


Abstract
The aqueous phase photochemistry of a series of amphiphilic α-keto acids with differing linear alkyl chain lengths was investigated, demonstrating the ability of sunlight-initiated reactions to build molecular complexity under environmentally relevant conditions. We show that the photochemical reaction mechanisms for α-keto acids in aqueous solution are robust and generalizable across alkyl chain lengths. The organic radicals generated during photolysis are indiscriminate, leading to a large mixture of photoproducts that are observed using high-resolution electrospray ionization mass spectrometry, but these products are identifiable following literature photochemical mechanisms. The alkyl oxoacids under study here can undergo a Norrish Type II reaction to generate pyruvic acid, increasing the diversity of observed photoproducts. The major products of this photochemistry are covalently bonded dimers and trimers of the starting oxoacids, many of which are multitailed lipids. The properties of these oligomers are discussed, including their spontaneous self-assembly into aggregates.
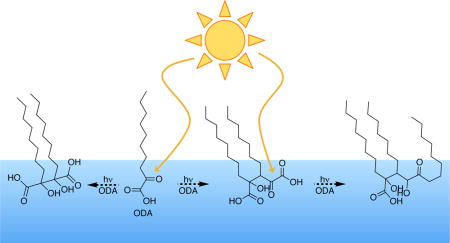
Graphical Abstract
INTRODUCTION
The reactivity and synthesis of lipid-like molecules are of interest for environmental chemistry of both the modern and ancient Earth.1–9 In the environment, lipid-like molecules selectively partition to air–water interfaces. Sunlight can drive photoinitiated chemistry of organic species at air–water interfaces, such as the sea-surface microlayer (SML).10–16 The SML contains high concentrations of amphiphilic organic molecules and, as the boundary layer between the ocean and atmosphere, connects processes at the sea surface with the atmosphere, including the generation of sea spray aerosol. Recent work suggests that oligomers formed from aqueous phase photochemistry of small organic molecules may contribute significantly to the formation and development of secondary organic aerosol (SOA).17–25 The surface-active molecules found in the SML are often biological in origin,14,26,27 but abiotic photochemical sources may be additional, important contributors.11,12,23,28–30 Such sunlight-driven reactions are critical in the understanding of the formation of marine-derived primary organic aerosol and SOA,31–37 influencing visibility, human health, cloud formation, and radiative forcing.17,19,38–46
The present study investigates the photochemistry of a series of amphiphilic α-keto acids, also known as 2-oxoacids that will be referred to as oxoacids here, with varying alkyl tail lengths. Such oxoacids have been identified at the ocean surface and are significant contributors to the environment today14,47,48 and likely would have been found on the early Earth.49–52 In this work, we describe the general photochemical mechanism for α-keto acids in aqueous solution under acidic conditions, investigating the effect of the alkyl tail length. By demonstrating this mechanistic understanding of the photochemistry of α-keto acids, we show its generalizability across a class of molecules, widening the library of compounds whose photolysis may contribute to the reactivity found in natural environments.
Previously, the photochemistry of the simplest of α-keto acid, pyruvic acid, has been studied in detail in both the gas53–60 and the aqueous phases,28,61–69 due to its atmospheric rele-vance.48,70–78 Like all α-keto acids, pyruvic acid absorbs light in the near-UV within the spectral range of wavelengths of sunlight that are available near the surface of the Earth, and direct photolysis is its main atmospheric sink.28,57,58 In the aqueous phase, pyruvic acid readily forms oligomers,28,63,64,67,79 including in atmospherically relevant, multiphase studies using an environmental simulation chamber.68 These covalently bonded dimers and trimers80 are generated in relatively high yields from the recombination of photochemically generated organic radicals and are the major observed photoproducts, especially under oxygen-limited reaction conditions.28,63,64,67
In this study, we examine the photoreactivity in aqueous solution at unadjusted pH of four oxoacids (shown in Chart 1) with differing saturated, linear alkyl chains, ranging from the four-carbon butyl tail of 2-oxohexanoic acid to the 10-carbon decyl tail of 2-oxododecanoic acid, and compare these results to the known photochemistry of pyruvic acid, possessing a single-carbon methyl tail. We demonstrate that the pyruvic acid photochemical mechanism is transferrable to molecules with the same reactive functional groups, including those compounds with longer alkyl chains that are relevant to the generation of the large, surface-active oligomer species that contribute to the SML. These longer-tailed species also follow additional reactive pathways that are not available to the short-tailed pyruvic acid, increasing the complexity of the mixture of photoproducts generated.
Chart 1.

Structures of Pyruvic Acid and the Four Oxoacids Studied in This Investigation: 2-Oxohexanoic Acid (OHA), 2-Oxooctanoic Acid (OOA), 2-Oxodecanoic Acid (ODA), and 2-Oxododecanoic Acid (ODDA)
Organic photochemistry, as investigated here, also has significant implications for the study of prebiotic chemical evolution and the generation of biomolecules in the absence of enzymatic assistance,4 including the abiotic synthesis of lipid molecules.4,52 The synthesis of lipid molecules is interesting, not only because of the formation of a carbon–carbon bond in the absence of biology, but also because of the importance of such molecules in the formation of membranous enclosures. Such enclosures form the basis for protocells and are recognized as vital to developing and sustaining life.9,81–87
EXPERIMENTAL SECTION
2-Oxooctanoic acid (OOA, ≥99.0%, Sigma-Aldrich) was diluted with 18.2 MΩ water (3 ppb TOC) to obtain solutions of 6, 3, and 1 mM and sonicated until fully dissolved. Solutions were filtered before use with a 0.1 µM syringe filter (MillexVV). The remaining alkyl oxoacids, 2-oxohexanoic acid (OHA), 2-oxodecanoic acid (ODA), and 2-oxododecanoic acid (ODDA), were synthesized as described below and diluted in the same manner as OOA to obtain 1 mM solutions. The 6 mM OHA and 3 mM ODA solutions were also used. Solutions consisting of mixtures of 1:1 OOA:ODA were obtained in the same manner with either 3 or 1.5 mM concentrations of each oxoacid, giving a total amphiphile concentration of either 6 or 3 mM, respectively. It has recently been suggested that solutions of OOA at concentrations similar to those used here can result in the formation of vesicles.88 We do not observe this under our experimental procedure. Dynamic light scattering of the prephotolysis solutions (OHA, OOA, ODA, and OOA/ODA) confirmed that aggregates were not detectable in our solutions prior to irradiation.
The solutions were used without adjustment from their natural pH, meaning all photolyses were conducted under acidic conditions. There is a slight variance in the pH of the solutions as a function of the concentration of ROA. For example, the 6 mM OOA solutions are pH ≈ 2.5, increasing slightly to ~2.8 and ~3.3 for the solutions at 3 and 1 mM, respectively. The pH of the solutions at a given concentration of ROA does not change significantly as the identity of the ROA is changed. The pH of 3 mM ODA is also ~2.8, and the observed pH for each ROA at 1 mM varies only slightly, between ~3.3 and ~3.7. These pH ranges are in keeping with the previously observed pH for solutions of pyruvic acid at similar concentrations (10 mM ≈ 2.4 and 0.5 mM ≈ 3.5).67 This is suggestive that the pKa of each ROA does not change significantly as alkyl tail length is varied. The effective pKa of OOA has recently been determined to be 2.78,88 which is similar to the well-known effective pKa of pyruvic acid of 2.49.89 The term effective is used to describe these pKa values because the titration method of determining pKa necessarily includes contributions from both the keto and the geminal-diol conformers in solution (discussed below), which each also have individual values of pKa.90
For each photolysis, 100 mL solutions were prepared, saving 10 mL as a prephotolysis control, and photolyzing the remaining 90 mL of solution for 5 h, using a 450 W ozone-free Xe arc lamp (Newport). Solution temperature was controlled by a temperature-stabilized water bath. For the 1 mM solutions of each oxoacid, the water bath was held at 20 °C because of solubility concerns for ODDA. The higher concentrations of OHA, ODA, and OOA were also conducted at 4 °C. No difference was observed in products with changes in the water bath temperature. Unless otherwise stated, each solution was purged with N2, to displace dissolved O2. Solutions were purged for the duration of photolysis, beginning for 1 h prior to the start of illumination. After photolysis, the solutions were brought to room temperature before any further analyses were conducted.
The absorption maximum of the excitation from the ground state to the S1, 1(n, π*), state for each of the oxoacids examined here occurs at λmax ≈ 315 nm. This is a slight blue shift from that of pyruvic acid (λmax ≈ 320 nm),28,67 but there is not a significant change in the absorption maximum as a function of changing alkyl tail length as for the four oxoacids investigated here (spectra shown in Figure S1).
The spectral output of the unfiltered Xe arc lamp used in these studies extends into the UV to about 220 nm as shown in Figure S1. Therefore, it is likely that, under our experimental conditions, some excitation to the S2, 1(π, π*), state also occurs in addition to excitation to the S1 state. This is unlikely to alter any reaction pathways, as it is known that, upon excitation to higher excited states, such as the S2 state, systems often rapidly undergo internal conversion to the first excited (S1) state.91,92 This process is expected to occur here, allowing the normal photochemical pathway of intersystem crossing and internal conversion from the S1 state to the reactive T1 state to follow. It has been previously observed that the photochemical products generated from the photolysis of pyruvic acid using an unfiltered Xe arc lamp67 are in good agreement with those obtained when the Xe lamp is filtered to remove wavelengths λ < 300 nm;28 however, the rate of photolysis is slowed when the Xe lamp is filtered, as would be expected when fewer photons are present. The S2 state cannot be excited with λ > 300 nm, suggesting the same reactive photochemical pathways are followed when the lamp remains unfiltered, without additional reactive pathways stemming from excitation to the S2 state. This is consistent with other observations in the literature, which have shown, for example, that the photochemistry of nonanoic acid in aqueous solution is faster but does not result in the formation of different products when an unfiltered Xe arc lamp is used.12
Synthesis of Alkyl Oxoacids
The same general procedure was used to synthesize each of 2-oxohexanoic acid, 2-oxodecanoic acid, and 2-oxododecanoic acid. The general synthetic procedure is outlined below. Detailed information and characterization of each synthesized oxoacid are given in the Supporting Information, along with 1H and 13C NMR spectra (Figures S2–4). A solution of the Grignard reagent, prepared from the appropriate 1-bromoalkane and the suspension of magnesium in THF, was added dropwise under N2 atmosphere to a cold (−78 °C) solution of diethyloxalate in THF. After the addition was complete, the reaction mixture was stirred at −78 °C for at least an additional hour. The reaction was quenched with 2 N HCl, and the aqueous layer was extracted with either ether or hexanes. The combined organic layers were washed with saturated NaCl, dried over MgSO4, and evaporated. The crude product was then purified either by distillation under reduced pressure to give a colorless oil (OHA) or by recrystallization in hexanes to give a white solid (ODA and ODDA).
Product Analysis
Detailed information and instrument parameters for the analytical techniques used to characterize the photolysis solutions (dynamic light scattering, UV–vis and NMR spectroscopy, and high-resolution negative mode electrospray ionization mass spectrometry) are given in the Supporting Information. In the ESI−MS, both [M − H]− and singly charged adduct ions are observed for the analytes of interest under the ionization conditions used here. The observed noncovalent adduct ions are formed from the coordination of two or more deprotonated organic species with a metal ion and a net charge of −1. We observe, primarily in the prephotolysis controls, adduct ions consisting of multiple deprotonated oxoacid molecules coordinated with a positive counterion, usually Na+ or Ca2+. All photolyses were conducted in 18.2 MΩ water without the addition of salt to minimize the presence of such adduct ions, and any metal ions are assumed to be present only in trace quantities in solution.
The ESI− MS analysis used here was not designed to be absolutely quantitative. As expected for solutions containing mixtures of analytes, the observed intensities of [M − H]− ions vary considerably due to variations in ionization efficiency as well as differences in concentration of species. To avoid incorrect ion assignments to noise peaks, a conservative intensity threshold of 104 counts was imposed for analyte identifications; the average noise threshold across runs is about 1000 counts. To allow for qualitative comparisons, categories of relative signal intensity were defined as follows: for given monoisotopic ions, those with intensities greater than 106 counts are “strong”, those with intensities greater than 105 counts are “medium”, and those with intensities greater that 104 counts are “weak”. These intensity categories are used only for relative comparisons because they do not necessarily directly correlate to absolute analyte concentrations.
RESULTS AND DISCUSSION
Here, we report on the generation of surface-active oligomers from the aqueous photochemistry of a series of four α-keto acids with differing alkyl tail lengths (2-oxohexanoic acid (OHA), 2-oxooctanoic acid (OOA), 2-oxodecanoic acid (ODA), and 2-oxododecanoic acid (ODDA) shown in Chart 1). The photochemical mechanisms governing this chemistry follow those that have been previously reported in the literature,52,93–95 primarily from detailed studies of the aqueous photolysis of pyruvic acid.28,62,64,65,67,96,97 Here, we perform a detailed high-resolution negative mode electrospray ionization mass spectrometry (ESI− MS) analysis, supported by NMR studies, to show that this chemistry is robust and broadly transferable to a whole class of molecules with the same reactive functionality and generates multitailed lipids.
The aqueous photolysis of each oxoacid we study here follows the same general mechanism, regardless of the alkyl tail length, detailed descriptions of which can be found in previous work on pyruvic acid.28,64,67 For clarity, here we will describe briefly the mechanistic pathways of this chemistry for the general case of an oxoacid with an arbitrary alkyl tail, R, abbreviated as ROA. The photochemistry holds for each of the specific alkyl tails examined (R = (CH2)2CH3, (CH2)4CH3, (CH2)6CH3, and (CH2)8CH3). Similarities and differences between the known photochemical behavior of pyruvic acid (R = H) will be discussed when relevant.
The solubility of the molecules investigated decreases as a function of chain length, so to facilitate comparison across alkyl tail lengths, photolyses for each oxoacid were conducted at concentrations of ~1 mM and at 20 °C. These experimental conditions approached the solubility limit of ODDA. Photolyses of 6 and 3 mM OOA and 3 mM 2-oxodecanoic acid ODA were also conducted at 4 °C. There were no observable differences in the products generated during photolysis when the water bath temperature was changed from 4 to 20 °C for OOA and ODA.
Despite the low concentrations required, photochemical products were still readily observed for each oxoacid. Averaged MS results for OOA are given in Table 1 with representative mass spectra for both the pre- and the postphotolysis solutions of OOA given in Figure 1. A summary of the MS results for the other alkyl oxoacids, OHA, ODA, and ODDA, is given in Table 2. Detailed MS results for each oxoacid are also given in Figures S5– 7 and Tables S1– 4.
Table 1.
Select Compiled 2-Oxooctanoic Acid Photolysis Mass Spectra Dataa
| assigned formula [M − H]− | assigned structure | average experimental m/zb | theoretical m/z | mass diff. (ppm) | prephotolysis | postphotolysis |
|---|---|---|---|---|---|---|
| C3H3O3 | pyruvic acid | 87.0092 ± 0.0006 | 87.0082 | 10.9 | below threshold | weak |
| C8H13O3 | 2-oxooctanoic acid | 157.0880 ± 0.0005 | 157.0865 | 9.6 | strong | strong |
| C8H13O4 | 2,2-DHOAc (OOA diol) | 173.0826 ± 0.0004 | 173.0814 | 7.0 | medium | medium |
| C6H9O6 | dimethyltartaric acid | 177.0411 ± 0.0005 | 177.0400 | 6.6 | below threshold | weak |
| C12H21O5 | R-DHO acid | 245.1400 ± 0.0005 | 245.1389 | 4.4 | below threshold | medium |
| C11H19O6 | methylhexyl-tartaric acid | 247.1193 ± 0.0005 | 247.1182 | 4.6 | below threshold | strong |
| C13H21O7 | R-DHO diacid | 289.1301 ± 0.001 | 289.1288 | 4.7 | below threshold | medium |
| C17H31O5 | R,R-DHO acid | 315.2181 ± 0.0006 | 315.2172 | 3.0 | below threshold | strong |
| C16H29O6 | dihexyltartaric acid | 317.1973 ± 0.0005 | 317.1965 | 2.8 | below threshold | strong |
| C18H31O7 | R,R-DHO diacid | 359.2071 ± 0.0007 | 359.2070 | 0.29 | below threshold | medium |
| C22H41O5 | R,R,R-DHO acid | 385.2958 ± 0.0003 | 385.2954 | 0.83 | below threshold | medium |
| C23H41O7 | R,R,R-DHO diacid | 429.2855 ± 0.0008 | 429.2860 | −1.2 | below threshold | weak |
Chemical formulas are assigned as the ionized [M − H]− species; structures are assigned as the neutral species.
The experimental m/z is the observed average across experiments, and the uncertainty given is the 95% confidence interval.
2,2-DHOA = 2,2-dihydroxyoctanoic acid, the gem-diol form of OOA.
Figure 1.

Representative MS of 3 mM OOA pre (black, counts multiplied by –1 for ease of presentation) and post (red) 5 h of photolysis in N2; high molecular weight products are not observed.
Table 2.
Summary of Key MS Data for OHA, ODA, and ODDAa
| 2-oxohexanoic acid, R = (CH2)2CH3
|
2-oxodecanoic acid, R = (CH2)6CH3
|
2-oxododecanoic acid, R = (CH2)8CH3 |
||||||
|---|---|---|---|---|---|---|---|---|
| identity | [M − H]− | avg exp m/zb (ppm) | [M − H]− | avg exp m/zb (ppm) | [M − H]− | avg exp m/zb (ppm) | pre-hνc | post-hνc |
| ROA | C6H9O3 | 129.0550 (−1.5) | C10H17O3 | 185.1186 (4.5) | C12H21O3 | 213.1502 (5.2) | S | S |
| R-DHO acid | C10H17O5 | 217.1077 (0.3) | C14H25O5 | 273.1708 (1.9) | C16H29O5 | 301.2025 (3.2) | B.T. | M |
| R,M-TAd | C9H15O6 | 219.0868 (−0.5) | C13H23O6 | 275.1500 (1.8) | C15H27O6 | 303.1820 (4.0) | B.T. | M |
| R,R-DHO acid | C13H23O5 | 259.1546 (−1.5) | C21H39O5 | 371.2805 (1.9) | C25H47O5 | 427.3450 (6.1) | B.T. | M |
| R,R-TAe | C12H21O6 | 261.1339 (0.0) | C20H37O6 | 373.2595 (1.2) | C24H45O6 | 429.3215 (−0.3) | B.T. | S |
| R,R,R-DHO acid | C16H29O5 | 301.2015 (0.6) | C28H53O5 | 469.3890 (−0.7) | B.T. | B.T. | M | |
Chemical formulas are assigned as the ionized [M − H]− species; structures are assigned as the neutral species.
The experimental m/z is the average observed mass across experiments. Values in parentheses are the averaged mass differences from the theoretical m/z given in ppm.
Relative intensities of the observed MS species are given as S = strong, M = medium, B.T. = below threshold.
R,M-TA = R,methyl-tartaric acid.
R,R-TA = R,R-tartaric acid.
In aqueous solution, the ketone group of ROA can be hydrated to the geminal diol form of the acid, R,gem-diolalkanoic acid (RGDA).28,64,67 These 2,2-dihydroxyalkanoic acids, unlike the ketone form, do not absorb light within the solar spectrum. Unlike many α-dicarbonyl species, which are almost completely hydrated in aqueous solution, oxoacids retain a significant amount of ketonic functionality.90,98,99 It has been shown for pyruvic acid that the extent of hydration is concentration-, pH-, and temperature-dependent,67,90,99–101 with percentages of diol conformer ranging from ~65% at 100 mM to ~50% at 10 mM concentrations.28,63,67 The longertailed oxoacid species under study here retain significantly more ketonic functionality in aqueous solution than pyruvic acid. The 6 mM OOA at ambient temperature exists as roughly 80% keto and 20% diol in solution as characterized by NMR (Figure S8). Both conformers are also observed in the MS data (Figure 1 and Table 1). The ratio of keto to diol conformers does not appear to change as a function of tail length for any of the longer-tailed species we investigate here. For example, ODA, which has two more carbons than OOA in its alkyl chain, also exists as an approximately 80% keto conformer in solution under our reaction conditions (Figure S9).
Additionally, even under our acidic reaction conditions, because the effective pK a of alkyl oxoacids is about ~2.6,88,89 a significant percentage of ROA are deprotonated in solution, with preferential deprotonation of the photoactive, keto conformer.90 This reduces the number of photoactive protonated species available for excitation, and at higher pH the rate of photolysis for pyruvic acid has been observed to slow. 65 However, under our acidic reaction conditions, the photochemistry still readily proceeds.
In aqueous solution, photolysis begins when the oxoacid, ROA, is excited by a UV photon from the ground state to the S1, 1(π, π*), state (λmax ≈ 315 nm), which is followed by intersystem crossing and internal conversion to the T1, 3(n, π*), state (reaction 1 of Scheme 1). ROA in the excited T1 state then forms a radical species by abstracting a hydrogen from another oxoacid, which can occur via two different pathways (Scheme 1),67 depending on the site of hydrogen abstraction. In the first (reaction 2 of Scheme 1), hydrogen abstraction occurs at the carboxyl group of either ROA or its geminal diol form (RGDA) and is followed by decarboxylation to form two radical species, one with hydroxy-acid functionality (denoted RHA•) and one with geminal diol functionality (denoted RGD•). For pyruvic acid, abstraction from the diol form is favored energetically, but it has been shown that abstraction from the keto conformer can also occur.64 Even with the lower amounts of diol observed for ROA as compared to pyruvic acid, it is clear from the products observed by ESI− MS that hydrogen abstraction at the carboxyl group readily occurs for all of the oxoacids under consideration here and is likely the dominant site of abstraction.
Scheme 1.

Formation Pathways of Radical Species during the Aqueous Photolysis of Oxoacids102
In the second possible pathway (reaction 3 of Scheme 1), the excited ROA in the T1 state abstracts a hydrogen from the alkyl tail of another, ground-state ROA. Calculations have shown that hydrogen abstraction from the methyl group of pyruvic acid is likely to be competitive with hydrogen abstraction from the carboxyl group.67 This competitiveness of this pathway is also likely increased in dilute solutions where pH is slightly higher because the methyl group becomes the only site available for abstraction when the keto form of pyruvic acid is deprotonated.67 It is reasonable, then, to assume that abstraction from the alkyl tail for ROA is likewise energetically competitive, occurring to some extent during photolysis, yielding both a radical that retains oxoacid functionality (ROA•) and RHA•. We have drawn, throughout our discussion, the hydrogen abstraction to form ROA• as occurring at the β-CH2 group. While abstraction from this site may be favored, it can, in principle, occur anywhere along the alkyl chain.103 Products that are generated from subsequent reactions of ROA• are observed in the MS of postphotolysis solutions, but ESI− MS alone cannot differentiate where along the alkyl chain hydrogen abstraction has occurred. It is likely that the observation of a given chemical formula in the MS data corresponds to a mixture of constitutional isomers.
The three radical species (RHA•, RGD•, and ROA•) generated by these two hydrogen abstraction processes can recombine following a variety of pathways, explaining the majority of the observed photoproducts for each of the oxoacids under study here (Tables 1 and 2, with detailed MS results for each ROA in Tables S1–4). The products observed from further reactions of the radical pair, RHA• and RGD•, generated by hydrogen abstraction from the carboxyl group, are outlined in Scheme 2. Tartaric acid derivatives with two alkyl chains (R,R-tartaric acid), are formed from the recombination of two RHA• radicals, as shown by reaction 4 of Scheme 2. R,R-Tartaric acid (e.g., dihexyltartaric acid for OOA) appears to be one of the major photochemical products formed during photolysis for each of the oxoacids under study here. It is observed as the photoproduct with the highest intensity in the MS data in each of the postphotolysis solutions analyzed, regardless of whether the photolysis was conducted in air or under N2. However, this is only a relative comparison as our ESI− MS analysis was not designed to be absolutely quantitative. The “R’s” used in the names of product species throughout our discussion simply denote the presence of an alkyl chain; the “R’s” do not represent an attempt at assigning the stereochemistry of these molecules, even though these tartaric acid derivatives do contain chiral centers (the implications of which are discussed further below).
Scheme 2.

General Photochemical Pathways of Alkyl Oxoacids Following Carboxyl Hydrogen Abstraction
Further reactions stemming from the recombination of RHA• and ROA• generated by hydrogen abstraction from the alkyl tail of ROA account for several of the other main photoproducts observed by MS. As shown in reaction 2 of Scheme 3, the recombination of RHA• and ROA• forms a derivative of parapyruvic acid with two alkyl chains, R,R-parapyruvic acid (R,R-PPA), which is observed in the MS for each of the ROAs under study here (Tables S1–4). The reactions in Scheme 3 have been drawn showing hydrogen abstraction occurring at the β-CH2 group, but it is possible for the hydrogen abstraction that generates ROA• to occur anywhere along the alkyl tail.
Scheme 3.

Potential Recombination Pathways for Alkyl Oxoacids Following Alkyl Hydrogen Abstraction To Form ROA•, Here Shown at the β-CH2 Site102
In addition to the R,R-PPA and R,R-tartaric acid species, we have tentatively detected dioxoacid species, R,R-dioxodiacid (R,R-DODA), as photoproducts for most of ROA under study. Generated from the recombination of two ROA• radicals, as shown in reaction 4 of Scheme 3, R,R-DODA likely exists in equilibrium with the enol in aqueous solution. In the postphotolysis solutions for OHA, ODA, and ODDA, we observe ions corresponding to the chemical formulas of R,R-DODA in the MS (Tables S2–4). However, because the intensities of the observed ions are at the cutoff threshold for detection for all three oxoacids, these assignments are tentative. However, these tentative detections help answer questions about the photochemical mechanism underlying the production of R,R-PPA.67 The recombination of ROA• and RHA• that generates R,R-PPA cannot occur immediately after the two radicals are generated by hydrogen abstraction along the alkyl tail (reaction 1 of Scheme 3) because the radical pair generated has an overall net triplet character, preventing intracage geminate recombination. Instead, before the radicals can react further, they must either undergo intersystem crossing back to the singlet state or undergo cage escape, either of which is possible. If intersystem crossing back to the singlet state is favored, then only R,R-PPA species should be detected; however, if the radicals undergo cage escape, then products from each of the potential recombination processes for the RHA• and ROA• radicals should also be observed. As shown in reaction 3 of Scheme 3, the recombination of RHA• radicals formed from this channel can also contribute to the formation of R,R-tartaric acid, while the recombination of ROA• radicals leads to R,R-DODA as mentioned above. In the previous study of pyruvic acid that initially proposed this mechanism, the dioxoacid, 2,5-dioxohexanedioic acid, was not observed in the MS,67 possibly because of its own ability to react photochemi-cally. This means that there was no way of differentiating between cage escape and intersystem crossing back to the singlet state, which would only generate parapyruvic acid as a product.67 Here, the presence of R,R-DODA species suggests that cage escape is the favored route by which the generated radical pair can recombine, rather than intersystem crossing back to the singlet state.
Regardless of the path by which they are formed, the R,R-PPA species created by the recombination of RHA• and ROA• are themselves photoactive. They are also α-keto acids and can, therefore, undergo the same photochemistry as other alkyl oxoacids, as shown in Scheme 4. Photoexcited R,R-PPA can then encounter another ROA, or the corresponding RGDA in solution, which then decarboxylates, following the pathway outlined in reaction 2 of Scheme 2, generating both a RGD• radical and a R,R-PPA radical (R,R-PPA•), as shown in reaction 1 of Scheme 4. These two radicals can then combine, following reactions 2 and 3 of Scheme 4, generating a dihydroxyox-odialkanoic acid with three alkyl chains (R,R,R-DHO diacid) and, following decarboxylation, a dihydroxyoxoalkanoic acid (R,R,R-DHO acid). The R,R,R-DHO acid is observed in the MS for each of the ROA except for ODDA (Tables 1 and 2). The intermediate, R,R,R-DHO diacid, is, however, observed for ODDA (Table S4), as it is for ODA and OOA. For OHA, the R,R,R-DHO diacid is observed readily at 6 mM, but it is below the threshold for detection in the 1 mM solutions.
Scheme 4.

Photochemistry of R,R-PPA (Here Shown As Formed from a β-CH2 ROA•) To Generate Trimer Species of ROA102
In addition to the photochemical pathways discussed above where organic radicals are generated from bimolecular interactions, there is also a unimolecular photochemical reaction pathway.52 For the four ROA species investigated here, photoexcitation can lead to an intramolecular Norrish Type II reaction as shown in Scheme 5. This is in contrast to pyruvic acid, which does not have an alkyl chain with the γ-carbon that is necessary for this chemistry to proceed. There has been some debate in the literature as to whether the Norrish Type II reaction happens from the singlet or the triplet state,93,94 with Davidson et al. presenting evidence for its occurrence on the singlet state.94 Regardless of the manifold on which it occurs, the Norrish Type II reaction generates a biradical on the oxoacid (reaction 1, Scheme 5) that then leads to homolytic bond cleavage (reaction 2, Scheme 5).
Scheme 5.

General Norrish Type II Reaction Scheme for ROA
This additional pathway adds considerably to the complexity of the photoproducts observed for longer-tailed alkyl oxoacids as compared to pyruvic acid. The 2-hydroxy-3-propenoic acid generated by bond cleavage of the ROA in reaction 2 of Scheme 5 can rearrange to form pyruvic acid, as shown in reaction 3 of Scheme 5. This photochemically synthesized pyruvic acid can, of course, undergo all of its normal aqueous photochemistry,28,64,67 including generating dimethyltartaric acid and 2,4-dihydroxy-2-methyl-5-oxohexanoic acid (DMOHA). Both of these species are observed in post-photolysis solutions for each ROA. The intensities of these species in the MS are near the threshold for detection, as would be expected given the probability of two minor species encountering each other in solution.
It is far more likely that the photochemically generated pyruvic acid would encounter an ROA molecule, leading to a cross-reaction between the two species. The most direct reaction involves the encounter of two radicals with hydroxy-acid functionality HA• from pyruvic acid and RHA• from ROA. The recombination of these two species forms a tartaric acid derivative with one methyl and one alkyl chain, R,methyl-tartaric acid (reaction 5 of Scheme 5). The R,methyl-tartaric acid species generated by this process was also observed clearly by MS as one of the main photoproducts for each of the four oxoacids.
As the radical species under study here are promiscuous and because of the formation of pyruvic acid from ROA via the Norrish Type II reaction, the photolysis of ROA leads to complex mixtures of products. Intermediate species, such as R,R-PPA and pyruvic acid, that can go on themselves to do further photochemistry, widely expand the photoproducts that can be formed. The formation of pyruvic acid in solution with ROA means that not only can R,R-PPA be formed from two ROA• radicals recombining, but R-PPA from the recombination of one ROA• and the pyruvic OA• and even parapyruvic acid itself from the recombination of two OA• can be formed. R-PPA species are observed for each of the oxoacids, although not for 1 mM OHA. The presence of parapyruvic acid is not observed by MS, but its concentration would be expected to be low due to the relatively low statistical probability of its formation.
R,R-PPA, R-PPA, and parapyruvic acid are oligomeric intermediate species that can go on to interact with either another ROA or pyruvic acid molecule in solution, following the reactions outlined in Scheme 4. This leads to the possible formation of several DHO diacid and DHO acid species, a few of which are shown in Scheme 6 for OOA. Even assuming the only site of hydrogen abstraction on the alkyl tail of ROA is the β-CH2 group, there are a number of constitutional isomers possible for the R-DHO acid and R,R-DHO acids. Scheme 6 shows two such R,R-DHO acids, one generated by the reaction of R,R-PPA with a pyruvic acid molecule and one generated by the reaction of one of the potential isomers of R-PPA with OOA. Clearly there are other possible reactions that generate species with the same chemical formula and reactive functionality but different tail positions. Considering the potential for hydrogen abstraction from multiple sites along the alkyl tail considerably expands the pool of potential constitutional isomers generated by this chemistry as well. R,R-DHO acid and R-DHO acids are observed in the MS of the postphotolysis solutions for each of the ROA studied here, as are the corresponding diacid intermediates, with each observable peak likely corresponding to a mixture of constitutional isomers.
Scheme 6.

Select Possible Reaction Pathways for ROA• Derived from Hydrogen Abstraction from the β-CH2 of OOA To Form R,R,R-DHO Acid Species, Where R = (CH2)4CH3102
Pathways that generate reactive intermediate species, such as the formation of R,R-PPA and pyruvic acid, ensure that even an initial solution of a single ROA develops into a complex, interconnected network of reactions that generate a rich mixture of oligomeric photoproducts upon photolysis. The generated radicals by the photolysis of oxoacids are capable of interacting not just with another identical oxoacid molecule but also with other radical species present in solution. Here, we show that the ability for cross-reactions carries through to the reaction between two different ROAs in solution, photolyzing a mixed solution of OOA and ODA. A representative postphotolysis mass spectrum is shown in Figure 2, with key results in Table 3 (a representative prephotolysis spectrum is given in Figure S10).
Figure 2.

Representative MS of 3 mM OOA and 3 mM ODA after 5 h of photolysis under N2.
Table 3.
New Photoproducts from the Mixed Photolysis of OOA and ODAa
| assigned formula [M − H]− | assigned structure | avg exp m/zb | mass diff. (ppm) | prephotolysis | postphotolysis |
|---|---|---|---|---|---|
| C18H31O6 | R,R′-PPA | 343.2113 | −2.5 | medium | medium |
| C19H35O5 | R,R′-DHO acid | 343.2490 | 1.5 | below threshold | medium |
| C18H33O6 | hexyloctyl-tartaric acid | 345.2280 | 0.75 | below threshold | strong |
| C20H35O7 | R,R′-DHO diacid | 387.2380 | −0.80 | below threshold | medium |
| C24H45O5 | R,R,R′-DHO acid | 413.2370 | 0.65 | below threshold | medium |
| C26H49O5 | R,R′,R′-DHO acid | 441.3575 | −1.2 | below threshold | medium |
| C25H45O7 | R,R,R′-DHO diacid | 457.3175 | 2.0 | below threshold | weak |
| C27H49O7 | R,R′,R′-DHO diacid | 485.3480 | 0.27 | below threshold | weak |
Chemical formulas are assigned as the ionized [M − H]− species; structures are assigned as the neutral species. Here, R = (CH2)4CH3 from OOA and R′ = (CH2)6CH3 from ODA.
The experimental m/z is the average observed mass across experiments.
As shown in Scheme 5, when both OOA and ODA are in solution, the R,R-tartaric acid species generated are not only the corresponding dihexyltartaric acid (DHTA) and dioctyltartartic acid (DOTA), but also the tartaric acid species with differing tail lengths, hexyloctyltartaric acid (HOTA). As shown in Figure 2, when equal amounts of OOA and ODA are present in solution, the amount of HOTA generated is comparable to both DOTA and DHTA, suggesting that there is no great preference for like radicals to recombine with each other. This observation is likely because, to form the R,R-tartaric acid, RHA• must undergo cage escape, and so there is no particular preference for either the partner in the solvent cage or the radicals they encounter following cage escape.
In addition to the mixed R,R′-tartaric acid, HOTA, the mixed DHO acid and DHO diacid products are also observed, as shown in Table 3. The R,R′-PPA species is formed from the recombination of either a ROA• and a R′HA• or a R′OA• and a RHA•. Any ROA• radical may, in principle, have the radical anywhere along the alkyl tail, as well, which further expands the number of possible isomers. Each of R,R-PPA, R′,R′-PPA, R-PPA, R′-PPA, parapyruvic acid, and this new species R,R′-PPA can go on to react with either OOA, ODA, or pyruvic acid in solution. Of the many possible recombination products, the mixed trimer species, R,R′,R′-DHO acid, R,R,R′-DHO acid, and the R,R′-DHO acid, are newly observed photoproducts, along with the corresponding DHO diacid intermediate species.
Clearly, the introduction of even one other starting species greatly adds to the already complex mixture of photoproducts that may be generated from alkyl oxoacids. However, it is worth noting that, while there are many possible oligomeric photoproducts that may be generated, each with several potential constitutional isomers, the photochemistry under study here can be explained using variants on well-understood mechanistic motifs,28,52,64,67 identifying a clear majority of the observed photochemical products, even for the mixed ROA solutions. Indeed, even the generation of R,R-DHO acid from the further reactions of R,R-PPA is, essentially, an equivalent reaction pathway to that which forms hydroxyalkanone (acetoin for pyruvic acid64) from the decarboxylation of the β-keto acid, R,R-2-hydroxy, 3-oxoalkanoic acid, which is generated from the recombination of RHA• and RGD•, as shown in reaction 6 of Scheme 2.67 The R,R-2-hydroxy, 3-oxoalkanoic acids are observed by MS for each ROA (Tables S1–4), and for OOA the hydroxyalkanone is also observed as a minor photoproduct. However, this species is not observed above our detection threshold for any of the other ROA.
Simple fatty acid species (alkanoic acid) are also generated as a minor product from the photolysis of ROA, following reaction 5 of Scheme 2. The alkanoic acid (e.g., heptanoic acid for OOA) is detected both before and after photolysis. It is likely that fatty acids are a common contaminant of the initial ROA starting material, just as acetic acid is a known contaminant of pyruvic acid. Interestingly, the corresponding 2-hydroxyalkanoic acid is not observed by MS for the ROA examined here. It has been previously shown that simple carboxylic acids do not have a strong intensity in the ESI− MS under the experimental conditions used here,67 and, given the presence of many other more surface-active photoproducts, the intensity of the MS signal is not quantitatively reflective of their concentration in solution. However, this may also indicate that the branching ratio of products observed for the longer-tailed, alkyl ROA differs somewhat from pyruvic acid. While both acetic acid and lactic acid generated from pyruvic acid following reaction 5 of Scheme 2 are minor photoproducts under oxygen-limited conditions, they are readily observed by NMR,28,64,67 and lactic acid has been observed by MS as well.67
Small changes in product branching ratios between pyruvic acid and the longer-tailed ROA, such as OOA, would not be surprising. It also appears as though OOA has a slower rate of reaction than pyruvic acid. Quantifying the amount of OOA consumed during photolysis is more difficult than for pyruvic because of the additional signal in the NMR due to the hydrogens on the alkyl chains, but we estimate that for the 3 mM OOA solutions approximately 30% of the OOA is consumed after 5 h of photolysis. This is a smaller yield than for pyruvic acid (~90% consumed) under similar reaction conditions.67 It is possible that we are underestimating the consumption of OOA, as the signal from some H atoms may be lost through the production of volatile species that partition to the gas phase, the formation of water, or due to the large aggregates we observe, which will have low NMR response. The aggregates formed by OOA photolysis are discussed further below.
There are several likely contributing factors for this apparent decrease in reactivity for the longer-tailed oxoacid. Hydrogen abstraction is favored energetically from the diol form of the oxoacid rather than the keto form; therefore, the chemistry might be less favorable because the ratio of diol to keto shifts for the longer-tailed species. For solubility reasons, we used lower concentrations of OOA than PA (~3–6 mM OOA vs 10 mM PA). Because the chemistry used to generate the oligomeric products relies on radical species encountering each other, the kinetics of reaction likely decrease for lower concentrations of oxoacid.
While the product yields and the exact branching ratios may differ slightly between pyruvic acid and the ROA studied here, the chemistry retains its sensitivity to environmental conditions even when longer alkyl chains are added to the oxoacids. The ESI− MS experiments used here were not designed to be quantitative, but these results can be used to infer qualitative differences in product yields when comparing very similar samples, especially if the effect is large. As shown in Figure 3, the difference between the postphotolysis samples that were irradiated in air versus in nitrogen is stark. While the oligomeric dihexyltartaric acid is the major observed product under both conditions, considerably more of the oligomeric products are formed in a nitrogen environment. Interestingly, while this effect is also seen in the NMR, the overall decrease in OOA concentration is only modestly increased under a nitrogen atmosphere (Figure S11), shifting from approximately 20% consumption in air to 30% in nitrogen following 5 h of photolysis. As mentioned above, this relatively small shift could be due to the loss of signal when the generated products are tied up into larger aggregates. Additionally, because the products generated by photolysis are oligomers of OOA, even a relatively modest change in overall consumption can have a large effect on the formation and yield of photoproducts. For example, there is a noticeable decrease in the relative amounts of the oligomeric photoproducts that are formed from the further reactions of species generated by photolysis, such as methylhexyltartaric acid and the DHO acid species. This result suggests that those processes that require two photochemical steps are less favored in the presence of oxygen, which can act as a radical quencher.
Figure 3.

Representative MS of 3 mM OOA after 5 h of photolysis under N2 (red) and open to air (blue, counts multiplied by −1 for ease of presentation).
We have observed here, for the first time, that environmental conditions such as the concentration of oxygen in the photolysis reactor affect the longer-tailed oxoacids in the same manner as was demonstrated for pyruvic acid.28 The increased yield of such oligomers in the absence of oxygen is to be expected, given both that oxygen is known to quench triplet state chemistry and that oxoacids may be regenerated through reactions with oxygen (reaction 3A in Scheme 2). However, it is worth noting that, while the formation of oligomeric products is reduced when oxygen is present in solution, oligomers are still formed and are the major observable product. This is consistent with previous studies on pyruvic acid that show that oligomers are formed in the presence of oxygen,28,69 including under conditions that closely replicate the modern atmos-phere.68 Nevertheless, the ability to shift the reaction branching ratios by shifting environmental conditions has significant implications, especially for longer, lipid-like oxoacids.
Anoxic conditions favor the formation of more complex oligomeric species, many of which have two or three alkyl chains. This dependence is intriguing because the prebiotic atmosphere, unlike that of today, contained very little O2 and ozone, allowing more UV light to reach the Earth’s surface.4,104 The absence of oxygen in the prebiotic atmosphere would render the common, modern photo-oxidation reactions with hydroxyl radical negligible, and makes it likely that direct photochemistry of carbonyl-containing compounds would be a more significant process on the ancient Earth than it is today. This points to the potential of such chemistry to generate more complex, double- and triple-tailed amphiphiles under prebioti-cally relevant conditions. This chemistry is among one of the only processes by which multitailed lipids from single-tailed precursors can be formed simply and in relatively high yields under prebiotically plausible conditions and using prebiotically plausible precursors.4,52
The generation of a large library of photoproducts, including that of mixed oligomeric species from two different ROA, shows the specificity of the photochemistry governing the formation of the radicals from oxoacids, while also demonstrating their ability to react promiscuously with other species in solution. The reactivity of mixtures and the ways in which such mixtures produce molecular complexity is inherently more applicable to environmental chemistry whether on the modern or ancient Earth than single molecule systems.
The photochemical pathways followed by oxoacids are robust and readily generalizable to the series of molecules under study here. While, for example, the Norrish Type II reaction pathway is accessible for the longer-tailed oxoacids, it is remarkable how consistent the generated photoproducts are across alkyl tail lengths. Although, within the photochemical framework there are many constitutional isomers possible for the R,R-DHO acids generated by Scheme 4 for the longer-tailed species, which we cannot distinguish within the scope of this study, it is remarkable how well a relatively simple mechanistic framework can describe the observed photochemistry. For each ROA under study, almost all significant ions that are observed in the MS corresponding to oligomeric species can be identified in this way, with only a few minor ions escaping classification. One might expect that as the molecules under study are increased in size, the complexity of the observed products would also increase. This is, however, not the case. If anything, the observed MS are “cleaner” than those of pyruvic acid under similar reaction conditions.67 This could be because of the surface partitioning of the longer-tailed species during mass spectrometry analysis, which is biased toward observation of the more surface-active species, a bias that increases with the longer alkyl tail of the starting oxoacid. Or, it could be because of the steric effects of the oligomerization process cutting off further reaction channels that produce minor products.
Properties of Generated Photoproducts
The major products, the tartaric acid derivatives and DHO acids, are interesting because they are molecules with chiral centers that have been produced from achiral starting materials. The tartaric acid derivatives produced, R,R-tartaric acid and the R-methyl-tartaric acid, have two chiral centers. For the double-tailed, R,R-tartaric acid molecules, one of the three stereoisomers produced is a meso isomer; however, in the case of the R,methyl-tartaric acid molecules and the mixed HOTA molecule, there is no meso isomer. In the case of the DHO acids, for oxoacids larger than pyruvic acid, there are always at least two chiral centers on each molecule, with the potential for three. The molecules generated are almost certainly a racemic mixture of all of the possible isomers. However, as has been noted in the literature, enantioselective environments may be able to generate an optically active product mixture.105–108
The abiotic production of chiral molecules under environmentally relevant conditions points to the importance of understanding the sources of chiral molecules in the modern environment. The SML and the surfaces of aerosols have been shown to contain chiral molecules, including lipids,109 but these are almost always assumed to be biotic in origin.14,26,27 However, as demonstrated here, it is possible to make chiral molecules in high yields from very simple, achiral precursors, suggesting that the potential for abiotic environmental sources of chirality should not be discounted.
During photolysis, oligomeric photoproducts with two and three alkyl chains are generated from single-chained ROA species. These multitailed amphiphilic products will likely have more surface activity than starting material, as has been shown for OOA.52 The increase in surface activity during photolysis also translates to a reduction in the critical aggregation concentration. Each of the oxoacids is observed to undergo self-assembly, forming aggregates that scatter light and turn the solutions visibly cloudy. For OHA, OOA, and the mixed 760 solutions of OOA and ODA, the aggregates were characterized 761 using DLS, and found to be monodisperse and uniform, with 762 sizes ranging from ~150 to ~250 nm in diameter, depending 763 on the ROA (Table 4). This aggregation behavior has been previously reported for OOA, with microscopy indicating a spherical morphology.52 However, even the very short-chained OHA is observed to assemble into such aggregates, with an average size of ~250 nm in diameter, larger than the ~160 nm observed for OOA. Prior to photolysis, the precursor oxoacids do not form aggregates, as confirmed by DLS, and are below their critical aggregation concentration (except for, perhaps, ODDA), but as the photolysis generates multitailed oligomeric species aggregation is observed, even at 1 mM initial starting concentrations, for all oxoacids. The generated oligomers, therefore, have much lower critical aggregation concentrations than the starting material, as would be expected for multitailed species.110
Table 4.
Average Sizes of Aggregates Observed by Dynamic Light Scattering
| ROA | avg observed diameter (nm)a |
|---|---|
| OHA | 250 ± 20 |
| OOA | 160 ± 20 |
| mixed OOA/ODA | 150 ± 10 |
Uncertainty is expressed as a 95% confidence interval.
The diameters of the aggregates observed here for OHA, OOA, and the mixed OOA:ODA are too large to be simple micelles, and the monodispersity in their size indicates ordered aggregation. It has previously been speculated that the aggregates observed for OOA may be vesicles,52 and the observed decrease in size as a function of increasing tail length is also suggestive that these aggregates are vesicles. The ratio of headgroup size to alkyl tail length is quite large for the oligomeric species generated from the photolysis of OHA, corresponding to a larger packing parameter111 than for the longer tail-length oligomers. The packing parameter for the multitailed lipids generated from the photolysis of each of the alkyl oxoacids studied here is likely near one, a regime that generally is associated with bilayer formation.111 If bilayers are formed, species with a higher packing parameter generally assemble into lower curvature, larger structures, which is consistent with the trend in size of aggregate observed here.
CONCLUSIONS
We have shown that the photochemical reaction mechanisms for α-keto acids in aqueous solution are readily generalizable to a series of molecules with differing alkyl chain lengths. Differences in reactivity due to chain length might be expected, but for all of the oxoacids under study here the chemical reactivity between molecules is surprisingly consistent, although a new intramolecular Norrish Type II pathway is accessible for alkyl oxoacids with a γ-CH2 group that is not available to the shorter pyruvic acid.
Even a simple starting solution of a single alkyl oxoacid species becomes a complex mixture of oligomeric species upon photolysis. This is due in large part to the formation of reactive intermediate species with the same α-keto functionality, including pyruvic acid and parapyruvic acid derivatives. These species also undergo photochemistry, and the organic radicals generated from each of the oxoacids can react indiscriminately with each other in aqueous solution. For processes that form radicals on the alkyl chain of an oxoacid, it is likely that hydrogen abstraction occurs at multiple sites along the alkyl chain, allowing for the formation of several constitutional isomers with the same chemical formula that cannot be distinguished from each other via the MS analysis conducted here. However, despite this, the products are not intractable as the majority of the observed species can be readily identified using simple mechanistic schemes.
The oligomers formed from this photochemistry are amphiphiles, many of which have two or three alkyl chains. The generation of chiral, multitailed species with increased surface activity and their subsequent self-assembly into monodisperse, spherical aggregates has important environmental implications both for today and for the early Earth. In the modern environment, this chemistry may contribute to the abiotic processes by which surface-active species are formed at the sea surface microlayer. Prebiotically, the photochemical reactions of alkyl oxoacids are one of the only demonstrated processes by which multitailed lipids can be formed simply and in relatively high yields from prebiotically relevant starting materials. The properties of these multitailed lipids, as well as their propensity to self-assemble into ordered aggregates shown here, demonstrate their potential importance in the generation of primitive enclosures.
Acknowledgments
We thank Dr. Jeremy L. Balsbaugh and the University of Colorado at Boulder Central Analytical Laboratory Mass Spectrometry Core Facility (partially funded by NIH S10 RR026641) for mass spectrometry measurements and advice about analysis. H.Y. was supported by the University of Colorado Boulder. G.M.M. is grateful for support from the University of Colorado Boulder and the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM119702. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Financial support for R.J.R., R.J.P., and V.V. was provided by the National Science Foundation (CHE 1306386), the National Aeronautics and Space Administration under grant no. NNX15AP20G issued through the Habitable Worlds Program, and a CIRES Innovative Research Proposal. R.J.P. acknowledges support by the NIH/CU Molecular Biophysics Training Program. R.J.R. also acknowledges support by NASA Headquarters under the NASA Earth and Space Science Fellowship Program – Grant NNX13AP85H and partial support from both the University of Colorado Center for the Study of Origins and a CIRES Graduate Student Research Award.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01707.
Detailed descriptions of the synthesis of alkyl oxoacids, instrument parameters for the analytical techniques (dynamic light scattering, UV–vis and NMR spectroscopy, and high-resolution negative mode electrospray ionization mass spectrometry), additional UV–vis, NMR, and high-resolution ESI mass spectra, and detailed tables of mass spectrometry analysis for each alkyl oxoacid (PDF)
The authors declare no competing financial interest.
References
- 1.Miyazaki Y, Sawano M, Kawamura K. Biogeosciences. 2014;11(16):4407–4414. [Google Scholar]
- 2.Rinaldi M, Decesari S, Finessi E, Giulianelli L, Carbone C, Fuzzi S, Dowd CD, Ceburnis D, Facchini MC. Advances in Meteorology. 2010;2010:310682. [Google Scholar]
- 3.Thornton DCO, Brooks SD, Chen J. Front. Mar. Sci. 2016;3:135. [Google Scholar]
- 4.Rapf RJ, Vaida V. Phys. Chem. Chem. Phys. 2016;18(30):20067–20084. doi: 10.1039/c6cp00980h. [DOI] [PubMed] [Google Scholar]
- 5.Donaldson DJ, Vaida V. Chem. Rev. 2006;106:1445–1461. doi: 10.1021/cr040367c. [DOI] [PubMed] [Google Scholar]
- 6.Griffith EC, Tuck AF, Vaida V. Acc. Chem. Res. 2012;45(12):2106–2113. doi: 10.1021/ar300027q. [DOI] [PubMed] [Google Scholar]
- 7.Deamer D, Weber AL. Cold Spring Harbor Perspect. Biol. 2010;2:1–16. doi: 10.1101/cshperspect.a004929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stano P, D’Aguanno E, Bolz J, Fahr A, Luisi PL. Angew. Chem., Int. Ed. 2013;52(50):13397–13400. doi: 10.1002/anie.201306613. [DOI] [PubMed] [Google Scholar]
- 9.Monnard P-A, Walde P. Life. 2015;5(2):1239–1263. doi: 10.3390/life5021239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cunliffe M, Engel A, Frka S, Gasparovic B, Guitart C, Murrell JC, Salter M, Stolle C, Upstill-Goddard R, Wurl O. Prog. Oceanogr. 2013;109:104–116. [Google Scholar]
- 11.Ciuraru R, Fine L, Pinxteren Mv, D’Anna B, Herrmann H, George C. Environ. Sci. Technol. 2015;49:13199–13205. doi: 10.1021/acs.est.5b02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossignol S, Tinel L, Bianco A, Passananti M, Brigante M, Donaldson DJ, George C. Science. 2016;353(6300):699–702. doi: 10.1126/science.aaf3617. [DOI] [PubMed] [Google Scholar]
- 13.Chiu R, Tinel L, Gonzalez L, Ciuraru R, Bernard F, George C, Volkamer R. Geophys. Res. Lett. 2017;44(2):1079–1087. [Google Scholar]
- 14.Cochran RE, Laskina O, Jayarathne T, Laskin A, Laskin J, Lin P, Sultana C, Lee C, Moore KA, Cappa CD. Environ. Sci. Technol. 2016;50(5):2477–2486. doi: 10.1021/acs.est.5b04053. [DOI] [PubMed] [Google Scholar]
- 15.Tervahattu H, Hartonen K, Kerminen VM, Kupiainen K, Aarnio P, Koskentalo T, Tuck AF, Vaida VJ. Geophys. Res. 2002;107:4053–4060. [Google Scholar]
- 16.Tervahattu H, Juhanoja J, Vaida V, Tuck AF, Niemi JV, Kupiainen K, Kulmala M, Vehkamaki H. J. Geophys. Res. 2005;110:D06207. [Google Scholar]
- 17.Ervens B, Turpin BJ, Weber RJ. Atmos. Chem. Phys. 2011;11(21):11069–11102. [Google Scholar]
- 18.Carlton AG, Wiedinmyer C, Kroll JH. Atmos. Chem. Phys. 2009;9(14):4987–5005. [Google Scholar]
- 19.Hallquist M, Wenger JC, Baltensperger U, Rudich Y, Simpson D, Claeys M, Dommen J, Donahue NM, George C, Goldstein AH, Hamilton JF, Herrmann H, Hoffmann T, Iinuma Y, Jang M, Jenkin ME, Jimenez JL, Kiendler-Scharr A, Maenhaut W, McFiggans G, Mentel TF, Monod A, Prévôt ASH, Seinfeld JH, Surratt JD, Szmigielski R, Wildt J. Atmos. Chem. Phys. 2009;9(14):5155–5236. [Google Scholar]
- 20.Herrmann H. Chem. Rev. 2003;103(12):4691–4716. doi: 10.1021/cr020658q. [DOI] [PubMed] [Google Scholar]
- 21.Herrmann H, Schaefer T, Tilgner A, Styler SA, Weller C, Teich M, Otto T. Chem. Rev. 2015;115(10):4259–4334. doi: 10.1021/cr500447k. [DOI] [PubMed] [Google Scholar]
- 22.Brégonzio-Rozier L, Giorio C, Siekmann F, Pangui E, Morales S, Temime-Roussel B, Gratien A, Michoud V, Cazaunau M, DeWitt H. Atmos. Chem. Phys. 2016;16(3):1747–1760. [Google Scholar]
- 23.Renard P, Reed Harris AE, Rapf RJ, Rainer S, Demelas C, Coulomb B, Quivet E, Vaida V, Monod A. J. Phys. Chem. C. 2014;118:29421–29430. [Google Scholar]
- 24.Renard P, Siekmann F, Gandolfo A, Socorro J, Salque G, Ravier S, Quivet E, Clement JL, Traikia M, Delort AM, Voisin D, Vuitton V, Thissen R, Monod A. Atmos. Chem. Phys. 2013;13(13):6473–6491. [Google Scholar]
- 25.Vaida V. J. Phys. Chem. A. 2009;113:5–18. doi: 10.1021/jp806365r. [DOI] [PubMed] [Google Scholar]
- 26.Alves CA. Quim. Nova. 2014;37:1382–1400. [Google Scholar]
- 27.Wang X, Sultana CM, Trueblood J, Hill TCJ, Malfatti F, Lee C, Laskina O, Moore KA, Beall CM, McCluskey CS, Cornwell GC, Zhou Y, Cox JL, Pendergraft MA, Santander MV, Bertram TH, Cappa CD, Azam F, DeMott PJ, Grassian VH, Prather KA. ACS Cent. Sci. 2015;1(3):124–131. doi: 10.1021/acscentsci.5b00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reed Harris AE, Ervens B, Shoemaker RK, Kroll JA, Rapf RJ, Griffith EC, Monod A, Vaida V. J. Phys. Chem. A. 2014;118(37):8505–8516. doi: 10.1021/jp502186q. [DOI] [PubMed] [Google Scholar]
- 29.Vaida V. Science. 2016;353(6300):650–650. doi: 10.1126/science.aah4111. [DOI] [PubMed] [Google Scholar]
- 30.Vaida V. ACS Cent. Sci. 2015;1(3):112–114. doi: 10.1021/acscentsci.5b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoque MMM, Kawamura K, Uematsu M. Atmos. Res. 2017;185:158–168. [Google Scholar]
- 32.Mochida M, Kitamori Y, Kawamura K, Nojiri Y, Suzuki K. J. Geophys. Res. Atmos. 2002;107:4325. [Google Scholar]
- 33.Kawamura K, Umemoto N, Mochida M, Bertram T, Howell S, Huebert BJ. J. Geophys. Res. Atmos. 2003;108:8639. [Google Scholar]
- 34.Long M, Keene W, Kieber D, Frossard A, Russell L, Maben J, Kinsey J, Quinn P, Bates T. Geophys. Res. Lett. 2014;41(7):2661–2670. [Google Scholar]
- 35.Galloway MM, Powelson MH, Sedehi N, Wood SE, Millage KD, Kononenko JA, Rynaski AD, De Haan DO. Environ. Sci. Technol. 2014;48(24):14417–14425. doi: 10.1021/es5044479. [DOI] [PubMed] [Google Scholar]
- 36.George C, Ammann M, D’Anna B, Donaldson DJ, Nizkorodov SA. Chem. Rev. 2015;115(10):4218–4258. doi: 10.1021/cr500648z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rudich Y. Chem. Rev. 2003;103(12):5097–5124. doi: 10.1021/cr020508f. [DOI] [PubMed] [Google Scholar]
- 38.Perraud V, Bruns EA, Ezell MJ, Johnson SN, Yu Y, Alexander ML, Zelenyuk A, Imre D, Chang WL, Dabdub D, Pankow JF, Finlayson-Pitts B. Proc. Natl. Acad. Sci. U. S. A. 2012;109(8):2836–2841. doi: 10.1073/pnas.1119909109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison RM, Yin J. Sci. Total Environ. 2000;249(1):85–101. doi: 10.1016/s0048-9697(99)00513-6. [DOI] [PubMed] [Google Scholar]
- 40.Finlayson-Pitts BJ, Pitts JN. Chemistry of the Upper and Lower Atmosphere. Academic Press; San Diego, CA: 1999. [Google Scholar]
- 41.Hinds WC. Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles. 2. Wiley; New York: 1999. p. 483. [Google Scholar]
- 42.Ervens B. Chem. Rev. 2015;115(10):4157–4198. doi: 10.1021/cr5005887. [DOI] [PubMed] [Google Scholar]
- 43.Lohmann U, Feichter J. Atmos. Chem. Phys. 2005;5:715–737. [Google Scholar]
- 44.Hansen J, Sato M, Ruedy R. J. Geophys. Res. Atmos. 1997;102(D6):6831–6864. [Google Scholar]
- 45.Seinfeld JH, Pandis SN. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. 2. Wiley; Hoboken, NJ: 2006. p. 1203. [Google Scholar]
- 46.Seinfeld JH, Pandis SN. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. John Wiley & Sons, Inc; New York: 1998. [Google Scholar]
- 47.Chebbi A, Carlier P. Atmos. Environ. 1996;30(24):4233–4249. [Google Scholar]
- 48.Ho K, Lee S, Cao J, Kawamura K, Watanabe T, Cheng Y, Chow JC. Atmos. Environ. 2006;40(17):3030–3040. [Google Scholar]
- 49.Cooper G, Reed C, Nguyen D, Carter M, Wang Y. Proc. Natl. Acad. Sci. U. S. A. 2011;108(34):14015–14020. doi: 10.1073/pnas.1105715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pizzarello S. Rend. Lincei. 2011;22(2):153–163. [Google Scholar]
- 51.McCollom TM, Ritter G, Simoneit BRT. Origins Life Evol. Biospheres. 1999;29(2):153–166. doi: 10.1023/a:1006592502746. [DOI] [PubMed] [Google Scholar]
- 52.Griffith EC, Rapf RJ, Shoemaker RK, Carpenter BK, Vaida V. J. Am. Chem. Soc. 2014;136(10):3784–3787. doi: 10.1021/ja5006256. [DOI] [PubMed] [Google Scholar]
- 53.Plath KL, Takahashi K, Skodje RT, Vaida V. J. Phys. Chem. A. 2009;113:7294–7303. doi: 10.1021/jp810687t. [DOI] [PubMed] [Google Scholar]
- 54.Vesley GF, Leermakers PA. J. Phys. Chem. 1964;68(8):2364–2366. [Google Scholar]
- 55.Takahashi K, Plath KL, Skodje RT, Vaida V. J. Phys. Chem. A. 2008;112(32):7321–7331. doi: 10.1021/jp803225c. [DOI] [PubMed] [Google Scholar]
- 56.Yamamoto S, Back RA. Can. J. Chem. 1985;63(2):549–554. [Google Scholar]
- 57.Mellouki A, Mu YJ. J. Photochem. Photobiol., A. 2003;157(2–3):295–300. [Google Scholar]
- 58.Horowitz A, Meller R, Moortgat GK. J. Photochem. Photobiol., A. 2001;146(1):19–27. [Google Scholar]
- 59.Schreiner PR, Reisenauer HP, Ley D, Gerbig D, Wu C-H, Allen WD. Science. 2011;332(6035):1300–1303. doi: 10.1126/science.1203761. [DOI] [PubMed] [Google Scholar]
- 60.Reed Harris AE, Doussin J-F, Carpenter BK, Vaida V. J. Phys. Chem. A. 2016;120(51):10123–10133. doi: 10.1021/acs.jpca.6b09058. [DOI] [PubMed] [Google Scholar]
- 61.Rincon AG, Guzman MI, Hoffmann MR, Colussi AJ. J. Phys. Chem. A. 2009;113(39):10512–10520. doi: 10.1021/jp904644n. [DOI] [PubMed] [Google Scholar]
- 62.Closs GL, Miller RJ. J. Am. Chem. Soc. 1978;100(11):3483–3494. [Google Scholar]
- 63.Guzman MI, Colussi AJ, Hoffmann MR. J. Phys. Chem. A. 2006;110(10):3619–3626. doi: 10.1021/jp056097z. [DOI] [PubMed] [Google Scholar]
- 64.Griffith EC, Carpenter BK, Shoemaker RK, Vaida V. Proc. Natl. Acad. Sci. U. S. A. 2013;110(29):11714–11719. doi: 10.1073/pnas.1303206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leermakers PA, Vesley GF. J. Org. Chem. 1963;28:1160–1161. [Google Scholar]
- 66.Larsen MC, Vaida V. J. Phys. Chem. A. 2012;116:5840–5846. doi: 10.1021/jp2087972. [DOI] [PubMed] [Google Scholar]
- 67.Rapf RJ, Perkins RJ, Carpenter BK, Vaida V. J. Phys. Chem. A. 2017 doi: 10.1021/acs.jpca.7b03310. under review. [DOI] [PubMed] [Google Scholar]
- 68.Reed Harris AE, Pajunoja A, Cazaunau M, Gratien A, Pangui E, Monod A, Griffith EC, Virtanen A, Doussin JF, Vaida V. J. Phys. Chem. A. 2017 doi: 10.1021/acs.jpca.7b01107. [DOI] [PubMed] [Google Scholar]
- 69.Eugene AJ, Guzman MI. J. Phys. Chem. A. 2017;121:2924. doi: 10.1021/acs.jpca.6b11916. [DOI] [PubMed] [Google Scholar]
- 70.Kawamura K, Kasukabe H, Barrie LA. Atmos. Environ. 1996;30(10–11):1709–1722. [Google Scholar]
- 71.Sempere R, Kawamura K. Atmos. Environ. 1994;28(3):449–459. [Google Scholar]
- 72.Nguyen TB, Bateman AP, Bones DL, Nizkorodov SA, Laskin J, Laskin A. Atmos. Environ. 2010;44(8):1032–1042. [Google Scholar]
- 73.Veres PR, Roberts JM, Cochran AK, Gilman JB, Kuster WC, Holloway JS, Graus M, Flynn J, Lefer B, Warneke C, de Gouw J. Geophys. Res. Lett. 2011;38:L17807. [Google Scholar]
- 74.Warneck P. J. Atmos. Chem. 2005;51(2):119–159. [Google Scholar]
- 75.Altieri KE, Carlton AG, Lim H-J, Turpin BJ, Seitzinger SP. Environ. Sci. Technol. 2006;40(16):4956–4960. doi: 10.1021/es052170n. [DOI] [PubMed] [Google Scholar]
- 76.Andreae MO, Talbot RW, Li SM. J. Geophys. Res. 1987;92(D6):6635–6641. [Google Scholar]
- 77.Talbot R, Andreae M, Berresheim H, Jacob DJ, Beecher K. J. Geophys. Res. 1990;95(D10):16799–16811. [Google Scholar]
- 78.Veres P, Roberts JM, Burling IR, Warneke C, de Gouw J, Yokelson RJ. J. Geophys. Res. 2010;115:D23. [Google Scholar]
- 79.Perkins RJ, Shoemaker RK, Carpenter BK, Vaida V. J. Phys. Chem. A. 2016;120(51):10096–10107. doi: 10.1021/acs.jpca.6b10526. [DOI] [PubMed] [Google Scholar]
- 80.The terms “dimer” and “trimer” are used to refer to covalently bonded oligomeric species, unless otherwise specified.
- 81.Hargreaves WR, Mulvihill SJ, Deamer DW. Nature. 1977;266(5597):78–80. doi: 10.1038/266078a0. [DOI] [PubMed] [Google Scholar]
- 82.Rao M, Eichberg J, Oro J. J. Mol. Evol. 1982;18(3):196–202. doi: 10.1007/BF01733046. [DOI] [PubMed] [Google Scholar]
- 83.Stano P, D’Aguanno E, Bolz J, Fahr A, Luisi PL. Angew. Chem., Int. Ed. 2013;52(50):13397–13400. doi: 10.1002/anie.201306613. [DOI] [PubMed] [Google Scholar]
- 84.Chen IA, Walde P. Cold Spring Harbor Perspect. Biol. 2010;2:7. doi: 10.1101/cshperspect.a002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pohorille A, Deamer D. Res. Microbiol. 2009;160(7):449–456. doi: 10.1016/j.resmic.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 86.Hanczyc M, Fujikawa S, Szostak J. Science (Washington, DC, U. S.) 2003;302:618–22. doi: 10.1126/science.1089904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mansy SS, Szostak JW. Cold Spring Harbor Symp. Quant. Biol. 2009;74:47–54. doi: 10.1101/sqb.2009.74.014. [DOI] [PubMed] [Google Scholar]
- 88.Xu H, Du N, Song Y, Song S, Hou W. Soft Matter. 2017;13:2246–2252. doi: 10.1039/c6sm02665f. [DOI] [PubMed] [Google Scholar]
- 89.Pedersen KJ. Acta Chem. Scand. 1952;6(2):243–256. [Google Scholar]
- 90.Pocker Y, Meany JE, Nist BJ, Zadorojny C. J. Phys. Chem. 1969;73(9):2879–2882. [Google Scholar]
- 91.Leermakers PA, Vesley GF. J. Chem. Educ. 1964;41(10):535. [Google Scholar]
- 92.Turro NJ, Ramamurthy V, Scaiano JC. Modern Molecular Photochemistry of Organic Molecules. University Science Books; Sausalito, CA: 2010. [Google Scholar]
- 93.Evans TR, Leermakers PA. J. Am. Chem. Soc. 1968;90(7):1840–1842. doi: 10.1021/ja01027a001. [DOI] [PubMed] [Google Scholar]
- 94.Davidson RS, Goodwin D, de Violet PF. Tetrahedron Lett. 1981;22(26):2485–2486. [Google Scholar]
- 95.Davidson RS, Goodwin D. J. Chem. Soc., Perkin Trans. 1982;2(12):1559–1564. [Google Scholar]
- 96.Davidson RS, Goodwin D, De Violet PF. Chem. Phys. Lett. 1981;78(3):471–474. [Google Scholar]
- 97.Leermakers PA, Vesley GF. J. Am. Chem. Soc. 1963;85(23):3776–3779. [Google Scholar]
- 98.Axson JL, Takahashi K, De Haan DO, Vaida V. Proc. Natl. Acad. Sci. U. S. A. 2010;107(15):6687–6692. doi: 10.1073/pnas.0912121107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buschmann HJ, Dutkiewicz E, Knoche W. Ber. Bunsen-Ges. Phys. Chem. 1982;86:129–134. [Google Scholar]
- 100.Maron MK, Takahashi K, Shoemaker RK, Vaida V. Chem. Phys. Lett. 2011;513(4):184–190. [Google Scholar]
- 101.Buschmann HJ, Füldner HH, Knoche W. Ber. Bunsen-Ges. Phys. Chem. 1980;84(1):41–44. [Google Scholar]
- 102.Here, ROA• is shown with the radical on the β-CH2 group for ease of presentation. Hydrogen abstraction can, in principle, occur anywhere along the alkyl chain.
- 103.Brocks JJ, Beckhaus H-D, Beckwith AL, Rüchardt C. J. Org. Chem. 1998;63(6):1935–1943. [Google Scholar]
- 104.Ranjan S, Sasselov DD. Astrobiology. 2016;16(1):68–88. doi: 10.1089/ast.2015.1359. [DOI] [PubMed] [Google Scholar]
- 105.Albrecht M, Borba A, Barbu-Debus KL, Dittrich B, Fausto R, Grimme S, Mahjoub A, Nedić M, Schmitt U, Schrader L, Suhm MA, Zehnacker-Rentien A, Zischang J. New J. Chem. 2010;34(7):1266–1285. [Google Scholar]
- 106.Dong H, Ignés-Mullol J, Claret J, Pérez L, Pinazo A, Sagués F. Chem. - Eur. J. 2014;20(24):7396–7401. doi: 10.1002/chem.201400248. [DOI] [PubMed] [Google Scholar]
- 107.Toxvaerd S. Int. J. Mol. Sci. 2009;10(3):1290–1299. doi: 10.3390/ijms10031290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pizzarello S, Weber AL. Origins Life Evol. Biospheres. 2010;40(1):3–10. doi: 10.1007/s11084-009-9178-1. [DOI] [PubMed] [Google Scholar]
- 109.Martinez IS, Peterson MD, Ebben CJ, Hayes PL, Artaxo P, Martin ST, Geiger FM. Phys. Chem. Chem. Phys. 2011;13(26):12114–12122. doi: 10.1039/c1cp20428a. [DOI] [PubMed] [Google Scholar]
- 110.Tanford C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. 2. J. Wiley; New York: 1980. [Google Scholar]
- 111.Gradzielski Ma. J. Phys.: Condens. Matter. 2003;15(19):R655. [Google Scholar]