACACTCCTG‐3′, Smad4 reverse: 5′‐AAGTGTTGTCAGCGCCCATA‐3′. The NBCe1 promoter sequence was retrieved using EnsEMBL, and conserved Smad4 binding sequence was identified by sequence alignment using ClustalW (EMBL‐EBI).
3. RESULTS
Previous data have shown that treatment of acute hippocampal slices with 4‐aminopyridine (4AP), an in vitro model for epilepsy (Gonzalez‐Sulser et al., 2011), significantly upregulates NBCe1 protein abundance (Schrödl‐Häußel et al., 2015). It is also well established that TGF‐βs play a pivotal role in epileptogenesis, both in vitro and in vivo (Heinemann et al., 2012). We have asked whether TGF‐βs might affect expression of NBCe1. Therefore, acute hippocampal slices were treated with 100 µM 4AP in the presence or absence of TGF‐βs, and subsequently NBCe1 protein abundance was determined by immunoblot analysis (Figure 1a). Using an antibody raised against NBCe1‐B/C, an immunoreactive band at ∼130 kDa was detected in controls (Figure 1a), and treatment of the slices with 100 µM 4AP for 20 min significantly upregulated NBCe1 abundance, confirming previous observations (1.14 ± .02 fold, n = 3 **p < .01, using the Student's t‐test). This upregulation, however, was prevented when slices were incubated with 4AP in the presence of 20 µg/mL anti‐TGF‐β1,2,3 (α‐TGF‐β; 0.68 ± .07; ## p < .01, using one‐way ANOVA and Bonferroni post hoc test; n = 3), a function‐blocking antibody potent to neutralize all endogenous expressed TGF‐βs. In contrast, in control slices, NBCe1 protein showed no changes following α‐TGF‐β treatment (0.90 ± 0.10 fold, n = 3, not significant, using the Student's t‐test). Similar results were also obtained following classical disinhibition using 20 µM bicuculline/0 Mg2+ for 10 min (Khalilov, Holmes, & Ben‐Ari, 2003). NBCe1 was significantly upregulated following treatment of the slices (Figure 1b; 1.16 ± .04 fold, n = 5, **p < .01, using the Student's t‐test) but this effect was again prevented in the presence of α‐TGF‐β1,2,3 (0.89 ± .08 fold n = 5, # p < .05).
Figure 1.
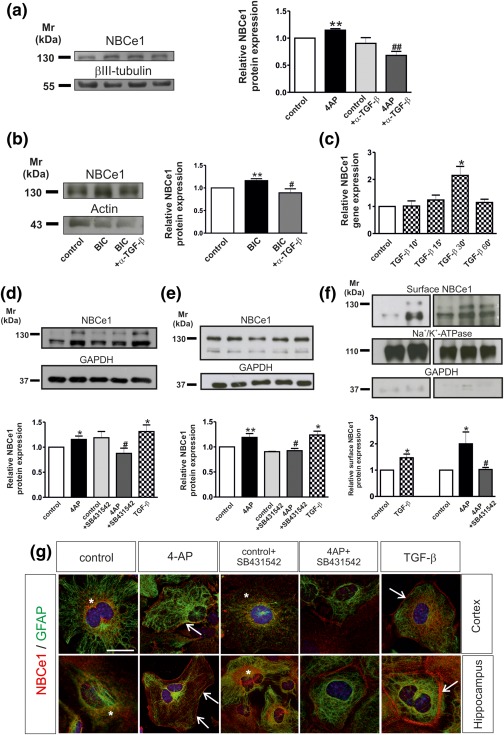
(a, b) Neutralization of endogenous TGF‐βs abolish 4‐AP‐ and bicuculline‐dependent NBCe1 upregulation in acute hippocampal slices. Immunoblotting for NBCe1 in control slices and following treatment with either 100 µM 4AP (a) or 20 µM bicuculline/0 Mg2+ (BIC) in the presence or absence of 20 µg/mL anti‐TGF‐β1,2,3 (α‐TGF‐β). Protein of 50 µg was loaded per lane, n = 3. (c) Time dependency of NBCe1 mRNA regulation in controls and following treatment with TGF‐β (2 ng/mL). Quantitative RT‐PCR analysis of cDNA from primary cortical astrocytes (*p < .05 for significant upregulation compared to controls, using the Student's t‐test, n = 3). (d, e) NBCe1 protein in cortical (d), and hippocampal (e) astrocytes following application of 4AP, 4AP in combination with the inhibitor of TGF‐β receptor II (10 µM, SB431642), or with 2 ng/mL recombinant TGF‐β. The blots are representative of six different experiments. Protein of 10 µg and 15 µg were loaded per lane for cortical and hippocampal astrocytes, respectively. (f) Immunoblot analysis of biotinylated cell surface proteins from cortical astrocytes for NBCe1. The blots are representative for three different experiments. Protein of 80 µg was loaded per lane. *p < .05, **p < .01 for significant increase, after densitometric analysis of the signal ratio NBCe1: βIII‐tubulin, NBCe1: actin, NBCe1: GAPDH, or NBCe1:Na+/K+‐ATPase and Student's t‐test, compared to the controls. # p < .05 and ## p < .01 for significant decrease compared to 4AP treatment using one‐way ANOVA and Bonferroni post hoc test. (g) Double immunofluorescence for NBCe1 and GFAP in primary cortical and hippocampal astrocytes. Nuclei are stained with DAPI. Arrows point to membrane NBCe1 and * indicates intracellular NBCe1 distribution. Scale bar: 10 µm
4‐AP effects on NBCe1 in hippocampal slices can be attributed to astrocytes (Schrödl‐Häußel et al., 2015). Since TGF‐βs have been shown to act on astrocytes (Heinemann et al., 2012) we have tested whether activation of TGF‐β signaling pathway might regulate NBCe1 expression in cortical astrocytes. Cortical astrocytes were treated with recombinant TGF‐β (2 ng/mL) for different time periods. Subsequently, NBCe1 transcript expression was determined by QRT‐PCR. Treatment of cortical astrocytes with TGF‐β for 10 min and 15 min had no effect on NBCe1 expression (Figure 1c; 1.02 ± 0.18 fold and 1.24 ± 0.18 fold, respectively, n = 3, not significant using the Student's t‐test). However, following treatment for 30 min, NBCe1 gene expression was significantly upregulated (2.14 ± 0.33 fold, n = 3, *p < .05, using the Student's t‐test), compared to controls. After 60 min TGF‐β treatment NBCe1 expression returned to baseline levels (1.15 ± 0.11 fold, n = 3).
3.1. TGF‐β is required for 4AP‐dependent NBCe1 regulation of transcript and protein
Based on these data, we next investigated TGF‐β requirement for 4AP‐dependent NBCe1 upregulation in cortical (Figure 1d) and hippocampal astrocytes (Figure 1e). Cultures were treated for 20 min either with 100 µM 4AP alone, or in the presence of SB431542 (10 µM) an Alk4, Alk5, and Alk7 inhibitor (Inman, 2002), and therefore inhibitor of TGF‐β receptor II‐, or with 2 ng/mL exogenous TGF‐β. Initial experiments have ensured that SB431542 has the same effect on cortical astrocytes as α‐TGF‐β. 4AP treatment significantly upregulated NBCe1 protein abundance in both primary cortical (Figure 1d) and hippocampal astrocytes (Figure 1e) (1.15 ±.06 fold and 1.19 ± .07 fold, respectively, n = 6, **p < .01 and *p < .05, using the Students t‐test), confirming previous observations. TGF‐β application significantly upregulated NBCe1 protein as well (1.31 ± 0.13 fold and 1.23 ± .07 fold, for cortical and hippocampal astrocytes, respectively, n = 6, **p < .01, using the Student's t‐test), whereas SB431542 abolished the 4AP‐dependent upregulation of NBCe1 protein (0.87 ± 0.10 fold and 0.92 ± .04 fold, n = 6, ## p < .01, using one way ANOVA and Bonferroni post hoc test). SB431542 under control conditions had no effect on NBCe1 protein as compared to untreated cultures (1.18 ± 0.12 and 0.90 ± .00 fold, n = 3). To test whether TGF‐β‐dependent upregulation of NBCe1 was accompanied by upregulation of NBCe1 surface expression as well, biotinylation of surface proteins was performed, followed by immunoblotting (Figure 1f). Treatment of the cultures with either TGF‐β or 4AP significantly upregulated NBCe1 surface expression (1.47 ± 0.14 fold and 2.00 ± 0.45 fold for TGF‐β (n = 3) and 4AP, respectively, *p < .05, using the Student's t‐test), compared to the controls and SB431542 abolished the 4AP‐dependent upregulation of NBCe1 surface protein (1.02 ± .07 fold, # p < .05).
These data have been confirmed by double immunofluorescence for NBCe1 and the astrocytic marker GFAP in cortical and hippocampal astrocytes and subsequent confocal microscopy (Figure 1g). NBCe1 immunoreactivity in control astrocytes was moderate and predominantly intracellularly distributed (asterisks). After 4AP treatment, intensity of intracellular NBCe1 immunolabeling was considerably increased, together with clear sharp labeling at the periphery of the cells (arrows), demonstrating plasma membrane labeling, as previously reported (Schrödl‐Häußel et al., 2015). NBCe1 immunolabeling was comparable in control cultures and those treated with SB431542, but were markedly reduced in cells treated with 4AP in the presence of SB431542 as compared to 4AP treatment alone. Application of exogenous TGF‐β caused a robust upregulation of both intracellular and membrane (arrow) NBCe1 immunolabeling, compared to the untreated controls.
Taken together, these data demonstrate that activation of TGF‐β signaling regulates NBCe1 transcript, protein, and surface expression in astrocytes, and that TGF‐β is required for 4AP‐dependent NBCe1 upregulation.
3.2. TGF‐β is required for 4AP‐dependent regulation of NBCe1 activity
After having shown that TGF‐β is required for 4AP‐dependent NBCe1 upregulation, we next investigated whether this effect is accompanied by changes on transport activity of NBCe1 by using intracellular [H+] recordings. [H+]i was monitored in cultured cortical astrocytes loaded with the H+‐selective dye BCECF. After calibration of the system (Figure 2a), NBCe1 activity was challenged by reducing the pH value and [ ] of the external solution from 7.4 and 26 mM to 7.1 and 13 mM, respectively. This activates NBCe1 to transport Na+‐ out of the cells, which results in an increase of [H+]i (Figure 2b,c) (Theparambil, Naoshin, Thyssen, & Deitmer, 2015). This increase in [H+]i, was determined in control (Ctr; not treated) and 4AP‐treated astrocytes in the presence or absence of SB431542. Using this experimental design, the rate of acidification in pH 7.1 (Figure 2b,c), the rate of alkalinisation upon returning from pH 7.1 to pH 7.4 (Figure 2b,d), and the amplitude of the acidification in pH 7.1 (Figure 2e) was measured in controls (30.96 ± 2.52 nM/min, −26.82 ± 2.4 nM/min and 117.6 ± 7.26 nM). A significant increase in all three parameters was induced by 4‐AP (57.86 ± 3.08 nM/min, −77.70 ± 3.46 nM/min and 194.1 ± 11.41 nM), confirming previous observations (Schrödl‐Häußel et al., 2015). The increase was largely suppressed by SB431542 (35.91 ± 3.24 nM/min, −46.26 ± 4.31 nM/min and 140.6 ± 10.7 nM). In contrast, application of SB431542 in control astrocytes had no effect on all parameters (31.07 ± 1.81 nM/min, −28.20 ± 1.36 nM/min, and 96.47 ± 8.87), compared to the untreated controls (Figure 2b–e).
Figure 2.
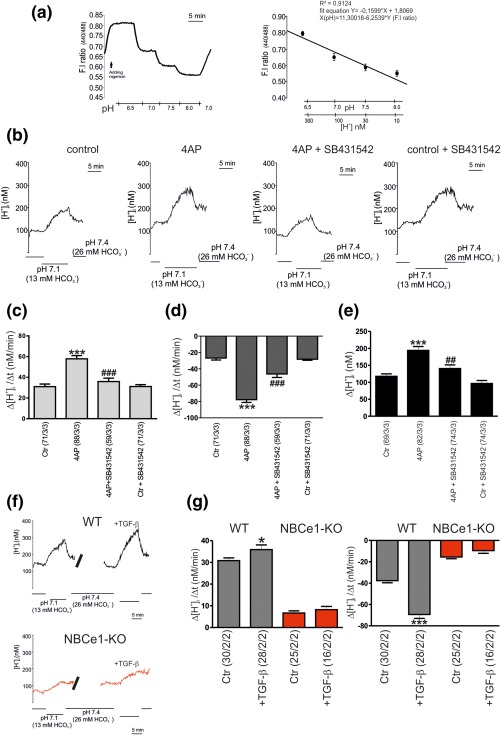
Evaluation of NBCe1 transport activity. Calibration of the H+ ‐sensitive fluorescence signal in cultured cortical astrocytes. Calibration of the fluorescence intensity (F.I) 440/488 ratio signal in the presence of nigericin to equilibrate H+ across the cell membrane, monitored at different extracellular pH values, 6.5, 7.0, 7.5, 8.0, and plotted against the pH value and the respective H+ concentration. The coefficient of correlation (R2) and the fit equation are indicated in the plot. (b): Original recordings of intracellular [H+] ([H+]i) in cultured cortical astrocytes during reduction of external pH and [ ] from 7.4 and 26 mM to 7.1 and 13 mM, respectively, to challenge outwardly directed NBCe1 activity, before and after incubation with 4‐AP (100 µM, 20 min), in control, and in the presence of the TGF‐β receptor II inhibitor SB431542 (10 µM) (b–e). Bar plots of the rate of acidification (c), the rate of alkalinisation (d), and the amplitude (e) as measured upon changing external pH and [ ] to 7.1 and 13 mM, respectively, and back to pH 7.4 and 26 mM [ ]. (f): Original recordings of intracellular [H+] ([H+]i) in cultured cortical astrocytes from wild type (black; WT) and Slc4a4 deficient mice (red; NBCe1‐KO) during reduction of external pH and [ ] from 7.4 and 26 mM to 7.1 and 13 mM, respectively, before and after incubation with 2 ng/mL TGF‐β for 60 min. (g): Bar plots of the rate of acidification, and the rate of alkalinisation as measured upon changing external pH and [ ] to 7.1 and 13 mM, respectively, and back to pH 7.4 and 26 mM [ ]. *p < .05, ***p < .001, for the significant increase, compared to the untreated controls, using the Student's t‐test and ## p < .01, ### p < .001 for the significant decrease, compared to 4AP treatment, using one‐way ANOVA and Bonferroni post hoc test. The number of cells/cultures/animals used in the experiments is indicated
3.3. Activation of TGF‐β signaling regulates NBCe1 activity
The data presented in Figure 1c–e implicate regulation of NBCe1 transcript and protein following activation of TGF‐β signaling. To test whether activation of TGF‐β signaling specifically regulates NBCe1 activity in cortical astrocytes, we treated wild type (WT) and Slc4a4‐deficient (NBCe1‐KO) astrocytes with 2 ng/mL exogenous TGF‐β and subsequently, NBCe1 activity was determined (Figure 2f,g). The rate of acidification in pH 7.1 and the rate of alkalinisation upon returning from pH 7.1 to pH 7.4 were 30.8 ± 1.26 nM/min, and −37.64 ± 1.98 nM/min for the WT astrocytes and 6.694 ± 0.98 nM/min, and −15.49 ± 1.58 nM/min for the NBCe1 KO astrocytes, respectively. After application of TGF‐β both the rate of acidification and rate of alkalinisation were significantly increased in WT (35.89 ± 2.17 nM/min, and −69.37 ± 3.63 nM/min). In NBCe1‐KO astrocytes, all parameters were greatly reduced as compared to WT cells (8.22 ± 1.50 nM/min and −9.47 ± 2.57 nM/min).
3.4. TGF‐β increases NBCe1 activity in cortical astrocytes through the canonical pathway and JNK signaling pathway
TGF‐βs can exert their actions through activation of the canonical (Smad‐dependent), and/or noncanonical signaling pathways (Akhurst & Hata, 2012). We have addressed the question whether activation of the canonical pathway is linked to the observed 4AP‐ and TGF‐β‐dependent increase of NBCe1 protein (Figure 1d). Moreover, based on our previous data where 4AP‐dependent NBCe1 upregulation can be mediated through JNK pathway (Schrödl‐Häußel et al., 2015), we asked whether inhibition of JNK pathway might affect TGF‐β‐dependent upregulation of NBCe1 protein. To that end, cortical astrocytes were treated with TGF‐β (2 ng/mL) for 60 min in the presence of 3 µM of the specific Smad3 inhibitor SIS3 or the JNK inhibitor SP600125 (10 µM). Inhibition of either Smad3 or of JNK pathway had no effect on NBCe1 protein abundance in control cortical astrocytes (Figure 3a; 1.04 ± .05 fold and 1.17 ± 0.17 fold for SIS3 and SP600125, respectively), but significantly decreased NBCe1 protein in the presence of TGF‐β (0.9 ± .06 fold and 0.84 ± .07 fold for SIS3 and SP600125, respectively, *p < .05 using the Student's t‐test and # p < .05, using one way ANOVA and Bonferroni post hoc test, n = 4). These results were confirmed by double immunofluorescence for NBCe1 and ALDH1 (Figure 3b). Following incubation of control cortical or hippocampal astrocytes with SIS3, NBCe1 immunoreactivity was comparable to the untreated cells and showed cytosolic distribution (asterisks). Following TGF‐β treatment, intensity of NBCe1 immunolabeling was increased and restricted to the periphery of the astrocytes, suggesting plasma membrane staining (arrows). This labeling pattern was prevented when hippocampal or cortical astrocytes were treated with TGF‐β in the presence of SIS3. 4AP‐dependent NBCe1 upregulation in cortical astrocytes was also prevented in the presence of SIS3 (Figure 3c; 1.16 ± .02 fold and 0.88 ± .08 fold for 4AP and 4AP in the presence of SIS3, respectively, *p < .05 using the two tailed unpaired Student's t‐test and # p < .05, using one way ANOVA and Bonferroni post hoc test, n = 3).
Figure 3.
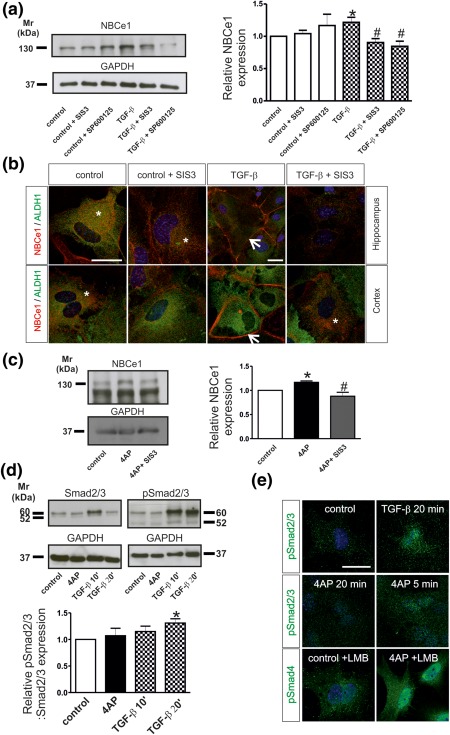
(a) NBCe1 protein expression in primary cortical astrocytes following application of the inhibitor of Smad3 SIS3 (3 µM), or the inhibitor of JNK signaling, SP600125 (10 µM) in the presence or absence of recombinant TGF‐β (2 ng/mL). The blot is representative of four different experiments. Protein of 20 µg was loaded per lane. (b) Double immunofluorescence for NBCe1 and ALDH1 in primary hippocampal and cortical astrocytes. Arrows point to membrane NBCe1 and * indicates intracellular NBCe1 distribution. Scale bar: 25 µm (c) NBCe1 protein expression in cortical astrocytes after 4AP treatment in the presence of the inhibitor of Smad3 SIS3 (3 µM). The blot is representative of three different experiments. Protein 50 µg was loaded per lane. (d) Activation of canonical TGF‐β signaling in primary astrocytes. Immunoblotting for Smad2/3 and phosphorylated Smad2/3 (pSmad2/3) in control cortical astrocytes and following treatment with 100 µM AP for 20 min, or 2 ng/mL TGF‐β for 10 or 20 min. Protein of 10 µg was loaded per lane, n = 3. (*p < .05 for significant increase after densitometric analysis of the signal ratio NBCe1:GAPDH or ratio (pSmad2/3: GAPDH): (Smad2/3:GAPDH) and Student's t‐test, compared to controls, # p < .05 for significant decrease using one‐way ANOVA and Bonferroni post hoc test, compared to TGF‐β or 4AP treatment. (e) Immunofluorescence for pSmad2/3 in control cortical astrocytes and after treatment with 4AP for 5 min and 20 min or TGF‐β for 20 min. Immunofluorescence for pSmad4 in control astrocytes and after treatment with 4AP in the presence of the nuclear export inhibitor leptomycin B (LMB). Nuclei are stained with DAPI. Scale bar: 25 µm
Activation of canonical pathway is shown in Figure 3d. Cortical astrocytes were treated either with 2 ng/mL TGF‐β (10 min or 20 min) or with 4AP (20 min) and subsequently total Smad2/3 and phosphorylated‐Smad2/3 (pSmad2/3) protein abundance were assessed by immunoblotting. Total Smad2/3 protein was detected as a double immunoreactive band at ∼52 kDa and ∼60 kDa, and the intensity of the band was comparable in all experimental groups. In contrast, pSmad2/3/Smad2/3 protein expression was found significantly increased after exposure of the cells to TGF‐β for 20 min (1.31 ± .08 fold) but not after 10 min (1.15 ± 0.10 fold) or after 4AP for 20 min (1.07 ± 0.14), as compared to untreated control astrocytes (*p < .05, using the Student's t‐test n = 3), demonstrating activation of the canonical pathway. Immunofluorescence for pSmad2/3 revealed intracellular distribution in controls and translocation to the nucleus following either TGF‐β treatment for 20 min or 4AP treatment for 5 min (Figure 3e). Treatment with 4AP for 20 min had no effect on pSmad2/3 localization but considerably increased labeling intensity and nuclear translocation of pSmad4 in the presence of leptomycin B (LMB; Chapnick & Liu, 2010), known to prevent nuclear export of Smad4.
As a next step, we studied whether the observed TGF‐β‐dependent regulation of NBCe1 protein through Smad and JNK pathways is associated with corresponding changes of the transporter activity. As shown in Figure 4a, using intracellular [H+] recordings as described above, we next tested regulation of NBCe1 activity by TGF‐β through the canonical and JNK signaling pathway. The rate of acidification in pH 7.1 (Figure 4a,b), the rate of alkalinisation upon returning from pH 7.1 to pH 7.4 (Figure 4a,c) and the amplitude of the acidification in pH 7.1 (Figure 4d) in control astrocytes was 27.68 ± 0.91 nM/min, −31.92 ± 1.16 nM/min, and 125.3 ± 4.29 nM, respectively. After application of exogenous TGF‐β, all three parameters were significantly increased (35.72 ± 1.19 nM/min, −50.11 ± 1.76 nM/min, and 149.7 ± 6.01 nM). The TGF‐β‐dependent NBCe1 activation was suppressed in the presence of SIS3 (25.53 ± 1.5 nM/min, −44.11 ± 2.45 nM/min and 119.8 ± 6.62 nM), whereas inhibition of JNK signaling with SP600125 had no effect on the rate of acidification (33.47 ± 1.90 nM/min), but significantly decreased the rate of alkalinisation (–38.50 ± 1.43 nM/min), and the amplitude (109.3 ± 4.93 nM), compared to TGF‐β treatment.
Figure 4.
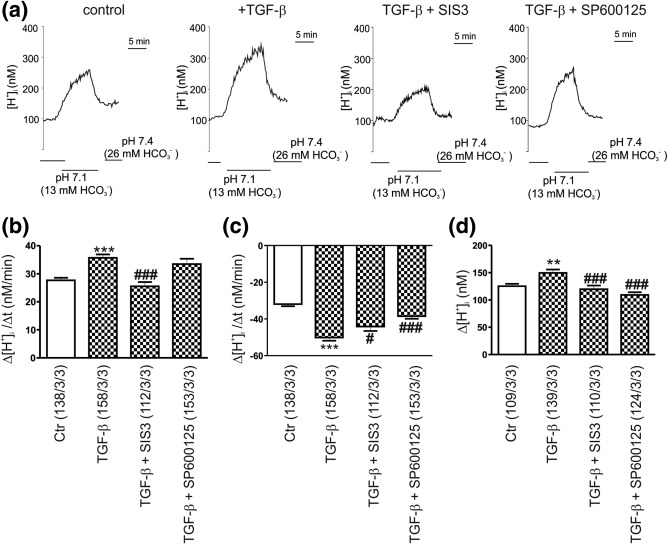
Evaluation of NBCe1 transport activity. (a) Original recordings of intracellular [H+] ([H+]i) in cultured mouse cortical astrocytes during reduction of external pH and [ ] from 7.4 and 26 mM to 7.1 and 13 mM, respectively, to challenge outwardly directed NBCe1 activity, before and after application of TGF‐β (2 ng/mL, 60 min), in control, and in the presence of the Smad3 inhibitor SIS3 (3 µM) or the JNK inhibitor SP600125 (10 µM). Bar plots of the rate of acidification (b), the rate of alkalinisation (c), and the amplitude (d) as measured by changing external pH and [ ] to 7.1 and 13 mM, respectively, and back to pH 7.4 and 26 mM [ ], before and after incubation with TGF‐β in controls, and in the presence of SIS3 or SP600125. **p < .01 and ***p < .001, for the significant increase, compared to the untreated controls, using the Student's t‐test and # # p < .05, and ### p < .001 for the significant decrease, compared to TGF‐β, using one way ANOVA and Bonferroni post hoc test. The number of cells/cultures/animals used in the experiments is indicated
We have previously shown that the isoform 2 of TGF‐β (TGF‐β2) is able to increase trafficking and functional expression of K+/Cl‐ cotransporter 2 in mouse hippocampal neurons (Roussa et al., 2016). We therefore asked whether TGF‐β2 is the isoform required for 4AP‐dependent regulation of NBCe1 in cortical astrocytes. Using intracellular [H+] recordings we have examined regulation of NBCe1 transport activity in cortical astrocytes from WT and Tgf‐β2 deficient mice (TGF‐β2‐KO). The results are presented in Figure 5a–d. The rate of acidification in pH 7.1 (Figure 5a,b) and the rate of alkalinisation (Figure 5a,c) upon returning from pH 7.1 to pH 7.4 and the amplitude of the acidification in pH 7.1 (Figure 5d) were 23.71 ± 2.06 nM/min, −32.04 ± 2.61 nM/min and 169.4 ± 7.78 nM for the WT astrocytes and 54.24 ± 3.82 nM/min, −64.36 ± 4.81 nM/min and 249.7 ± 14.31 for the TGF‐β2‐KO astrocytes, respectively. After application of 4AP, both the rate of acidification and rate of alkalinisation and amplitude were significantly increased in WT (58.5 ± 3.4 nM/min, −66.52 ± 4.54 nM/min and 268.7 ± 12.25 nM) and in TGF‐β2‐KO (88.96 ± 5.72 nM/min, −97.37 ± 6.09 nM/min and 309.8 ± 9.89). Because the rate of acidification and the rate of alkalinisation in TGF‐β2‐KO astrocytes were significantly increased as compared to WT (Figure 5b,c), we next examined putative subsequent changes in baseline pHi. As shown in Figure 5e, baseline pHi was comparable between WT and in astrocytes derived from Tgf‐β2 deficient mice.
Figure 5.
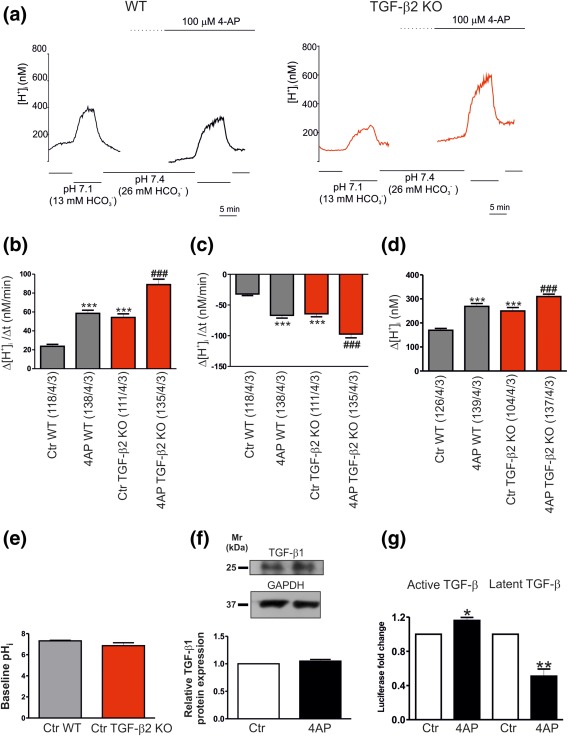
4AP activates extracellular TGF‐βs. (a) Original recordings of intracellular [H+] ([H+]i) in cultured wild type (WT) and Tgf‐β2‐deficient (TGF‐β2‐KO) mouse cortical astrocytes during reduction of external pH and [ ] from 7.4 and 26 mM to 7.1 and 13 mM, respectively, to challenge outwardly directed NBCe1 activity, before and after application of 4AP. Bar plots of the rate of acidification (b), the rate of alkalinisation (c), and the amplitude (d) as measured by changing external pH and [ ] to 7.1 and 13 mM, respectively, and back to pH 7.4 and 26 mM [ ], before and after incubation with 4AP in WT and TGF‐β2‐KO astrocytes (***p < .001, for significant increase, compared to the untreated (Ctr) WT astrocytes, ### p < .001 for significant increase, compared to untreated TGF‐β2‐KO astrocytes, using the Student's t‐test, n = 3). (e) Quantification of baseline pHi in untreated (Ctr) cortical astrocytes derived from either WT or Tgf‐β2 deficient mice (not significant, using Student's t‐test, n = 3). (f) Immunoblotting for endogenous TGF‐β1 protein in control and 4AP‐treated cortical astrocytes (not significant after densitometric analysis of the signal ratio TGF‐β1: GAPDH and Student's t‐test, n = 6). Protein of 50 µg was loaded per lane. (g) The amount of active and latent TGF‐β in the extracellular environment was quantified with the MLEC/PAI‐luciferase assay. Data are given as relative amounts of TGF‐β in the supernatant following 4AP treatment, as compared to the untreated controls (*p < .05 and **p < .01, using the Student's t‐test, n = 3)
Since cortical astrocytes endogenously express TGF‐β1 (Vivien et al., 1998), we next investigated whether 4AP might increase endogenous TGF‐β1 expression. TGF‐β1 (dimer) protein was detected in control astrocytes as an immunoreactive band at ∼25 kDa, and the intensity of the band was comparable in controls and 4‐AP treated cells (Figure 5f; 1.05 ± .03 fold; n = 6).
We further addressed whether TGF‐β secretion was changed following 4AP treatment of cortical astrocytes. Therefore, the conditioned media from 20 min 4AP‐treated, as well as nontreated astrocytes were harvested, and the MLEC assay was performed to monitor TGF‐ β secretion (Figure 5g). Quantification of TGF‐β‐induced intensity of luciferase shows that cortical astrocytes secreted TGF‐β under basal conditions. 4AP treatment significantly increased active TGF‐β (1.16 ± .03 fold, n = 3 *p < .05) and significantly decreased latent TGF‐β (0.50 ± .08 fold, **p < .01, n = 3, using the Student's t‐test) present in the medium.
3.5. TGF‐β directly activates NBCe1 transcription via Smad4
To provide conclusive evidence that TGF‐β can directly upregulate NBCe1 via the canonical pathway, we performed a search for conserved Smad binding sequence in the promoter region of NBCe1 (Figure 6a). Indeed, the NBCe1 promoter on chromosome 5: 88886260–88887360 contains one conserved Smad4 binding site. To demonstrate that Smad4 binds to the NBCe1 promoter sequence, chromatin immunoprecipitation (ChIP) was performed in control mouse primary cortical astrocytes and after treatment with exogenous TGF‐β (2 ng/mL) for 30 min. Smad4 was immunoprecipitated using an antibody against Smad4, and subsequently PCR was performed to amplify DNA fragments bound to Smad4. Therefore, primers flanking the Smad4 binding sequence at position 88886675–88886690 were used, as shown in Figure 6a. The results are shown in Figure 6b. Bands at the expected size of 89 bp were detected in input of controls (lane 1) and treated cortical astrocytes (lane 4). Moreover, a band was also detected for Smad4 binding sequence in the NBCe1 promoter region following TGF‐β treatment (lane 5) that was not found in the control astrocytes (lane 2). H3 (lane 8) and IgG (lane 7) were used as positive and negative control for the immunoprecipitation, respectively. These results demonstrate that TGF‐β transcriptionally activates NBCe1 via canonical pathway.
Figure 6.
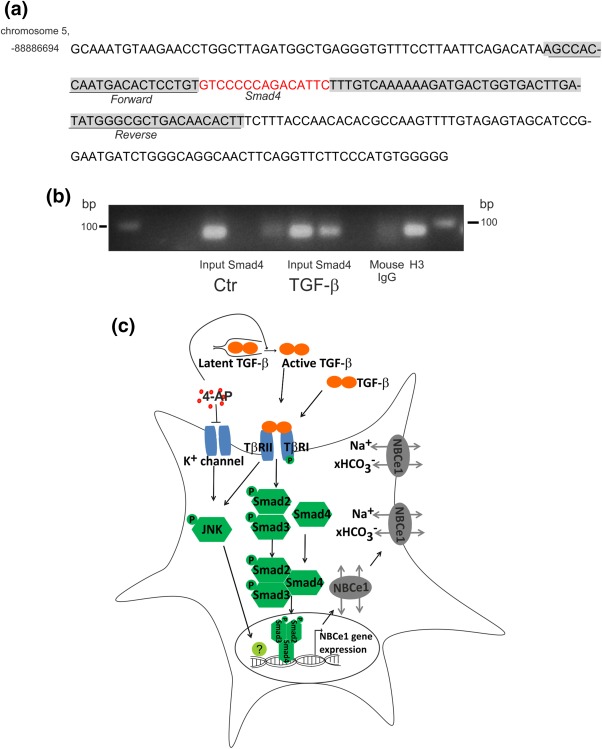
Smad4 specifically binds to the promoter of NBCe1 in mouse primary cortical astrocytes after TGF‐β treatment. (a) Smad4 binding site (red) to the NBCe1 promoter region. Underlined sequences represent primer sequences used to detect Smad4 binding. (b) PCR following chromatin immunoprecipitation assay of control and TGF‐β‐treated mouse primary cortical astrocytes demonstrates increased binding of Smad4 to the predicted binding sequence in the promoter region of NBCe1. Predicted PCR product size 89 bp. Histone 3 (H3), and mouse IgG represent positive and negative control, respectively. The figure is a representative of n= 2. (c) Proposed model for TGF‐β‐dependent NBCe1 regulation
The results of the present work are graphically summarised in Figure 6c.
4. DISCUSSION
Intracellular pH (pHi) regulation is a homeostatic process fundamental for cellular function and survival. In astrocytes, regulation of pHi is of particular importance because astrocytes control the ionic environment of the neuropil, thereby contributing to modulation of neuronal signaling and synaptic transmission (Deitmer & Rose, 2010; Haydon & Carmignoto, 2006). Hence, one of the fundamental questions is to identify regulatory signals that orchestrate expression, trafficking, and activity of acid‐base transporters, among them of NBCe1.
In the present work, we have hypothetised that TGF‐β regulates NBCe1. This hypothesis was based on following observations: First, the role of TGF‐βs in epileptogenesis and a TGF‐β‐dependent activation of astrocytes with subsequent changes in expression and trafficking of channels and transporters is well established (Heinemann et al., 2012). Second, in an in vitro model of epilepsy (4AP treatment), NBCe1 expression was upregulated in both hippocampal slices and astrocytes (Schrödl‐Häußel et al., 2015). And third, in other cellular paradigms, TGF‐β regulates transporters that are functionally coupled to NBCe1 (Roux et al., 2010; Yi et al., 2013).
We show that blocking of TGF‐βs in acute hippocampal slices prevented 4AP‐ and bicuculline‐dependent NBCe1 upregulation (Figure 1a,b). NBCe1 is highly expressed in astrocytes and responsible for the bulk of bicarbonate movement across the astrocytic membrane (Deitmer & Rose, 1996, 2010). However, neurons express also NBCe1 (Rickmann et al., 2007; Svichar, Esquenazi, Chen, & Chesler, 2011), and NBCe1 has even been considered to act by an unknown mechanism as communication molecule between neurons and astrocytes (Salameh et al., 2017). Moreover, TGF‐β is activity‐dependent released by neurons (Lacmann, Hess, Gohla, Roussa, & Krieglstein, 2007), highlighting the complexity of the network. Although we cannot exclude that the effects observed in acute slices may partly result from neuron/glia interactions, we have previously shown that 4AP has its own effect on astrocytes which is totally independent of neurons (Schrödl‐Häußel et al., 2015). We have therefore focused on astrocytes to analyse the impact of TGF‐β on NBCe1.
NBCe1 transcript, protein, and activity can be regulated by Src/ERK signaling (Namkoong et al., 2015; Schrödl‐Häußel et al., 2015), and surface expression and activity by phosphorylation (Hong et al., 2013; Thornell & Bevensee, 2015), JNK signaling (Schrödl‐Häußel et al., 2015), and intracellular bicarbonate concentration (Theparambil et al., 2015). However, the question whether intrinsic stimuli, by means of signaling molecules and growth factors might initiate changes in NBCe1 expression has not been addressed so far. Here, we show that the previously observed 4AP‐dependent NBCe1 regulation of NBCe1 functional expression in astrocytes requires TGF‐β: (1) inhibition of TGF‐β signaling prevented 4AP‐dependent regulation of NBCe1 protein (Figure 1d,e) and decreased 4AP‐dependent surface expression of NBCe1 in cortical and hippocampal astrocytes (Figure 1f,g); and (2) inhibition of TGF‐β signaling significantly decreased 4AP‐dependent upregulation of NBCe1 transport activity (Figure 2b–d). This effect is specific for 4AP, since inhibition of TGF‐β signaling in control astrocytes had no effect on NBCe1 (Figure 2b–d). We have used SB431542 to block TGF‐β signaling (Inman, 2002). Because this inhibitor inhibits Alk4, 5, and 7, we have performed initial experiments with α‐TGF‐β1,2,3 and ensured that α‐TGF‐β1,2,3 and SB31542 have the same effects on 4AP‐dependent NBCe1 regulation, suggesting that the results of this work indeed derive from inhibition of TGF‐β signaling.
A further main finding of this work is that activation of TGF‐β signaling regulates transcript (Figure 1c), protein (Figure 1d,e), surface expression, (Figure 1f,g) and transport activity (Figure 4) of NBCe1 in astrocytes. TGF‐β had however no effect on transport activity of NBCe1 in astrocytes obtained from NBCe1‐KO mice (Figure 2f,g). To our knowledge, this is the first demonstration of a growth factor dependency of NBCe1. TGF‐β has been shown to regulate several transport proteins in astrocytes (Rathore, Redensek, & David, 2012). TGF‐βs exert their effects in a cell‐ and context‐dependent manner through several signaling pathways (Akhurst & Hata, 2012). In the present study, TGF‐β regulates NBCe1 through at least two signaling pathways: the canonical, Smad‐dependent, and the JNK pathway (Figures 3 and 4). Treatment of cortical astrocytes with TGF‐β in the presence of either SIS3, a specific Smad 3 inhibitor (Jinnin, Ihn, & Tamaki, 2006) or SP600125 prevented the TGF‐β effects on NBCe1 protein (Figure 3a), surface expression (Figure 3b) and transport activity (Figure 4). We could also detect a link between 4AP treatment of the astrocytes and activation of Smad2/3‐ signaling (Figure 3c,e), as well as enrichment of Smad4 in the nucleus in the presence of leptomycin B, an inhibitor of nuclear export (Figure 3e). Together with previous observations (Schrödl‐Häußel et al., 2015), we conclude that TGF‐β is required for 4AP‐dependent NBCe1 regulation acting via the canonical and/or JNK pathway.
Since astrocytes express TGF‐β1 and TGF‐β2 (Vivien et al., 1998), we asked which isoform is responsible for the regulation of NBCe1. The phenotypes resulting from the knockout of the mammalian TGF‐β isoforms are very distinct (reviewed in Dünker & Krieglstein, 2000). We have recently shown that TGF‐β2 regulates functional expression of the neuronal K+/Cl‐ cotransporter 2 (KCC2) (Roussa et al., 2016), a molecular component increasingly appreciated in the context of epilepsy (Kelley et al., 2016; Kourdougli, Varpula, Chazal, & Rivera, 2015). We have hypothesized that TGF‐β2 is the isoform required for 4AP‐dependent NBCe1 regulation. In both WT and Tgf‐β2‐/‐ astrocytes NBCe1 transport activity was significantly increased (Figure 5b–d), while baseline pHi was comparable between WT and TGF‐β2‐KO (Figure 5e). These data imply that other molecular mechanisms are activated and upregulate NBCe1 activity in order to compensate for TGF‐β2 loss and to maintain pHi homeostasis. These data suggest that TGF‐β2 is necessary but not sufficient for 4AP‐dependent NBCe1 regulation.
What are the mechanisms that lead to 4AP‐dependent activation of TGF‐β signaling in astrocytes? 4AP does not increase intracellular TGF‐β concentration in astrocytes (Figure 5f). 4AP does not increase TGF‐β release in the extracellular space either, because after 4AP treatment, the amount of latent extracellular TGF‐β was significantly decreased (Figure 5g). Our results rather support the notion that 4AP activates extracellular latent TGF‐β. The underlying mechanisms for this effect are not clear but may include 4AP‐dependent acidification of the extracellular space, or increased expression of metalloproteinases, required to cleave latent TGF‐β. Finally, we investigated whether TGF‐β directly regulates transcriptional activity of NBCe1. Promoter analysis of NBCe1 revealed conserved Smad4 binding site and ChIP experiments have demonstrated increased Smad4 binding to the promoter of NBCe1 upon treatment of cortical astrocytes with TGF‐β (Figure 6), ultimately demonstrating that NBCe1 is a direct target of TGF‐β.
What could be the biological significance of our results? Various membrane channels and transporters are altered in the epileptic brain, and experimental evidence increasingly highlights the crucial role of glia in the context of epileptogenesis, (reviewed in Bedner & Steinhäuser, 2016). NBCe1 may act as an acid extruder, enhance depolarization‐induced alkalinisation of astrocytes, and extracellular acidification, and thereby reduce susceptibility to epileptic seizures. However, NBCe1 may act as an acid‐loader as well (Schrödl‐Häußel et al., 2015 and Figure 2), hence potent to modulate synaptic pH (Theparambil et al., 2015).
Our data provide first insights into the TGF‐β‐dependent transcriptional control and regulation of NBCe1 functional expression. We propose a model (Figure 6c) in which, under pathophysiological conditions, here 4AP, TGF‐β in the extracellular space becomes activated and acts at the autocrine and/or paracrine mode to the astrocytes by binding to the TGF‐β receptor and activating TGF‐β and JNK signaling pathways, leading to regulation of NBCe1 functional expression. TGF‐β acts directly on NBCe1 by binding of Smad4 to the NBCe1 promoter. NBCe1 is a novel direct target of TGF‐β and maybe one of the molecular players in the pathophysiology of epilepsy. In this context, further studies need to elucidate the regulation mode of NBCe1 in neurons and TGF‐β‐ and NBCe1‐dependent possible neuron/glia interactions together with in vivo validation of the model.
ACKNOWLEDGMENT
We thank Ellen Gimbel for excellent technical assistance, Shefeeq Theparambil for his advice on functional experiments, and Gary E. Shull, Cincinnatti, USA, for providing NBCe1‐KO mice. This work was supported by the Deutsche Forschungsgemeinschaft to ER (KR 1477/15‐1).
Khakipoor S, Ophoven C, Schrödl‐Häußel M, et al. TGF‐β signaling directly regulates transcription and functional expression of the electrogenic sodium bicarbonate cotransporter 1, NBCe1 (SLC4A4), via Smad4 in mouse astrocytes. Glia. 2017;65:1361–1375. https://doi.org/10.1002/glia.23168
REFERENCES
- Abe, M. , Harpel, J. G. , Metz, C. N. , Nunes, I. , Loskutoff, D. J. , & Rifkin, D. B. (1994). An assay for transforming growth factor‐b using cells transfected with a plasminogen activator inhibitor‐1 promotor‐luciferase construct. Analytical Biochemistry, 216, 276–284. [DOI] [PubMed] [Google Scholar]
- Akhurst, R. J. , & Hata, A. (2012). Targeting the TGFβ signalling pathway in disease. Nature Reviews Drug Discovery, 11, 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedner, P. , & Steinhäuser, C. (2016). Neuron‐glia interaction in epilepsy. Journal of Neuroscience Research, 94, 779–780. [DOI] [PubMed] [Google Scholar]
- Brandes, A. , Oehlke, O. , Schumann, A. , Heidrich, S. , Thevenod, F. , & Roussa, E. (2007). Adaptive redistribution of NBCe1‐A and NBCe1‐B in rat kidney proximal tubule and striated ducts of salivary glands during acid‐base disturbances. American Journal of Physiology, 293, R2400–R2411. [DOI] [PubMed] [Google Scholar]
- Brookes, N. , & Turner, R. J. (1994). K(+)‐induced alkalinization in mouse cerebral astrocytes mediated by reversal of electrogenic Na(+)‐ cotransport. American Journal of Physiology, 267, C1633–C1640. [DOI] [PubMed] [Google Scholar]
- Brune, T. , Fetzer, S. , Backus, K. H. , & Deitmer JW. (1994). Evidence for electrogenic sodium‐bicarbonate cotransport in cultured rat cerebellar astrocytes. European Journal of Physiology, 429, 64–71. [DOI] [PubMed] [Google Scholar]
- Chapnick, D. A. , & Liu, X. (2010). Analysis of ligand‐dependent nuclear accumulation of Smads in TGF‐β signalling In Higgins P. J. (Ed.), Transcription factors: Methods and protocols (Vol. 647, pp. 95–111). New York, NY: Humana Press, c/o Springer Science+Business Media, LLC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer, J. W. (1991). Electrogenic sodium‐dependent bicarbonate secretion by glial cells of the leech central nervous system. The Journal of General Physiology, 98, 637–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer, J. W. , & Rose, C. R. (1996). pH regulation and proton signalling by glial cells. Progress in Neurobiology, 48, 73–103. [DOI] [PubMed] [Google Scholar]
- Deitmer, J. W. , & Rose, C. R. (2010). Ion changes and signalling in perisynaptic glia. Brain Research Reviews, 63, 113–129. [DOI] [PubMed] [Google Scholar]
- Derynck, R. , & Miyazono, K. (2008). TGF‐β and The TGF‐β Family In Derynck R. & Miyazono K. (Eds.), The TGF‐ β family (Vol. 50, pp. 25–40). New York, N.Y: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Dünker, N. , & Krieglstein, K. (2000). Targeted mutations of transforming growth factor‐β genes reveal important roles in mouse development and adult homeostasis. European Journal of Biochemistry, 267, 6982–6988. [DOI] [PubMed] [Google Scholar]
- Gawenis, L. R. , Bradford, E. M. , Prasad, V. , Lorenz, J. N. , Simpson, J. E. , Clarke, L. L. , … Shull, G. E. (2007). Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/ cotransporter. Journal of Biological Chemistry, 282, 9042–9052. [DOI] [PubMed] [Google Scholar]
- Giffard, R. G. , Papadopoulos, M. C. , van Hooft, J. A. , Xu, L. , Giuffrida, R. , & Monyer, H. (2000). The electrogenic sodium bicarbonate cotransporter: Developmental expression in rat brain and possible role in acid vulnerability. Journal of Neuroscience, 20, 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Sulser, A. , Wang, J. , Motamedi, G. K. , Avoli, M. , Vicini, S. , & Dzakpasu, R. (2011). The 4‐aminopyridine in vitro epilepsy model analysed with a perforated multi‐electrode array. Neuropharmacology, 60, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon, P. G. , & Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiological Reviews, 86, 1009–1031. [DOI] [PubMed] [Google Scholar]
- Heinemann, U. , Kaufer, D. , & Friedman, A. (2012). Blood‐brain barrier dysfunction, TGFβ signaling, and astrocyte dysfunction in epilepsy. Glia, 60, 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, J. H. , Yang, D. , Shcheynikov, N. , Ohana, E. , Shin, D. M. , & Muallem, S. (2013). Convergence of IRBIT, phosphatidylinositol (4,5) bisphosphate, and WNK/SPAK kinases in regulation of the Na+‐ cotransporters family. Proceedings of the National Academy of Sciences of the United States of America, 110, 4105–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman, G. J. (2002). SB‐431542 is a potent and specific inhibitor of transforming growth factor‐beta superfamily type i activin receptor‐like kinase (ALK) receptors ALK4, ALK5, and ALK7. Molecular Pharmacology, 62, 65–74. [DOI] [PubMed] [Google Scholar]
- Jinnin, M. , Ihn, H. , & Tamaki, K. (2006). Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor‐beta1‐induced extracellular matrix expression. Molecular Pharmacology, 69, 597–607. [DOI] [PubMed] [Google Scholar]
- Jung, Y. W. , Choi, I. J. , & Kwon, T. H. (2007). Altered expression of sodium transporters in ischemic penumbra after focal cerebral ischemia in rats. Neuroscience Research, 59, 152–159. [DOI] [PubMed] [Google Scholar]
- Kang, T. C. , An, S. J. , Park, S. K. , Hwang, I. K. , Suh, J. G. , Oh, Y. S. , … Won, M. H. (2002). Alterations in Na+/H+ exchanger and Na+/ cotransporter immunoreactivities within the gerbil hippocampus following seizure. Molecular Brain Research, 109, 226–232. [DOI] [PubMed] [Google Scholar]
- Kelley, M. R. , Deeb, T. Z. , Brandon, N. J. , Dunlop, J. , Davies, P. A. , & Moss, S. J. (2016). Compromising KCC2 transporter activity enhances the development of continuous seizure activity. Neuropharmacology, 108, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov, I. , Holmes, G. L. , & Ben‐Ari, Y. (2003). In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nature Neuroscience, 6, 1079–1085. [DOI] [PubMed] [Google Scholar]
- Kourdougli, N. , Varpula, S. , Chazal, G. , & Rivera, C. (2015). Detrimental effect of post Status Epilepticus treatment with ROCK inhibitor Y‐27632 in a pilocarpine model of temporal lobe epilepsy. Frontiers in Cellular Neuroscience, 9, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein, K. , Richter, S. , Farkas, L. , Schuster, N. , Dünker, N. , Oppenheim, R. W. , & Unsicker, K. (2000). Reduction of endogenous transforming growth factors beta prevents ontogenetic neuron death. Nature Neuroscience, 3, 1085–1090. [DOI] [PubMed] [Google Scholar]
- Krieglstein, K. , Zheng, F. , Unsicker, K. , & Alzheimer, C. (2011). More than being protective: Functional roles for TGF‐β/activin signaling pathways at central synapses. Trends in Neurosciences, 34, 421–429. [DOI] [PubMed] [Google Scholar]
- Lacmann, A. , Hess, D. , Gohla, G. , Roussa, E. , & Krieglstein, K. (2007). Activity‐dependent release of transforming growth factor‐beta in a neuronal network in vitro. Neuroscience, 150, 647–657. [DOI] [PubMed] [Google Scholar]
- Lein, P. J. , Barnhart, C. D. , & Pessah, I. N. (2011). Acute hippocampal slice preparation and hippocampal slice cultures In Costa L. G., Giordano G., & Guizzetti M., (Eds.), In vitro neurotoxicology, methods and protocols (Vol. 758, pp. 115–134). New York, NY: Humana Press, c/o Springer Science+Business Media, LLC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar, D. , & Bevensee, M. O. (2010). Na‐coupled bicarbonate transporters of the solute carrier 4 family in the nervous system: Function, localization, and relevance to neurologic function. Neuroscience, 171, 951–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, K. D. , & de Vellis, J. (1980). Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. Journal of Cell Biology, 85, 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa, K. , Shinozaki, M. , Hara, T. , Furuya, T. , & Miyazono, K. (2002). Two major Smad pathways in TGF‐beta superfamily signalling. Genes to Cells, 7, 1191–1204. [DOI] [PubMed] [Google Scholar]
- Moustakas, A. , & Heldin, C. H. (2009). The regulation of TGFbeta signal transduction. Development, 136, 3699–3714. [DOI] [PubMed] [Google Scholar]
- Namkoong, E. , Shin, Y. H. , Bae, J.S. , Choi, S. , Kim, M. , Kim, N. , … Park, K. (2015). Role of sodium bicarbonate cotransporters in intracellular pH regulation and their regulatory mechanisms in human submandibular glands. PLoS One 10:e0138368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlke, O. , Schlosshardt, C. , Feuerstein, M. , & Roussa, E. (2012). Acidosis‐induced V‐ATPase trafficking in salivary ducts is initiated by cAMP/PKA/CREB pathway via regulation of Rab11b expression. The International Journal of Biochemistry & Cell Biology, 44, 1254–1265. [DOI] [PubMed] [Google Scholar]
- Perillan, P. R. , Chen, M. , Potts, E. A. , & Simard, J. M. (2002). Transforming growth factor‐beta 1 regulates Kir2.3 inward rectifier K+ channels via phospholipase C and protein kinase C‐delta in reactive astrocytes from adult rat brain. Journal of Biological Chemistry, 277, 1974–1980. [DOI] [PubMed] [Google Scholar]
- Rathore, K. I. , Redensek, A. , & David, S. (2012). Iron homeostasis in astrocytes and microglia is differentially regulated by TNF‐α and TGF‐β1. Glia, 60, 738–750. [DOI] [PubMed] [Google Scholar]
- Rickmann, M. , Orlowski, B. , Heupel, K. , & Roussa, E. (2007). Distinct expression and subcellular localization patterns of Na+/ cotransporter (SLC 4A4) variants NBCe1‐A and NBCe1‐B in mouse brain. Neuroscience, 146, 1220–1231. [DOI] [PubMed] [Google Scholar]
- Roussa, E. , Speer, J. M. , Chudotvorova, I. , Khakipoor, S. , Smirnov, S. , Rivera, C. , & Krieglstein, K. (2016). The membrane trafficking and functionality of the K+‐Cl‐ co‐transporter KCC2 is regulated by TGF‐β2. Journal of Cell Science, 129, 3485–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, J. , Carles, M. , Koh, H. , Goolaerts, A. , Ganter, M. T. , Chesebro, B. B. , … Pittet, J. F. (2010). Transforming growth factor β1 inhibits cystic fibrosis transmembrane conductance regulator‐dependent cAMP‐stimulated alveolar epithelial fluid transport via a phosphatidylinositol 3‐kinase‐dependent mechanism. Journal Biological Chemistry, 285, 4278–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruminot, I. , Gutierrez, R. , Pena‐Munzenmayer, G. , Anazco, C. , Sotelo‐Hitschfeld, T. , Lerchundi, R. , … Barros, L. F. (2011). NBCe1 mediates the acute stimulation of astrocytic glycolysis by extracellular K+. Journal of Neuroscience, 31, 14264–14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salameh, A. I. , Hubner, C. A. , & Boron, W. F. (2017). Role of Cl‐‐ exchanger AE3 in intracellular pH homeostasis in cultured murine hippocampal neurons, and in crosstalk to adjacent astrocytes. The Journal of Physiology, 595, 93–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford, L. P. , Ormsby, I. , Gittenberger‐de Groot, A. C. , Sariola, H. , Friedman, R. , Boivin, G. P. , … Doetschman, T. (1997). TGFbeta2 knockout mice have multiple developmental defects that are non‐overlapping with other TGF beta knockout phenotypes. Development, 124, 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödl‐Häußel, M. , Theparambil, S. M. , Deitmer, J. W. , & Roussa, E. (2015). Regulation of functional expression of the electrogenic sodium bicarbonate cotransporter 1, NBCe1 (SLC4A4), in mouse astrocytes. Glia, 63, 1226–1239. [DOI] [PubMed] [Google Scholar]
- Sohn, Y. , Yoo, K. Y. , Park, O. K. , Kwon, S. H. , Lee, C. H. , Choi, J. H. , … Won, M. H. (2011). Na+/ cotransporter immunoreactivity changes in neurons and expresses in astrocytes in the gerbil hippocampal CA1 region after ischemia/reperfusion. Neurochemical Research, 36, 2459–2469. [DOI] [PubMed] [Google Scholar]
- Svichar, N. , Esquenazi, S. , Chen, H. Y. , & Chesler, M. (2011). Preemptive regulation of intracellular pH in hippocampal neurons by a dual mechanism of depolarization‐induced alkalinization. Journal of Neuroscience, 31, 6997–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Dijke, P. , & Arthur, H. M. (2007). Extracellular control of TGFβ signalling in vascular development and disease. Nature Reviews Molecular Cell Biology, 8, 857–869. [DOI] [PubMed] [Google Scholar]
- Theparambil, S. M. , & Deitmer, J. W. (2015). High effective cytosolic H+ buffering in mouse cortical astrocytes attributable to fast bicarbonate transport. Glia, 63, 1581–1594. [DOI] [PubMed] [Google Scholar]
- Theparambil, S. M. , Naoshin, Z. , Thyssen, A. , & Deitmer, J. W. (2015). Reversed electrogenic sodium bicarbonate cotransporter 1 is the major acid loader during recovery from cytosolic alkalosis in mouse cortical astrocytes. The Journal of Physiology, 16, 3533–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theparambil, S. M. , Ruminot, I. , Schneider, H. P. , Shull, G. E. , & Deitmer, J. W. (2014). The electrogenic sodium bicarbonate cotransporter NBCe1 is a high‐affinity bicarbonate carrier in cortical astrocytes. Journal of Neuroscience, 34, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theparambil, S. M. , Weber, T. , Schmälzle, J. , Ruminot, I. , & Deitmer, J. W. (2016). Proton fall or bicarbonate rise. Glycolytic rate in mouse astrocytes is paved by intracellular alkalinisation. The Journal of Biological Chemistry, 291, 19108–19117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornell, I. M. , & Bevensee, M. O. (2015). Phosphatidylinositol 4,5‐bisphosphate degradation inhibits the Na+/bicarbonate cotransporter NBCe1‐B and ‐C variants expressed in Xenopus oocytes. The Journal of Physiology, 593, 541–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivien, D. , Bernaudin, M. , Buisson, A. , Divoux, D. , MacKenzie, E. T. , & Nouvelot, A. (1998). Evidence of type I and type II transforming growth factor‐beta receptors in central nervous tissues: Changes induced by focal cerebral ischemia. Journal of Neurochemistry, 70, 2296–2304. [DOI] [PubMed] [Google Scholar]
- Yang, Y. C. , Piek, E. , Zavadil, J. , Liang, D. , Xie, D. , Heyer, J. , … Bottinger, E. P. (2003). Hierarchical model of gene regulation by transforming growth factor beta. Proceedings of the National Academy of Sciences of the United States of America, 100, 10269–10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, H. , Azad, P. , Zhao, H. W. , Wang, J. , Poulsen, O. , Freitas, B. C. , … Haddad, G. G. (2016). The Na+/ co‐transporter is protective during ischemia in astrocytes. Neuroscience, 339, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, S. , Pierucci‐Alves, F. , & Schultz, B. D. (2013). Transforming growth factor‐β1 impairs CFTR‐mediated monolayer via the p38 MAPK pathway. American Journal of Physiology, 305, C867–C876. [DOI] [PMC free article] [PubMed] [Google Scholar]