

Abstract
Pancreatic polypeptide (PP) is a satiety-inducing gut hormone targeting predominantly the Y4 receptor within the neuropeptide Y multiligand/multireceptor family. Palmitoylated PP-based ligands have already been reported to exert prolonged satiety-inducing effects in animal models. Here, we suggest that other lipidation sites and different fatty acid chain lengths may affect receptor selectivity and metabolic stability. Activity tests revealed significantly enhanced potency of long fatty acid conjugates on all four Y receptors with a preference of position 22 over 30 at Y1, Y2 and Y5 receptors. Improved Y receptor selectivity was observed for two short fatty acid analogues. Moreover, [K30(E-Prop)]hPP2–36 (15) displayed enhanced stability in blood plasma and liver homogenates. Thus, short chain lipidation of hPP at key residue 30 is a promising approach for anti-obesity therapy because of maintained selectivity and a sixfold increased plasma half-life.
Keywords: lipidation, pancreatic polypeptide, receptors, selectivity, therapeutic peptides
Introduction
The pancreatic polypeptide (PP) is a hormone that is secreted by the pancreatic islets in response to meal ingestion proportionally to caloric intake.[1] It mediates its biological function by a rhodopsin-like G protein-coupled receptor (GPCR), the human Y4 receptor (hY4R). This peripheral gut-derived peptide can act either indirectly by gut vagal signals to higher centers in the brainstem or directly at its native target, which is expressed in the hypothalamus and brainstem of the central nervous system. Circulating PP can enter parts of the blood–brain barrier in the area postrema that is located in the dorsal vagal complex within the brainstem.[2] Activation of the hY4R, which is mainly expressed in the brain and gastrointestinal tract[3] leads to suppression of appetite and food intake as well as delayed gastric emptying and motility[2b] It has been shown that peripheral administration of PP reduces appetite and food intake in mice[4] as well as in healthy[5] and morbidly obese humans.[6] Accordingly, PP represents an important regulator of feeding behavior and energy homeostasis.
PP belongs to the neuropeptide Y (NPY) hormone/receptor-family, which consists of three 36 amino acid peptide ligands, named NPY, peptide YY (PYY) and PP that interact with four receptors in humans, hY1R, hY2R, hY4R and hY5R.[7] These structurally related Y receptors are differently expressed throughout the human body and bind the ligands with different preferences. PP predominantly targets the hY4R, whereas hY1R and hY2R show low PP binding, and the hY5R exhibits only moderate affinity for PP. NPY and PYY address hY1R, hY2R and hY5R with nanomolar affinity. Different effects have been reported for hYR with respect to appetite regulation. While orexigenic effects (stimulation of hunger) are conveyed by central hY1R and hY5R, anorexigenic effects (induction of satiety) are transmitted by peripheral and central hY2R and hY4R.[2a, 8] Thus, exploiting the satiety hormone PP by selective activation of either the hY2R or the hY4R is a favorable approach for anti-obesity therapy[9] in the context of the epidemically growing health problem of obesity and overweight, as well as associated disorders such as cardiovascular diseases and diabetes mellitus.[10]
Apart from many advantages of peptide drugs like their high specificity and activity, rare toxic degradation and in vivo predictability, they are therapeutically restricted by their low bioavailability owing to rapid proteolytic degradation and short circulation times.[11] Hence, for effective medication, the short-acting pharmacokinetic properties of PP[1, 12] have to be improved. Among the versatile established strategies to modulate plasma half-lives of potential peptide drugs,[13] covalent modification with fatty acids (lipidation) has come into focus during the last years.[14] Hydrophobic moieties lead to reversible, high-affinity binding to human serum albumin, which shields and transports the acylated compounds throughout the body leading to prolonged activity profiles.[15]
Up to now, only few studies investigated the critical size of the fatty acid or the influence of positioning.[16] Both factors, however, are important for efficient development of peptide drugs with high selectivity and increased bioavailability. Hence, individual residues that might not contribute to receptor interaction were selectively replaced and modified according to the comparative model of hY4R binding PP,[17] and two positions were chosen for systematic modification with various saturated fatty acids. Independent of position and lipid, all analogues maintained high activity for hY4R, which confirms the suitability of the ligand–receptor model used for this type of experiments. Interestingly, not only the position but also the length of the fatty acid dramatically influenced receptor selectivity and partly led to an increase in potency for other receptor subtypes. Moreover, pronounced resistance to proteolytic degradation was observed for a highly hY4R-preferring agonist equipped with a propanoic acid, which is comparable to the related palmitoyl variant and suggests that also short-chain fatty acid peptide conjugates might be likely drug candidates in the future.
Results
Identification of best position for modification
With the help of a recently described comparative 3D model of hY4R bound to PP,[17] accessible residues that might not contribute to ligand–receptor interaction were identified for modification (Figure 1). Positions in close proximity to the C-terminal RPRY-NH2 sequence that is important for binding and activation[18] as well as residues, which are not proposed to be part of the peptide binding pocket, were chosen for modification with lysine-γ-glutamyl fatty acid moieties in order to protect the peptide from fast degradation. Lipopeptides were accessible by solid-phase peptide synthesis (SPPS)[11b, 19] applying the 9-fluorenylmethoxycarbonyl (Fmoc)/tert-butyl (tBu) orthogonal protecting group strategy. This method allowed the selective on-resin modification at distinct amino acid positions, which have been substituted with Lys, a γ-glutamyl linker and the desired saturated fatty acids.[20] In a first step, the influence of the acylation site on hY4R affinity was validated. Therefore, hPP was modified with palmitic/hexadecanoic acid (Pam) at Gln16, Ala22 and Met30 inside the reported a-helix of the peptide.[21] In addition, Tyr7 located within the poly-l-proline type II helix as well as Glu4 in the N-terminal part of hPP represented further modification sites (Figure 1). Since the N terminus does not contribute to receptor binding,[18b] it was labeled with 5(6)-carboxyfluorescein (CF) for this first set of peptides, enabling the possibility of supplementary biological experiments. All CF-tagged palmitoylated hPP analogues were prepared in sufficient amounts and purities (see Supporting information, Table 1). In order to identify the most suitable modification sites for peptide lipidation, CF-labeled hPP variants palmitoylated at position 4 (2), 7 (3), 16 (4), 22 (5), 30 (6) as well as CF-hPP (1b) as wild-type control were tested in radioligand binding assays. As shown in Figure 2a, high specific binding was observed for control peptide 1b (90%), but also compounds 5 (84%) and 6 (83%) displayed high specific binding that was not statistically different from CF-hPP (1b). In contrast, the remaining three compounds, acylated at positions 4 (2), 7 (3) and 16 (4) exhibited significantly reduced specific [3H]-hPP binding (2: 37%, 3: 67%, 4: 36%). Compounds 3, 5 and 6 were further tested for competitive displacement using an appropriate concentration range of cold ligand in presence of radioligand (Figure 2b). The determined IC50 values (1b: 3.0 nm, 3: 401 nm, 5: 8.6 nm, 6: 14.9 nm) gave evidence that residues 22 and 30 were most suitable for acylation with fatty acids with only modestly elevated IC50 values. In contrast, 3 showed more than 100-fold loss in affinity in comparison to control peptide 1b.
Figure 1.
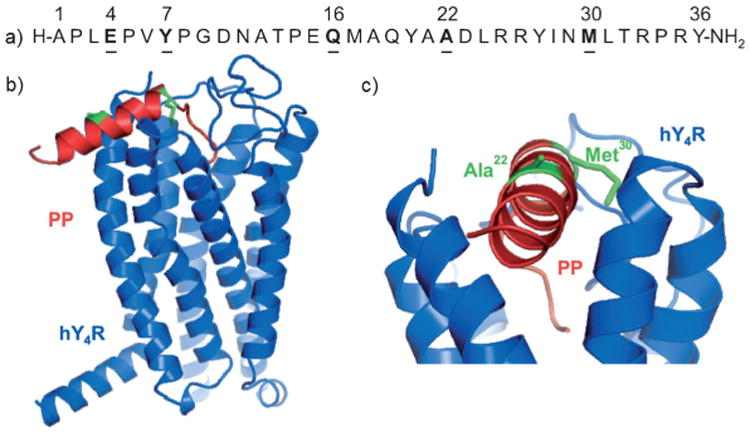
a) Amino acid sequence of hPP; residues to be modified are underlined. b) Side view of the comparative model of human Y4R (blue) with docked bovine PP (red; PDB code: 1LJV);[21b] lipidated positions are indicated in green.[17] c) Enlarged section of the same model obtained by horizontal rotation by approximately 90°; lipidated residues Ala22 and Met30 appear green.
Table 1.
Analytical and conformational characterization of hPP2–36 compounds with modifications at position 22 and 30 and their respective controls.
| No. | Compound | Purity [%][a] | HPLC-Elution [%] of B in A | Chemical formula | MALDI-TOF MS | Quantity [mg][d] | Yield [%][e] | % α-Helix ±SEM[f] | ||
|---|---|---|---|---|---|---|---|---|---|---|
| C18 90 A[b] | C18 300 A[c] | Mcalcd [Da] | [M+H]+ [Da] | |||||||
| 7 | pNPY | > 99 | 44.1 | 46.3 | C190H287N55O57 | 4251.1 | 4252.1 | 17.5 | 19.2 | |
| 1a | hPP | > 99 | 44.3 | 46.0 | C185H287N53O54S2 | 4179.1 | 4180.0 | 16.0 | 17.9 | (59±1) |
| 8 | [K22]hPP2–36 | > 99 | 43.4 | 40.5 | C185H289N53O53S2 | 4165.1 | 4166.0 | 15.0 | 16.8 | 59±1 |
| 9 | [K22(E-Prop)]hPP2–36 | > 98 | 44.8 | 41.6 | C193H300N54O57S2 | 4350.2 | 4351.5 | 13.0 | 13.9 | 53±2 |
| 10 | [K22(E-Capr)]hPP2–36 | > 98 | 46.9 | 43.7 | C198H310N54O57S2 | 4423.1 | 4424.1 | 18.0 | 19.0 | 49±10 |
| 11 | [K22(E-Laur)]hPP2–36 | > 99 | 52.6 | 49.5 | C202H318N54O57S2 | 4479.2 | 4480.6 | 17.0 | 17.7 | 39±7 |
| 12 | [K22(E-Pam)]hPP2–36 | > 99 | 57.1 | 53.4 | C206H326N54O57S2 | 4535.4 | 4536.8 | 13.0 | 13.4 | 28±10 |
| 13 | [K22(E-Ara)]hPP2–36 | > 99 | 61.0 | 57.1 | C210H334N54O57S2 | 4588.4 | 4589.4 | 4.2 | 8.5 | 2±0(60±0) |
| 14a | [K30]hPP2–36 | > 98 | 42.6 | 39.6 | C183H285N53O53S1 | 4105.1 | 4106.0 | 11.2 | 12.7 | 70±10 |
| 15a | [K30(E-Prop)]hPP2–36 | > 98 | 44.2 | 41.3 | C191H296N54O57S1 | 4290.2 | 4291.6 | 17.0 | 18.5 | 54±2 |
| 16a | [K30(E-Capr)]hPP2–36 | > 98 | 46.9 | 43.9 | C196H306N54O57S1 | 4360.2 | 4361.6 | 12.0 | 12.9 | 57±1 |
| 17 | [K30(E-Laur)]hPP2–36 | > 99 | 52.8 | 44.0 | C200H314N54O57S1 | 4419.1 | 4420.3 | 15.0 | 15.8 | 61 ±2 |
| 18a | [K30(E-Pam)]hPP2–36 | > 91 | 57.0 | 53.6 | C204H322N54O57S1 | 4475.2 | 4476.0 | 13.0 | 13.6 | 51 ±6 |
| 19 | [K30(E-Ara)]hPP2–36 | > 99 | 61.3 | 70.7 | C208H330N54O57S1 | 4528.4 | 4529.3 | 5.0 | 10.3 | 2 ± 0 (60 ± 1) |
Peptide purity was quantified by RP-HPLC elution at 220 nm using two different columns ([b] and [c]) and linear gradients of eluent B (CH3CN with 0.08% (v/v) TFA) in eluent A (H2O with 0.1 % (v/v) TFA). Analytical HPLC columns used:
Jupiter 4u Proteo RP-C18 column (90 Å, 4 μm, 250×4.6 mm, Phenomenex),
Jupiter 5u Proteo RP-C18 column (300 Å, 5 μm, 250×4.6 mm, Phenomenex).
Amounts correspond to observed masses without TFA salts.
Yield of synthesized compounds is referred to maximal resin loading capacity of 15 μmolg−1.
α-Helical content of 10 μm hPP analogues was estimated by two independent CD spectra obtained either in 10 mm sodium phosphate buffer (pH 7.0) or in buffer containing 20 mM SDS (given in parentheses) applying Dichroweb with K2D-analysis.[22]
Statistical analysis using one way-ANOVA, followed by Dunnett's post-hoc test (GraphPad Prism 5.0) revealed no significantly reduced fraction of α-helix for the lipidated hPP analogues compared to respective [K]hPP2–36, except of the E-Ara compounds (13: **; 19: ***) in buffer not supplemented with SDS.
P ≤ 0.01,
P ≤ 0.001, referred to [K22]hPP2–36 for 13 and [K30]hPP2–36 for 19.
Figure 2.
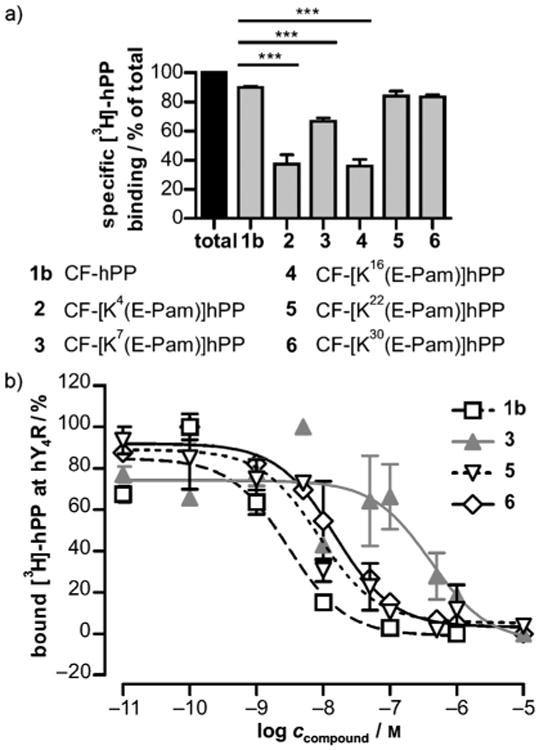
a) First evaluation of the binding capacity by 1 μm CF-labeled hPP analogues 1b, 2–6 in transiently hY4R-expressing COS-7 cells towards [3H]-hPP after a 90 min stimulation period. Total binding was set to 100% and corresponds to binding of radioligand in presence of H2O in 1 % (w/v) BSA. Statistical analysis was performed by one-way ANOVA, followed by Dunnett's post-hoc test (GraphPad Prism 5.0). ***P ≤ 0.001, as compared to 1b (horizontal lines). b) For competition binding experiments, [3H]-hPP was displaced by increasing concentrations of most promising analogues 1, 3, 5 and 6. Assays were performed in triplicate; mean values ±SEM of independently examined experiments are shown.
Comparison of different fatty acids
In the next step, truncated hPP2–36 lacking a dipeptidyl peptidase IV (DPP-IV) cleavage site[18b] was modified at Ala22 or Met30 (Figure 1b, 1c), respectively, with propanoic acid (Prop), caprylic/octanoic acid (Capr), lauric/dodecanoic acid (Laur), Pam or arachidic/eicosanoic acid (Ara). A slightly altered synthesis strategy was applied (Scheme 1a).[20] Table 1 shows the full analytical characterization of all lipidated hPP2–36 conjugates. High purities and an increase in hydrophobicity attributed to fatty acid length were examined by two independent reversed-phase high-performance liquid chromatography (RPHPLC) systems, while their identity was confirmed applying matrix-assisted laser/desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). Preparation on solid support in 15 μmol or 7.5 μmol scale resulted in appropriate quantities (Table 1). Subsequently, the lipidated hPP2–36 conjugates were analyzed with respect to their biological functionality and selectivity. Signal transduction experiments were performed with COS-7 cells stably co-expressing one of the four hYR and a chimeric Gi,q protein[23] to allow robust signal readout via [3H]-inositol phosphates (IP). A summary of the obtained concentration–response curves for hY4R activation by the modified hPP2–36 compounds is illustrated in Figure 3, while numerical data can be found in Table 2. The native ligand hPP has an inherent activity in the low-nanomolar range (1a: EC50=1.3 nm), which is in accordance with the literature.[17] More importantly, the high potency is not affected by any lipidation. This is reflected in similar EC50 values and full efficacies (Figure 3, Table 2) for all analogues acylated at position 22 and 30. So, the fatty acid chain length neither has beneficial nor detrimental effects on hY4R activation.
Scheme 1.
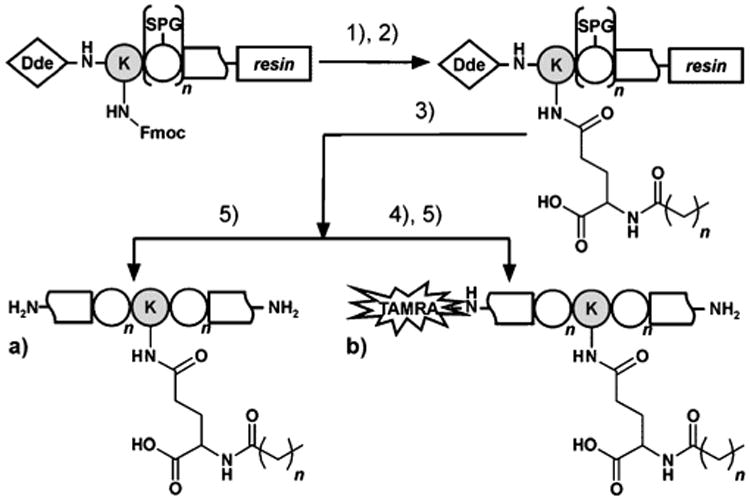
Synthesis of a) [K(E-Lip)]hPP2–36 and b) TAMRA-[K(E-Lip)]hPP2–36. Peptides were assembled by automated SPPS up to modification site (22 or 30), substituted with Lys (grey) that was protected by Fmoc at the γ-amino group and by Dde at the N terminus. 1) Fmoc removal and coupling of Fmoc-l-Glu-OtBu. 2) Fmoc deprotection and acylation with fatty acids (Lip): C2H5COOH (Prop) for 9, 15a, 15b; C7H15COOH (Capr) for 10, 16a, 16b; C11H23COOH (Laur) for 11, 17; C15H31COOH (Pam) for 12, 18a, 18b; C19H39COOH (Ara) for 13, 19. 3) Dde removal and automated elongation to desired peptide sequence. 4) TAMRA-labeling of peptide N terminus. 5) Cleavage from resin including deprotection of all acid-labile side chain protecting groups (SPG) and methionine reduction.
Figure 3.
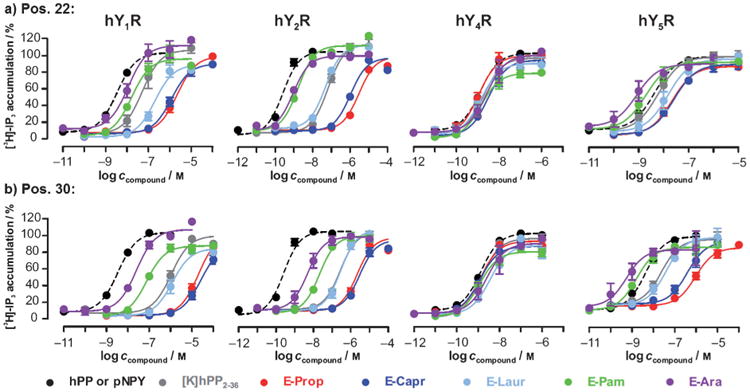
Functional characterization of hPP compounds fatty acid acylated at a) position 22 and b) residue 30 at COS-7 cells stably expressing the anorexigenic hY4R and hY2R as well as the orexigenic hY1R and hY5R. Co-expression of hYR and a chimeric Gi/q protein allowed concentration-dependent radioactive inositol phosphate accumulation. Mean concentration–response curves of at least two independent experiments, fitted by non-linear regression (GraphPad Prism 5.0) are shown with ±SEM. Dashed black lines correspond to the respective native ligands (hPP for hY4R and porcine NPY (pNPY) for hY1R, hY2R and hY5R).
Table 2.
Summary of potency (EC50) and efficacy (Emax) profiles of all hPP2–36 conjugates lipidated at position 22 and 30. IP accumulation assays were performed in COS-7 cells stably co-expressing the respective hYR subtypes and a chimeric Gi/q protein.
| No. | Modification | EC50 [nM][a] (pEC50) [b] |
hY1R EC50 ratio[c] |
Emax [%] [d] | EC50 [nM] (pEC50) |
hY2R EC50 ratio |
Emax [%] | EC50 [nM] (pEC50) |
hY4R EC50 ratio |
Emax [%] | EC50 [nM] (pEC50) |
hY5R EC50 ratio |
Emax [%] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 3.5 (8.5±0.04) | 1 | 96±2 | 0.3 (9.6±0.1) | 1 | 100 ±3 | 44.7 (7.4±0.3) | 34 | 82 ±9 | 4.3 (8.4±0.1) | 1 | 88±2 | |
| 1a | 76.0 (7.1±0.1) | 22 | 95±3 | 341.4(6.5±0.1) | > 1000 | 96±5 | 1.3(8.9±0.1) | 1 | 93±2 | 11.4(7.9±0.1) | 3 | 90±5 | |
| [K22]hPP2–36 | |||||||||||||
| 8 | 51.4 (7.3±0.2) | 15 | 101 ±7 | 67.1 (7.2±0.1) | 260 | 107 ±4 | 1.9 (8.7±0.1) | 1 | 91 ±3 | 6.1 (8.2±0.1) | 1 | 89±5 | |
| 9 | E-Prop | > 1000 (5.7±0.1) | 618 | 92 ±4 | > 1000 (5.5±0.1) | > 1000 | 88 ±6 | 1.1 (9.0±0.1) | 1 | 96±6 | 21.9 (7.7±0.1) | 5 | 83±2 |
| 10 | E-Capr | > 1000 (5.9±0.1) | 339 | 84 ±4 | > 1000 (6.0±0.2) | > 1000 | 88 ±7 | 3.1 (8.5±0.1) | 2 | 91 ±4 | 26.0 (7.6±0.1) | 6 | 84±4 |
| 11 | E-Laur | 204.0 (6.7±0.1) | 57 | 87 ±5 | 49.6 (7.3±0.2) | 205 | 101 ±7 | 1.5 (8.8±0.1) | 1 | 86±5 | 14.9 (7.8±0.1) | 4 | 87±5 |
| 12 | E-Pam | 16.5 (7.8±0.1) | 5 | 93 ±6 | 1.7 (8.8±0.2) | 6 | 103 ± 12 | 1.5 (8.8±0.1) | 1 | 78±4 | 1.9 (8.7±0.1) | 0.4 | 81 ±6 |
| 13 | E-Ara | 11.3 (8.0±0.1) | 3 | 101 ±6 | 0.9 (9.0±0.1) | 3 | 90±5 | 2.7 (8.6±0.1) | 2 | 93±6 | 0.8 (9.1 ±0.2) | 0.2 | 75 ±6 |
| [K30]hPP2–36 | |||||||||||||
| 14a | > 1000 (6.0±0.1) | 297 | 93 ±5 | 270.0 (6.6±0.1) | 782 | 93 ±3 | 1.4 (8.9±0.1) | 1 | 95 ±4 | 23.9 (7.6±0.1) | 6 | 79±4 | |
| 15a | E-Prop | >1000 (4.7±0.1) | > 1000 | 99 ±4 | > 1000 (5.7±0.1) | > 1000 | 88 ±6 | 1.3 (8.9±0.1) | 1 | 88±4 | 937.2 (6.0±0.1) | 244 | 70 ±3 |
| 16a | E-Capr | > 1000 (4.6±0.1) | > 1000 | 84 ±6 | > 1000 (5.5±0.1) | > 1000 | 85 ±5 | 1.2 (8.9±0.1) | 1 | 82 ±4 | 386.5 (6.4±0.1) | 99 | 75 ±5 |
| 17 | E-Laur | > 1000 (5.9±0.1) | 344 | 81 ±4 | 296.4 (6.5±0.1) | 875 | 98 ±4 | 3.6 (8.5±0.2) | 3 | 83±5 | 38.8 (7.4±0.1) | 9 | 81 ±5 |
| 18a | E-Pam | 76.2 (7.1 ±0.1) | 22 | 85 ±3 | 21.1 (7.7±0.1) | 66 | 92 ±3 | 1.2 (8.9±0.1) | 1 | 78±4 | 1.6 (8.8±0.1) | 0.4 | 75 ±5 |
| 19 | E-Ara | 25.1 (7.6±0.1) | 7 | 98 ±4 | 4.9 (8.3±0.1) | 22 | 88 ±5 | 2.5 (8.6±0.2) | 2 | 84±7 | 0.6 (9.3±0.3) | 0.1 | 69±10 |
The concentration of half-maximal response (EC50) was obtained from concentration-response curves and fitted by non-linear regression with GraphPad Prism 5. EC50 values are given as total mean of at least two independently repeated experiments, measured in duplicate.
pEC50 values are the mean ± SEM negative decadic logarithm of each normalized IP accumulation curve.
Comparison of potency was performed by calculation of EC50 shifts between ligand and respective wild type (pNPY for hY1R, hY2R, hY5R and hPP for hY4R) using GraphPad Prism 5.0.
The effect at the highest concentration of wild type was set to 100%, normalized to each further tested compound and given as maximal response (Emax) ±SEM.
Loss of subtype preference by long fatty acid modification
Next, IP accumulation assays with hY1R-expressing COS-7 cells were performed in order to evaluate the activity potential of the lipopeptides at the structurally related Y receptor subtype that exhibits opposing effects (stimulation of food intake). As shown in Figure 3, both non-lipidated precursor peptides [K22]hPP2–36 (8: EC50 = 51.4 nm) and [K30]hPP2–36 (14a: EC50 > 1000 nm) exhibit remarkably little activation of hY1R compared to the native ligand NPY (7: EC50 = 3.5 nm). Interestingly however, introduction of lysine at position 22 led to increased inherent hY1R potency of hPP2–36 analogues with EC50 values of [K30]hPP2–36 being 15-fold lower than for [K22]hPP2–36 (Table 2). At both locations, a substantial decrease in activity occurred for the short fatty acid conjugates (E-Prop, E-Capr), whereas from a chain length of 12 carbon atoms (E-Laur) potency was regained. Interestingly, palmitoylation and arachidoylation led to considerably more active analogues compared to their respective predecessors. Additionally, the potencies for compounds 12 and 13 as well as 18a and 19 are only 5- to 20-fold reduced compared to the native ligand NPY (7) at this receptor. Overall, the hY1R activity of [K30]hPP2–36 lipidated conjugates was lower compared to the compounds modified at position 22 (Figure 3, Table 2).
Signal transduction studies at anorexigenically acting hY2R (Figure 3, Table 2) revealed a similar pattern as observed for the hY1R. However, albeit the potency of the native ligand NPY (7: EC50 = 0.3 nm) was about one order of magnitude higher than for the hY1R, the precursor peptides yielded potencies comparable to hY1R. Again, a shift from lower to higher potency with extension of fatty acid length was observed (Table 2), with short fatty acids increasing the specificity of the ligand for the hY4R, and long fatty acids increasing activity on hY receptors in general. Most notably, [K22]hPP2–36 lipidated with palmitic acid (12: EC50= 1.7 nm) and arachidic acid (13: EC50 = 0.9 nm) were identified as highly active ligands at the hY2R, showing potencies in the range of the native ligand NPY (7) and being 40- and 75-fold, respectively, more potent than the parent peptide 8.
Finally, activity tests at the hY5R exhibited a marginally different activation profile of the acylated conjugates (Figure 3, Table 2). This receptor subtype seemed to be less selective towards its native ligand NPY (7: EC50 = 4.3 nm) since both precursor peptides [K]hPP2–36 possessed high activities in the low-nanomolar range (8: EC50 = 6.1 nm, 14a: EC50 = 23.9 nm). While modification with short fatty acids again provoked potency decrease, activity was enhanced with longer fatty acids as observed for hY1R and hY2R and even resulted in the first hPP-based hY5R agonists with even better potencies than NPY (12: EC50 = 1.9 nm, 18a: EC50 = 1.6 nm, 13: EC50 = 0.8 nm, 19: EC50 = 0.6 nm). Moreover, activity comparison of the shorter fatty acid hPP analogues (E-Prop, E-Capr) at the two distinct positions reflected about 10-fold difference in hY5R activation compared to any other peptide, indicating again a preference of the receptor for position 22 over 30 (Table 2).
Structural impairments are not the reason for the altered activity pattern
Since long-chain fatty acid analogues differed in their behavior towards hYR selectivity relative to short- or medium-chain fatty acid conjugates, we hypothesized that conformational changes could be the reason. We performed circular dichroism (CD) spectroscopy in order to determine potential structural influences or changes induced by different fatty acid moieties. Nearly all hPP analogues lipidated at position 22 or 30 displayed typical features of an α-helix in CD spectra (see Supporting information, Figure 1a,b). Negative Cotton effects at 208 and 222 nm as well as a large maximum at around 190 nm were observed. Surprisingly, compounds acylated with the longest fatty acid (E-Ara) revealed disturbed structures with more β-sheet-like characteristics.[24] The amounts of secondary structure were quantified by Dichroweb using K2D analysis (Table 1).[22] These calculations suggested that the decrease of α-helical proportion is stronger with increase in fatty acid length at position 22 opposed to residue 30. Exclusively, arachidoylated hPP compounds were not capable to form the classical PP structure in solution. In order to prove the possibility that these compounds fold only upon approaching the cell membrane, CD spectra were recorded in the presence of 20 mm sodium dodecylphosphate (SDS). Indeed, hPP analogues 13 and 19 revealed an increased fraction of α-helix comparable to wild type in this membrane-mimicking environment (see Supporting information, Figure 1c, and Table 1). These results indicated that in solution only fatty acids of very long chain length bias the α-helicity of the ligand, probably due to changes in hydrophobicity of the peptide microenvironment. Capturing the hydrophobic moiety in a detergent or membrane environment can thus restore secondary structure. In general, however, these data suggested that the global peptide backbone orientation was not impaired by lipidation, and, thus, direct receptor interaction seems to affect receptor recognition.
In vitro stability
To underline the proposed therapeutic potential of the new hY4R-preferring agonists by the fatty acid moieties, degradation was investigated in human blood plasma (Figure 4a) and porcine liver homogenates (Figure 4b). Two selective hY4R ligands [K30(E-Prop)]hPP2–36 (15b) and [K30(E-Capr)]hPP2–36 (16b), the palmitoylated variant 18b and the control peptide 14b were equipped with an additional N-terminal fluorescent dye, 6-carboxytetramethylrhodamine (TAMRA) (Scheme 1 b), allowing peptide-specific detection of degradation by RP-HPLC. Analogue 20, which was fluorescently labeled at the side chain of Lys30, served to detect N-terminally degraded peptides (Table 3).
Figure 4.
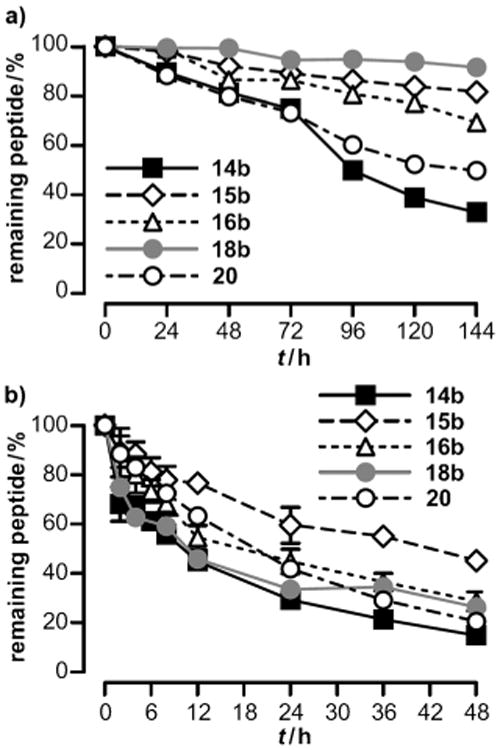
In vitro stability tests of TAMRA-labeled hY4R-selective lipidated hPP conjugates (15b, 16b) along with the Pam-variant (18b) and relevant control peptides (14b, 20) performed in a) human blood plasma and b) 50 mgmL−1 porcine liver extract homogenates. 10 μm peptide solutions were incubated at 37°C and 500 rpm. Degradation was followed using RP-HPLC at indicated time points by peptide-specific fluorescence monitoring and referred to control at 0 h (100%). Values are the mean ±SEM of two independent experiments.
Table 3.
Analytical data of TAMRA-modified hPP analogues.
| No. | Compound | Purity [%][a] | HPLC Elution [%] of B in A | Chemical formula | MALDI-TOF MS | Quantity [mg] [d] | Yield [%] [e] | ||
|---|---|---|---|---|---|---|---|---|---|
| C18 90 Å [b] | Polym. 200 Å [c] | Mcalcd [Da] | [M+H]+ [Da] | ||||||
| 14b | TAMRA-[K30]hPP2 36 | > 98 | 40.7 | 53.0 | C208H305N55O57S1 | 4517.2 | 4518.3 | 9.9 | 32.1 |
| 15b | TAMRA-[K30(E-Prop)]hPP2–36 | > 96 | 41.9 | 48.0 | C216H316N56O61S1 | 4702.3 | 4703.3 | 4.7 | 31.3 |
| 16b | TAMRA-[K30(E-Capr)]hPP2–36 | > 98 | 44.2 | 50.1 | C221H326N56O61S1 | 4772.4 | 4773.3 | 10.0 | 35.7 |
| 18b | TAMRA-[K30(E-Pam)]hPP2–36 | > 98 | 55.0 | 55.1 | C229H342N56O61S1 | 4887.7 | 4888.5 | 1.9 | 11.7 |
| 20 | [K30(TAMRA)]hPP2–36 | > 97 | 40.0 | 40.7 | C208H305N55O57S1 | 4520.0 | 4520.2 | 10.0 | 17.5 |
Purities of peptides were quantified by HPLC elution at 220 nm using two different columns ([b] and [c]) and linear gradients of eluent B in eluent A. Analytical HPLC columns applied:
Jupiter 4u Proteo RP-C18 column (90 Å, 4 μm, 250×4.6 mm; Phenomenex),
VariTide RPC column (200 Å, 6 μm, 250×4.6 mm, Varian); Polym.: polymer-based RPC-like column material.
Amounts correspond to observed masses without TFA salts.
Yield of synthesized compounds is referred to maximal resin loading capacity of 15 μmolg−1.
As depicted in Figure 4a, all investigated compounds displayed a decay of intact peptide in human blood plasma over 144 h. Both precursor peptides 14b and 20 revealed similar degradation with comparable half-lives of around 100 h (Table 4). The similar amount of N- and side chain fluorescently labeled precursor peptide underlines the suitability of the experimental setup and implies that peptide stability is not affected by the fluorophore TAMRA itself. As expected, the long-chain lipopeptide 18b showed most pronounced resistance to proteolytic degradation with more than 10-fold increased half-life (18b: t1/2 = 1088.1 ± 282.4 h) compared to the control peptide (14b: t1/2 = 84.7 ± 2.3 h). Surprisingly, degradation rates were faster for the longer E-Capr variant (16b: t1/2 = 281.3 ± 26.7 h) relative to the shorter E-Prop compound (15b: t1/2 = 473.6 ± 17.3 h, see also Table 4). To evaluate the stability in high enzyme and low albumin concentration, metabolism in homogenized liver extracts was monitored over 48 h. Again, all analogues were degraded over time (Figure 4b), and with respect to the control peptides 14b and 20, no significant differences between their half-lives were observed (Table 4). Notably, for the peptide modified with the shortest fatty acid (15b) a considerably slower degradation was determined, which is also reflected in significantly increased half-life (15b: t1/2 = 46.7 ± 6.6 h) compared to the parent peptide (14b: t1/2 = 11.0 ± 0.4 h). In contrast, no improved in vitro stability was determined for the E-Capr (16b) and the E-Pam (18b) peptide analogues although they contained a longer fatty acid moiety (Figure 4b, Table 4).
Table 4.
Half-lives of fluorescently labeled peptides as determined by degradation profiles in human blood plasma and porcine liver extract homogenates.
| No. | Modification | Blood plasma | Liver homogenate | |||
|---|---|---|---|---|---|---|
| N-terminal | Side chain | t1/2±SEM [h] | P value[a] | t1/2±SEM [h] | P value[a] | |
| 14b | TAMRA | – | 84.7 ± 2.3 | – | 11.0 ± 0.4 | – |
| 15b | TAMRA | E-Prop | 473.6 ± 17.3 | ≤ 0.001 | 46.7 ± 6.6 | ≤0.01 |
| 16b | TAMRA | E-Capr | 281.3 ± 26.7 | ≤0.01 | 14.6 ± 2.8 | ns |
| 18b | TAMRA | E-Pam | 1088.1 ± 282.4 | ≤ 0.001 | 11.8± 0.8 | ns |
| 20 | – | TAMRA | 136.0 ± 3.1 | ns | 19.7 ± 0.4 | ns |
Statistical significance of t1/2 values was determined by one-way ANOVA, followed by Dunnett's post-hoc test (GraphPad Prism 5.0) with comparison to control peptide 14b; ns: not significant.
In summary, the E-Pam hPP variant 18b was the most stable lipopeptide in plasma followed by the E-Prop and E-Capr compounds. Thus, the E-Prop-modified hPP variants 15a and 15b could be identified as a hY4R selective ligand with improved half-lives both in human blood plasma as well as in porcine liver homogenates.
Discussion
Lipidation has already been highlighted as a successful and prospective strategy to modulate plasma half-life and to improve bioavailability of several peptides[14] including hPP.[20] In a previous study, an hPP ligand has been developed that contained palmitic acid at lysine residue 13 equipped with a γ-glutamyl spacer. This analogue specifically targeted not only the hY4R but also the hY2R[25] and showed anorexigenic effects in mice as well as increased circulation times. More recently, hY2R/hY4R-targeting hPP analogues with modification of Prop, Capr and Pam at position 22 were investigated with respect to their selectivity, stability and most notably, for their internalization. In this study, merely the long-chain Pam variant revealed significantly improved plasma stability, whereas shorter-chain lipopeptides showed only slightly slower metabolic degradation.[20b] However, the development of ligands with a preference of the hY4R over the hY2R is desirable because activation of the latter in central and diverse peripheral regions might lead to unwanted side effects in therapy. Y2R targeting could worsen retinopathy in diabetic patients[26] or induce cancer growth and vascularization.[27] Furthermore, the hY2R-preferring PYY3–36, which is the naturally cleaved form of PYY, is also reported to cause nausea and fullness, especially at high dosages.[28]
In the present study, the therapeutically interesting gut hormone hPP was chemically modified by lipidation with albumin-binding fatty acids of different chain length at diverse positions in order to improve selectivity and stability features. To overcome potential side effects and cross-reactivity, the impact of the peptide substitution site and fatty acyl chain length on target specificity and neuropeptide Y receptor subtype selectivity was investigated by signal-transduction as well as conformational studies. All designed peptides were accessible by SPPS and could be obtained in high purities and good yields.
To find an appropriate acylation position, five hPP analogues were generated by palmitoylation at different parts of their secondary structure. To date, the model of a hairpin-like secondary structure of PP (PP-fold) composed of an N-terminal type II poly-l-proline helix (residues 1-8), followed by a β-turn (residues 9–12), an extended amphiphatic α-helix (residues 13–30) and a relatively flexible C-terminal region (residues 31–36) is well established in literature.[21a,29] It is known that the characteristic PP-fold leads to correct orientation of the C-terminal hexapeptide and the N-terminal amino acids of PP, thereby mediating receptor recognition.[18a,21a\ The location of the chosen residues for modification ranged from the poly-l-proline type II helix to the α-helix.[18b,21b\ For analogues acylated at position 22 (5) and 30 (6), located at the center and the C-terminal end of the PP-α-helix, respectively, high-affinity binding was determined. On the contrary, lipidation at residues 4, 7 and 16 showed a loss in affinity. Interestingly, position 4 was not suitable for modification although for NPY it is known as a well-tolerated derivatization site.[30] These observations suggest diverse active structures and binding modes of both neuropeptides. Residues Tyr7 and Gln16, found in more central parts of the peptide, might also be important for stabilization of the biologically important PP-fold, which is reflected in low hY4R-binding capacities. The 3D comparative model of the hY4R binding hPP[17] suggested C-terminal residues to be appropriate for substitution, although some positions in this region (Tyr27, Arg33 and Arg35) are described to actively interact with transmembrane residues of the hY4R.[17,31] The retained hY4R affinity of peptides with modification at position 30 confirmed this proposed orientation in the binding pocket.
The predecessor for the following studies, hPP2–36,[32] lacks the first amino acid Ala1, thus representing a more stable peptide compared to hPP1–36 that is a substrate of the aminopeptidase DPP-IV.[18b] Likewise, it has been reported that it constituted an improved selectivity window since the hY1R required the first amino acid for activation.[18b] Most importantly, a single subcutaneous administration of hPP2–36 was shown to significantly suppress food intake in mice over an 8 h period, demonstrating its high anorexigenic potential.[18b] Hence, novel peptides substituted with Lys-γGlu and natural lipophilic building blocks containing two (Prop), seven (Capr), 11 (Laur), 15 (Pam) or 19 (Ara) hydrocarbon moieties at the most promising amino acid positions 22 and 30 were prepared. CD studies confirmed that hPP and the related peptides PYY and NPY bear an intense α-helical character.[20a,33[ CD spectroscopy of the diverse lipidated analogues revealed no significantly altered helix formation in accordance to a previous report,[20b] except for derivatization with Ara, although modifications were located in the a-helix of PP. These results suggested that the lipophilic groups are oriented away from the hydrophobic peptide core[21a] without altering the global peptide backbone conformation. For the investigated analogues, α-helix formation was somewhat more impaired upon modification with increasing fatty acid length at position 22 in contrast to position 30. This reflects an influence of the location of the substitution on secondary structure formation. However, especially the long-chain fatty acids might influence single side chain orientation or intra- and intermolecular interactions, which has been published previously for a short NPY analogue containing a hydrophobic modification.[34] Interestingly, the longest hydrophobic modification (Ara) exhibited strongly reduced helicity in solution, which was not observed in the presence of detergent-containing buffer. Thus, arachidoyl compounds may fold into their bioactive secondary structure once they approach membranes. Lerch et al.[21b] investigated bovine PP (bPP) by NMR studies in membrane-bound and solution state. They showed that the slightly flexible peptide N terminus was folded back in solution, while in a membrane-mimicking environment that region interacted with phospholipids.[21b] These data support the relatively flexible and dynamic conformation of the individual folded compartments, when the peptide changes from solution to a membrane-bound state. However, we could not find a general stabilization of the peptide secondary structure by acylation as shown by Poschner et al.[35]
Extensive signal transduction studies of all hPP2–36 conjugates lipidated at position 22 or 30, respectively, disclosed an overall altered receptor selectivity profile depending on the used acylation moiety. While all lipopeptides retained hY4R recognition, those modified with short-chain fatty acids revealed higher, but those with longer-chain fatty acids lower subtype specificity. Furthermore, these observations were more pronounced for modifications at residue 30 opposed to 22 since the inherent hYR selectivity profile of the [K30]hPP2–36 was higher compared to [K22]hPP2–36 As a consequence, on the one hand, two novel short-chain fatty acid analogues with improved hY4R preference in comparison to the natural ligand hPP (15a, 16a) were identified. On the other hand, the first hPP-based peptide conjugates acylated with long-chain fatty acids (12, 13, 18a, 19) were found that have the ability to recognize and activate all four hYR subtypes at low- to sub-nanomolar potency. Interestingly, conformational differences of the lipidated compounds were not found to be significantly affected by the variably sized hydrophobic entities. Thus, an altered global backbone orientation might not be the reason for the unexpected receptor selectivity profiles. So far, it is known that NPY and PP possess different preferences at distinct hYR, although hY4R and hY1R share high sequence identities.[7] Additionally, diverse docking of neuropeptide YR ligands to hY1/4R opposed to hY2/5R has been proposed.[31] Different membrane-bound structures of the native YR ligands NPY and PP[21b] suggest diverse binding modes at the hYR that might be influenced by lipophilic groups at the ligand site. For modification with long fatty acids, hydrophobic peptide conjugates might increase local peptide concentrations at the receptor or membrane proximity, accumulate and impart superior activities, as concluded from former studies.[16c] However, this assumption would also speak for improved hY4R activation for longer fatty acid ligands, which was not observed in our experiments. In view of the literature, many authors report about enhanced agonistic effects towards diverse targets[16c,36] but also slightly disturbed or unaffected efficacy in correlation to conjugation with fatty acids.[16a,d,e] Thus, the reason for the unaltered activity towards the native hY4R could be that the hPP ligands have high affinities leading to fast target binding independent of whether the agonists are in solution or attached to the cell membrane. Steady receptor occupancy may thus prevent membrane-accumulated ligands from receptor interaction. In contrast, hY2R and hPP have a reduced affinity, leading to an increased influence of the elevated local ligand concentration in proximity to the receptor due to membrane binding. Another possibility to explain the diversely improved potency might be that hydrophobic positions within the individual binding pockets in the hY1R, hY2R or hY5R that are normally not addressed by the native ligands become accessible by long-chain fatty acid analogues and contribute to binding. Apart from that, all hPP2–36 peptides lipidated with E-Prop (9, 15a), E-Capr (10, 16a) or E-Laur (11, 17) showed acylation-site-dependent high activity towards hY5R. Nevertheless, these compounds might still be favorable hY4R-selective therapeutics with respect to their application as drugs since the hY5R is solely expressed in the central nervous system, which may not be accessible for peripherally administered hPP.[18b]
The best hY4R-preferring hPP agonists [K30(E-Prop)]hPP2–36 (15a) and [K30(E-Capr)]hPP2–36 (16a) along with the E-Pam analogue (18a) were further examined for their in vitro stability in human blood plasma and porcine liver homogenates. In contrast to hY2R/hY4R-addressing hPP peptides lipidated at position 22,[20b] all lipopeptides of the current study significantly improved plasma half-lives that did not directly correlate to the fatty acid length. Interestingly, protection was not observed for the longer-chain fatty acid conjugates in concentrated liver homogenates, whereas the Prop variant revealed increased metabolic stabilization related to the precursor. Although PP constitutes several structural features such as C-terminal amidation[18b] that are beneficial in terms of degradation through enzymes, its half-life in the circulation was determined to be less than 7 min.[1] This might be due to fast excretion via the kidneys[18b,20a] or rapid proteolytic cleavage.
Basically, fatty acid acylation goes along with prolonged actions and circulation times that facilitate reversible serum albumin binding in a fatty acid length-cooperative manner.[15,37] This successful method has broad application[14] and already led to effective long-acting peptide drugs that are launched for diabetes treatment (Levemir and Victoza).[11b] Nevertheless, these results reveal that the optimal length for binding albumin might be palmitic acid, but also the very short propanoic acid revealed remarkable protection at residue 30. Interestingly, stability towards liver enzymatic digestion was only observed for the shortest lipopeptide. This could originate from low serum albumin concentrations in the liver, where albumin is just produced. However, the propanoyl entity might extend the half-life not only due to hydrophobic but also by possibly more pronounced electrostatic interactions of the carboxylate anion that is also known to contribute to albumin binding.[38]
Conclusions
In summary, two novel human pancreatic polypeptide (hPP)-derived human Y4 receptor (hY4R)-preferring analogues were discovered by lipidation with short-chain fatty acids. Strikingly, the propylated conjugate was more stable in blood plasma and liver homogenates compared to the precursor peptide indicating its prospective therapeutic potential as anti-obesity drug. Moreover, within the substituted amino acid residues, position 22 exhibited a general preference over position 30 at hY1R, hY2R and hY5R. The impact of variably sized covalently conjugated fatty acids on the biological profile of hPP uncovered a general selectivity pattern. For this multiligand/multireceptor system, a universal duality between selectivity and membrane targeting is proposed by using peptide lipidation. With the increase in fatty acid chain length, the ligands become more concentrated on membranes leading to faster recognition and earlier transitions to the respective receptor binding pockets.[39] In contrast, shorter fatty acids might lack appropriate lipophilicity in order to significantly accumulate in the membrane environment and are thus less active. Hence, for general applications, one has to distinguish between desired drugability/membrane targeting or selectivity of chemically engineered hPP. Long-acting and highly active analogues that target receptors or other membrane proteins without the need of being subtype selective can be generated with long alkyl chains such as palmitic acid. Instead, high target specificity and consequently lower side effects might be achieved with shorter hydrocarbon chains with moderate protection at suitable modification sites.
Experimental Section
Solid-phase peptide synthesis (SPPS)
Multiple and automated SPPS was performed with a Syro I or Syro II peptide synthesizer from MultiSynTech (Witten, Germany) using plastic syringes equipped with teflon frits. Peptides that have been cleaved from the resin by trifluoroacetic acid (TFA) were identified by MALDI-TOF MS using a Microflex or Ultraflex III TOF/TOF device from Bruker Daltonics and the software FlexControl/-Analysis (version 3.0). Purity and quantity of synthesized peptides was analyzed by RP-HPLC in analytical scale by applying a Merck HPLC system equipped with a Jupiter 4u Proteo (90 Å, 4 μm, 250×4.6 mm; Phenomenex), a Jupiter 5u Proteo (300 Å, 5 μm, 250×4.6 mm; Phenomenex), a Vydac RP-18 (300 Å, 5 μm, 250×4.6 mm; Grace Vydac), or a Varian-VariTide RPC column (200 Å, 6 μm, 250×4.6 mm). Solvents were 0.1 % (v/v) TFA (Sigma–Aldrich) in H2O (eluent A) and 0.08% (v/v) TFA in CH3CN (Prolabo; eluent B). Purification of raw peptides was performed with a Shimadzu preparative RP-HPLC by using the same eluents and a Jupiter 10u Proteo RP-C18 (90 Å, 7.78 μm, 250×21.2 mm; Phenomenex) or a Vydac RP-C18 HPLC column (300 Å; 10 μm, 250×22 mm; Grace Vydac), respectively.
Robot-assisted SPPS was performed using the Fmoc/tBu orthogonal protecting group strategy. Amino acid side chain protecting groups were as follows: trityl (Trt) for Asn, Cys and Gln; tBu for Asp, Glu, Thr and Tyr; tert-butyloxycarbonyl (Boc) for Trp; and pentamethyl-2,3-dihydrobenzofuran-5-sufonyl (Pbf) for Arg (Fmoc-amino acids purchased from Iris Biotech, Marktredwitz, Germany). Automated Fmoc deprotection was carried out with 40% (v/v) piperidine (Sigma–Aldrich) in N,N-dimethylformamide (DMF; Biosolve, Valkenswaard, The Netherlands) for 3 min and 20% (v/v) piperidine in DMF for 10 min. In situ activation and coupling of Fmoc-amino acids (0.12 μmol) that were dissolved to 0.5 m in DMF was performed with OxymaPure (0.12 μmol in DMF, 2 min pre-incubation on resin; Iris Biotech) and N,N′-diisopropylcarbodiimide (DIC; 0.12 μmol in DMF; Iris Biotech) twice for 30 min.
CF-hPP (1 b), CF-[K4(E-Pam)]hPP (2), CF-[K7(E-Pam)]hPP (3), CF-[K16(E-Pam)]hPP (4), CF-[K22(E-Pam)]hPP (5), CF-[K30(E-Pam)]hPP (6)
For synthesis of the full-length 5(6)-carboxyfluorescein (CF)-labeled peptide, automated SPPS was performed on Rink amide AM resin (Iris Biotech; 7.5 μmol scale) including a 3 h manual coupling step of Fmoc-l-Lys(Dde)-OH (20 mg, 0.038 μmol) with 1-hydroxy-benzotriazole (HOBt; 5.8 mg, 0.038 μmol; Novabiochem) and DIC (5.9 μL; 0.038 μmol) in 100 μL DMF at position 4 (2), 7 (3), 16 (4), 22 (5) or 30 (6). The N termini of the peptides were modified by reaction with CF (14.1 mg, 0.075 μmol; Sigma–Aldrich) using HOBt (5.8 mg, 0.038 μmol) and DIC (5.9 μL, 0.038 μmol) in 100 μL DMF overnight. CF-polymers were cleaved by incubation with 20% (v/v) piperidine in 500 μL DMF for 1 h, followed by an overnight Trt-protection of CF-hydroxyl groups with trityl chloride (8.4 mg, 0.03 μmol; Merck) and N,N-diisopropylethylamine (DIPEA; 5.3 μL, 0.03 μmol; Sigma–Aldrich) diluted in 250 μL CH2Cl2 (Biosolve). The 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl (Dde)-group was removed by applying 2% (v/v) hydrazine in 1 mL DMF (10×; Sigma–Aldrich) for 10 min including DMF washing steps in between. Success of deprotection was determined by UV measurements of wash solutions at 301 nm. After 3 h manual coupling of Fmoc-l-Glu-OtBu (16.0 mg, 0.38 μmol) applying HOBt (5.8 mg, 0.038 μmol) and DIC (5.9 μL, 0.038 μmol) in 100 μL DMF, and Fmoc removal by incubation with 20% (v/v) piperidine in DMF (2×20 min), palmitic acid (Sigma–Aldrich) coupling (9.6 mg, 0.038 μmol) was performed using HOBt (5.8 mg, 0.038 μmol) and DIC (5.9 μL, 0.038 μmol) in 100 μL N-methyl-2-pyrrolidone (NMP; Biosolve) for 3 h. Cleavage from the resin was performed with TFA (450 μL), and a scavenger mixture of 1,2-ethanedithiol (EDT; 35 μL; Sigma–Aldrich) and thioanisole (TA; 15 μL; Sigma–Aldrich) for 3 h. Peptides were precipitated from ice-cold Et2O (10 mL), washed 5× and dried in vacuo. In order to reduce partially oxidized methionine residues, compounds were re-dissolved in TFA (1 mL) and incubated with EDT (16 μL) and bromotrimethylsilane (TMSBr; 12 μL; Sigma–Aldrich) for 30 min, precipitated and washed with Et2O (5×10 mL), dissolved in H2O/tBuOH (1:3, v/v) and finally lyophilized. Purification was achieved with a preparative RP-C18 HPLC column (300 Å; 10 μm, 250×22 mm; Grace Vydac) and linear gradients of eluent B in A: 20% to 60% (v/v) in 40 min for 1b, 40% to 80% (v/v) in 40 min for peptides 2–6. Incorporation of the fluorophore and all following steps were performed in the dark. Analytical data and yields of all compounds are summarized in the Supporting information, Table 1. Calculated masses refer to the averaged isotope pattern. High deviations of measured versus calculated masses resulted from measurement in the linear mode instead of reflector mode.
pNPY (7), hPP (1a), [K22]hPP2–36 (8) and [K30]hPP2–36 (14a)
Peptides were synthesized automatically using a Rink amide AM resin (15 μmol scale), cleaved from resin with TFA (900 μL) with a scavenger mixture of either TA (50 μL) and p-thiocresol (50 μL; Sigma–Aldrich) for 7, or EDT (70 μL) and TA (30 μL) for 1a, 8 and 14a within 3 h. After precipitation from ice-cold Et2O (10 mL), peptides were washed 5× and dried in vacuo. hPP sequences containing oxidized methionines were reduced as mentioned above, dissolved in H2O/tBuOH (3:1, v/v) and finally lyophilized. Purification was performed with a preparative Jupiter 10u Proteo RP-C18 HPLC column (90 Å, 7.78 μm, 250×21.2 mm; Phenomenex) using a linear gradient of 20% to 60% (v/v) of eluent B in A over 40 min. Yield, purities, RP-HPLC and MALDI-TOF characterization can be found in Table 1.
[K22(E-Prop)]hPP2–36 (9), [K22(E-Capr)]hPP2–36 (10), [K22(E-Laur)]hPP2–36 (11), [K22(E-Pam)]hPP2–36 (12), [K22(E-Ara)]hPP2–36 (13), [K30(E-Prop)]hPP2–36 (15a), [K30(E-Capr)]hPP2–36 (16a), [K30(E-Laur)]hPP2–36 (17), [K30(E-Pam)]hPP2–36 (18a) and [K30(E-Ara)]hPP2–36 (19)
SPPS was carried out automatically up to hPP21–36 (9–13) or hPP31–36 (15a, 16a, 17–19) on a Rink amide AM (9–12, 15a, 16a, 17, 18a) or NovaSyn TGR R (Novabiochem) (13, 19) resin (15 μmol scale). Dde-l-Lys(Fmoc)-OH (40.0 mg, 0.075 μmol) was coupled to the peptide sequence with HOBt (11.5 mg, 0.075 μmol) and DIC (11.7 μL; 0.075 μmol) in 200 μL DMF for 3 h, followed by Fmoc deprotection with 10% 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU; Sigma–Aldrich; v/v) and 10% (v/v) piperidine in 1 mL DMF (2 and 10 min, respectively), and a 3 h manual coupling of Fmoc-l-Glu-OtBu (31.9 mg, 0.75 μmol) applying again HOBt (11.5 mg, 0.075 μmol) and DIC (11.7 μL, 0.075 μmol) in 200 μL DMF. Subsequent Fmoc removal with 10% (v/v) DBU and 10% (v/v) piperidine in 1 mL DMF (2 and 10 min, respectively) allowed acylation with fatty acids within 3 h. Propanoic acid (9, 15a; 5.6 μL, 0.075 μmol; Sigma–Aldrich), caprylic/octanoic acid (10, 16a; 11.9 μL, 0.075 μmol; Sigma–Aldrich) and lauric/dodecanoic acid (11, 17; 15 mg, 0.075 μmol; Sigma–Aldrich) were reacted with HOBt (11.5 mg, 0.075 μmol) and DIC (11.7 μL; 0.075 μmol) in 200 μL DMF, while palmitic/hexadecanoic (12, 18a; 19.2 mg, 0.075 μmol) and arachidic/eicosanoic acid (13, 19; 11.7 mg, 0.075 μmol; Sigma–Aldrich) were incubated with reactants in NMP. Dde removal was performed as described above, followed by automated peptide elongation to [K22(E-Lip)]hPP2–36 or [K30(E-Lip)]hPP2–36 respectively. Finally, lipidated compounds were cleaved from the resin with TFA (900 μL) and a scavenger mixture of EDT (70 μL) and TA (30 μL) within 3 h. Methionine reduction and subsequent dilution was performed as reported for 1a, 8 and 14a. For arachidoyl-modified compounds (13, 19), the scale was halved to 7.5 μmol effecting all described amounts of reagents. Purification was performed with a preparative Jupiter 10u Proteo RP-C18 HPLC column (90 Å, 7.78 μm, 250×21.2 mm; Phenomenex) using linear gradients of eluent B in A: 20% to 60% (v/v) in 40 min for 9, 10, 15a, 16a, 20% to 70% (v/v) in 40 min for 11, 12, 17, 18a and 30% to 80% (v/v) in 40 min for 13 and 19. Yield, purities, RP-HPLC and MALDI-TOF data are shown in Table 1.
TAMRA-[K30]hPP2–36 (14b)
After automated SPPS to full sequence on a Rink amide AM (14b) resin (7.5 μmol scale), the peptide was N-terminally modified with TAMRA (9.7 mg, 0.023 μmol; emp Biotech, Berlin, Germany) by reaction of O-(7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyluronium-hexafluorophosphate (HATU; 8.6 mg, 0.023 μmol; Novabiochem) and DIPEA (3.9 μL, 0.023 μmol) in 100 μL DMF for 3 h in the dark. The peptide was cleaved from the resin, reduced as described for CF-labeled compounds, dissolved in H2O/tBuOH (1:3, v/v) and lyophilized. Purification was performed with a preparative Jupiter 10u Proteo RP-C18 HPLC column (90 Å, 7.78 μm, 250×21.2 mm; Phenomenex) using a linear gradient of 20% to 60% (v/v) of eluent B in A over 40 min. Steps after incorporation of the fluorophore were performed in the dark. Yield, purities, RP-HPLC and MALDI-TOF characterization are listed in Table 3.
K30(TAMRA)]hPP2–36 (20)
The peptide was fully synthesized by robot-assisted peptide synthesis on a NovaSyn TGR R resin (15 μmol scale) including a 3 h manual coupling step of Fmoc-l-Lys(Dde)-OH (40 mg, 0.075 μmol) with HOBt (11.5 mg, 0.075 μmol) and DIC (11.7 μL; 0.075 μmol) in 200 μL DMF at position 30. The N-terminal l-Ala was Boc-protected allowing selective lysine side chain modification. After Dde removal as mentioned above, lysine was modified with TAMRA (19.4 mg, 0.045 μmol) applying HATU (17.1 mg, 0.045 μmol) and DIPEA (7.7 μL, 0.045 μmol) in 200 μL DMF for 3 h in the dark. Resin cleavage and methionine reduction was carried out as described for 1a. Finally, the peptide was dissolved in H2O/tBuOH (1:3, v/v) and lyophilized. Purification was performed with a preparative Jupiter 10u Proteo RP-C18 HPLC column (90 Å, 7.78 μm, 250×21.2 mm; Phenomenex) using linear gradients of 20% to 60% (v/v) of eluent B in A over 40 min. Steps after incorporation of the fluorophore were performed in the dark. Yield, purity, RP-HPLC and MALDI-TOF characterization are listed in Table 3.
TAMRA-[K30(E-Prop)]hPP2–36 (15b), TAMRA-[K30(E-Prop)]hPP2–36 (16b) and TAMRA-[K30(E-Pam)]hPP2–36 (18b)
SPPS was performed automatically up to hPP31–36 on a Rink amide AM resin (7.5 μmol scale). Dde-l-Lys(Fmoc)-OH (20.0 mg, 0.038 μmol) was coupled to the peptide using HOBt (5.8 mg, 0.038 μmol) and DIC (5.9 μL; 0.038 μmol) in 100 μL DMF for 3 h, followed by Fmoc removal with 10% (v/v) DBU and 10% (v/v) piperidine in DMF (1 mL; 2 and 10 min, respectively) as well as a 3 h manual coupling of Fmoc-l-Glu-OtBu (16.0 mg, 0.038 μmol) with the same HOBt/DIC-activation mixture in DMF (100 μL). Fmoc deprotection with 10% (v/v) DBU and 10% (v/v) piperidine enabled acylation with fatty acids within 3 h. Propanoic acid (15b; 2.8 μL, 0.038 μmol), caprylic acid (16b; 6.0 μL, 0.038 μmol) or palmitic acid (18b; 9.6 mg, 0.038 μmol) were reacted with HOBt (5.8 mg, 0.038 μmol) and DIC (5.9 μL; 0.038 μmol) in 100 μL DMF. Dde removal was performed as described above, followed by automated peptide elongation to full sequence. Lipidated compounds were N-terminally labeled with TAMRA (9.7 mg, 0.023 μmol) by reaction of HATU (8.6 mg, 0.023 μmol) and DIPEA (3.9 μL, 0.023 μmol) in 100 μL DMF for 3 h in the dark, cleaved from resin, reduced as described for CF-labeled compounds, dissolved in H2O/tBuOH (1:3, v/v) and lyophilized. Purification was performed with a preparative Jupiter 10u Proteo RP-C18 HPLC column (90 Å, 7.78 μm, 250×21.2 mm; Phenomenex) using a linear gradient of 20% to 60% (v/v) of eluent B in A over 40 min. Steps after incorporation of the fluorophore were performed in the dark. Yield, purities, RP-HPLC and MALDI-TOF characterization can be found in Table 3.
Radioligand binding studies
African green monkey kidney (COS-7) cells were cultured in Dulbecco's modified Eagle's medium (DMEM; PAA) with 4.5 gL−1 glucose and l-glutamine supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS; PAA), 100 units ml−1 penicillin (Invitrogen) and 100 μg mL−1 streptomycin (Invitrogen) in a humidified atmosphere at 37°C and 5% CO2. For transfection, cells were grown to 60–70% confluence in 25 cm[2] cell culture flasks and treated with 13 μg hY4R-CFP-N1 cDNA[40] and 13 μL Lipofectamine2000 (Invitrogen) in OptiMEM reduced serum medium (2.5 mL; Gibco) for 1 h. After aspirating DNA/lipid solutions, cells were maintained in normal culture medium (5 mL). 24 h after transient transfection, COS-7 cells were collected and resuspended in minimal essential medium (MEM; PAA) without l-glutamine, containing 1% (w/v) bovine serum albumin (BSA; PAA) and 5 mm pefabloc (Sigma–Aldrich). For the radioligand binding assay, 70000 of transiently transfected COS-7 cells (200 μL per tube) were treated for 90 min at RT with 1 nm [3H]-propionylated hPP ([3H]-hPP) (25 μL per tube) solution and either 1 % (v/v) BSA in H2O (for total binding), or 1 μm cold hPP or peptide analogue (25 μL per tube; for unspecific binding), respectively. [3H]-hPP with a specific activity of 95 Ci mmol−1 was obtained by selective radiolabeling as published.[41] Incubation was terminated by centrifugation (4°C, 3200 rpm, 5 min). Cell pellets were washed twice with ice-cold phosphate buffered saline (PBS; PAA) (400 μL per tube), resuspended in PBS (100 μL per tube) and finally mixed with 3 mL scintillation cocktail (PerkinElmer). Radioactivity was measured using a betacounter (Tri-Carb 2910 TR, PerkinElmer). In order to examine IC50 values (concentration of half-maximal inhibition) of selected analogues, dilution series were prepared in a range of 10−4–10−10 m and incubated and processed as described above. IC50 values were calculated by nonlinear regression assuming one-site competition using GraphPad Prism 5.0. Experiments were performed in triplicate and repeated three times. Statistical analysis was performed by one-way ANOVA, followed by Dunnett's post-hoc test and referred to total binding (*P ≤ 0.05, **P ≤ 0.01, *** P ≤ 0.001) using GraphPad Prism 5.0.
Inositol phosphate accumulation assays
For signal transduction assays, COS-7 cells stably expressing the respective hYR subtypes and a chimeric Gi/q protein (kindly provided by E. Kostenis, Universität Bonn) were generated as follows. COS-7 cells were co-transfected with linearized hY1/2/4/5-EYFP-pVitro2-hygro-mcs vector (2 μg) and linearized GΔ6qi4myr-pVitro2-neo-mcs[23] (2 μg) using 12 μL Metafectene (Biontex) transfection reagent according to the manufacturer's protocol. Three days post-transfection, selection was started using 1.5mgmL−1 G418-sulfate (amresco) and 146 μgmL−1 hygromycin B (Invivogen). Cell lines were raised from single colonies. Cultivation of stable COS-7-hYR-GαΔ6qi4myr cells was achieved in a humidified atmosphere at 37°C and 5% CO2. Cells were maintained in DMEM with 4.5 gL−1 glucose and l-glutamine supplemented with 10% (v/v) heat-inactivated FCS, 100 units ml−1 penicillin, 100 μg mL−1 streptomycin, 1.5 mgmL−1 G418-sulfate and 146 μgmL−1 hygromycin B. To determine ligand-induced IP accumulation, stably transfected COS-7 cells were seeded into 48-well plates (70000 cells in 500 μL per well) and grown for 24 h. Subsequently, cells were labeled with 2 μCimL−1 myo-[2-3H]-inositol (PerkinElmer) in culture medium (150 μL per well) without penicillin and streptomycin for at least 16 h, washed (250 μL per well) and stimulated for 1 h at 37°C (150 μL per well) with peptides at concentrations ranging from 10−4 to 10−12 m (depending on expected potency) in FCS-free DMEM with 4.5 gL−1 glucose and l-glutamine containing 10 mm LiCl (Sigma–Aldrich). Next, cells were lysed with NaOH (0.1 n, 100 μL per well; Grossing, Filsum, Germany) for 5 min, neutralized by addition of formic acid (0.13 m, 50 μL per well; Grossing) and finally diluted in sodium borate (5 mm; Merck)/ethylenediaminetetraacetic acid (EDTA; 0.5 mm; Applichem) buffer (750 μL per well). Cell debris was removed, and samples were loaded on a fresh or regenerated anion exchange resin (AG 1-X8 formate, BIO-RAD; 500 mg per column). After column washing with glycerophosphate elution buffer (5 mm sodium borate, 60 mm sodium formate (Sigma–Aldrich); 5 mL per column) and H2O (5 mL per column), radioactive phosphoinositides were eluted with 1 mm ammonium formate (Paul Lohmann) buffer containing 0.1 m formic acid (1.25 mL per well), mixed with 3 mL scintillation cocktail and measured using a betacounter (Tri-Carb 2910 TR). Decay-per-minute values at the highest used analogue concentration were normalized to the mean value of the respective wild type at full activity concentration. Collected normalized data were analyzed by nonlinear regression of all datasets using GraphPad Prism 5.0 revealing global mean EC50 and pEC50 values as well as mean efficacies given with ±SEM. All compounds were investigated in duplicate by at least two independent experiments.
Circular dichroism (CD) spectroscopy
CD spectra of 10 μm peptide solutions were recorded on a Jasco-715 spectropolarimeter with a constant nitrogen stream at 22 °C. 10 mm sodium phosphate (Sigma–Aldrich) buffer supplemented with or without 20 mm SDS (Carl Roth) served as buffer solutions (pH 7.0). Measurements in the far UV region between 190 nm and 250 nm were conducted with a cuvette of 2 mm path length and the following parameters: 100 mdeg sensitivity, 0.5 nm data pitch, continuous scanning mode, 50 nm min−1 scanning speed, 4 s response, 2 nm bandwidth and 6 accumulations. Compound concentration was calculated from aromatic absorption of the examined peptide aliquots in aqueous solution using the molar extinction coefficient at 280 nm (ε = 5960 m−1cm−1). Obtained CD spectra were baseline corrected in order to subtract buffer effects and converted into mean residue molar ellipticity [Θ] given in deg cm2 dmol−1 by the equation: [Θ] = (Θ×M)/(10×c×l×n). Θ corresponds to the ellipticity in mdeg, M is the compound molar mass in gmol−1, c is the concentration in mgmL−1, l is the path length of the cuvette in cm and n is the number of peptide bond residues. All experiments were performed twice independently. α-Helical contents were calculated by Dichroweb applying K2D estimation.[22] Statistical significance was determined by one way-ANOVA, followed by Dunnett's post-hoc test and referred to respective [K]hPP2–36 lead compounds (**P ≤ 0.01, ***P ≤ 0.001) using GraphPad Prism 5.0.
Stability tests in human blood plasma
In order to follow enzymatic degradation of TAMRA-modified analogues, the peptides (15 nmol) were freshly reduced by dilution in 100 μL TFA and treatment with 3 μL TMSBr and 3 μL EDT for 30 min. After precipitation from 1 mL ice-cold Et2O for 20 min, they were washed and re-suspended further three times with Et2O and dried in vacuo. Then, peptide aliquots were diluted to 10 μm in human blood plasma and incubated at 37°C with mechanical shaking at 500 rpm. Individual samples (150 μL) were taken after 0, 24, 48, 72, 96, 120 and 144 h, and precipitation of proteins was performed with 150 μL CH3CN/EtOH (1:1, v/v; Applichem) at −20°C for 3 h. After centrifugation (RT, 14000 rpm, 30 s), supernatants were processed in Costar Spin-X tubes (0.22 μm membrane pore size) for HPLC analysis. A Varian-VariTide RPC column (200 Å, 6 μm, 250×4.6 mm) with fluorescence detection (λex: 525 nm, λem: 572 nm) was used with a linear gradient of 5% to 60% (v/v) of eluent B in A over 45 min. Quantity of intact peptide-specific fluorescence was calculated as proportion of control at 0 h (100%). The first data points revealed an exponential decay that allowed determination of half-lives (t1/2) according to an enzymatic degradation of first order. For each compound, every single time point was analyzed independently at least twice and is presented as the mean ±SEM. Statistical significance of t1/2 values was determined by one-way ANOVA, followed by Dunnett's post-hoc test with comparison to control peptide 14b (**P ≤ 0.01, ***P ≤ 0.001) using GraphPad Prism 5.0.
Metabolic stability in porcine liver homogenates
Porcine liver (50 g) was hackled and homogenized in PBS (200 mL) and centrifuged (4°C, 5000 rpm, 30 min). The supernatant was aliquoted and frozen at −70°C. Degradation of fluorescently labeled peptides (10 μm final concentration) that were dissolved in 15 μL H2O/tBuOH (1:3, v/v) was initiated by addition of 50 mgmL−1 homogenized liver solution in PBS (1.5 mL final volume). Reaction tubes were kept at 37°C and mechanical shaking at 500 rpm. Samples (150 μL) were taken after 0, 2, 4, 6, 8, 12, 24, 36 and 48 h, processed and analyzed as described for the human blood plasma assay. For each compound, every single time point was analyzed independently at least twice and is shown as the mean ±SEM. Statistical analysis of t1/2 values was calculated by one-way ANOVA, followed by Dunnett's post-hoc test and referred to the parent peptide 14b (**P ≤ 0.01) using GraphPad Prism 5.0.
Acknowledgments
The authors thank Jan-Patrick Fischer and Andrea Kindermann for experimental assistance. Regina Reppich-Sacher, Kristin Löbner and Christina Dammann (Universität Leipzig) are kindly acknowledged for their excellent help in mass spectrometry, cell culture and peptide synthesis. The authors gratefully thank for financial support from the European Union, “Gastro-intestinal peptides in obesity” (GIPIO), FP7 (No. 223057), the US National Institutes of Health (NIH) (GM077561 and DK097376), the Deutsche Forschungsgemeinschaft (DFG) (BE1264/16) and the Leipzig Graduate School BuildMoNa. Helpful discussions with all GIPIO collaborators are kindly acknowledged.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201402235
References
- 1.Adrian TE, Greenberg GR, Besterman HS, Bloom SR. Gut. 1978;19:907–909. doi: 10.1136/gut.19.10.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Murphy KG, Bloom SR. Nature. 2006;444:854–859. doi: 10.1038/nature05484. [DOI] [PubMed] [Google Scholar]; b) Simpson K, Parker J, Plumer J, Bloom S. In: Handb Exp Pharmacol. Joost HG, editor. Vol. 209. Springer; Heidelberg: 2012. pp. 209–230. [DOI] [PubMed] [Google Scholar]
- 3.Bard JA, Walker MW, Branchek TA, Weinshank RL. J Biol Chem. 1995;270:26762–26765. doi: 10.1074/jbc.270.45.26762. [DOI] [PubMed] [Google Scholar]
- 4.Asakawa A, Inui A, Ueno N, Fujimiya M, Fujino MA, Kasuga M. Peptides. 1999;20:1445–1448. doi: 10.1016/s0196-9781(99)00155-2. [DOI] [PubMed] [Google Scholar]
- 5.a) Asakawa A, Inui A, Yuzuriha H, Ueno N, Katsuura G, Fujimiya M, Fujino MA, Niijima A, Meguid MM, Kasuga M. Gastroenterology. 2003;124:1325–1336. doi: 10.1016/s0016-5085(03)00216-6. [DOI] [PubMed] [Google Scholar]; b) Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M, Frost GS, Ghatei MA, Bloom SR. J Clin Endocrinol Metab. 2003;88:3989–3992. doi: 10.1210/jc.2003-030630. [DOI] [PubMed] [Google Scholar]
- 6.Berntson GG, Zipf WB, O'Dorisio TM, Hoffman JA, Chance RE. Peptides. 1993;14:497–503. doi: 10.1016/0196-9781(93)90138-7. [DOI] [PubMed] [Google Scholar]
- 7.Pedragosa-Badia X, Stichel J, Beck-Sickinger AG. Front Endocrinol. 2013;4:5. doi: 10.3389/fendo.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenwood HC, Bloom SR, Murphy KG. Rev Diabet Stud. 2011;8:355–368. doi: 10.1900/RDS.2011.8.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yulyaningsih E, Zhang L, Herzog H, Sainsbury A. Br J Pharmacol. 2011;163:1170–1202. doi: 10.1111/j.1476-5381.2011.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lean MEJ. Proc Nutr Soc. 2000;59:331–336. doi: 10.1017/s0029665100000379. [DOI] [PubMed] [Google Scholar]
- 11.a) Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Discovery Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]; b) Mäde V, Els-Heindl S, Beck-Sickinger AG. Beilstein J Org Chem. 2014;10:1197–1212. doi: 10.3762/bjoc.10.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu JH, Kim MS. Diabetes Metab J. 2012;36:391–398. doi: 10.4093/dmj.2012.36.6.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahrens VM, Bellmann-Sickert K, Beck-Sickinger AG. Future Med Chem. 2012;4:1567–1586. doi: 10.4155/fmc.12.76. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Bulaj G. Curr Med Chem. 2012;19:1602–1618. doi: 10.2174/092986712799945003. [DOI] [PubMed] [Google Scholar]
- 15.Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Larsen UD, Ribel U, Markussen J. Biochem J. 1995;312:725–731. doi: 10.1042/bj3120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Dasgupta P, Singh A, Mukherjee R. Biol Pharm Bull. 2002;25:29–36. doi: 10.1248/bpb.25.29. [DOI] [PubMed] [Google Scholar]; b) Wang J, Wu D, Shen WC. Pharm Res. 2002;19:609–614. doi: 10.1023/a:1015397811161. [DOI] [PubMed] [Google Scholar]; c) Todorovic A, Holder JR, Bauzo RM, Scott JW, Kavanagh R, Abdel-Malek Z, Haskell-Luevano C. J Med Chem. 2005;48:3328–3336. doi: 10.1021/jm0490843. [DOI] [PubMed] [Google Scholar]; d) Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thøgersen H, Wilken M, Johansen NL. J Med Chem. 2007;50:6126–6132. doi: 10.1021/jm070861j. [DOI] [PubMed] [Google Scholar]; e) Zhang L, Robertson CR, Green BR, Pruess TH, White HS, Bulaj G. J Med Chem. 2009;52:1310–1316. doi: 10.1021/jm801397w. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Agnihotri G, Crall BM, Lewis TC, Day TP, Balakrishna R, Warshakoon HJ, Malladi SS, David SA. J Med Chem. 2011;54:8148–8160. doi: 10.1021/jm201071e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedragosa-Badia X, Sliwoski GR, Dong Nguyen E, Lindner D, Stichel J, Kaufmann KW, Meiler J, Beck-Sickinger AG. J Biol Chem. 2014;289:5846–5859. doi: 10.1074/jbc.M113.502021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gehlert DR, Schober DA, Beavers L, Gadski R, Hoffman JA, Smiley DL, Chance RE, Lundell I, Larhammar D. Mol Pharmacol. 1996;50:112–118. b) T. Schwartz, US2008/0269114A1, 30.10.2008.
- 19.Merrifield RB. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]
- 20.a) Bellmann-Sickert K, Elling CE, Madsen AN, Little PB, Lundgren K, Gerlach LO, Bergmann R, Holst B, Schwartz TW, Beck-Sickinger AG. J Med Chem. 2011;54:2658–2667. doi: 10.1021/jm101357e. [DOI] [PubMed] [Google Scholar]; b) Mäde V, Babilon S, Jolly N, Wanka L, Bellmann-Sickert K, Diaz Gimenez LE, Mörl K, Cox HM, Gurevich VV, Beck-Sickinger AG. Angew Chem Int Ed. 2014 doi: 10.1002/anie.201403750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Schwartz TW, Fuhlendorff J, Kjems LL, Kristensen MS, Vervelde M, O'Hare M, Krstenansky JL, Bjørnholm B. Ann N Y Acad Sci. 1990;611:35–47. doi: 10.1111/j.1749-6632.1990.tb48920.x. [DOI] [PubMed] [Google Scholar]; b) Lerch M, Gafner V, Bader R, Christen B, Folkers G, Zerbe O. J Mol Biol. 2002;322:1117–1133. doi: 10.1016/s0022-2836(02)00889-6. [DOI] [PubMed] [Google Scholar]
- 22.http://dichroweb.cryst.bbk.ac.uk/html/home.shtml (last accessed: May 2014).
- 23.Kostenis E, Degtyarev MY, Conklin BR, Wess J. J Biol Chem. 1997;272:19107–19110. doi: 10.1074/jbc.272.31.19107. [DOI] [PubMed] [Google Scholar]
- 24.Greenfield NJ. Nat Protoc. 2007;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz T. 7Tm Pharma A/S; Hoersholm, Denmark: US Pat Appl US20080261871A1. Y2/Y4 Selective Receptor Agonists for Therapeutic Interventions. 2008 Oct;
- 26.Koulu M, Movafagh S, Tuohimaa J, Jaakkola U, Kallio J, Pesonen U, Geng Y, Karvonen MK, Vainio-Jylhä E, Pöllönen M, Kaipio-Salmi K, Seppälä H, Lee EW, Higgins RD, Zukowska Z. Ann Med. 2004;36:232–240. doi: 10.1080/07853890410031236. [DOI] [PubMed] [Google Scholar]
- 27.Lu C, Everhart L, Tilan J, Kuo L, Sun CCJ, Munivenkatappa RB, Jonsson-Rylander AC, Sun J, Kuan-Celarier A, Li L, Abe K, Zukowska Z, Toretsky JA, Kitlinska J. Oncogene. 2010;29:5630–5642. doi: 10.1038/onc.2010.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Degen L, Oesch S, Casanova M, Graf S, Ketterer S, Drewe J, Beglinger C. Gastroenterology. 2005;129:1430–1436. doi: 10.1053/j.gastro.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Bjørnholm B, Jørgensen FS, Schwartz TW. Biochemistry. 1993;32:2954–2959. doi: 10.1021/bi00063a005. [DOI] [PubMed] [Google Scholar]
- 30.a) Zwanziger D, Khan IU, Neundorf I, Sieger S, Lehmann L, Friebe M, Dinkelborg L, Beck-Sickinger AG. Bioconjugate Chem. 2008;19:1430–1438. doi: 10.1021/bc7004297. [DOI] [PubMed] [Google Scholar]; b) Ahrens VM, Frank R, Stadlbauer S, Beck-Sickinger AG, Hey-Hawkins E. J Med Chem. 2011;54:2368–2377. doi: 10.1021/jm101514m. [DOI] [PubMed] [Google Scholar]
- 31.Merten N, Lindner D, Rabe N, Rompler H, Mörl K, Schöneberg T, Beck-Sickinger AG. J Biol Chem. 2007;282:7543–7551. doi: 10.1074/jbc.M608902200. [DOI] [PubMed] [Google Scholar]
- 32.Walker MW, Smith KE, Bard J, Vaysse PJ, Gerald C, Daouti S, Weinshank RL, Branchek TA. Peptides. 1997;18:609–612. doi: 10.1016/s0196-9781(97)00070-3. [DOI] [PubMed] [Google Scholar]
- 33.Haack M, Enck S, Seger H, Geyer A, Beck-Sickinger AG. J Am Chem Soc. 2008;130:8326–8336. doi: 10.1021/ja8004495. [DOI] [PubMed] [Google Scholar]
- 34.Hofmann S, Frank R, Hey-Hawkins E, Beck-Sickinger AG, Schmidt P. Neuropeptides. 2013;47:59–66. doi: 10.1016/j.npep.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Poschner BC, Langosch D. Protein Sci. 2009;18:1801–1805. doi: 10.1002/pro.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.a) Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thøgersen H, Wilken M, Agersø H. J Med Chem. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]; b) Ward BP, Ottaway NL, Perez-Tilve D, Ma D, Gelfanov VM, Tschöp MH, DiMarchi RD. Mol Metab. 2013;2:468–479. doi: 10.1016/j.molmet.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashbrook JD, Spector AA, Santos EC, Fletcher JE. J Biol Chem. 1975;250:2333–2338. [PubMed] [Google Scholar]
- 38.Spector AA. J Lipid Res. 1975;16:165–179. [PubMed] [Google Scholar]
- 39.Zou C, Kumaran S, Markovic S, Walser R, Zerbe O. ChemBioChem. 2008;9:2276–2284. doi: 10.1002/cbic.200800221. [DOI] [PubMed] [Google Scholar]
- 40.Böhme I, Stichel J, Walther C, Mörl K, Beck-Sickinger AG. Cell Signalling. 2008;20:1740–1749. doi: 10.1016/j.cellsig.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 41.Koglin N, Lang M, Rennert R, Beck-Sickinger AG. J Med Chem. 2003;46:4369–4372. doi: 10.1021/jm0341251. [DOI] [PubMed] [Google Scholar]