

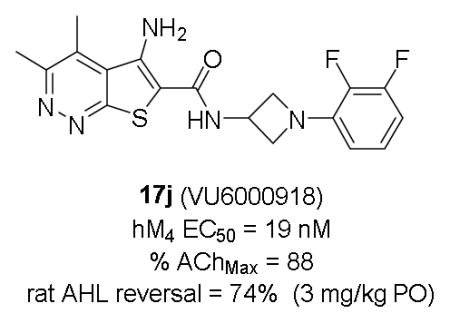
Abstract
This letter details the continued chemical optimization of a novel series of M4 positive allosteric modulators (PAMs) based on a 5-amino-thieno[2,3-c]pyridazine core by incorporating a 3-amino azetidine amide moiety. The analogs described within this work represent the most potent M4 PAMs reported for this series to date. The SAR to address potency, clearance, subtype selectivity, CNS exposure, and P-gp efflux are described. This work culminated in the discovery of VU6000918, which demonstrated robust efficacy in a rat amphetamine-induced hyperlocomotion reversal model at a minimum efficacious dose of 0.3 mg/kg. 2009 Elsevier Ltd. All rights reserved.
Keywords: M4, Muscarinic acetylcholine receptor, Positive allosteric modulator (PAM), Schizophrenia, Azetidine
Graphical Abstract
Positive allosteric modulators (PAMs) of the muscarinic acetylcholine receptor (M4) (1-4) have emerged as an exciting potential strategy for the treatment of numerous CNS disorders, including schizophrenia,1–20 Huntington’s disease,21 and Alzheimer’s disease.22 Previous reports from our laboratory have described the discovery and characterization of VU0152100 (ML108, 1), an in vivo tool compound which demonstrated efficacy in rodent models of anti-psychotic efficacy.3,4 We subsequently reported related congener VU0467154 (2), based on a 5-amino-thieno[2,3-c]pyridazine core, which, despite its robust in vivo activity in multiple preclinical rodent models and a favorable pharmacokinetic (PK) profile, suffered from considerably lower potency at the human M4 receptor as compared to rat.11,19 In the course of our medicinal chemistry campaign to identify a compound with improved potency at the human M4 receptor while maintaining suitable DMPK properties for a clinical candidate, we encountered steep SAR not only in potency at M4, but in multiple DMPK properties as well.13,14,19,20 Herein, we describe our efforts to replace the benzylic linker present in compounds 1-4 with substituted 3-amino azetidines.
Observing that small cyclic amides afforded potent analogs in both Eli Lilly’s and our M4 PAM programs, we wished to examine the introduction of a cyclic linker between the 5-aminothieno[2,3-c]pyridazine amide core and the appended aryl ring. Such a change may serve to decrease the planarity of the molecule, thus reducing its ability to form pi-stacking interactions and thereby improve solubility, restrict the conformations available for the aryl ring to adopt, and remove the benzylic methylene as a potential metabolic soft spot. Diamine linkers would provide a convenient synthetic handle by which to introduce substituents on the cyclic linker. Several potential linkers were examined, including monocyclic and bicyclic diamines; however, 3-amino substituted azetidines yielded the most potent analogs (Figure 2).
Figure 2.
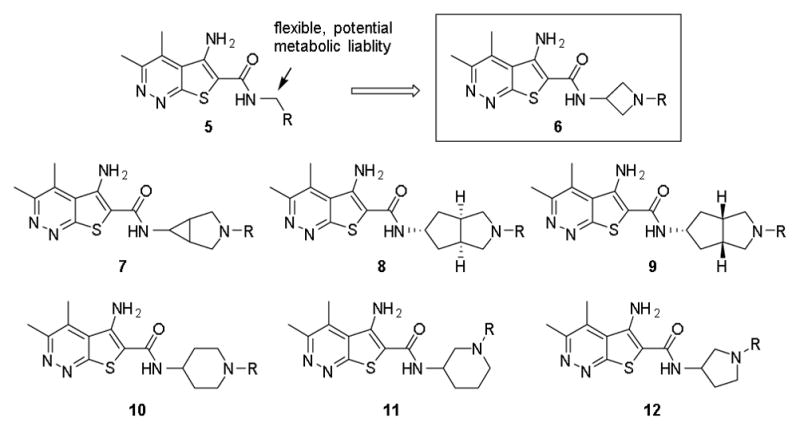
Cyclic diamines examined as alternative amide linkers to thieno[2,3-c]pyridazine core.
Analogs were readily prepared following functionalization of commercially available 3-(Boc-amino)-azetidine via nucleophilic substitution or Buchwald-Hartwig23,24 cross-coupling reactions, followed by Boc deprotection and amide coupling to the thieno[2,3-c]pyridazine core (Scheme 1). Our initial library examined the effect of tertiary carbamates, sulfonamides, and amides (Table 1). Basic tertiary azetidine amines were poorly tolerated and led to a sharp decrease in human M4 (hM4) potency (data not shown). Carbamates proved to be the most potent compounds in this class, with analog 6b displaying an EC50 of 23 nM. However, upon further profiling, 6b was found to have weak activity at human M2 (hM2, EC50 = 2.65 μM) and a short elimination half-life in vivo in rat (t1/2 < 30 min) due to facile hydrolysis of the carbamate, which proved to be the case in general for the carbamate series and thus precluded their advancement. Azetidine sulfonamides (6e), ureas (6d), and amides (6f-h) were also tolerated, albeit with lower potency as compared to the carbamates. Compound 6h was selected for further assessment, which gratifyingly found an improved profile compared to the carbamate series with reduced activity at hM2 (EC50 > 10 μM) and low in vivo clearance (rat CLp = 3.1 mL/min/kg). Unfortunately, 6h was found to have low CNS exposure (rat brain:plasma Kp = 0.03, Kp,uu = 0.37 at 0.25 hr post-IV cassette dose) likely due to P-gp efflux (MDCK-MDR1 ER = 96).
Scheme 1.
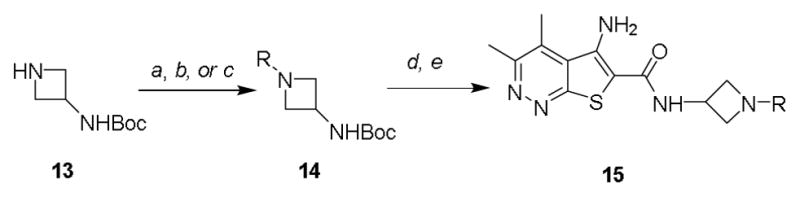
Synthesis of M4 PAM analogs 6, 16, 17. Reagents and conditions: (a) R-X, DCM, DIPEA, rt. (b) R-Het-X, Cs2CO3, DMF, heat (c) Ar-X, Pd2(dba)3, rac-BINAP, Cs2CO3, toluene, 100 °C (d) TFA, DCM, rt, 3 hr (e) 5-amino-3,4-dimethylthieno[2,3-c]pyridazine-6-carboxylic acid, HATU, DIPEA, DMF, 2 hr.
Table 1.
Structures and activities for M4 PAM analogs 6.
| |||
|---|---|---|---|
| Cmpd | R | hM4 EC50 (nM)a [% ACh Max ±SEM] | hM4 pEC50 (±SEM) |
| 6a | CO2Bn | 30 [81±8] | 7.62±0.19 |
| 6b | CO2Ph | 23 [96±3] | 7.65±0.03 |
| 6c | CO2(3-Me)Ph | 67 [85±9] | 7.23±0.17 |
| 6d | C(O)NHPh | 217 [89±6] | 6.66±0.03 |
| 6e | SO2Ph | 268 [70±8] | 6.58±0.07 |
| 6f | C(O)Ph | 773 [85±6] | 6.22±0.25 |
| 6g | C(O)2-pyridyl | 564 [91±5] | 6.25±0.03 |
| 6h | C(O)4-pyridyl | 179 [85±7] | 6.75±0.03 |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n=3) independent experiments performed in triplicate.
Encouraged by our initial results within this series, we sought to further improve the azetidine amides by examining N-aryl azetidines. Initial efforts sought to mimic the carbamate functionality by incorporating 1,3-heteroaryl substituents (16a-c). Compound 16c maintained reasonable hM4 potency (EC50 = 106 nM), exhibited moderate in vitro clearance (predicted rat and human CLhep = 41 and 12 mL/min/kg, respectively, based on hepatic microsomal CLint), and achieved moderate CNS exposure (rat brain:plasma Kp = 0.17, Kp,uu = 1.3). Despite lacking mAChR subtype selectivity (hM2 EC50 = 220 nM), 16c was advanced to a rat amphetamine hyperlocomotion (AHL) reversal study where it exhibited marginal efficacy (16% reversal following 10 mg/kg PO) but provided initial proof-of-concept for the azetidine amide class of compounds. Broadening our scope of N-heteroaryl azetidines led us to identify numerous potent analogs (16d-q). A wide variety of heterocycles was tolerated, with pyridyl, pyrimidyl, and pyrazinyl substituents providing EC50s < 100 nM. Substitution of the heteroaryl ring proved capable of imparting dramatic shifts in potency. While the parent 4-pyridyl azetidine analog possessed micromolar PAM activity (data not shown), introduction of halogen substituents on the 4-pyridyl ring provided exquisitely potent analogs (16p, 16q). Substitution at the meta-position was also tolerated on 3-pyridyl analogs, with methoxy (16l) and fluorine (16m) providing analogs of comparable potency to 16e.
While we were able to achieve excellent in vitro potencies within this series, obtaining both reasonable CNS exposure and metabolic stability proved more challenging. A number of compounds (16e-h, 16l) failed to achieve acceptable CNS exposure (rat brain:plasma Kp < 0.05) and/or were found to be substrates for human P-gp efflux (16e, 16i, 16q,). Additionally, rat in vivo PK studies revealed evidence for extrahepatic non-CYP450 metabolism in certain cases (16o, 16p; possibly due to aldehyde oxidase-mediated metabolism alpha to the 4-pyridyl nitrogen). Increasing the lipophilicity of analogs by incorporation of halogen atoms generally led to modest increases in rat CNS exposure and reduced P-gp efflux. Compound 16j was selected for further DMPK profiling, which revealed low predicted clearance (human and rat predicted CLhep = 5.6 and 18 mL/min/kg, respectively, based on hepatic microsomal CLint) and low potential for CYP450 inhibition (3A4, 2D6, 2C9, 1A2 IC50 > 30 μM). In a rat (male, Sprague-Dawley; n = 2) in vivo PK study, 16j demonstrated low clearance (CLp = 8.8 mL/min/kg) with a small volume of distribution (Vss = 0.89 L/kg) and moderate elimination half-life (t1/2 = 1.3 hr). Total and unbound distribution of 16j to the brain was moderate in rat (brain:plasma Kp = 0.12, Kp,uu = 0.33 at 0.25 hr post-IV cassette dose), and, while it was still a substrate for human P-gp (ER = 8.5 in MDCK-MDR1 cells), its efflux was attenuated compared to related analogs. Given this favorable profile, it was advanced to a dose-response amphetamine hyperlocomotion (AHL) study in rat where it demonstrated robust reversal of AHL (Figure 3). An oral dose of 1 mg/kg provided a 44% reversal of AHL, and a maximal effect of 55% AHL reversal was achieved from 10 and 30 mg/kg dose levels. This level of in vivo efficacy was encouraging for the series and comparable to that previously reported for benchmark compounds 3 and 4. However, due to 16j’s P-gp liability and potentiation of hM2 (EC50 = 0.96 μM, AChMax = 43%), it was deemed not suitable for further development.
Figure 3.
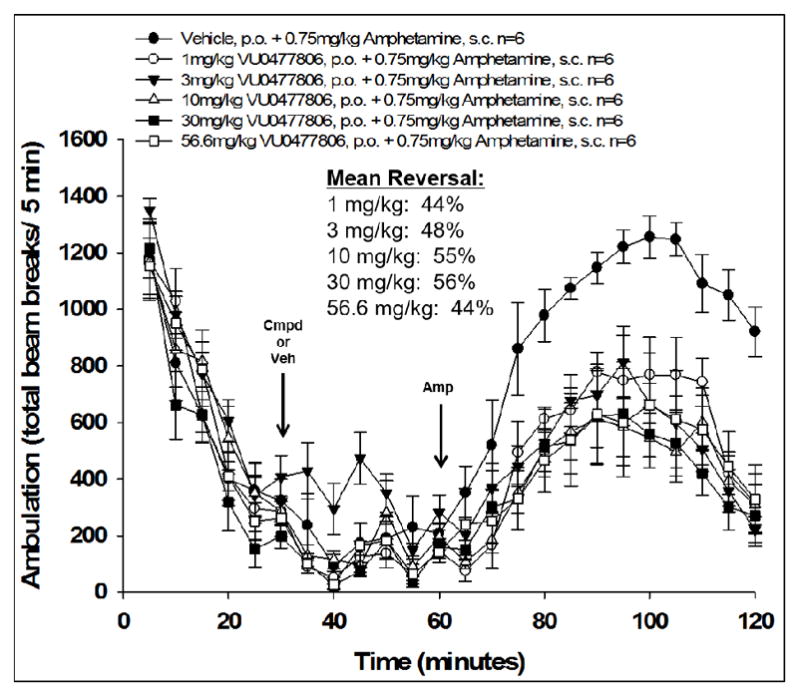
Reversal of amphetamine-induced hyperlocomotion in rat (male, Sprague Dawley, n = 6 per dose group) by 16j (VU0477806). M4 PAM or vehicle (10% tween-80 90% water [v/v]) was administered orally 30 min after habituation in the chamber, and then 0.75 mg/kg amphetamine was administered subcutaneously 30 min later (t = 60 min). Total ambulations were measured over the subsequent 1 hr interval (t = 60–120 min) and used to calculate %reversal of AHL for each dose group. Data represent means ± SEM.
Expanding on the scope of azetidine analogs we had already investigated, we synthesized a library of N-aryl analogs (Table 3). Phenyl congener 17a retained the excellent hM4 potency that could be attained with N-heteroaryl analogs. While 17a did suffer from higher in vivo clearance (rat CLp = 45 mL/min/kg), a consequence of phenyl hydroxylation based on in vitro metabolic soft-spot experiments (data not shown), as well as moderately potent potentiation of hM2 (EC50 = 1.78 μM, 58% AChMax), it showed a promising divergence from the N-heteroaryl azetidine SAR with improved CNS exposure (rat brain:plasma Kp = 0.57, Kp,uu = 0.73). Fluorine substitution(s) on the phenyl ring (17b, 17h-j) generally maintained good hM4 potency and reduced in vivo clearance (17i, 17j; rat CLp = 4.4 and 16 mL/min/kg, respectively). Notably, the azetidine congeners (17f, 17g) of benzylic-linked M4 PAMs previously reported by our group19,20 exhibited significantly weaker hM4 activity compared to that of 17j (~10-fold and 40-fold, respectively). In light of its favorable potency and rat PK, compound 17j, bearing a 2,3-difluorophenyl substituent, was selected for additional characterization. Operational model parameters were determined for 17j from ACh CRC fold-shift experiments (Ca2+ mobilization assays), which revealed a rat M4 KB of 120 nM and αβ of 63, a cynomolgus monkey M4 KB of 890 nM and αβ of 120, and a human M4 KB of 2000 nM and αβ of 380. Gratifyingly, 17j displayed broadly attractive properties including an absence of P-gp efflux (MDCK-MDR1 ER = 1.3), acceptable selectivity versus hM2 (~230-fold based on potency), and high brain distribution (rat brain:plasma Kp = 0.77, Kp,uu = 0.86 at 0.25 hr post-IV cassette dose).
Table 3.
Structures and activities for M4 PAM analogs 17.

| |||
|---|---|---|---|
| Cmpd | R | hM4 EC50 (nM)a [% ACh Max ±SEM] | hM4 pEC50 (±SEM) |
| 17a |
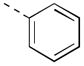
|
25 [85±8] | 7.61±0.08 |
| 17b |

|
28 [82±9] | 7.56±0.03 |
| 17c |
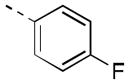
|
282 [77±8] | 6.55±0.04 |
| 17d |

|
141 [77±11] | 6.88±0.11 |
| 17e |
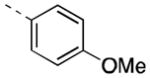
|
1289 [81±8] | 5.90±0.06 |
| 17f |
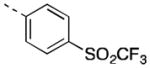
|
934 [86±1] | 6.03±0.04 |
| 17g |
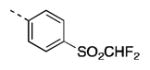
|
325 [75±13] | 6.63±0.29 |
| 17h |
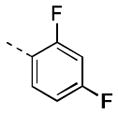
|
37 [94±9] | 7.49±0.12 |
| 17i |
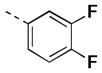
|
30 [84±5] | 7.54±0.06 |
| 17j |
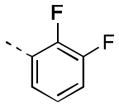
|
19 [88±5] | 7.73±0.06 |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n=3) independent experiments performed in triplicate.
Based on these findings, compound 17j was then evaluated in a rat AHL dose response study (Figure 4) where it demonstrated statistically significant AHL reversal (18%) from a low oral dose of 0.03 mg/kg and reached maximal reversal (74%) from a 3 mg/kg dose, with a resulting in vivo plasma EC50 of 74 nM (0.66 nM unbound) based on terminal concentrations measured in the study animals (1.5 hr post-administration of 17j).
Figure 4.
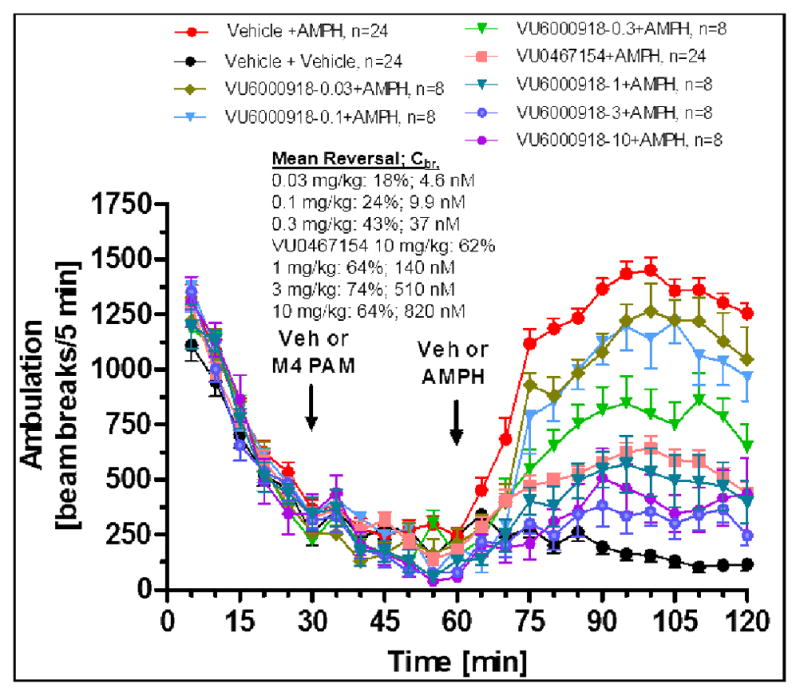
Reversal of amphetamine-induced hyperlocomotion in rat (male, Sprague Dawley, n = 8–24 per dose group) by 17j (VU6000918). M4 PAM, control M4 PAM (VU0467154) or vehicle (10% tween-80 90% water [v/v]) was administered orally 30 min after habituation in the chamber, and then 0.75 mg/kg amphetamine or vehicle (100% water) was administered subcutaneously 30 min later (t = 60 min). Total ambulations were measured over the subsequent 1 hr interval (t = 60–120 min) and used to calculate % reversal of AHL for each dose group. Data represent means ± SEM.
Compound 17j was further studied to evaluate its suitability for progression into IND-enabling studies. An Ames test found no evidence of mutagenesis, a large secondary pharmacology panel (Cerep) revealed a fairly clean profile (all IC50s/EC50s >25–30 μM except for human DAT binding IC50 = 0.45 μM), and CYP450 inhibition was acceptable (1A2 IC50 = 25 μM, 3A4 IC50 = 11 μM, 2D6 IC50 = 14 μM, 2C9 IC50 = 3.8 μM, 2C19 IC50 = 6.3 μM; with no evidence for time-dependent inhibition). IV and PO PK studies with 17j in rat (male, Sprague Dawley, n = 1–3) and dog (female, mongrel, n = 1–2) found the compound to possess low to moderate in vivo clearance (rat and dog CLp = 16 and 17 mL/min/kg, respectively) with a small to moderate volume of distribution (rat and dog Vss = 0.67 and 1.6 L/kg, respectively), and moderate oral bioavailability (rat and dog F = 38% and 20%, respectively, from a 2–3 mg/kg solution dose). However, lower oral bioavailability in dog was observed (11%) from a suspension dose (2 mg/kg) in a low-excipient vehicle. These findings, coupled with a low kinetic solubility (solubility in FaSSIF at pH 6.5 after 1 hr = 0.015 mg/mL; aqueous solubility at pH 7.4 = 1.8 μM), as well as suboptimal predicted human PK (~1–2 hr t1/2 based on moderate and small predicted CL and Vss, respectively) led us to deprioritize 17j for further evaluation as a preclinical candidate and focus efforts on overcoming the evident solubility-limited absorption.
In summary, substitution of the benzyl linker with a 3-aminoazetidine moiety afforded facile entry into an extremely potent series of M4 PAMs. By varying the substitution pattern of N-aryl and N-heteroaryl groups, we were able to optimize subtype selectivity, clearance, CNS exposure, and P-gp efflux. Compound 17j demonstrated robust AHL reversal in a rodent model; however, an unacceptably short projected human half-life and low oral bioavailability (due in part to solubility-limited absorption) precluded its advancement as a clinical candidate. Further study and optimization within this series is ongoing, and will be reported in due course.
Figure 1.
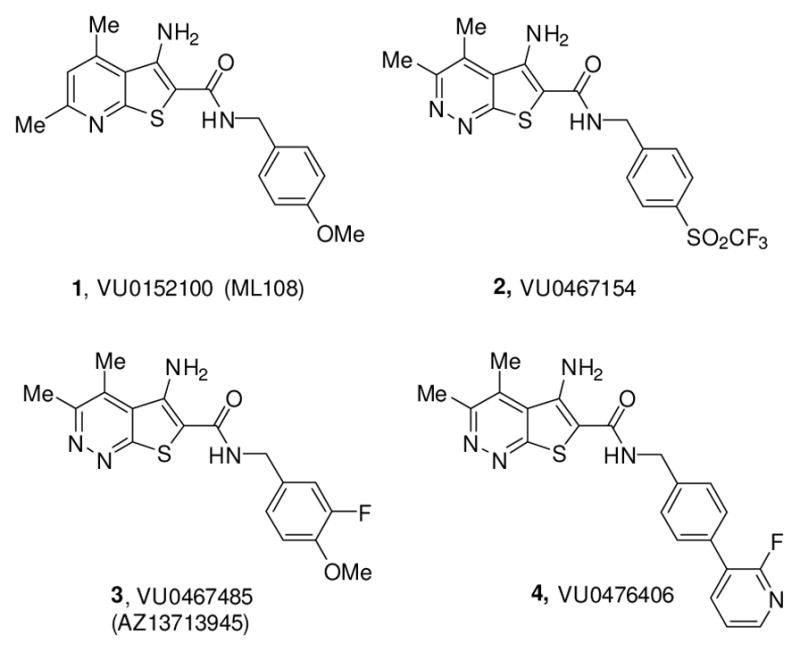
Structures of representative M4 PAMs 1-4, highlighting the optimized rodent in vivo tool M4 PAM, VU0467154 (2), the clinical candidate VU0467485/AZ13713945 (3) and the non-human primate in vivo tool VU0476406 (4).
Table 2.
Structures and activities for M4 PAM analogs 16.
| |||
|---|---|---|---|
| Cmpd | R | hM4 EC50 (nM)a [% ACh Max ±SEM] | hM4 pEC50 (±SEM) |
| 16a |
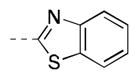
|
439 [87±4] | 6.36±0.04 |
| 16b |

|
419 [89±6] | 6.45±0.19 |
| 16c |
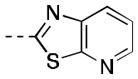
|
106 [81±4] | 7.04±0.16 |
| 16d |
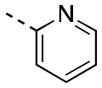
|
124 [81±9] | 6.99±0.18 |
| 16e |
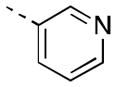
|
34 [96±5] | 7.48±0.09 |
| 16f |
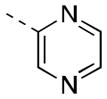
|
78 [80±11] | 7.13±0.11 |
| 16g |
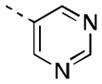
|
41 [89±11] | 7.41±0.11 |
| 16h |
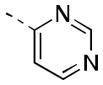
|
38 [83±10] | 7.48±0.17 |
| 16i |
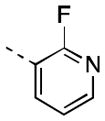
|
29 [82±5] | 7.74±0.22 |
| 16j |
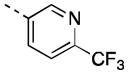
|
76 [75±6] | 7.12±0.08 |
| 16k |
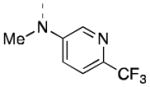
|
Inactive | Inactive |
| 16l |
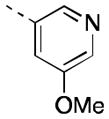
|
17 [88±11] | 7.77±0.01 |
| 16m |
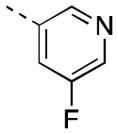
|
16 [90±7] | 7.81±0.03 |
| 16n |
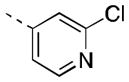
|
185 [87±4] | 6.80±0.19 |
| 16o |
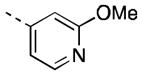
|
51 [86±6] | 7.30±0.00 |
| 16p |
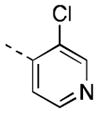
|
8 [86±3] | 8.09±0.05 |
| 16q |
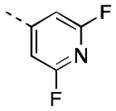
|
11 [84±5] | 7.97±0.09 |
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n=3) independent experiments performed in triplicate.
Acknowledgments
The authors would like to thank NIH (U01MH087965, Vanderbilt, NCDDG). We also thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K Warren, Jr. Chair in Medicine (to C.W.L.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Chan WY, McKinize DL, Bose S, Mitchell SN, Witkins JM, Thompson RC, Christopoulos A, Birdsall NJ, Bymaster FP, Felder CC. Proc Natl Acad Sci USA. 2008;105:10978–10983. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leach K, Loiancono RE, Felder CC, McKinize DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. Neuropsychopharmacology. 2010;35:855–869. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brady A, Jones CK, Bridges TM, Kennedy PJ, Thompson AD, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey J, Conn PJ, Lindsley CW. J Pharm & Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byun NE, Grannan M, Bubser M, Barry RL, Thompson A, Rosanelli J, Gowrishnakar R, Kelm ND, Damon S, Bridges TM, Melancon BJ, Tarr JC, Brogan JT, Avison MJ, Deutch AY, Wess J, Wood MR, Lindsley CW, Gore JC, Conn PJ, Jones CK. Neuropsychopharmacology. 2014;39:1578–1593. doi: 10.1038/npp.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrell M, Roth BL. Neuropsychopharmacology. 2010;35:851–852. doi: 10.1038/npp.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones CK, Byun N, Bubser M. Neuropsychopharmacology. 2012;37:16–42. doi: 10.1038/npp.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. Nat Chem Bio. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- 8.Le U, Melancon BJ, Bridges TM, Utley TJ, Lamsal A, Vinson PN, Sheffler DJ, Jones CK, Morrison R, Wood MR, Daniels JS, Conn PJ, Niswender CM, Lindsley CW, Hopkins CR. Bioorg Med Chem Lett. 2013;23:346–350. doi: 10.1016/j.bmcl.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kennedy JP, Bridges TM, Gentry PR, Brogan JT, Brady AE, Shirey JK, Jones CK, Conn PJ, Lindsley CW. ChemMedChem. 2009;4:1600–1607. doi: 10.1002/cmdc.200900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salovich JM, Sheffler DJ, Vinson PN, Lamsal A, Utley TJ, Blobaum AL, Bridges TM, Le U, Jones CK, Wood MR, Daniels JS, Conn PJ, Niswender CM, Lindsley CW, Hopkins CR. Bioorg Med Chem Lett. 2012;22:5084–5088. doi: 10.1016/j.bmcl.2012.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bubser M, Bridges TM, Thorbeck DD, Gould RW, Grannan M, Noetzel MJ, Niswender CM, Daniels JS, Melancon BJ, Tarr JC, Wess J, Duggan ME, Brandon NJ, Dunlop J, Wood MW, Wood MR, Lindsley CW, Conn PJ, Jones CK. ACS Chem Neurosci. 2014;5:920–942. doi: 10.1021/cn500128b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith E, Chase P, Niswender CM, Conn PJ, Lindsley CW, Madoux F, Acosta M, Scampavia L, Spicer T, Hodder P. J Biomol Screening. 2015;20:858–868. doi: 10.1177/1087057115581770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood MR, Noetzel MJ, Tarr JC, Rodriguez AL, Lamsal A, Chang S, Foster JJ, Smith E, Hodder PS, Engers DW, Niswender CM, Brandon NJ, Wood MW, Duggan ME, Conn PJ, Bridges TM, Lindsley CW. Bioorg Med Chem Lett. 2016;26:4282–4286. doi: 10.1016/j.bmcl.2016.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood MR, Noetzel MJ, Engers JL, Bollinger KA, Melancon BJ, Tarr JC, Han C, West M, Gregro AR, Lamsal A, Chang S, Ajmera S, Smith E, Chase P, Hodder PS, Bubser M, Jones CK, Hopkins CR, Emmitte KA, Niswender CM, Wood MW, Duggan ME, Conn PJ, Bridges TM, Lindsley CW. Bioorg Med Chem Lett. 2016;26:3029–3033. doi: 10.1016/j.bmcl.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo M, Huynh T, Valant C, Lane JR, Sexton PM, Christopoulos A, Capuano B. MedChemComm. 2015;6:1998–2003. [Google Scholar]
- 16.Huynh T, Valant C, Crosby IT, Sexton PM, Christopoulos A, Capuano B. ACS Chem Neurosci. 2015;6:1592–1599. doi: 10.1021/acschemneuro.5b00035. [DOI] [PubMed] [Google Scholar]
- 17.Croy CH, Schober DA, Xiao H, Quets A, Christopoulos A, Felder CC. Mol Pharmacol. 2014;86:106–115. doi: 10.1124/mol.114.091751. [DOI] [PubMed] [Google Scholar]
- 18.Huynh T, Valant C, Crosby IT, Sexton PM, Christopoulos A, Capuano B. J Med Chem. 2013;56:1592–1599. doi: 10.1021/jm401032k. [DOI] [PubMed] [Google Scholar]
- 19.Wood MR, Noetzel MJ, Poslusey MS, Melancon BJ, Tarr JC, Lamsal A, Chang S, Luscombe VB, Weiner RL, Cho HP, Bubser M, Jones CK, Niswender CM, Wood MW, Engers DW, Brandon NJ, Duggan ME, Conn PJ, Bridges TM, Lindsley CW. Bioor Med Chem Lett. 2017;27:171–175. doi: 10.1016/j.bmcl.2016.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Wood MR, Noetzel J, Melancon BJ, Poslusney MS, Nance KD, Hurtade MA, Luscombe VB, Weiner RL, Rodriguez AL, Lamsal A, Chang S, Bubser M, Blobaum AL, Engers DW, Niswender CM, Jones CK, Brandon NJ, Wood MW, Duggan ME, Conn PJ, Bridges TM, Lindsley CW. ACS Med Chem Lett. 2017;8:233–238. doi: 10.1021/acsmedchemlett.6b00461. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Melancon BJ, Wood MR, Noetzel J, Nance KD, Engelberg EM, Han C, Lamsal A, Chang S, Bubser M, Blobaum AL, Engers DW, Niswender CM, Jones CK, Brandon NJ, Wood MW, Duggan ME, Conn PJ, Bridges TM, Lindsley CW. Bioor Med Chem Lett. submitted. [Google Scholar]
- 21.Pancani T, Foster DJ, Bichell T, Bradley E, Bridges TM, Klar R, Daniels JS, Jones CK, Bowman AB, Lindsley CW, Xiang Z, Conn PJ. Proc Natl Acad Sci USA. 2015;112:14078–14083. doi: 10.1073/pnas.1512812112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen W, Plotkin JL, Francardo V, Ko WKD, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, Conn PJ, Greengrad P, Bezard E, Cenci MA, Surmeier DJ. Neuron. 2015;88:762–773. doi: 10.1016/j.neuron.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolfe JP, Wagaws S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805. [Google Scholar]
- 24.Hartwig JF. Acc Chem Res. 1998;31:852. [Google Scholar]