

Abstract
Targeted next‐generation‐sequencing (NGS) panels have largely replaced Sanger sequencing in clinical diagnostics. They allow for the detection of copy‐number variations (CNVs) in addition to single‐nucleotide variants and small insertions/deletions. However, existing computational CNV detection methods have shortcomings regarding accuracy, quality control (QC), incidental findings, and user‐friendliness. We developed panelcn.MOPS, a novel pipeline for detecting CNVs in targeted NGS panel data. Using data from 180 samples, we compared panelcn.MOPS with five state‐of‐the‐art methods. With panelcn.MOPS leading the field, most methods achieved comparably high accuracy. panelcn.MOPS reliably detected CNVs ranging in size from part of a region of interest (ROI), to whole genes, which may comprise all ROIs investigated in a given sample. The latter is enabled by analyzing reads from all ROIs of the panel, but presenting results exclusively for user‐selected genes, thus avoiding incidental findings. Additionally, panelcn.MOPS offers QC criteria not only for samples, but also for individual ROIs within a sample, which increases the confidence in called CNVs. panelcn.MOPS is freely available both as R package and standalone software with graphical user interface that is easy to use for clinical geneticists without any programming experience. panelcn.MOPS combines high sensitivity and specificity with user‐friendliness rendering it highly suitable for routine clinical diagnostics.
Keywords: clinical diagnostics, copy‐number variation, deletion, duplication, panel sequencing, targeted next‐generation sequencing
1. INTRODUCTION
For many disease genes, comprehensive mutation analysis in clinical diagnostics includes the detection of (1) small sequence alterations, such as substitutions, deletions, duplications, and insertions of one or a few nucleotides, and (2) copy‐number variations (CNVs), which are often defined as deletions or duplications larger than 50 bp (Alkan, Coe, & Eichler, 2011). CNVs may affect one or more genes of interest where either the whole gene or only one or more exons thereof are altered. Previously, Sanger sequencing was used for routine detection of small sequence alterations. If no disease‐causing mutation was found by these means, multiplex ligation‐dependent probe amplification (MLPA) (Schouten et al., 2002), quantitative PCR (Charbonnier et al., 2000), or array‐based techniques (Komura et al., 2006) were applied in a second step to identify (intragenic) CNVs. Targeted next‐generation‐sequencing (NGS) panels have now replaced Sanger sequencing in many diagnostic laboratories. They provide several advantages over whole‐genome sequencing (WGS) and whole‐exome sequencing (WES): since only a limited number of genes is targeted, data can be generated and stored on smaller machines, which makes NGS panels more flexible as well as time‐ and cost‐effective. Furthermore, higher accuracy is achieved due to deeper coverage (de Leeneer et al., 2015; Lin et al., 2012; Meder et al., 2011; Weiss et al., 2013).
Enrichment‐based targeted NGS panels in particular allow not only for the detection of small sequence alterations, but also of CNVs. However, as all currently available computational methods for detecting CNVs in targeted NGS panel data have shortcomings, clinical copy‐number (CN) analysis continues to be largely based on MLPA or other techniques. Current CNV detection methods suffer from one or more of the following problems:
-
1.
Compromise between sensitivity and specificity: Although the sensitivity of CNV detection methods is improving, this is often achieved at the expense of a high number of false‐positive findings (Pugh et al., 2016).
-
2.
Poor quality control (QC): Variations of the DNA quality, the library preparation, or the sequencing process itself can lead to differences in read count (RC) characteristics between samples. Therefore, the RCs of the affected samples cannot be compared. Further, the coverage of regions of interest (ROIs) can be low due to high GC content or sequence characteristics that affect either enrichment or mappability of reads. RCs and gene assignment can also be highly variable in ROIs with a highly homologous pseudogene. These issues influence the read depth and prevent reliable CN detection in these ROIs. QC that filters out low‐quality samples and ROIs is therefore an essential step, especially in clinical diagnostics (Johansson et al., 2016). However, if too many samples or ROIs are excluded, a method may cease to be useful.
-
3.
Low sensitivity in the detection of small or large CNVs: Methods perform well at detecting either small CNVs that affect only (part of) one ROI or large CNVs that encompass all ROIs of a gene. However, in a clinical setting, both is required.
-
4.
Risk of incidental findings: Since many gene panels are designed to cover several different disease types at once, incidental findings become problematic in clinical interpretation and reporting. Current CNV detection methods do not provide a satisfactory solution that limits the analysis report to genes of interest without diminishing the ability to detect large CNVs that cover all ROIs of the one gene to be analyzed.
-
5.
Low user‐friendliness: While commercial products usually provide graphical user interfaces to facilitate analysis, most noncommercial methods require at least some basic programming experience, which often discourages clinical laboratories from using them.
With the aim to address all these shortcomings, we developed panelcn.MOPS, a pipeline for detecting CNVs in targeted NGS panel data that builds upon cn.MOPS (Copy Number estimation by a Mixture Of Poissons) (Klambauer et al., 2012) (see Fig. 1). The superior performance of cn.MOPS on WGS and WES data has already been demonstrated (Guo et al., 2013; Klambauer et al., 2012). Specifically, cn.MOPS has been used to detect CNVs in the Taiwanese Han and Qatari populations (Fakhro et al., 2015; Lin, Tseng, Jeng, & Sun, 2014), in individuals with intellectual disability (Schuurs‐Hoeijmakers et al., 2013), and in patients with early‐onset neuropsychiatric disorders (Brand et al., 2014). cn.MOPS builds a local model that captures the read characteristics of each ROI. Thus, problems such as the bias induced by the targeting procedure are circumvented making it the prime candidate for CNV detection in targeted NGS panel data.
Figure 1.

panelcn.MOPS analysis pipeline. The input to panelcn.MOPS is a BAM file for every sample and the corresponding BED file. The final output is a table of results and boxplots of the normalized RCs for user‐selected genes of interest
In this study, we present the newly developed panelcn.MOPS pipeline and compare it against five state‐of‐the‐art CNV detection methods: ExomeDepth (Plagnol et al., 2012), CoNVaDING (Johansson et al., 2016), VisCap (Pugh et al., 2016), NextGENe (Softgenetics, State College, PA), and SeqNext (JSI Medical Systems GmbH, Kippenheim, Germany).
2. MATERIALS AND METHODS
2.1. panelcn.MOPS pipeline
2.1.1. Input
The panelcn.MOPS pipeline uses BAM files as input. A BED file is needed to specify the ROIs that constitute the count windows. Alternatively, a matrix of RCs can be used directly as input to the panelcn.mops algorithm. For our analyses, each ROI consisted of a coding exon ±31 nucleotides of flanking intronic sequences leading to ROIs with a size between 67 and 6,636 bp. Although it was not used for the reported results, larger exons can be split into multiple overlapping ROIs in order to increase the resolution of the CNV detection algorithm.
2.1.2. Read counting
We adapted the read counting procedure of the R package exomeCopy (Love et al., 2011) for NGS panel data: all reads that overlap with the current ROI are counted with paired reads being handled separately. A fixed read length of 150 bp was assumed throughout the analysis because in tests on the validation set this achieved more robust results than the true read length for each read. However, the read length is a parameter that needs to be set by the user according to the specifications of the data used.
2.1.3. QC
panelcn.MOPS includes several QCs: ROIs are excluded if their median RC across all samples does not exceed a user‐defined threshold (default: 30). Additionally, after control sample selection and normalization, ROIs are marked as low quality if their RCs show a high variation across all remaining samples (test sample and selected controls). There are also two QC criteria for samples, failure in which leads to the exclusion of control samples and to a warning if the failing sample is the test sample. Samples with a low total number of RCs are generally of lower quality. Therefore, samples with a median RC across all ROIs that is lower than 0.55 times the median of all samples fail the first step of the sample QC. The second step is performed after control sample selection and normalization. For each ROI, the ratio between the normalized RCs of each sample and the median across all remaining samples (test sample and selected controls) is calculated. Samples that show a high variation in these RC ratios fail the second QC step.
2.1.4. Control sample selection
In order to limit the variance of the RC characteristics of the control samples, we use only control samples with a high correlation of RCs to the RCs of the test sample. ROIs that are part of a gene of interest for a specific test sample are excluded from the correlation calculation. If fewer than eight control samples exceed the correlation threshold of 0.99, the eight samples with the highest correlation are used. If more than 25 samples exceed the threshold, only the best 25 are kept. This lowers the variance further and additionally reduces computation time. We recommend to use at least eight high quality samples as controls samples; however, if more control samples are used, the results typically become more robust.
2.1.5. Normalization
Each sample's RCs are scaled such that the total number of reads are comparable across samples. The best performance was achieved by normalizing to the third quartile, which is equivalent to the upper quartile normalization (Bullard, Purdom, Hansen, & Dudoit, 2010), and rescaling with the first quartile of the scaling factors.
2.1.6. CN detection
The raw CNV detection algorithm of the cn.mops R package is applied to each ROI separately. In order to increase sensitivity, we adapted the expected fold change of the CN classes. According to our results on the validation set (see section “TSC panel validation set”), we set the parameter to 0.57 for CN1 (deletion) and to 1.46 for CN3 (duplication).
2.1.7. Segmentation
We tried various segmentation algorithms. However, for the pipeline to be able to detect CNVs that affect only a single ROI or even just part of a ROI, omitting the segmentation step leads to better results.
2.1.8. Filtering for genes of interest
In clinical settings, the analysis of only a subset of genes covered by the panel is frequently requested. Therefore, any results concerning the remaining genes should be masked to avoid incidental findings. Accordingly, we implemented a function that presents results only for user‐specified genes while using the RCs of all genes to compute the CNs, which offers several advantages (see section “Whole‐gene CNVs”). If all genes of the panel are of interest, no filtering is performed and results for all genes are displayed.
2.1.9. Output
In addition to a table of results, panelcn.MOPS creates boxplots of the normalized RCs of each ROI of a user‐specified gene for visual inspection (see Fig. 2).
Figure 2.
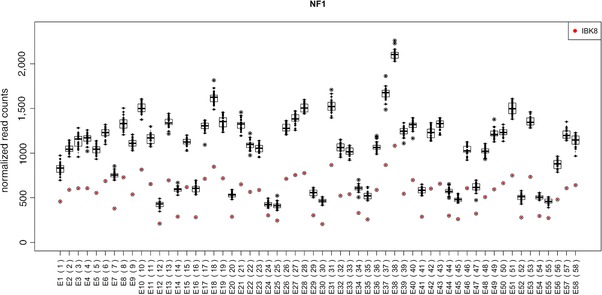
Boxplot of normalized RCs. The normalized RCs of the test sample and all controls used are displayed as boxplots for each ROI (exons numbered in consecutive order according to BED file) of the NF1 gene. The RCs of each control sample are symbolized by black dots, whereas the RCs of the test sample are highlighted by red dots. The deletion of all ROIs is clearly visible by the red dots that are distinctly below the boxes and whiskers
2.1.10. Graphical user interface
We do not only offer panelcn.MOPS as an R package, but also provide a simple GUI with an easy‐to‐use installer. As the implementation is based on a combination of R shiny (Chang, Cheng, Allaire, Xie, & McPherson, 2016) and R portable (Redd & Huber, 2010), it does not require any additional programs to be installed by the user.
2.2. Samples
This study was approved by the ethics review board of the Medical University Innsbruck (MUI). Written informed consent to analyze the genes of interest was obtained from all participants.
Our cohort consisted of 180 samples that were analyzed with an NGS panel in the Division of Human Genetics of the MUI. All CNVs (17 single‐exon deletions or duplications, 22 multiexon deletions or duplications, five deletions of an entire gene, and five deletions or duplications spanning only part of a ROI) have previously been assessed with MLPA and, therefore, can be considered as confirmed (see Supp. Tables S1 and S2 for more details).
One hundred seventy samples were processed using the TruSightTM Cancer (TSC) panel (Illumina, San Diego, CA), which targets 94 genes associated with a predisposition towards cancer. One hundred fifty of these samples were diagnostic cases previously analyzed at MUI. The remaining 20 samples were provided by the Medical Genomics Laboratory of the University of Alabama at Birmingham (UAB) and were enriched for NF1 deletions/duplications. All TSC panel samples were divided into a validation (n = 25, TSC panel validation set) and test set (n = 145, TSC panel test set). The validation set was used to optimize the parameter settings for the different methods. The test set was used to fully evaluate the methods in terms of sensitivity, specificity, and no‐call‐rate as described below. A “blind” analysis was performed on the test set: the true CNs were made available only after the analyses with all programs were completed. Four samples of the test set were evaluated separately because they had CNVs smaller than one ROI (small CNVs dataset). Three samples were excluded from further analysis because they contained de novo ALU insertions (Wimmer, Callens, Wernstedt, & Messiaen, 2011). Nevertheless, all seven samples were used as controls for the rest of the samples.
In order to extend our study to a different NGS panel, we created a set of 10 additional cases processed on a customized panel (Nextera Rapid Capture; Illumina) targeting 117 genes in the following referred to as Custom Panel.
2.3. Library preparation and sequencing
Adapter‐tagged libraries were prepared according to the TruSightTM Rapid Capture workflow (Illumina) using a transposase‐based method (Nextera) (Marine et al., 2011). Sequencing was performed on a MiSeq instrument following the Illumina 300 sequencing cycle program with paired‐end reads. For more details, see Supp. Methods.
2.4. Sequence alignment
Only the commercial tools NextGENe and SeqNext use FASTQ files directly as input. For methods, which require BAM files as input, sequence alignment to human reference genome build b37 with BWA version 0.7.12 (Li & Durbin, 2009) was followed by removal of duplicates and fixing of mate information with SAMtools version 0.1.19 (Li et al., 2009). Afterwards, local realignment around indels and base quality score recalibration was performed with the Genome Analysis Tool Kit (GATK) version 3.5 (McKenna et al., 2010). Supp. Table S3 shows the total number of raw and mapped reads for each sample. For VisCap, DepthOfCoverage files were also generated with GATK version 3.5. The BED files used throughout the analysis comprised all coding exons ±31 nucleotides of flanking intronic sequences.
We performed additional tests with changes in the alignment process to check how panelcn.MOPS depends on these settings. Omitting the postprocessing steps of GATK did not affect the RCs of the genes of interest and therefore panelcn.MOPS reported exactly the same CNs. Using BWA‐MEM (Li & Durbin, 2009) for alignment showed that the RCs differ only slightly. Also, with BAM files produced by NextGENe, the performance of panelcn.MOPS did not change as long as all samples were processed the same way. This indicates that the choice of alignment algorithm does not have a large influence on the results. For more details, see Supp. Methods.
2.5. Controls
Except for NextGENe, which can handle only 10 control samples, the same set of controls was used, which consisted of all samples from the validation and test sets that did not have a clinical indication that matched or overlapped that of the test sample. This ensured that the control samples were devoid of CNVs in the analyzed genes of interest that would interfere with CNV detection in the test sample (see section “Controls”). For NextGENe, samples from the same sequencing run—or at least recent runs—were used, ideally from a different indication. For the custom panel, all control samples used for NextGENe also formed the pool of control samples for the other methods. Again, only samples with a different clinical indication were used.
2.6. Evaluation criteria
In each sample, each ROI was classified as positive if it was affected by a CNV, and as negative otherwise. ROIs marked as low quality by a method were considered as no‐call for that method and removed prior to sensitivity and specificity calculation. For a low‐quality sample, all ROIs of the genes of interest were classified as no‐call. A ROI was classified as true positive (TP) if it contained a CNV that was detected by the method and not classified as no‐call. ROIs with no known CNV that were detected as normal and without no‐call were called true negatives (TNs). Falsely detected CNVs and missed CNV calls were classified as false positive (FP) and false negative (FN), respectively, if they did not contain a no‐call. Finally, sensitivity was calculated as #TP/(#TP+#FN) and specificity as #TN/(#TN+#FP). The no‐call rate was defined as the number of ROIs with no‐call divided by the total number of ROIs.
2.7. Methods compared
We compared panelcn.MOPS against the following five state‐of‐the art methods for CNV detection in NGS panel data: ExomeDepth version 1.1.8, CoNVaDING version 1.1.6, VisCap, NextGENe version 2.4.1.1, and SeqNext version 4.3.0. Details about the methods, including the parameters that were optimized on the validation set, can be found in Supp. Methods.
2.8. Restriction to genes of interest
To avoid incidental findings, we automatically filtered the results for ROIs of genes of interest. This was done through computational scripts that processed the result files of the different methods and only made results for genes of interest available. Since the commercial tools NextGENe and SeqNext do not support the filtering of results before they are displayed, we restricted the whole analysis with these tools to the genes of interest.
2.9. Availability of code and materials
panelcn.MOPS is freely available as an R package at https://github.com/bioinf-jku/panelcn.mops. The standalone software with GUI and Windows installer can be obtained from http://www.bioinf.jku.at/software/panelcnmops/.
Sequence data have been deposited at the European Genome‐phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the EBI, under ENA accession number PRJEB18961.
3. RESULTS
3.1. TSC panel validation set
The parameter settings of all methods were optimized on the 25 samples of the validation set. The aim was to find an optimal trade‐off between highest sensitivity and a low number of FPs as well as good QC settings. One sample (IBK42) was labeled as low quality by all methods. Other samples were marked as low quality by only one or two methods (see Supp. Table S1). For panelcn.MOPS and CoNVaDING, we were able to adjust the parameters such that both methods achieved 100% sensitivity and specificity. panelcn.MOPS reported only IBK42 as low quality, whereas CoNVaDING excluded one‐fifth of the samples. ExomeDepth reported one false positive deletion in exon 15 of the PMS2 gene (IBK47) and no additional low‐quality samples. Even with optimized parameters, VisCap performed relatively poorly on the validation set, since it missed parts of multiexon deletions and duplications in several samples and marked three of 25 samples as low quality. panelcn.MOPS, ExomeDepth, CoNVaDING, and VisCap could be optimized to detect an 80‐bp deletion that affected only part of a ROI (IBK9).
An inherent weakness of the tested version of NextGENe and SeqNext is the failure to report deletions of all ROIs of a single gene of interest, for example, the deletion of the whole NF1 gene in three samples where NF1 was the only gene analyzed. Furthermore, neither CNV detection program was able to detect the above‐mentioned small deletion (IBK9). Additionally, we were unable to define parameters for NextGENe that avoided one false positive duplication (IBK25) without creating FNs, and hence losing sensitivity.
3.2. TSC panel test set
Using the best parameters as determined on the validation set (see Supp. Methods), we evaluated the performance of all six methods on a test set of 138 samples as described in the section “Evaluation criteria.” Table 1 shows the sensitivities, specificities, and no‐call rates of all methods. panelcn.MOPS achieved 100% sensitivity and specificity while the no‐call rate was 0.04. This means that 4% of ROIs were marked as low quality because either the whole sample or the ROI was of low quality. Both CoNVaDING and SeqNext missed only a single positive ROI with CoNVaDING additionally reporting one FP (see Supp. Table S1). Both had no‐call rates similar to that of panelcn.MOPS. As ExomeDepth lacks a QC criterion for ROIs, it did not flag any ROI as low quality. Consequently, ExomeDepth had a high number of FPs, and thus low specificity. Also, NextGENe achieved a low no‐call rate at the expense of low specificity. VisCap exhibited low specificity, although it had the highest no‐call rate.
Table 1.
Sensitivity, specificity, and no‐call rate for each method analyzed
| panelcn.MOPS | ExomeDepth | CoNVaDING | VisCap | NextGENe | SeqNext | Optimal | |
|---|---|---|---|---|---|---|---|
| TP | 91 | 90 | 90 | 91 | 91 | 90 | 91 |
| TN | 7,889 | 8,203 | 7,705 | 7,279 | 8,141 | 7,954 | 8,222 |
| FP | 0 | 19 | 1 | 12 | 19 | 0 | 0 |
| FN | 0 | 1 | 1 | 0 | 0 | 1 | 0 |
| No‐call | 333 | 0 | 516 | 931 | 62 | 268 | 0 |
| Total | 8,313 | 8,313 | 8,313 | 8,313 | 8,313 | 8,313 | 8,313 |
| Sensitivity | 1.0000 | 0.9890 | 0.9890 | 1.0000 | 1.0000 | 0.9890 | 1.0000 |
| Specificity | 1.0000 | 0.9977 | 0.9999 | 0.9984 | 0.9977 | 1.0000 | 1.0000 |
| No‐call rate | 0.0401 | 0.0000 | 0.0621 | 0.1120 | 0.0075 | 0.0322 | 0.0000 |
TP: true positive; TN: true negative; FP: false positive; FN: false negative.
3.3. Small CNVs
In order to explore the limits of the methods compared, we tested whether CNVs smaller than a ROI could be detected (see Supp. Table S1). Although CNVs are often defined as deletions or duplications larger than 50 bp, we also consider smaller ones. Since we only used RCs of whole ROIs, CNVs smaller than a ROI were reported as deletions or duplications of the whole ROI. None of the methods was able to detect the small duplication of 21 bp (NF1 c.31_51dup) encompassing approximately 17% of the ROI. However, panelcn.MOPS, ExomeDepth, CoNVaDING, and VisCap detected the 20‐bp deletion (APC c.856_875del) encompassing only 12.5% of the ROI and the deletion of 80 bp of which only 37 bp overlap with the ROI of 142 bp (NF1 c.1642‐73_1648del). CoNVaDING, VisCap, and NextGENe correctly reported the 64‐bp deletion (NF1 c.1225_1260+29del) at the end of a ROI of 137 bp that was missed by the other methods. The sample with the 63‐bp duplication (APC c.532‐13_581dup) was flagged as low quality by panelcn.MOPS, CoNVaDING, and VisCap. The other methods missed this duplication that affected approximately 36% of the ROI. The CNV detection tool of SeqNext did not find any of the small CNVs, but all of them were identified by the variant calling algorithm of SeqNext. Similarly, the variant calling algorithm of NextGENe identified all CNVs that were missed by the CNV detection tool except for the 80‐bp deletion.
3.4. Custom panel
The methods’ performance results on the custom panel agree with those on the TSC panel. CoNVaDING and VisCap flagged three out of 10 samples as low quality, whereas panelcn.MOPS excluded one sample and the other methods excluded none. ExomeDepth called a false positive CNV in CYP21A2 (sample IH7), which is known to have a highly homologous pseudogene. NextGENe and ExomeDepth both missed part of the deletion in sample IH6. Additionally, NextGENe reported one false positive CNV (IH4). panelcn.MOPS, CoNVaDING, and SeqNext achieved 100% sensitivity and specificity, but only SeqNext and panelcn.MOPS had a reasonably low no‐call rate (see Supp. Table S1).
3.5. Runtime
The runtimes of the different programs were determined by running the CNV detection algorithm itself five times on all 170 TSC panel samples and calculating the median. The time for creating BAM files or calculating RCs was not considered since these procedures must be performed only once for each file. Subsequently, the precalculated RCs can be used directly by the programs. The runtime calculations were performed on an Intel i5‐4300U CPU with 8 GB of RAM. The median runtime of panelcn.MOPS was less than 9 min, followed closely by ExomeDepth with 13 min, whereas VisCap was considerably slower with 2.5 hr. CoNVaDING was by far the slowest with a median runtime of 7 hr and 50 min. Since NextGENe and SeqNext can only be used via the GUI, calculating the runtime is not straightforward. NextGENe cannot handle more than one sample at a time; however, since the analysis of a single sample takes more than 1 min, analyzing 170 samples takes several hours. As SeqNext performs variant calling and CNV detection in a single run, it is not possible to determine the runtime of the CNV detection algorithm alone. However, the whole analysis takes several hours.
4. DISCUSSION
Nearly all methods yielded similarly high sensitivity and specificity, but panelcn.MOPS led the field. However, the following aspects are also important (see Fig. 3).
Figure 3.

Strengths and weaknesses of all methods analyzed. In general, “+++” indicates the best possible performance, whereas “++” means the method is close to the best, “+” is acceptable, but not very good, and “−” signals failure or missing feature. For the two detection performance measures (i.e., sensitivity and specificity), 100% is indicated by “+++.” A single FN or FP is indicated by “++” in the corresponding sensitivity or specificity row. More than one false classification but an overall sensitivity or specificity greater than 95% is indicated by “+.” “No‐call rate” stands for the fraction of ROIs classified as low quality (see the section “Evaluation criteria”). “+++” means no‐call rate of 0, “++” means less than 0.01, “+” means less than 0.1, and “−” means no‐call rate larger than 0.1. Programs with QC for samples and ROIs are marked as “+++,” whereas programs with QC only for samples are marked as “+.” For “CNVs <1 ROI,” each plus sign symbolizes one successful detection of a CNV that affected only part of a ROI (see the section “Small CNVs”). The row “Whole‐gene CNVs” especially concerns CNVs that affect all ROIs that are within the gene of interest for a patient. “+++” indicates that all of them were detected, whereas “++” indicates that some of them were detected and others classified as low quality. If the CNVs affecting the entire gene of interest are not fully detected, this is indicated by “+,” whereas “−” means that these CNVs can only be detected while risking incidental findings. While “+” in the row “Incidental findings” indicates that incidental findings can be avoided, but only at the risk of missing CNVs that affect all ROIs analyzed, “+++” means that the method avoids incidental findings without loss of power. “−” reflects that the CNV tool offers no option to filter the results for genes of interest and, therefore, for avoiding incidental findings. The row “Runtime” indicates the runtime of the CNV detection algorithm measured as described in the section “Runtime.” The thresholds for “+++,” “++,” “+,” and “−” are less than 10 min, less than 1 hr, less than 6 hr, and more than 6 hr, respectively. Classes for the row “GUI” are: “+++” if there is an easy‐to‐use graphical user interface (GUI), “++” if the GUI is not easy to use, and “−” if there is no GUI at all. Since only two of the programs are commercial, they were marked “−” in the corresponding row, whereas all others have “+++”
4.1. QC
Differences in quality and amount of DNA, or measurement noise may render the RCs of affected samples incomparable and can lead to FPs. Sequence characteristics like a high GC content interfere with enrichment or mappability of reads and, consequently, lead to low and/or uneven coverage of certain ROIs. Additionally, RCs can be highly variable in ROIs with a highly homologous pseudogene sequence. These influences on the RCs prevent reliable CN calls in the affected ROIs and increase the risk for FPs and FNs. Particularly, in a clinical setting, it is crucial to find a good balance between (1) excluding samples/ROIs that are indeed of low quality and, therefore, bear a risk of false CN calls and (2) avoiding unnecessary exclusion of too many samples/ROIs because low‐quality samples need to be resequenced, and low‐quality ROIs often require additional tests.
While all programs have QCs for samples, only CoNVaDING, SeqNext, and panelcn.MOPS have additional QCs for ROIs. With a no‐call rate of more than 10% for validation and test set, VisCap excluded numerous samples that were correctly called by all other methods. Also, CoNVaDING flagged many samples as low quality, especially on the validation set and on the custom panel. ExomeDepth, NextGENe, and VisCap have no QC for ROIs, which gives rise to FPs, especially in ROIs that have highly homologous sequences in the genome or a high variation in RCs due to other reasons mentioned above. In most cases, the false positive calls affected PMS2 exons 12–15 known to be problematic due to the highly homologous pseudogene PMS2L (Ganster et al., 2010; van der Klift et al., 2010; Vaughn, Baker, Samowitz, & Swensen, 2013). For the custom panel, most FPs were caused by CYP21A2 and its pseudogene CYP21A1P (New & Wilson, 1999). These FPs are avoided by panelcn.MOPS, CoNVaDING, and SeqNext since the ROIs are recognized as low quality. Overall, the methods with the best balance between low no‐call rate and sufficient QC were panelcn.MOPS and SeqNext. All other methods either lacked QC for ROIs and/or excluded too many samples, which resulted in a low specificity and/or a high no‐call rate.
4.2. Small CNVs
We were interested in the methods’ limitations for CNVs smaller than one ROI. Remarkably, most of the methods identified a deletion of only 20 bp, which would technically not be considered a CNV. However, duplications are harder to identify and so not all of the small CNVs were detected. Our tests have shown that a sensitive variant calling program such as that of SeqNext is able to detect the CNVs smaller than one ROI, and in contrast to CNV detection methods can also define the exact breakpoints. Therefore, the CNV detection methods should be rather used as an additional confirmation tool and not as a primary identification tool for these small CNVs.
4.3. Whole‐gene CNVs
Some of the methods analyzed have problems with the detection of CNVs that extend over a whole gene. As NextGENe and SeqNext restrict their entire analysis to the gene(s) of interest, whole‐gene CNVs cannot be detected if only a single gene is analyzed, since the difference in RCs vanishes after normalization. The low/high RCs caused by a whole‐gene deletion or duplication are considered as normal (CN2) since they affect the majority of ROIs analyzed and consequently all RCs for this sample are rescaled. However, analyzing more genes than required bears the risk of incidental findings as discussed below. CoNVaDING flagged two samples with a whole‐gene deletion of NF1 as low quality. VisCap flagged only one of the two samples as low quality and missed many of the deleted ROIs in the other (see Supp. Table S1). Only panelcn.MOPS and ExomeDepth (with our additional filter for genes of interest) were able to find all whole‐gene deletions without risking incidental findings.
4.4. Incidental findings
Many gene panels contain genes that are relevant for different diseases. For each patient, only a specific set of these genes is of interest and no information concerning other genes should be reported. ExomeDepth, CoNVaDING, and VisCap do not provide an option for restricting the analysis to a specific set of genes. Writing a software for filtering the results like we did is an easy task for someone with programming experience, but not trivial for most clinical geneticists. For NextGENe and SeqNext, the user can provide a list of ROIs that should be analyzed. However, the whole CNV detection including normalization is then restricted to these ROIs. Consequently, both methods have problems when only a single gene is of interest, as discussed in the section “Whole‐gene CNVs.” panelcn.MOPS is the only method that uses all targeted ROIs for determining the correct CNs, but shows results only for the genes of interest for each sample.
4.5. Controls
Some of the methods compared are highly dependent on the controls. Since NextGENe accepts a maximum of only 10 controls, the developers suggest using samples of the same sequencing run as they are likely to show the same sequencing characteristics. However, the controls have to be chosen carefully, such that they do not harbor the same CNV as the test sample since that would conceal the CNV. This poses a challenge if most samples analyzed in a run have the same clinical indication. Additionally, the small number of control samples can be a source of false positive calls if the RC variation within these 10 samples is relatively high. For CoNVaDING, in contrast, at least 20 control samples are needed. Even then, samples are likely to fail one of the QC criteria if not enough control samples with RC characteristics similar to those of the test sample are provided. The control selection of ExomeDepth works well if, as in our analysis, only controls are used that do not have a clinical indication that matches or overlaps that of the test sample. In contrast to panelcn.MOPS, the control selection algorithm of ExomeDepth does not exclude the genes of interest, and it is possible that only one control sample is selected. A large CNV that is present in the test sample and one or more control samples would lead to a high correlation of RCs between these samples and consequently to their selection as best controls. The CNV detection algorithm would then regard the CNV as a normal CN2. This was confirmed in a test using all samples as controls where the deletion of all ROIs of the NF1 gene in samples IBK6 and IBK7 was not detected since only the other sample with the same CNV was chosen as control. For panelcn.MOPS, we tested how the results change if we provide as controls (1) only the 10 control samples used for NextGENe or (2) all TSC panel samples (see Supp. Table S4). Although more samples are excluded due to low quality when only 10 controls are used and CNVs smaller than one ROI are missed, our tests showed that in general for panelcn.MOPS the choice of control samples is less important than for other methods, such as NextGENe, CoNVaDING, and ExomeDepth.
4.6. Runtime
If only a small number of samples is analyzed, a runtime of a few minutes per sample does not pose a big problem. However, in a larger setting such as ours with more than 100 samples, this means hours rather than minutes. For NextGENe, there is an additional issue: it does not support the analysis of more than one sample at a time; therefore, in addition to a runtime of several hours, the hands‐on time is longer than for the other methods. Since calculating CNs for a single sample takes only seconds with panelcn.MOPS, large datasets with thousands of samples can be analyzed within hours rather than days.
4.7. Graphical user interface
ExomeDepth, CoNVaDING, and VisCap do not have a GUI, but are executed via the command line or an R console. Additionally, R or other programming tools must be installed. This is often a barrier for users without programming experience. Further, commercial software such as NextGENe and SeqNext cannot be incorporated into existing variant detection pipelines or even modified by bioinformaticians. While a GUI is especially helpful to clinical geneticists without any programming experience, an R package enables integration of the analysis in a larger pipeline. Hence, we provide panelcn.MOPS both as an R package and as standalone software with GUI and Windows installer to serve both needs.
4.8. Usage with different types of data
In this analysis, we only tested panelcn.MOPS with one sequencing library preparation type and one type of sequencing kit. For other enrichment types or sequencing kits, parameters such as the expected fold change for deletions and duplications should be adapted to the data at hand. Especially the fixed read length parameter should be set to the actual read length of the current data. Since the read counting procedure of panelcn.MOPS handles paired‐end reads separately, single‐read sequencing data can be used without any adaption of the read counting procedure. Additional tests have shown that pooling of different library preparations do not affect the performance of panelcn.MOPS if sufficient control samples from each library are available. Furthermore, our tests have shown that panelcn.MOPS is also suitable for small panels with only 10 genes as long as no sample harbors a CNV that spans the majority of ROIs of the panel.
5. CONCLUSIONS
In this first thorough comparison of six state‐of‐the‐art CNV detection methods for targeted NGS panels, we have demonstrated that panelcn.MOPS has not only the highest sensitivity and specificity, but also one of the best QCs. Furthermore, panelcn.MOPS uses reads from all ROIs to determine the CN, but exclusively presents results for user‐selected genes avoiding incidental findings. The freely available panelcn.MOPS can readily be used via the GUI by clinical geneticists without programming experience, or integrated as R package into existing variant detection pipelines.
Supporting information
Supplementary Methods
Table S1
Table S3
Table S4
ACKNOWLEDGMENTS
We thank Drs. S. Wenzel, E. Maurer, and M. Witsch‐Baumgartner for helpful discussions and providing precharacterized samples.
Disclosure statement
The authors declare no conflict of interest.
Povysil G, Tzika A, Vogt J, et al. panelcn.MOPS: Copy‐number detection in targeted NGS panel data for clinical diagnostics. Human Mutation. 2017;38:889–897. https://doi.org/10.1002/humu.23237
Katharina Wimmer, Peter‐Mayr‐Straße 1/1.OG, Innsbruck A‐6020, Austria. Email: katharina.wimmer@i-med.ac.at
Contract grant sponsors: Institute of Bioinformatics, Johannes Kepler University Linz; Division of Human Genetics, Medical University Innsbruck.
Communicated by Paolo M. Fortina
REFERENCES
- Alkan, C. , Coe, B. P. , & Eichler, E. E. (2011). Genome structural variation discovery and genotyping. Nature Review Genetics, 12, 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, H. , Pillalamarri, V. , Collins, R. L. , Eggert, S. , O'Dushlaine, C. , Braaten, E. B. , … Doyle, A. E. (2014). Cryptic and complex chromosomal aberrations in early‐onset neuropsychiatric disorders. The American Journal of Human Genetics, 95, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard, J. H. , Purdom, E. , Hansen, K. D. , & Dudoit, S. (2010). Evaluation of statistical methods for normalization and differential expression in mRNA‐Seq experiments. BMC Bioinformatics, 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, W. , Cheng, J. , Allaire, J. J. , Xie, Y. , & McPherson, J. (2016). Shiny: Web application framework for R. Retrieved from https://CRAN.R-project.org/package=shiny
- Charbonnier, F. , Raux, G. , Wang, Q. , Drouot, N. , Cordier, F. , Limacher, J. , … Frebourg, T. (2000). Detection of exon deletions and duplications of the mismatch repair genes in hereditary nonpolyposis colorectal cancer families using multiplex polymerase Chain Reaction of Short Fluorescent Fragments. Cancer Research, 60, 2760–2763. [PubMed] [Google Scholar]
- de Leeneer, K. , Hellemans, J. , Steyaert, W. , Lefever, S. , Vereecke, I. , Debals, E. , … Claes, K. (2015). Flexible, scalable, and efficient targeted resequencing on a benchtop sequencer for variant detection in clinical practice. Human Mutation, 36, 379–387. [DOI] [PubMed] [Google Scholar]
- Fakhro, K. A. , Yousri, N. A. , Rodriguez‐Flores, J. L. , Robay, A. , Staudt, M. R. , Agosto‐Perez, F. , … Crystal, R. G. (2015). Copy number variations in the genome of the Qatari population. BMC Genomics, 16, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganster, C. , Wernstedt, A. , Kehrer‐Sawatzki, H. , Messiaen, L. , Schmidt, K. , Rahner, N. , … Wimmer, K. (2010). Functional PMS2 hybrid alleles containing a pseudogene‐specific missense variant trace back to a single ancient intrachromosomal recombination event. Human Mutation, 31, 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Sheng, Q. , Samuels, D. C. , Lehmann, B. , Bauer, J. A. , Pietenpol, J. , & Shyr, Y. (2013). Comparative study of exome copy number variation estimation tools using array comparative genomic hybridization as control. BioMed Research International, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, L. F. , van Dijk, F. , de Boer, E. N. , van Dijk‐Bos, K. K. , Jongbloed, J. D. H. , van der Hout, A. H. , … Sikkema‐Raddatz, B. (2016). CoNVaDING: Single exon variation detection in targeted NGS data. Human Mutation, 37, 457–464. [DOI] [PubMed] [Google Scholar]
- Klambauer, G. , Schwarzbauer, K. , Mayr, A. , Clevert, D. , Mitterecker, A. , Bodenhofer, U. , & Hochreiter, S. (2012). cn.MOPS: Mixture of Poissons for discovering copy number variations in next‐generation sequencing data with a low false discovery rate. Nucleic Acids Research, 40, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komura, D. , Shen, F. , Ishikawa, S. , Fitch, K. R. , Chen, W. , Zhang, J. , … Aburatani, H. (2006). Genome‐wide detection of human copy number variations using high‐density DNA oligonucleotide arrays. Genome Research, 16, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , …1000 Genome Project Data Processing Subgroup . (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X. , Tang, W. , Ahmad, S. , Lu, J. , Colby, C. C. , Zhu, J. , & Yu, Q. (2012). Applications of targeted gene capture and next‐generation sequencing technologies in studies of human deafness and other genetic disabilities. Hearing Research, 288, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. , Tseng, J. T. , Jeng, S. , & Sun, H. S. (2014). Comprehensive analysis of common coding sequence variants in Taiwanese Han population. Biomarkers and Genomic Medicine, 6, 133–143. [Google Scholar]
- Love, M. I. , Myšičková, A. , Sun, R. , Kalscheuer, V. , Vingron, M. , & Haas, S. A. (2011). Modeling read counts for CNV detection in exome sequencing data. Statistical Applications in Genetics and Molecular Biology, 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine, R. , Polson, S. W. , Ravel, J. , Hatfull, G. , Russell, D. , Sullivan, M. , … Wommack, K. E. (2011). Evaluation of a transposase protocol for rapid generation of shotgun high‐throughput sequencing libraries from nanogram quantities of DNA. Applied and Environmental Microbiology, 77, 8071–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meder, B. , Haas, J. , Keller, A. , Heid, C. , Just, S. , Borries, A. , … Rottbauer, W. (2011). Targeted next‐generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circulation: Cardiovascular Genetics, 4, 110–122. [DOI] [PubMed] [Google Scholar]
- New, M. I. , & Wilson, R. C. (1999). Steroid disorders in children: Congenital adrenal hyperplasia and apparent mineralocorticoid excess. Proceedings of the National Academy of Sciences of the United States of America, 96, 12790–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagnol, V. , Curtis, J. , Epstein, M. , Mok, K. Y. , Stebbings, E. , Grigoriadou, S. , … Nejentsev, S. (2012). A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics, 28, 2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh, T. J. , Amr, S. S. , Bowser, M. J. , Gowrisankar, S. , Hynes, E. , Mahanta, L. M. , … Lebo, M. S. (2016). VisCap: Inference and visualization of germ‐line copy‐number variants from targeted clinical sequencing data. Genetics in Medicine, 18, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redd, A. , & Huber, W. (2010). R portable. Retrieved from https://sourceforge.net/projects/rportable/
- Schouten, J. P. , McElgunn, C. J. , Waaijer, R. , Zwijnenburg, D. , Diepvens, F. , & Pals, G. (2002). Relative quantification of 40 nucleic acid sequences by multiplex ligation‐dependent probe amplification. Nucleic Acids Research, 30, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurs‐Hoeijmakers, J. H. M. , Vulto‐van Silfhout, A. T. , Vissers, L. E. , van de Vondervoort, I. I. , van Bon, B. W. , de Ligt, J. , … de Brouwer, A. P. (2013). Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. Journal of Medical Genetics, 50, 802–811. [DOI] [PubMed] [Google Scholar]
- van der Klift, H. M. , Tops, C. M. J. , Bik, E. C. , Boogaard, M. W. , Borgstein, A. , Hansson, K. B. M. , … Wijnen, J. T. (2010). Quantification of sequence exchange events between PMS2 and PMS2CL provides a basis for improved mutation scanning of lynch syndrome patients. Human Mutation, 31, 578–587. [DOI] [PubMed] [Google Scholar]
- Vaughn, C. P. , Baker, C. L. , Samowitz, W. S. , & Swensen, J. J. (2013). The frequency of previously undetectable deletions involving 3′ exons of the PMS2 gene. Genes Chromosomes Cancer, 52, 107–112. [DOI] [PubMed] [Google Scholar]
- Weiss, M. M. , van der Zwaag, B. , Jongbloed, J. D. H. , Vogel, M. J. , Brüggenwirth, H. T. , Lekanne Deprez, R. H. , … van der Stoep, N. (2013). Best practice guidelines for the use of next‐generation sequencing applications in genome diagnostics: A national collaborative study of Dutch genome diagnostic laboratories. Human Mutation, 34, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Wimmer, K. , Callens, T. , Wernstedt, A. , & Messiaen, L. (2011). The NF1 gene contains hotspots for L1 endonuclease‐dependent de novo insertion. PLoS Genetics, 7, e1002371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Table S1
Table S3
Table S4