

Abstract
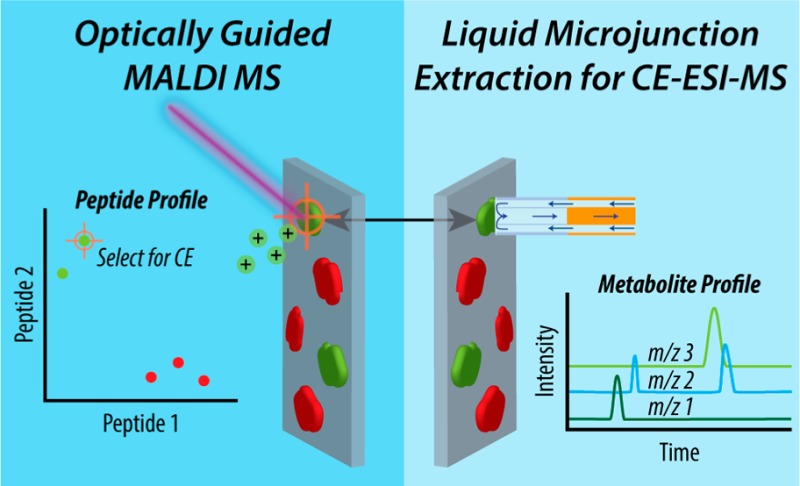
The ability to characterize chemical heterogeneity in biological structures is essential to understanding cellular-level function in both healthy and diseased states, but these variations remain difficult to assess using a single analytical technique. While mass spectrometry (MS) provides sufficient sensitivity to measure many analytes from volume-limited samples, each type of mass spectrometric analysis uncovers only a portion of the complete chemical profile of a single cell. Matrix-assisted laser desorption/ionization (MALDI) MS and capillary electrophoresis electrospray ionization (CE–ESI)-MS are complementary analytical platforms frequently utilized for single-cell analysis. Optically guided MALDI MS provides a high-throughput assessment of lipid and peptide content for large populations of cells, but is typically nonquantitative and fails to detect many low-mass metabolites because of MALDI matrix interferences. CE–ESI-MS allows quantitative measurements of cellular metabolites and increased analyte coverage, but has lower throughput because the electrophoretic separation is relatively slow. In this work, the figures of merit for each technique are combined via an off-line method that interfaces the two MS systems with a custom liquid microjunction surface sampling probe. The probe is mounted on an xyz translational stage, providing 90.6 ± 0.6% analyte removal efficiency with a spatial targeting accuracy of 42.8 ± 2.3 μm. The analyte extraction footprint is an elliptical area with a major diameter of 422 ± 21 μm and minor diameter of 335 ± 27 μm. To validate the approach, single rat pancreatic islet cells were rapidly analyzed with optically guided MALDI MS to classify each cell into established cell types by their peptide content. After MALDI MS analysis, a majority of the analyte remains for follow-up measurements to extend the overall chemical coverage. Optically guided MALDI MS was used to identify individual pancreatic islet α and β cells, which were then targeted for liquid microjunction extraction. Extracts from single α and β cells were analyzed with CE–ESI-MS to obtain qualitative information on metabolites, including amino acids. Matching the molecular masses and relative migration times of the extracted analytes and related standards allowed identification of several amino acids. Interestingly, dopamine was consistently detected in both cell types. The results demonstrate the successful interface of optical microscopy-guided MALDI MS and CE–ESI-MS for sequential chemical profiling of individual, mammalian cells.
Assessing the cellular chemical heterogeneity of biological tissues is an ongoing challenge in many research fields.1−4 Frequently, the analysis of bulk homogenates masks unique features of individual cells by averaging the molecular content of cell populations.5 While a biological organ or tissue requires many distinct cells to function properly, a malfunction can manifest from small cellular subpopulations or even a single cell.6,7 Furthermore, cells that are morphologically indistinguishable may possess unique intracellular chemistries and, therefore, physiologies.
Mass spectrometry (MS) is among the most commonly used analytical methods for nontargeted single-cell analysis.4 Matrix-assisted laser desorption/ionization (MALDI) MS,8,9 electrospray ionization (ESI)-MS,1,10−13 and secondary ion MS (SIMS)14−17 are well-suited for multiplexed analysis of a wide range of biological molecules.18 Measurements are typically label-free and often consume only a fraction of surface-available analytes. Page and Sweedler utilized radiography to demonstrate that, even after the MALDI MS signal is fully depleted, about 30% of a protein standard is removed.19 The recent progress in single-cell MS can be attributed to advances in sensitivity, mass resolution, and sample throughput of modern mass spectrometers, as well as hyphenation of MS to other approaches. For example, optical microscopy combined with single-cell MALDI MS allows rapid characterization of dispersed cell populations.20 By locating cells with optical microscopy, the analysis can proceed at an acquisition rate of approximately 1 Hz.21 Using this approach, differential peptide processing was detected in γ cells derived from islets of Langerhans located in the dorsal and ventral regions of the rat pancreas.22 This finding supports the view that the chemical cellular heterogeneity of different organs is neither well-understood nor well-characterized. As another example, flow cytometry and transcriptomic analyses of insulin-secreting β cells identified up to four β-cell subtypes in humans with significantly different glucose-stimulated insulin secretion.23 Motivated by these examples, islet cells were chosen as a single-cell sample system in an attempt to discover previously unknown heterogeneity or chemical messengers.
MALDI MS is well-suited to detecting peptides and lipids as their high molecular masses minimize interference from the MALDI matrix; however, many smaller metabolites are not detectable. A complementary method for single-cell analysis is capillary electrophoresis (CE)–ESI-MS, which is well suited for metabolomics measurements as it can quantitatively identify metabolites from individual cells.11,24,25 Sample preparation for CE–ESI-MS typically involves manual cell isolation, microfluidic cell sorting, or collection of cell cytoplasm using a patch-clamp pipette.26,27 Metabolite detection using CE–ESI-MS generally requires injection of the sample content from an entire cell.28 In contrast to MALDI MS, CE–ESI-MS has relatively low throughput and is limited to a few cells per hour, a time constraint that precludes the cell-by-cell analysis of even modestly sized populations.
Preliminary classification of the most informative individual cells in a population via MALDI MS facilitates targeted CE–ESI-MS analysis of rare and representative cells from among hundreds to thousands of cells. Previous attempts to combine MALDI MS and CE–ESI-MS utilized microfluidic29,30 and hydrodynamic31,32 interfaces. Although the same sample was analyzed with both instruments, the methods had relatively low throughput due to the lack of automated target collection, which therefore required excessive manual sample handling. To reveal chemical heterogeneity in large populations of cells, it is important that the interface method is capable of collecting small sample volumes with high efficiency.
With that goal in mind, we developed a semiautomated, microscopy-guided liquid microjunction probe system for collection of analytes from single cells that have been classified by their MALDI MS profiles. The first coupling of CE and MALDI worked in the reverse of the system described here in that the CE effluent was deposited onto a membrane or MALDI target for analyte detection after the CE separation.30,33,34 In our approach, the MALDI measurement is performed first, and then samples are collected from the target for CE separation and analysis. The collection probe utilizes two coaxial capillaries, similar to previous designs from our lab,32 and liquid microjunction surface sampling probes.35,36
Single-cell targeting is achieved with precise motion in three axes of linear freedom controlled by a lab-built graphical user interface that allows microscopy-guided cell targeting. The software, microMS,37 is an extension of software we originally developed for microscopy-guided MALDI MS22 and SIMS,38 which directly controls the extraction probe. While MALDI MS is not required for performing liquid extraction, it can complement the microscopy information by providing label-free classification of large populations. By interfacing two powerful analytical tools for small-volume samples, the combined data obtained from CE–ESI-MS and MALDI MS were used to successfully classify and analyze six α and five β cells. Each cell was identified by MALDI MS as a standard histological class by the detection of glucagon and insulin, respectively. Small molecules detected with CE–ESI-MS include 18 proteinogenic amino acids as well as dopamine. While the enzymes for dopamine synthesis suggest the presence of dopamine in β cells,39,40 it appears that dopamine has not been directly characterized at the single-cell level in either α or β cells.
Experimental Section
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification.
Isolation of Islets of Langerhans and Single-Cell Preparation
A 1.5 month old, male Sprague–Dawley outbred rat (Rattus norvegicus) was housed on a 12 h light cycle and fed ad libitum. Animal euthanasia was performed in accordance with the appropriate institutional animal care protocols (the Illinois Institutional Animal Care and Use Committee), and in full compliance with federal guidelines for the humane care and treatment of animals. Islets of Langerhans were manually isolated from an enzymatically digested and mechanically treated pancreas, as previously reported.22 Briefly, the pancreas is injected through the bile duct with 2 mL of 1.4 mg/mL collagenase P in modified Gey’s balanced salt solution (mGBSS) supplemented with 5 mM glucose and 1% (w/v) bovine serum albumin (BSA). The mGBSS contained 1.5 mM CaCl2, 4.9 mM KCl, 0.2 mM KH2PO4, 11 mM MgCl2, 0.3 mM MgSO4, 138 mM NaCl, 27.7 mM NaHCO3, 0.8 mM NaH2PO4, and 25 mM HEPES dissolved in Milli-Q water (Millipore, Billerica, MA), with the pH adjusted to 7.2. The pancreas was then surgically dissected and placed into 8 mL of the collagenase P solution. Solutions were incubated in a recirculating water bath for 20–30 min at 37 °C with agitation to dissociate bulk tissue. Excess collagenase P was washed from the resulting tissue with mGBSS containing glucose and BSA, and centrifuged for 3 min at 300g. The resulting tissue pellet was dispersed into mGBSS, and islets were manually isolated with a micropipette. Single islets were incubated in 20 μL of 40% (v/v) glycerol and 60% mGBSS with glucose, BSA, and 0.1 mg/mL Hoechst 33342. This step resulted in staining of cell nuclei while mechanically stabilizing cellular morphology.41 After 30 min, single cells were dissociated onto clean indium–tin oxide (ITO)-coated glass slides by gentle trituration in the staining solution and allowed to adhere to the slides overnight. Prior to imaging, excess glycerol was aspirated and the surface rinsed with 150 mM ammonium acetate (pH 10).
Optically Guided Single-Cell Profiling
The next step in the experimental workflow, as outlined in Scheme 1, is to locate cells by optical microscopy. ITO-coated glass slides were prepared for optically guided single-cell profiling by marking the perimeter of dissociated cells with ∼20 fiducial marks. Each mark consisted of an etched “x” (see Figure 3A), which remained visible during MALDI MS acquisition and liquid microjunction extraction. The locations of fiducials and cells were determined by whole-slide bright-field and fluorescence microscopy using an Axio Imager M2 (Carl Zeiss, Jena, Germany). Images were acquired with a 10× objective and tiled to cover the entire region of interest. Florescence imaging of Hoechst 33342 utilized an X-CITE 120 mercury lamp (Lumen Dynamics, Mississauga, Canada) and a 31000v2 DAPI filter set (Chroma Technology, Irvine, CA).
Scheme 1. Overview of the MALDI MS Guided Liquid Microjunction Extraction Approach.

Islets of Langerhans are isolated from a rat pancreas and dissociated onto an ITO-coated glass slide. MALDI MS is used to assay the hormone profile of individual cells from a large population to identify extraction targets. The liquid microjunction probe collects cell contents from specified locations on the ITO-coated glass slide for follow-up CE–ESI-MS analysis.
Figure 3.
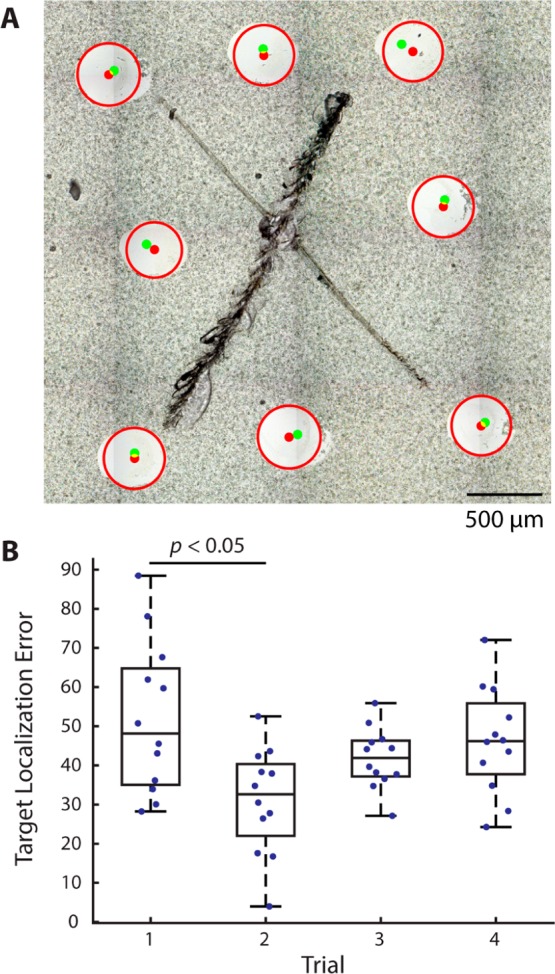
Representative extractions for determining target localization error. Target points were positioned around fiducial marks placed in the center of a glass slide. (A) Overlay of microscope image with the position of target locations (green) and extraction areas (red). (B) Box plot of the accuracies over four trials of fiducial registration. The registration trial was the only confounding variable found to significantly affect target localization error.
Whole-slide images were utilized for optically guided single-cell profiling with microMS.37 Before MALDI MS acquisition, the pixel locations of each fiducial were correlated to their physical positions in the mass spectrometer. A point-based similarity registration was then used to map cell locations on the image to their corresponding physical locations.
After optical imaging, samples were coated with MALDI matrix, using an artist’s airbrush, containing 50 mg/mL 2,5-dihydroxy-benzoic acid (DHB) in 1:1 (v/v) ethanol/water with 0.1% trifluoroacetic acid (TFA), nebulized with 40 psi nitrogen. Coating thickness was assessed optically during matrix application, with typical thicknesses of 0.2–0.4 mg/cm2. Samples were stored at room temperature (∼22 °C) in a nitrogen drybox until analyzed.
MALDI MS
Pancreatic cell populations were rapidly profiled with MALDI MS to stratify the population into traditional histological classes. Specifically, the α and β cells were identified based on the detection of glucagon (monoisotopic m/z 3481.6) or insulin-1 C peptide (m/z 3259.8). To prevent detection of the cellular content of several cells simultaneously, as well as collection of multicellular content during follow-up analyte extraction, cell coordinates were first passed through a 300 μm distance filter. From a single islet dispersed on an ITO-coated glass slide, approximately 200–400 pancreatic cells satisfied the sample analysis criteria.
Mass spectra were acquired on a ultrafleXtreme MALDI TOF/TOF mass spectrometer with a frequency tripled Nd:YAG solid-state laser (Bruker Daltonics, Billerica, MA). Each cell was profiled with 1000 shots using a 1 kHz laser repetition rate with the “Ultra” laser setting (spot diameter ∼100 μm). The resulting spectra were read into MATLAB 8.6.0 with the readbrukermaldi function (https://github.com/AlexHenderson/readbrukermaldi). Data from molecular mass windows containing signals from the peptide hormones of interest were extracted and signal intensities were plotted, as shown in Figure 1. Cells were classified based on their spectral profiles as α or β using signal intensities at m/z 3483.9 and m/z 3259.8, respectively. For each mass channel, a threshold value for signal intensity was manually determined to identify cell types with high confidence. Because of the stringent filter values, fewer than 100 cells were successfully classified by this approach for each islet. Images of classified cells were then examined to ensure the analyte extraction area contained no adjacent cells.
Figure 1.

MALDI MS classification of pancreatic islet cells. (A) A single dorsal pancreas islet is composed primarily of glucagon-containing α cells (blue) and insulin-containing β cells (red). Classifications are based on a threshold signal abundance to identify cell types for follow-up CE–ESI-MS analysis. Cell identities correspond to labeling of cell number for the α and β cells. (B) Spectra of single pancreatic cells identified in panel A, parts i and ii.
Liquid Microjunction Extraction Probe System
The liquid microjunction extraction probe utilizes two coaxial capillaries. The inner and outer fused-silica capillaries are 100 μm/170 μm and 250 μm/350 μm in diameter, respectively (Polymicro Technologies, Phoenix, AZ). The probe position was monitored in real time with a digital video camera (Sony, Park Ridge, NJ; P/N DFW-X700). Extraction liquid was delivered with a PHD 2000 syringe pump (Harvard Apparatus, Holliston, MA) and aspirated with 7–10 in Hg of vacuum, supplied with a diaphragm vacuum/pressure pump (Cole-Parmer, Vernon Hills, IL). The liquid microjunction was positioned with three linear stages (Zaber Technologies, Vancouver, BC, Canada) controlled with the in-house written software, microMS.37 Samples collected by the probe were dried using a Mi-Vac sample concentrator (SP Scientific, Warminster, PA) and stored at −20 °C prior to CE–ESI-MS analysis.
Radioactive Material and Radiation Detection
Tritiated (3H) angiotensin II, with the specific activity of 50 Ci/mmol at 1 mCi/mL, was purchased from American Radiolabeled Chemicals (St. Louis, MO). Radioactivity experiments were performed in accordance with the Illinois Radiation Protection Act under the University of Illinois at Urbana–Champaign Type A Broad Scope Radioactive Materials License issued by the Illinois Emergency Management Agency.
Radioactive material deposition and extraction was visually monitored using a Wild M3Z stereomicroscope (Leica, Buffalo Grove, IL). The pre- and postextraction radioactivity of the deposited sample was determined with a storage phosphor screen (BAS-IP TR 2025 E Tritium Screen, Sigma-Aldrich) exposed to the sample for 6 h. Developing the screen with a phosphorimager (Phosphorimager SI, Molecular Dynamics, Sunnyvale, CA) allowed for relative quantitation of the sample/analyte removal. Image processing was performed with custom MATLAB scripts. The fraction of material removed was determined by the background-corrected, normalized intensity at each pixel, before and after extraction.
CE–ESI-MS Analysis
Each cell extract was dried and resuspended in 1 μL of 1% formic acid in liquid chromatography–MS grade water. CE–ESI-MS was performed as reported previously using a micrOTOF mass spectrometer (Bruker Daltonics).27 Analyses were conducted in positive ion mode using a 70.7 cm long CE fused-silica capillary (Polymicro Technologies), a separation potential of 17 kV, and a sample injection volume of ∼15 nL. Extracted ion electropherograms were exported using custom scripts in Bruker DataAnalysis version 4.4. Compounds were identified from the electropherograms by matching the migration order and mass-to-charge (m/z) values with standards. In MATLAB, each extracted ion electropherogram was baseline-subtracted and smoothed with a seven point moving average filter. Analyte migration times were aligned to corresponding analyte migration times in a reference mass electropherogram (α1), as shown in Figure S1. The alignment used a linear regression between migration times of a set of amino acids found in each sample (i.e., glycine, alanine, threonine, leucine/isoleucine, histidine, phenylalanine). To confirm the presence of dopamine, a standard mix of 10 μM glycine, alanine, threonine, leucine, histidine, and phenylalanine in 1% formic acid in water was analyzed with a 68 cm long CE capillary at 10 kV with and without the addition of 10 μM dopamine.
Results and Discussion
Liquid Microjunction Extraction Probe System
As shown in Figure S2 and partially in Figure 2, the liquid microjunction extraction system consists of a lab-built, concentric capillary probe coupled to a three-axis linear actuator positioning system. The single-cell collection setup was designed to transfer cell metabolites from an ITO-coated glass slide into a 200 μL microcentrifuge tube. The basic operating principle is similar to a liquid microjunction surface sampling probe except the solution is aspirated by vacuum pressure instead of an electrospray. The diameters of the probe capillaries were selected to be larger than the diameter of individual pancreatic cells to ensure complete extraction, prevent clogging, and accommodate the stage accuracy. The sizes of the inner and outer capillaries were 100 μm/170 μm and 250 μm/350 μm in diameter, respectively; the diameter of pancreatic cells is ∼10–15 μm.42 Sample carryover may result in cross-contamination of samples; therefore, ∼5 mm of the polyimide coating was thermally removed at the ends of both capillaries.43 Following each sample collection, the probe was immersed in extraction solution to thoroughly wash out its interior.
Figure 2.
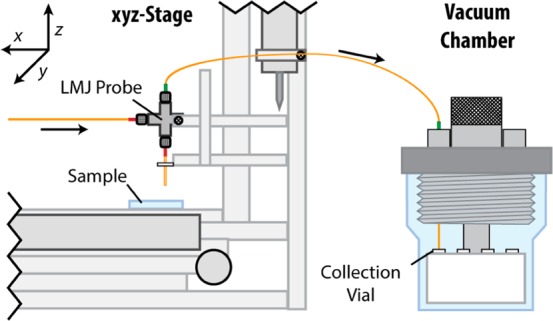
Partial schematic of the liquid microjunction analyte extraction system. Left: a system of three linear actuators positions the liquid microjunction probe above a targeted cell. Right: analyte extraction solution is pumped through the system to collect cellular content, which is transferred to the custom vacuum chamber containing collection vials.
The extraction solution consisted of 1:1 methanol/water with 0.5% acetic acid (v/v), which was previously shown to facilitate metabolite extraction and detection with CE–ESI-MS.27 As shown in Figure S3, a small meniscus forms at the probe tip during operation. Collections can be performed sequentially without having to open the vacuum chamber. The number of collections corresponds to the number of sample tubes the system can accommodate (our system holds eight tubes). Extraction liquid is delivered at 1.5 μL/min and aspirated with vacuum.
During system operation, the user moves the x,y-translation stage away from the sample area and lowers the probe to the surface. The software records the z-axis position at the slide surface to enable automatic analyte extraction. The probe position is monitored in real time with a digital video camera. Next, coordinates from the whole-slide image and linear actuator positions are correlated with a point-based similarity registration utilizing more than 18 etched fiducial marks. Choosing a targeted cell on the image activates the motion of the x,y-translation stage, moving it into position for analyte extraction. The user initiates semiautomatic extractions by signaling the microMS software with a key press. During extraction, the probe is lowered to the slide for 60 s and then retracted. Alternatively, analytes from a population of cells may be sequentially extracted and pooled into a single collection vial. Following either collection scheme, the probe is returned to the home position and submerged into a reservoir of extraction solution for 90 s to rinse the probe exterior, flush the inner capillary, and prevent carryover between samples. As seen in Figure S6, blanks acquired from locations adjacent to cells between extractions contained negligible background signal.
The cell content collected at each coordinate travels from the MALDI sample plate (e.g., ITO glass slide), through the inner capillary of the coaxial system, and into one of the microcentrifuge tubes contained in the vacuum chamber. Inside the vacuum chamber, the microcentrifuge tubes are covered with a thin strip of Parafilm M to prevent extraction solution from clinging on the capillary when moving between collection vials. The inner capillary is retracted from the current collection tube, the tube carousel is indexed to the next position, and the inner capillary is placed into the next collection tube without breaking vacuum in the chamber. Individual samples were dried and stored at −20 °C prior to CE–ESI-MS analysis.
Determination of Target Localization Error
To ensure that each analyte extraction is from the expected cell, it is imperative to determine the target localization error. Analyte extraction locations were visualized and characterized by the removal of MALDI matrix from the sample plate. Image registration of fiducial markers allowed the spatial correlation of requested target points and realized analyte extraction positions (Figure 3).
A glass slide was etched with 18 fiducial marks for point-based registration, similar to typical cell extractions. An additional six etched marks were placed within these fiducials to assist with image registration, as they remain visible after MALDI matrix application and extraction. Eight target locations were manually placed around each of the six, interior etched marks in pairs to assess the effect of repeated registrations. The slide was then coated with DHB and placed into the liquid extraction stage as before. Two users each performed two sets of extractions with 12 targets spread over the six etched marks. This experimental design allowed evaluation of the influence of the user, registration, and location on the target localization error. Each target was extracted for 5 s, and the probe was washed for 60 s after each set of 12 extractions.
Following extraction, the sample was imaged again to locate target etched marks and extraction locations. Extraction centers and diameters were manually annotated. A custom MATLAB script was utilized to assess the target localization error of each extraction. Regions surrounding each etched mark were cropped from the whole-slide image. Several locations on each mark were utilized to overlay the pre- and postextraction images. Target locations on the pre-extraction image were then mapped to the postextraction image with the same coordinate transformation. The pixel distances between the target and actual positions were scaled to micrometers. A three-way linear analysis of variance (ANOVA) was utilized to assess the effect of each confounding variable. While the operator and target spot location did not significantly influence the target localization error (p = 0.15 and 0.06, respectively), there was a significant effect from performing replicates with the same sample and images (p = 0.004). This highlights that the accurate determination of fiducial locations has the largest influence on target localization error. The overall target localization error was determined as 42.8 ± 2.3 μm (±SEM, n = 48; range 3.9–88.5 μm), which is well within the average extraction radius of 206.3 ± 1.7 μm, as determined by measurement of the size of the spot of removed DHB from the surface (Figure S4). Therefore, it is assumed that each extraction would contain only the target cell when a cell-to-cell distance filter is set to be larger than 250 μm. This approach ensures that the collection of single-cell samples is free from cross-contamination by neighboring cells.
Characterization of Analyte Removal Efficiency
3H-angiotensin II was spotted onto an ITO-coated glass slide to determine the extraction profile and analyte removal efficiency. Five spots of ∼1 μL of 1000 pCi of 3H-angiotensin II in mGBSS supplemented with 5 mg/mL Fast green were deposited onto the surface of an ITO-coated glass slide and allowed to dry for 24 h at room temperature (∼22 °C). Liquid microjunction extraction of the radioactive material was performed as described above, with minor adjustments to minimize the possibility of radioactive contamination of the equipment. To replicate single-cell extraction conditions, each 3H-angiotensin II spot was extracted for 60 s. The removal efficiency was estimated by fitting the two-dimensional distribution to a general Gaussian function, as described in Table S1.
Fitting results (Figures 4 and S5) provide an estimated removal efficiency of 90.6 ± 0.6% (Table S1). While the extraction efficiency may be dependent on the target analyte, the high removal efficiency found with angiotensin suggests that the solvent composition and extraction time are suitable for collecting small and intermediate-sized polar compounds, such as the amino acids. The extraction footprint was found to be elliptical, with a major diameter of 422 ± 21 μm and minor diameter of 335 ± 27 μm. The estimated diameter from optical measurements of DHB removal falls within the range of the minor and major diameters. The eccentricity of the extraction footprint is likely due to imperfect fabrication of the probe tip or stochastic wetting of the rough, matrix-covered surface.
Figure 4.
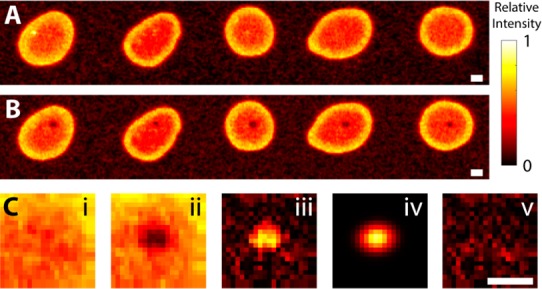
Measurement of removal efficiency of 3H-angiotensin. A phosphorimager was utilized to measure angiotensin distributions pre- and postextraction, shown in panels A and B, respectively. (C) Sample analysis of the left-most spot. Subregions surrounding each extraction (i and ii) are utilized to determine the distribution of the fraction of radioactivity removed (iii). The distribution is fit to a general two-dimensional Gaussian (iv) to determine the fraction removed. Residuals of the fit (v) are nonstructured, indicating the model is appropriate. All scale bars are 500 μm.
Profiles of Small Molecules
CE–ESI-MS complements MALDI MS analyses by identifying small molecules from a single cell. We present example extracted ion electropherograms with corresponding MALDI mass spectra in Figure 5. All collected electropherograms are provided in Figure S6.
Figure 5.

Single pancreatic islet cell analysis using MALDI MS and CE–ESI-MS interfaced with the off-line liquid microjunction extraction system. (A) Representative single-cell MALDI MS profiles of single α and β cells. (B) Corresponding CE–ESI-MS extracted ion electropherograms of the same cells showing signals of amino acids with high intensities and (C) signals of amino acids with lower signal intensities.
Detected compounds include the majority of the proteinogenic amino acids, precursor molecules, and endocrine signaling molecules. In contrast to characteristic peptide signatures, no obvious differences were found between α and β cells in their metabolite profiles. However, increasing the number of replicates and performing quantitative measurements (e.g., including a labeled standard for metabolites of interest) may allow identification of subtle heterogeneity between each population. Improvements in CE–ESI-MS sensitivity would facilitate detection of minor metabolites. An interesting observation was the presence of dopamine in all α and β cells (Figure 6; a separation with dopamine standard is shown in Figure S7). Previously, endogenous dopamine has been detected in single islets via an enzyme-linked immunosorbent assay (ELISA) assay,40 but to our knowledge, not in single cells. β cells are known to have the required enzymes for synthesis, metabolism, and storage of dopamine, such as tyrosine hydroxylase44 and vesicular monoamine transporter type 2;45 thus, it is generally accepted that dopamine is produced in β cells.39 Dopamine within α cells is less studied, and whether dopamine is endogenous to α cells has not yet been investigated. We report direct detection of dopamine in single α and β cells, illustrating the unique capabilities of the presented methodology and small-scale analyses.
Figure 6.
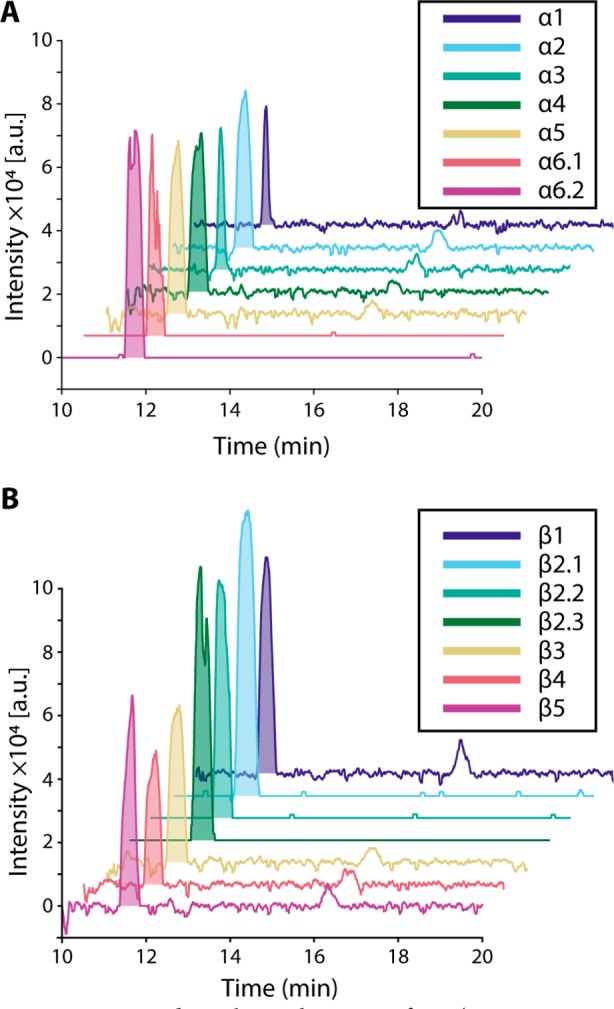
Extracted ion electropherograms for m/z 154.09 ± 0.01. The peak with m/z and migration time matching dopamine standard is shaded in each electropherogram: (A) α cells; (B) β cells. Dopamine was detectable in every cell analyzed. The peak at ∼16 min is attributed to sodiated leucine. Single-cell samples analyzed with technical replicates are annotated with decimals, e.g., α6.1.
Conclusions
We developed a semiautomated method that couples high-throughput single-cell chemical profiling with MALDI MS, followed by in-depth analyses of representative cellular types with CE–ESI-MS metabolomics. The approach leverages the low sample consumption of MALDI MS, which enables the follow-up analysis of the same sample by CE–ESI-MS. By hyphenating the two methods, we identified cell types by their peptide profiles, and detected most amino acids and the signaling molecule dopamine, a difficult task for either technique alone. While pancreatic islet cell types were the focus of this study, the methodology is suitable for a broad range of single-cell analyses of dissociated tissues. Future work will leverage the unique capabilities to examine heterogeneity within the nervous and endocrine systems.
Acknowledgments
We gratefully acknowledge Dr. Ta-Hsuan Ong for his help with initial testing and Dr. Meng Qi for assistance with data analysis. We also thank the UIUC School of Chemical Sciences machine shop for their expertise in designing and machining components for the vacuum chamber and stage. Research reported in this publication was supported by the National Institutes of Health, Award No. P30 DA018310 from the National Institute on Drug Abuse, and Award No. U01 MH109062 from the National Institute of Mental Health. T.J.C. acknowledges support from the National Science Foundation Graduate Research Fellowship Program, the Springborn Fellowship, and the Training Program at Chemistry-Interface with Biology (T32 GM070421). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01782.
Supporting Figures S1–S7 and Table S1 showing the following: analyte migration time alignment results, full liquid microjunction extraction probe schematic, microphotographs of the liquid microjunction extraction probe, montage of extraction areas, removal efficiency images, Gaussian fits of removal efficiency, complete set of single-cell MALDI mass spectra and CE–ESI-MS extracted ion electropherograms, and extracted ion electropherograms of dopamine and a number of amino acids standards (PDF)
Author Contributions
T.J.C., M.A.M., and M.C.P. contributed equally to this work. T.J.C. performed MALDI MS, data analysis, wrote control software, isolated islets, and assisted with extractions. M.A.M. designed and built the initial extraction system, developed the protocol for the extraction and transfer of individual cells, performed radiography, and obtained preliminary CE–ESI-MS data. M.C.P. performed single-cell CE–ESI-MS, single-cell extractions, and assisted with data analysis. All authors assisted with writing the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Onjiko R. M.; Moody S. A.; Nemes P. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6545–6550. 10.1073/pnas.1423682112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrecht L.; Dittrich P. S. Anal. Chem. 2017, 89, 2–21. 10.1021/acs.analchem.6b04255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malucelli E.; Fratini M.; Notargiacomo A.; Gianoncelli A.; Merolle L.; Sargenti A.; Cappadone C.; Farruggia G.; Lagomarsino S.; Iotti S. Analyst 2016, 141, 5221–5235. 10.1039/C6AN01091A. [DOI] [PubMed] [Google Scholar]
- Zenobi R. Science 2013, 342, 1243259. 10.1126/science.1243259. [DOI] [PubMed] [Google Scholar]
- Altschuler S. J.; Wu L. F. Cell 2010, 141, 559–563. 10.1016/j.cell.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D. A.; Bhakta N. R.; Kessenbrock K.; Prummel K. D.; Yu Y.; Takai K.; Zhou A.; Eyob H.; Balakrishnan S.; Wang C.-Y.; Yaswen P.; Goga A.; Werb Z. Nature 2015, 526, 131–135. 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell A. A.; Talasaz A. H.; Zhang H.; Coram M. A.; Reddy A.; Deng G.; Telli M. L.; Advani R. H.; Carlson R. W.; Mollick J. A.; Sheth S.; Kurian A. W.; Ford J. M.; Stockdale F. E.; Quake S. R.; Pease R. F.; Mindrinos M. N.; Bhanot G.; Dairkee S. H.; Davis R. W.; et al. PLoS One 2012, 7, e33788. 10.1371/journal.pone.0033788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez A. J.; Fagerer S. R.; Schmidt A. M.; Urban P. L.; Jefimovs K.; Geiger P.; Dechant R.; Heinemann M.; Zenobi R. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8790–8794. 10.1073/pnas.1209302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubakhin S. S.; Romanova E. V.; Nemes P.; Sweedler J. V. Nat. Methods 2011, 8, S20–S29. 10.1038/nmeth.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong T.-H.; Tillmaand E. G.; Makurath M.; Rubakhin S. S.; Sweedler J. V. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 732–740. 10.1016/j.bbapap.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes P.; Knolhoff A. M.; Rubakhin S. S.; Sweedler J. V. Anal. Chem. 2011, 83, 6810–6817. 10.1021/ac2015855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C.; Moody S. A.; Nemes P. Angew. Chem., Int. Ed. 2016, 55, 2454–2458. 10.1002/anie.201510411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H.; Tsuyama N.; Harada T.; Masujima T. J. Mass Spectrom. 2008, 43, 1692–1700. 10.1002/jms.1460. [DOI] [PubMed] [Google Scholar]
- Ostrowski S. G.; Kurczy M. E.; Roddy T. P.; Winograd N.; Ewing A. G. Anal. Chem. 2007, 79, 3554–3560. 10.1021/ac061825f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarelli M. K.; Ewing A. G.; Winograd N. Anal. Chem. 2013, 85, 2231–2238. 10.1021/ac303038j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S. In Mass Spectrometry Imaging: Principles and Protocols; Rubakhin S. S., Sweedler J. V., Eds.; Humana Press: Totowa, NJ, 2010; pp 113–130. [Google Scholar]
- Lanni E. J.; Dunham S. J. B.; Nemes P.; Rubakhin S. S.; Sweedler J. V. J. Am. Soc. Mass Spectrom. 2014, 25, 1897–1907. 10.1007/s13361-014-0978-9. [DOI] [PubMed] [Google Scholar]
- Li L. J.; Garden R. W.; Sweedler J. V. Trends Biotechnol. 2000, 18, 151–160. 10.1016/S0167-7799(00)01427-X. [DOI] [PubMed] [Google Scholar]
- Page J. S.; Sweedler J. V. Anal. Chem. 2002, 74, 6200–6204. 10.1021/ac025898k. [DOI] [PubMed] [Google Scholar]
- Comi T. J.; Do T. D.; Rubakhin S. S.; Sweedler J. V. J. Am. Chem. Soc. 2017, 139, 3920–3929. 10.1021/jacs.6b12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong T.-H.; Kissick D. J.; Jansson E. T.; Comi T. J.; Romanova E. V.; Rubakhin S. S.; Sweedler J. V. Anal. Chem. 2015, 87, 7036–7042. 10.1021/acs.analchem.5b01557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson E. T.; Comi T. J.; Rubakhin S. S.; Sweedler J. V. ACS Chem. Biol. 2016, 11, 2588–2595. 10.1021/acschembio.6b00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C.; Schug J.; Canaday P. S.; Russ H. A.; Tarlow B. D.; Grompe M. T.; Horton T.; Hebrok M.; Streeter P. R.; Kaestner K. H.; Grompe M. Nat. Commun. 2016, 7, 11756. 10.1038/ncomms11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski J. A.; Tracht S.; Sweedler J. V. TrAC, Trends Anal. Chem. 1995, 14, 170–176. 10.1016/0165-9936(95)98315-Y. [DOI] [Google Scholar]
- Lapainis T.; Rubakhin S. S.; Sweedler J. V. Anal. Chem. 2009, 81, 5858–5864. 10.1021/ac900936g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts J. T.; Louis K. R.; Crandall S. R.; Govindaiah G.; Cox C. L.; Sweedler J. V. Anal. Chem. 2014, 86, 3203–3208. 10.1021/ac500168d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes P.; Rubakhin S. S.; Aerts J. T.; Sweedler J. V. Nat. Protoc. 2013, 8, 783–799. 10.1038/nprot.2013.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecala C.; Sweedler J. V. Analyst 2012, 137, 2922–2929. 10.1039/c2an16211c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Tseng K.; Garcia B.; Lebrilla C. B.; Mukerjee E.; Collins S.; Smith R. Anal. Chem. 2001, 73, 2147–2151. 10.1021/ac001326t. [DOI] [PubMed] [Google Scholar]
- Rejtar T.; Hu P.; Juhasz P.; Campbell J. M.; Vestal M. L.; Preisler J.; Karger B. L. J. Proteome Res. 2002, 1, 171–179. 10.1021/pr015519o. [DOI] [PubMed] [Google Scholar]
- Page J. S.; Rubakhin S. S.; Sweedler J. V. Anal. Chem. 2002, 74, 497–503. 10.1021/ac0156621. [DOI] [PubMed] [Google Scholar]
- Fan Y.; Lee C. Y.; Rubakhin S. S.; Sweedler J. V. Analyst 2013, 138, 6337–6346. 10.1039/c3an01010d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisler J.; Foret F.; Karger B. L. Anal. Chem. 1998, 70, 5278–5287. 10.1021/ac9807823. [DOI] [PubMed] [Google Scholar]
- Tracht S. E.; Cruz L.; Stobba-Wiley C. M.; Sweedler J. V. Anal. Chem. 1996, 68, 3922–3927. 10.1021/ac9603901. [DOI] [PubMed] [Google Scholar]
- Van Berkel G. J.; Kertesz V.; King R. C. Anal. Chem. 2009, 81, 7096–7101. 10.1021/ac901098d. [DOI] [PubMed] [Google Scholar]
- ElNaggar M. S.; Barbier C.; Van Berkel G. J. J. Am. Soc. Mass Spectrom. 2011, 22, 1157–1166. 10.1007/s13361-011-0145-5. [DOI] [PubMed] [Google Scholar]
- Comi T. J.; Neumann E. K.; Do T. D.; Sweedler J. V.. J. Am. Soc. Mass Spectrom., in press, 2017; DOI: 10.1007/s13361-017-1704-1. [DOI] [Google Scholar]
- Do T. D.; Comi T. J.; Dunham S. J. B.; Rubakhin S. S.; Sweedler J. V. Anal. Chem. 2017, 89, 3078–3086. 10.1021/acs.analchem.6b04819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Barrado M. J.; Iglesias Osma M. C.; Blanco E. J.; Carretero Hernández M.; Sánchez Robledo V.; Catalano Iniesta L.; Carrero S.; Carretero J. PLoS One 2015, 10, e0123197. 10.1371/journal.pone.0123197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustione A.; Piston D. W. Mol. Endocrinol. 2012, 26, 1928–1940. 10.1210/me.2012-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker K. R.; Li Z.; Rubakhin S. S.; Sweedler J. V. J. Am. Soc. Mass Spectrom. 2012, 23, 1931–1938. 10.1007/s13361-012-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano E.; Cirulli V.; Bosco D.; Rouiller D.; Halban P.; Meda P. Am. J. Physiol. Cell Physiol. 1993, 265, C358–C364. [DOI] [PubMed] [Google Scholar]
- Mayer B. X. J. Chromatogr. 2001, 907, 21–37. 10.1016/S0021-9673(00)01057-8. [DOI] [PubMed] [Google Scholar]
- Borelli M. I.; Rubio M.; García M. E.; Flores L. E.; Gagliardino J. J. BMC Endocr. Disord. 2003, 3, 2. 10.1186/1472-6823-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffo A.; Hancock K.; Polito T.; Xie Y.; Andan G.; Witkowski P.; Hardy M.; Barba P.; Ferrara C.; Maffei A.; Freeby M.; Goland R.; Leibel R. L.; Sweet I. R.; Harris P. E. J. Endocrinol. 2008, 198, 41–49. 10.1677/JOE-07-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.