

Abstract
Ubiquitin linkage to cysteine is an unconventional modification targeting protein for degradation. However, the physiological regulation of cysteine ubiquitination is still mysterious. Here we found that ACAT2, a cellular enzyme converting cholesterol and fatty acid to cholesteryl esters, was ubiquitinated on cysteine 277 (C277) for degradation when lipid level was low. gp78/Insigs catalyzed K48-linked polyubiquitination on this C277. High concentration of cholesterol and fatty acid, however, induced cellular reactive oxygen species (ROS) that oxidized C277, rendering ACAT2 stabilization and subsequently elevated cholesteryl esters. Furthermore, ACAT2 knockout mice were more susceptible to HFD-associated insulin resistance. By contrast, expression of constitutively stable form of ACAT2 (C277A) resulted in higher insulin sensitivity. Together, these data indicate that lipid-induced stabilization of ACAT2 ameliorates lipotoxicity from excessive cholesterol and fatty acid. This unconventional cysteine ubiquitination of ACAT2 constitutes an important mechanism sensing lipid overload-induced ROS and fine-tuning lipid homeostasis.
Introduction
The ubiquitin-proteasome pathway plays a vital role in regulating protein homeostasis. Ubiquitin is often linked to lysine (Lys, K) residues and less frequently to the N-terminus of proteins via an amide bond. Some viral E3s can also attach ubiquitin to the SH moiety of cysteine (Cys, C) residues through a thioester bond1. This unconventional ubiquitin linkage similarly targets the modified proteins, such as myosin heavy chain I, for proteasomal degradation. However, little is known about the physiological significance of a Cys being an ubiquitination site.
Cholesterol and fatty acid (FA) are essential lipids involved in many biological processes. However, excessive polar lipids, such as free cholesterol (FC) and free FA (FFA), are the major risk factors for atherosclerosis and type 2 diabetes mellitus. One of the mechanisms by which lipid overload causes lipotoxicity and insulin resistance is the generation of reactive oxygen species (ROS)2. Cholesterol and oxysterols could cause mitochondria dysfunction and ROS production3, 4. Saturated fatty acids, but not unsaturated fatty acids, also efficiently induce ROS generation through activating NADPH oxidation and mitochondrial electron transport5, 6. However, how cells sense and adapt to lipid overload-induced oxidative stress remains elusive.
Acyl-CoA: cholesterol acyltransferase (ACAT) (EC 2.3.1.26, also known as sterol O-acyltransferase (SOAT)) is an ER membrane-spanning enzyme converting cholesterol and FA to cholesteryl esters (CEs)7. There are two ACATs. ACAT1 is ubiquitously expressed and regulates cholesterol homeostasis, whereas ACAT2 is selectively expressed in the small intestine and liver for efficient dietary cholesterol absorption and lipoprotein assembly8, 9, 10. Both ACATs can convert toxic polar lipids to less toxic neutral lipids. The enzymatic activity of ACATs is highly activated by cholesterol through allosteric mechanism11. But the regulation of ACATs protein amount is poorly understood.
Here, we show that ACAT2 is ubiquitinated on C277 when cellular lipid level is low. High levels of cholesterol and FA induce ROS, which oxidizes C277 and prevents ACAT2 from ubiquitination and degradation. Moreover, ACAT2 knockout (ACAT2−/−) mice are more susceptible to high-fat diet (HFD)-induced insulin resistance. Re-expressing a constitutively stable form of ACAT2 (C277A) in the liver of ACAT2−/− mice increases insulin sensitivity. Together, we conclude that ROS-mediated stabilization of ACAT2 provides a critical mechanism for fine-tuning lipid overload.
Results
ACAT2 is stabilized by sterols and saturated fatty acids
We first examined whether endogenous ACATs are responded to sterol stimulation. The protein level of ACAT2 but not ACAT1 dramatically increased following cholesterol and 25-hydroxycholesterol (25-HC) treatments in various cells (Fig. 1a). Notably, their mRNAs were unchanged (Fig. 1b). In lines with these in vitro findings, high-cholesterol-diet substantially increased ACAT2 protein but not mRNA in mouse liver and small intestine (Fig. 1c and 1d). We next determined the specificity of sterols on ACAT2 protein level. As shown in Supplementary Fig. 1, sterol intermediates of cholesterol biosynthesis (desmosterol, 7-dehydrocholesterol (7-DHC), zymosterol and lanosterol), oxysterols and cholestanol markedly increased ACAT2 expression. However, ergosterol and phytosterols including β-sitosterol, stigmasterol and brassicasterol failed to elevate ACAT2 (Fig. 1e).
Figure 1. ACAT2 is stabilized by sterols and saturated fatty acids.
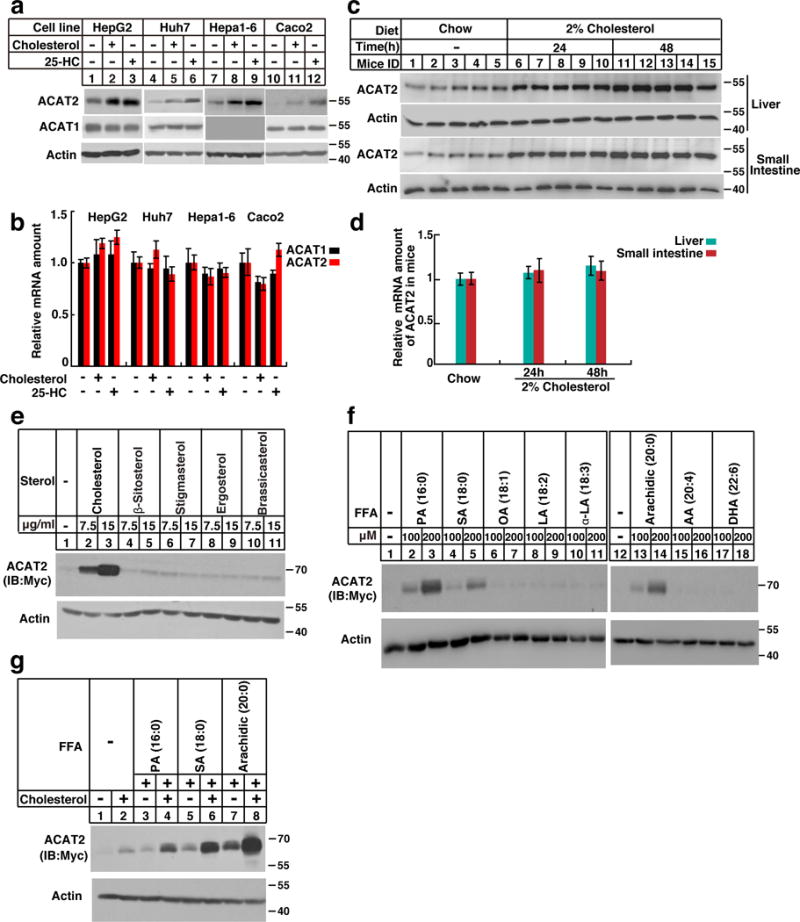
(a–b) Cells (HepG2, Huh7, Hepa1-6 and Caco2) were depleted of lipids by incubating in medium supplemented with 5% LPDS, 1 μM lovastatin, 50 μM mevalonate for 16 hrs. Then the cells were treated without (−) or with (+) sterols (15 μg/mL cholesterol and 3 μg/mL 25-HC) for 16 hrs. The cells were harvested for western blotting and RT-qPCR (n=3 independent experiments, mean±S.D.).
(c–d) Male C57/BL6 mice (8–12 weeks) were fed on chow diet or high cholesterol diet (HCD, chow diet supplemented with 2% cholesterol) for 24 and 48 hrs. The liver and small intestine samples were subjected to western blotting and RT-qPCR analysis (mean ± S.D, n=5 mice).
(e–g) The CHO/ACAT2-Myc cells were depleted of lipids as in Figure 1a. Then the cells were treated with different sterols (e), fatty acids (f) and cholesterol together with FAs (g) at indicated concentration. After incubation for 16 hrs, the cells were harvested for western blotting.
The immunoblots are representative of at least 3 independent experiments. Uncropped blots are shown in Supplementary Fig. 8. Statistics source data for b, d can be found in Supplementary Table 2.
Given the fact that ACATs utilizes both sterols and FAs as substrates, we next investigated the effects of FAs on ACATs. Palmitic acid (PA, 16:0), but not oleate acid (OA, 18:1), dramatically elevated the protein level of endogenous ACAT2 but not ACAT1 (Supplementary Fig. 1c). Neither ACAT1 nor ACAT2 mRNAs were altered (Supplementary Fig. 1d). Saturated FAs including PA, stearic acid (SA, 18:0) and arachidic acid (arachidic, 20:0) elevated ACAT2 in a dose-dependent manner (Fig. 1f). However, unsaturated FAs exerted no effects on ACAT2 (Fig. 1f). Importantly, the ACAT2 protein level was synergistically increased by cholesterol and saturated FAs (Fig. 1g).
Sterols and FAs prevent ACAT2 from ubiquitination on C277
Fig. 2a showed that inhibiting proteasome function by MG132 instead of lysosome function by NH4Cl blocked lipid-stimulated increase of ACAT2. ACAT2 was ubiquitinated at the lipid-depleted condition, which was considerably reduced following the treatment of 25-HC, PA or both (Fig. 2b). Ubiquitin is predominately conjugated to Ks of target proteins. To determine whether certain Ks serve as the ubiquitination sites, we constructed a mutant ACAT2 in which all seven Ks were replaced by arginines (Rs) (indicated as K-null). Surprisingly, both wild-type (WT) and K-null forms of ACAT2 were ubiquitinated and underwent sterol-regulated stabilization (Fig. 2c and 2d), suggesting that the Ks are not responsible for ACAT2 ubiquitination.
Figure 2. Lipid-regulated ACAT2 ubiquitination occurs on C277.
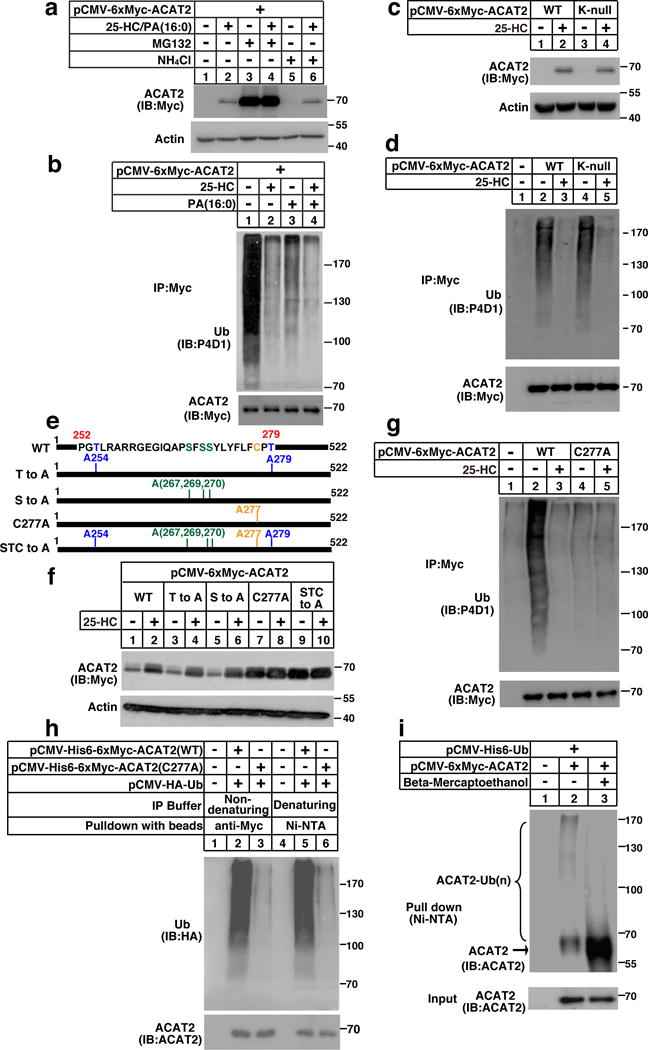
(a) CHO/ACAT2-Myc cells were depleted of lipids as depicted in Figure 1a. The cells were then treated with 25-HC (3 μg/mL) and PA (200 μM). Eight hrs later, 10 μM MG132 or 20 mM NH4Cl was added. After additional 8 hrs, cells were harvested for western blotting.
(b) CHO/ACAT2-Myc cells were depleted of lipids and treated with 25-HC, PA, or both as indicated. Eleven hrs later, cells were treated with 10 μM MG132 for another 5 hrs. Cells were then harvested for ubiquitination assay.
(c) CHO cells were transfected with plasmids expressing ACAT2 variants and depleted of lipids. Cells were then treated with 25-HC for 16 hrs and subjected to western blotting.
(d) CHO cells transfected with plasmids expressing ACAT2 variants were depleted of lipids and treated with 25-HC. Eleven hrs later, cells were treated with MG132. After another 5 hrs, cells were harvested for ubiquitination assay.
(e) Schematic of human ACAT2 protein.
(f) CHO cells transfected with plasmids expressing ACAT2 variants in (e) were depleted of lipids and treated with 25-HC. Cells were harvested 16 hrs later for western blotting.
(g) CHO cells transfected with plasmids expressing ACAT2 variants were depleted of lipids and treated with 25-HC. 11 hrs later, cells were treated with MG132. After another 5 hrs, cells were harvested for ubiquitination assay.
(h) CHO cells were transfected with plasmids expressing His6- and Myc- double-tagged ACAT2 variants together with HA-tagged ubiquitin and depleted of lipids. Cells were then treated with MG132 for another 5 hrs. Cells were harvested and lysed in the non-denaturing and denaturing IP buffers, and subsequently subjected to ubiquitination assays.
(i) CHO cells were transfected with indicated plasmids, depleted of lipids and incubated with MG132 for 5 hrs. Cells were lysed in the denaturing IP buffer, incubated with Ni-NTA beads and the precipitates were treated without (−) or with (+) BME (15 mg/mL) followed by western blotting.
The immunoblots are representative of 3 independent experiments. Uncropped blots are shown in Supplementary Fig. 8.
We next made a series of truncations to map the ubiquitination site(s). The region spanning aa 252–279 is essential for sterol-regulated ACAT2 stabilization and may contain critical ubiquitination site(s) (Supplementary Fig. 2a–2e). Since ubiquitination has also been shown to occur on serine (S), threonine (T) or C1, 12, we substituted the Ss, Ts or Cs for alanine (A)s in the aforementioned region (aa 252–279) (Fig. 2e). The alanine replacement of either S or T had no effects (Fig. 2f). However, C277A mutation significantly blocked ACAT2 degradation and ubiquitination (Fig. 2f and 2g). Moreover, substituting other Cs within a broader region (aa 227–328) barely affected ACAT2 stability (Supplementary Fig. 2f–2h). To exclude the possibility that ubiquitination may take place on ACAT2-associated proteins, we pulled down WT and C277A forms of ACAT2 using anti-Myc beads in the non-denaturing buffer or Ni-NTA beads in the denaturing buffer. The ubiquitination of WT but not mutant ACAT2 was detected in both conditions (Fig. 2h), suggesting that ACAT2 ubiquitination indeed occur on C277.
The formation of a thioester bond between ubiquitin and Cys could be broken down by the redox agent beta-mercaptoethanol (BME). To further validate that polyubiquitination takes place on C277, we pulled down ACAT2 with Ni-NTA beads in the denaturing buffer and compared the level of ubiquitination in the presence or absence of BME. As expected, we observed a downshift of the ubiquitinated ACAT2 to its original molecular weight in the presence of BME (Fig. 2i).
We next sought to determine the type of ubiquitin linkage on C277 of ACAT2. It has been known that the C-terminus of ubiquitin could be covalently attached to the ε-amino groups of any of the seven Ks (K6-linked, K11-linked, K27-linked, K29-linked, K33-linked, K48-linked and K63-linked) or to the α-amino group of the N-terminal methionine (M1-linked) of ubiquitin. By using various ubiquitin mutants, we found that the mutant ubiquitin containing only a K48 was efficiently conjugated to ACAT2, and that arginine substitution of K48 (K48R) largely abrogated ACAT2 ubiquitination (Fig. 3a and 3b).
Figure 3. ACAT2 is ubiquitinated by K48-specific ubiquitin linkages.
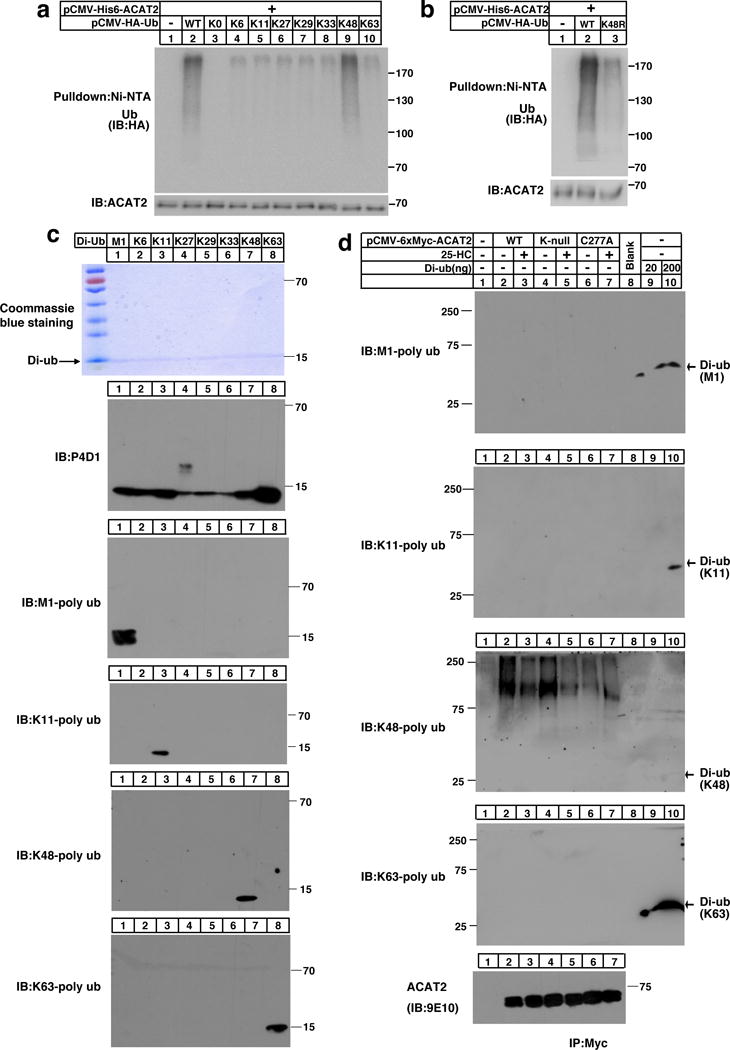
(a–b) CHO cells co-transfected with His6 tagged-ACAT2 and HA tagged-Ub ((WT, K0, K6-only, K11-only, K27-only, K29-only, K33-only, K48-only, or K63-only) (a) or (K48R) (b)) were depleted of lipids for 27 hrs. Cells were then treated with MG132 for another 5 hrs. Cells were harvested and lysed in the denaturing IP buffer, and subsequently subjected to ubiquitination analysis. The immunoblots are representative of 3 independent experiments.
(c) 200 ng of M1-linked (M1), K6-linked (K6), K11-linked (K11), K27-linked (K27), K29-linked (K29), K33-linked (K33), K48-linked (K48) or K63-linked (K63) ubiquitin dimers were separated by SDS-PAGE and probed with different anti-ubiquitin antibodies as indicated. The coommassie brilliant blue-stained gel is shown as a loading control. The independent repeat of these experiments is shown in Supplementary Fig. 3a.
(d) CHO cells were transfected with pCMV-HA-Ub together with the plasmid expressing Myc-tagged ACAT2 (WT), (K-null) or (C277A) and depleted of lipids for 16 hrs. Cells were treated with or without 25-HC for 11 hrs. Then MG132 was added into medium for another 5 hrs. Cells were harvested and lysed. The ACAT2 proteins were immunoprecipitated by anti-Myc antibodies coupled agarose and eluted with Myc peptides. Western blotting was carried out using different ubiquitin linkage antibodies or anti-Myc antibody as indicated. The di-ubiquitin was loaded about 30 min later than ACAT2 samples and served as controls. The independent repeat of these experiments is shown in Supplementary Fig. 3b.
Uncropped blots are shown in Supplementary Fig. 8.
We then analyzed ACAT2 ubiquitination using ubiquitin-linkage specific antibodies. Different types of di-ubiquitin were subjected to western blotting with antibodies against linkage specific ubiquitin including anti-M1, anti-K11, anti-K48, anti-K63, and P4D1 antibody (that detects all kinds of ubiquitin linkages), respectively. The results demonstrated that these antibodies are of high specificity (Fig. 3c and supplementary Fig. 3a). The ubiquitin linkage on C277 was then studied with these confirmed antibodies. Although anti-M1, anti-K11 or anti-K63 gave no ubiquitination signal on the ACAT2 proteins, anti-K48 ubiquitin antibody gave dramatic signal for both ACAT2 (WT) and ACAT2 (K-null) in a sterol-regulated manner. In contrast, ACAT2 (C277A) was barely ubiquitinated. The corresponding type of di-ubiquitin peptides were loaded on the same gel ~30 min later than the ACAT2 immunoprecipitation samples (due to the small molecular weight of di-ubiquitin) to serve as positive control (Fig. 3d and supplementary Fig. 3b).
Collectively, these data strongly support that C277 is the critical conjugation site for lipid-regulated ACAT2 ubiquitination. Interestingly, this Cys is highly conserved among ACAT2 proteins from different species, whereas in ACAT1 protein it is an Ala that occupies the position (Supplementary Fig. 2i).
Lipid-induced ACAT2 stabilization is mediated by ROS via C277 oxidation
Why does lipid-regulated degradation of ACAT2 require the ubiquitination at a Cys instead of a Lys? Why does lipid-regulated degradation of ACAT2 require the unbiquitination at C instead of K? Why does lipid-regulated degradation of ACAT2 require the unbiquitination at C instead of K?It It has been known that high levels of lipids cause oxidative stress and that Cys is readily oxidized. We thus hypothesized that sterols and FAs may induce ROS to oxidize C277 and hence prevent ACAT2 from ubiquitination and subsequent degradation. As shown in Fig. 4, cholesterol, sterol intermediates (7-DHC, lanosterol), oxysterols and saturated FAs substantially promoted the generation of ROS. However, unsaturated FAs and phytosterols (beta-sitosterol, stigmasterol) failed to elicit ROS. Tthe scattergraph showed a strong correlation between ROS production and ACAT2 protein level (Fig. 4c). We further determined critical concentrations of cholesterol (5 μg/mL), 25-HC (1 μg/mL) and PA (100 μM) that elicit ROS production and elevate ACAT2 protein level (Supplementary Fig. 4a–4i). Sterols and FAs induced ROS production in a time-dependent manner as early as 8 hours (Supplementary Fig. 4j–4r). The elevation of ACAT2 was detected from 12 hours since protein may take some time to accumulate.
Figure 4. The ubiquitination on C277 of ACAT2 is regulated by oxidative stress.
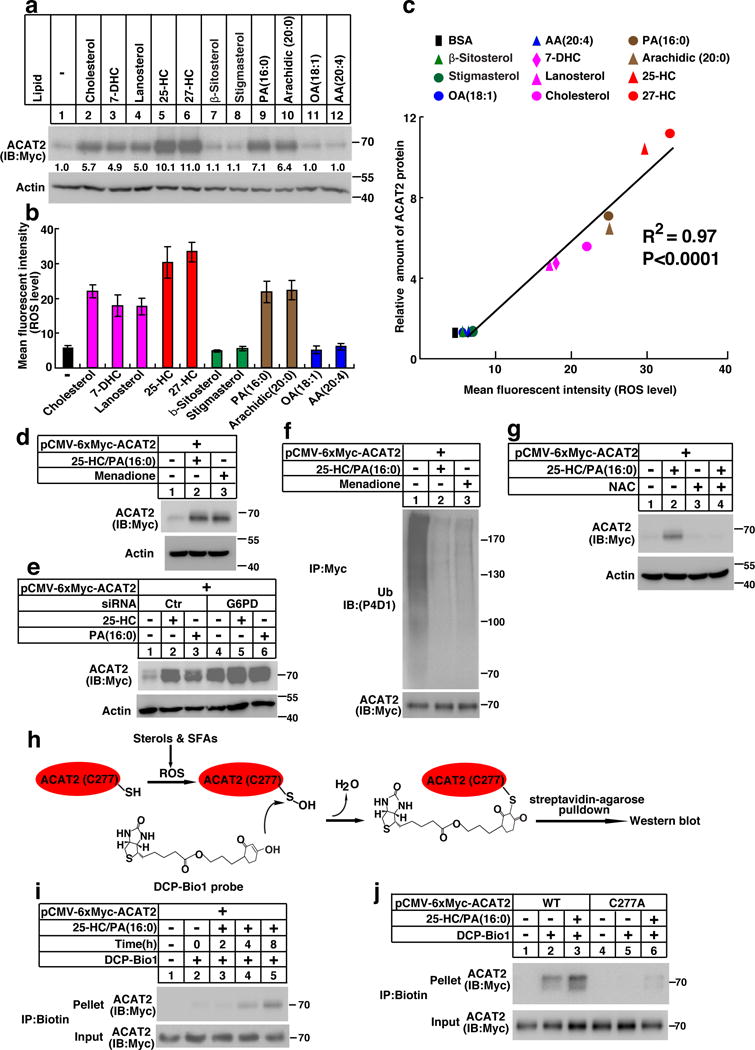
(a–c) CHO/ACAT2-Myc cells were depleted of lipids as depicted in Figure 1a and treated with vehicle or different kinds of sterols and fatty acids. Cells were harvested for western blotting and ROS measurement. (a) Blots were quantified and the mean intensity of ACAT2 normalized to beta-actin is indicated (from 3 independent experiments, control treatment is defined as 1). (b) ROS levels were presented as mean ± S.D., n=3 independent experiments. (c) A correlation (R2 =0.97, p<0.0001) between lipids-induced ROS production and ACAT2 protein level was analyzed by GraphPad Prism and demonstrated by a scatterdiagram.
(d) CHO/ACAT2-Myc cells were depleted of lipids and treated with 25-HC/PA or menadione (12.5 μM) as indicated. 16 hrs later, cells were harvested for western blotting.
(e) SV589 cells transfected with ACAT2 plasmid and indicated siRNAs were depleted of lipids and treated with 25-HC or PA. Cells were harvested 16 hrs later for western blotting.
(f) CHO/ACAT2-Myc cells were depleted of lipids and treated with 25-HC/PA or menadione as indicated. 11 hrs later, cells were treated with MG132. After incubation for another 5 hrs, cells were harvested for ubiquitination assay.
(g) CHO/ACAT2-Myc cells were depleted of lipids, pretreated with NAC (7.5 mM) for 1 hr and then incubated with vehicle or 25-HC/PA for 16 hrs. Then the cells were harvested for western blotting.
(h) A scheme showing how DCP-Bio1 probe detects the oxidation of C277 in ACAT2. (i) CHO/ACAT2-Myc cells were depleted of lipids and treated with 25-HC/PA for indicated durations. Cells were then harvested for sulfenic acids detection as described in Methods.
(j) CHO cells transfected with plasmids expressing ACAT2 variants were depleted of lipids and treated with 25-HC/PA for 8 hrs. Cells were then harvested for sulfenic acids detection.
The immunoblots are representative of 3 independent experiments. Uncropped blots are shown in Supplementary Fig. 8. Statistics source data for a, b, c can be found in Supplementary Table 2.
To assess whether ACAT2 stability is regulated by cellular redox, we treated cells with prooxidants menadione or H2O2 and found that both oxidative reagents markedly stabilized ACAT2 (Fig. 4d and Supplementary Fig. 5a). In addition, siRNA depletion of glucose-6-phosphate dehydrogenase (G6PD), a key enzyme in synthesizing the main intracellular reductant NADPH, also resulted in ACAT2 stabilization (Fig. 4e), in agreement with the notion that G6PD deficiency sensitizes cells to oxidative stress. Furthermore, menadione or H2O2 dramatically lessened ACAT2 ubiquitination (Fig. 4f and Supplementary Fig. 5b), whereas the antioxidant N-acetyl-L-cysteine (NAC) attenuated lipid-induced stabilization of ACAT2 (Fig. 4g).
We next tested whether C277 of ACAT2 could be oxidized by ROS. Cys oxidation may result in the formation of sulfenic acid (–SOH) which can be detected by DCP-Bio1 (Fig. 4h)13, 14, 15. Upon 25-HC/PA treatment, ACAT2 was time-dependently labeled by DCP-Bio1 (Fig. 4i). However, ACAT2 (C277A) was barely labeled by the probe (Fig. 4j). Collectively, it is concluded that sterols and FAs trigger ROS which then oxidizes C277 and impedes ubiquitination-mediated degradation of ACAT2.
gp78/Insigs mediates the ubiquitination and degradation of ACAT2
To identify the E3 of ACAT2, the ACAT2 complex was immunoprecipitated from membrane fraction of CHO/ACAT2-Myc cells and subjected to tandem-mass-spectrometry analysis. gp78, a membrane-anchored E3, was revealed. It has been reported that gp78 interacts with Insig1/2 and promotes sterol-regulated ubiquitination of HMG-CoA reductase (HMGCR), a rate-limiting enzyme in cholesterol biosynthesis16. We thus evaluated the role of gp78 and Insigs in ACAT2 degradation. Overexpression of WT gp78 remarkably promoted ACAT2 degradation. Conversely, the dominant-negative mutant gp78 increased the amount of ACAT2 (Supplementary Fig. 5c). Moreover, knockdown of gp78, but not Hrd1 (an independent ER membrane E3) or Sel1L (an ER protein involved in ER-associated degradation), dramatically increased ACAT2 level (Fig. 5a). These data suggest that gp78 specifically mediates ACAT2 ubiquitination and degradation. To confirm these in vitro results, we also measured ACAT2 level in gp78−/− CHO cells or primary hepatocytes from liver-specific gp78−/− mice. ACAT2 was constitutively stable regardless of sterol treatment when gp78 was absent (Fig. 5b and 5c). Consistently, ACAT2 level was significantly higher in the liver and intestine of gp78−/− mice compared with those of WT littermates (Fig. 5d).
Figure 5. The gp78/Insigs E3 complex mediates the ubiquitination and degradation of ACTA2.
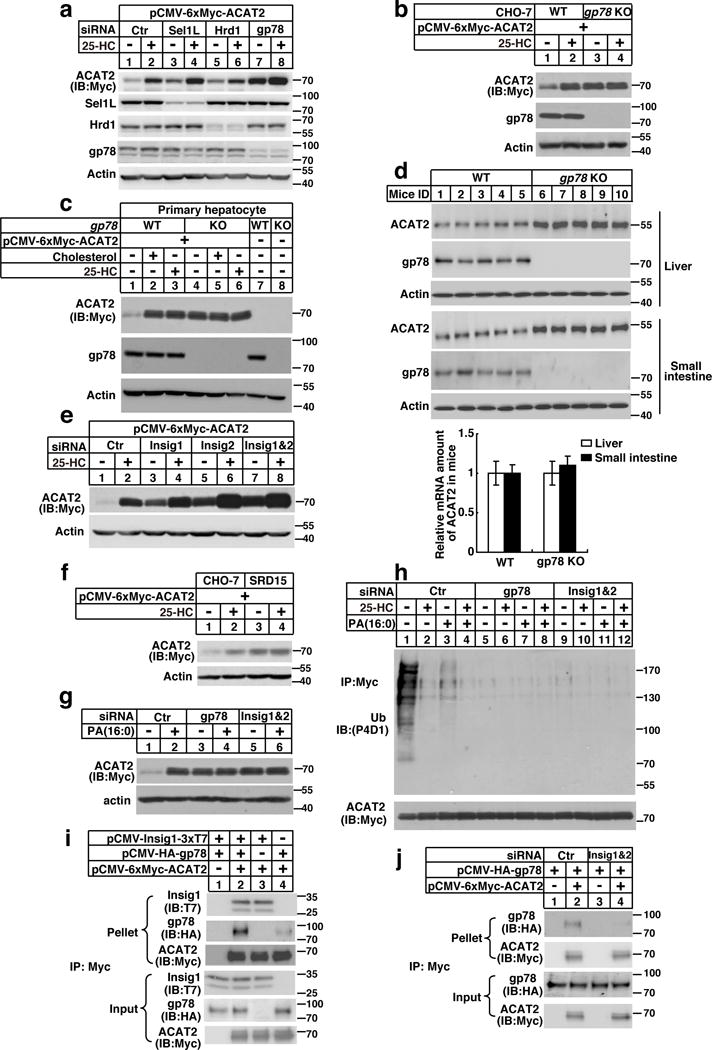
(a) SV589 cells transfected with ACAT2 plasmid and siRNAs were depleted of lipids and treated with 25-HC. Cells were harvested 16 hrs later for western blotting.
(b) CHO-7 (WT) and gp78−/− deficient CHO-7 cell line (gp78 KO) cells transfected with ACAT2 plasmid were depleted of lipids and treated with 25-HC. Cells were harvested 16 hrs later for western blotting.
(c) Primary hepatocytes from WT or liver-specific gp78−/− mice (male, 8–12 weeks) were transfected with ACAT2 plasmid, depleted of lipids and treated with sterols. 16 hrs later, cells were harvested for western blotting.
(d) The liver and small intestine samples from WT or gp78−/− mice (male, 8–12 weeks) were subjected to western blotting and RT-qPCR analysis (mean±S.D., n=5 mice).
(e) SV589 cells were transfected with ACAT2 expression plasmid and siRNAs, depleted of lipids and treated with 25-HC. 16 hrs later, cells were harvested for western blotting.
(f) CHO-7 and Insigs deficient CHO-7 cell line (SRD15) were transfected with ACAT2, depleted of lipids and treated with 25-HC. 16 hrs later, cells were harvested for western blotting.
(g) SV589 cells were transfected with ACAT2 expression plasmid and siRNAs, depleted of lipids and treated with PA. 16 hrs later, cells were harvested for western blotting.
(h) SV589 cells were transfected with ACAT2 expression plasmid and siRNAs, depleted of lipids and treated with 25-HC, PA, or both. 11 hrs later, cells were treated with MG132. After additional 5 hrs, cells were harvested for ubiquitination assay.
(i) CHO cells were transfected with indicated plasmids, depleted of lipids and treated with MG132 for 5 hrs. Cells were then harvested for co-IP analysis.
(j) SV589 cells were transfected with plasmids and siRNAs, depleted of lipids and treated with MG132 for 5 hrs. Cells were then harvested for co-IP analysis.
The immunoblots are representative of 3 independent experiments. Uncropped blots are shown in Supplementary Fig. 8. Statistics source data for d can be found in Supplementary Table 2.
Since Insig1/2 work as cofactors of gp78 in recruiting substrates, we asked whether Insigs were required for ACAT2 degradation. ACAT2 stabilization was detected in Insig1, Insig2 or Insig1/2 siRNA-depleted cells (Fig. 5e), as well as in SRD15 cells (Fig. 5f), a cell line deficient in both Insigs17. Moreover, knockdown of gp78 or Insigs abolished lipid-regulated ACAT2 ubiquitination and degradation (Fig. 5g and 5h).
We next performed co-immunoprecipitation (co-IP) experiments to evaluate the interactions. We found that ACAT2 directly bound Insig1 but not gp78 (Fig. 5i). Knockdown of Insigs abolished ACAT2-gp78 association (Fig. 5j). These results indicate that ACAT2, gp78 and Insigs form triplex and that Insigs mediate ACAT2 and gp78 interaction. Importantly, ACAT2-Insig1 association was independent of C277 (Supplementary Fig. 5d) or of redox state (Supplementary Fig. 5e–5g), ruling out the possibility that oxidation may regulate ACAT2-Insig-gp78 interaction. In addition, gp78 was neither oxidatively modified nor inactivated by ROS (Supplementary Fig. 6a and 6b).
ACAT2−/− mice are more susceptible to HFD-induced insulin resistance
To examine whether ACAT2 deletion results in FC and FFA over-accumulation in vivo, we fed WT and ACAT2−/− mice with HFD or chow diet (CD) for 12 weeks. We observed an increase in FFA (14% and 24% higher in the plasma and liver respectively) and a decrease in CEs content (15% and 96% lower in the plasma and liver respectively) in ACAT2−/− mice fed on HFD compared with WT controls (Fig. 6a). To confirm ACAT2 converts saturated FAs to CEs, we measured hepatic CEs composition of WT mice by LC/MS. The content of saturated FAs (16:0 and 18:0) was substantially higher in HFD-feeding mice than CD-feeding mice (Supplementary Fig. 7d). These data suggest that ACAT2 prevents against FFA over-accumulation by catalyzing CEs formation.
Figure 6. ACAT2−/− mice are more susceptible to HFD-induced insulin resistance.
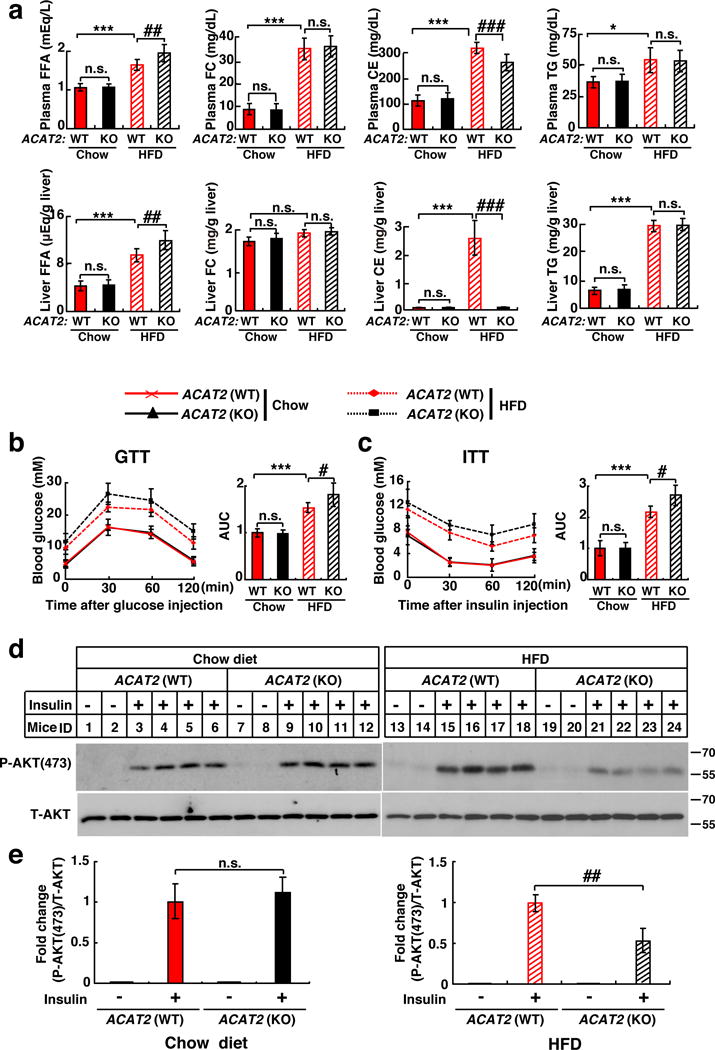
Male ACAT2−/− mice and their WT littermates (8–12 weeks) were randomly grouped (n=6 mice) and allowed ad libitum access to water and chow diet or HFD for 12 weeks.
(a) FFA, FC, CE, and TG contents in the plasma and liver.
(b–c) GTT and ITT were performed after 10-week treatment and area under the curve (AUC) was calculated.
(d) Representative immunoblots showing phosphorylated AKT (P-AKT) and total AKT (T-AKT) in the livers of WT and ACAT2−/− mice fed on chow diet or HFD under the basal (−Insulin) or in vivo acute insulin-stimulated (+Insulin) conditions. The livers were harvested at 10 min post insulin treatment.
(e) Quantification of fold change of P-AKT/T-AKT (n=4 mice).
Data are presented as mean ± S.D.. Asterisks indicate the differences between WT mice fed on chow diet and those on HFD, * p < 0.05, ** p < 0.01, *** p < 0.001; Pounds indicate the differences between WT and ACAT2−/− mice fed on HFD, # p < 0.05, ## p < 0.01, ### p < 0.001. n.s., no significance; two-way ANOVA followed by Tukey’s HSD post-hoc test.
Statistics source data for a, b, c, e can be found in Supplementary Table 2. Uncropped images of blots are shown in Supplementary Fig. 8.
To investigate whether ACAT2 deletion exacerbates HFD-induced insulin resistance, we performed glucose tolerance test (GTT) and insulin tolerance test (ITT) and found that ACAT2−/− mice were much less sensitive to insulin than WT littermates when fed on HFD. However, no differences were detected between WT and ACAT2−/− mice fed on CD (Fig. 6b and 6c). In addition, we injected insulin intraperitoneally and assayed the liver for AKT phosphorylation 10 min post-injection. Insulin-stimulated phosphorylation of AKT was much lower in ACAT2−/− mice compared with WT mice on HFD (Fig. 6d and 6e, right panel). However, AKT phosphorylation was similar in both groups fed on CD (Fig. 6d and 6e, left panel). Together, these data indicate that ACAT2−/− mice are more susceptible to HFD-induced insulin resistance than WT mice and that ACAT2 plays an important role in ameliorating lipotoxicity.
Stabilized ACAT2 protects from HFD-induced insulin resistance
To directly assess the effect of ACAT2 stabilization on metabolic parameters, we first asked whether C277 was required for enzyme activity. Microsomes were prepared from AC29 cells (ACAT-deficient CHO cells) stably expressing WT or C277A forms of ACAT2. The ACAT2 activity was measured in vitro. ACAT2 (C277A) demonstrated similar capability of catalyzing CEs formation to WT ACAT2 (Fig. 7a–7c). These results suggest that C277 is dispensable for the enzyme activity, consistent with the previous finding that H438 is the catalytic site of human ACAT218.
Figure 7. The stabilization of hepatic ACAT2 ameliorates lipotoxicity and protects HFD-induced insulin resistance in mice.
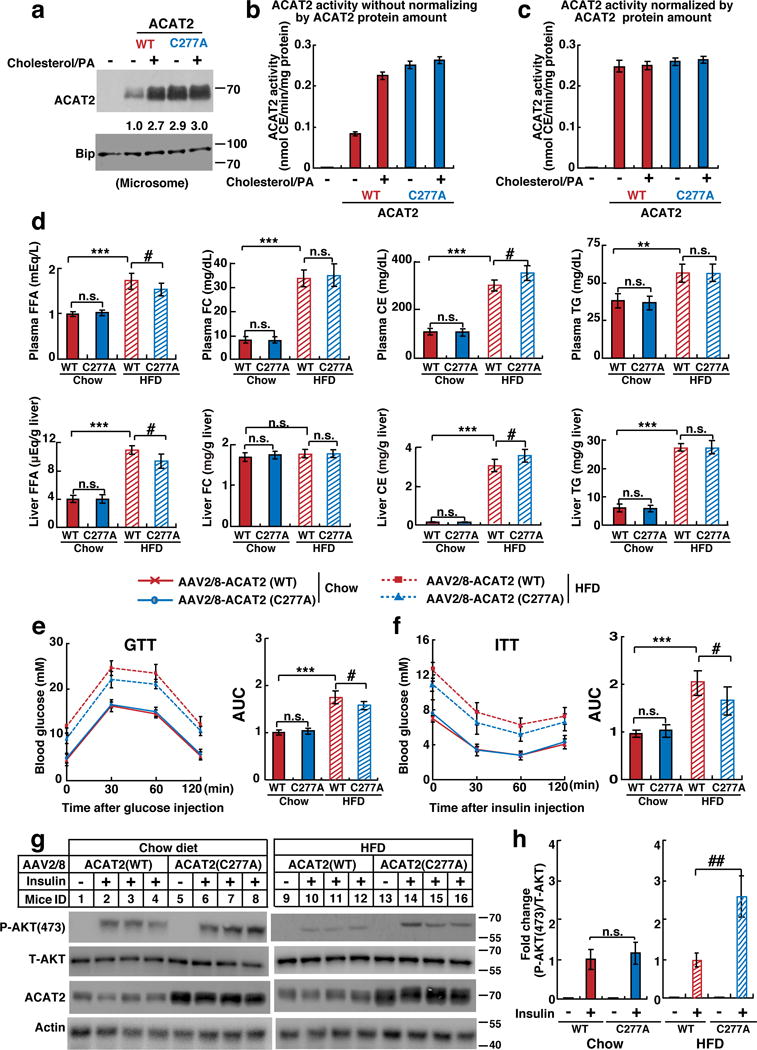
(a–c) AC29 cells stably expressing ACAT2 (WT) or (C277A) were depleted of lipids and treated with cholesterol/PA for 16 hrs. (a) Equal amount of microsomes (30 μg) were subject to western blotting. ACAT2 was quantified as in Figure 4a. (b–c) The ACAT2 activity monitored by measuring [14C] oleoyl CoA incorporation into [14C] steryl ester without (b) or with (c) normalization by ACAT2 protein amount (mean±S.D., n=3 independent experiments).
(d–h) Male ACAT2−/− mice (8–12 weeks) were randomly grouped (n=5 mice) and intravenously injected with AAV2/8-ACAT2 (WT) or (C277A) at a dose of 1×1012 vg per mouse. Mice were then allowed ad libitum access to water and chow diet or HFD for 12 weeks.
(d) FFA, FC, CE, and TG contents in the plasma and liver.
(e–f) GTT and ITT were performed after 10-week treatment and area under the curve (AUC) was calculated.
(g) Representative immunoblots showing phosphorylated AKT (P-AKT), total AKT (T-AKT) and ACAT2 in the livers of AAV2/8-ACAT2 (WT)- or (C277A)-injected mice fed on chow diet or HFD under the basal (−Insulin) or in vivo acute insulin-stimulated (+Insulin) conditions. The livers were harvested at 10 min post insulin treatment.
(h) Quantification of fold change of P-AKT/T-AKT following insulin stimulation in AAV2/8-ACAT2 (WT)- or (C277A)-injected mice fed on chow diet or HFD.
Data are presented as mean ± S.D.. Asterisks indicate the differences of ACAT2−/− mice injected with AAV2/8-ACAT2 (WT) fed on chow diet and those on HFD, * p < 0.05, ** p < 0.01, *** p < 0.001; Pounds indicate the differences between ACAT2−/− mice injected with AAV2/8-ACAT2 (WT) and (C277A) fed on HFD, # p < 0.05, ## p < 0.01, ### p < 0.001. n.s., no significance; two-way ANOVA followed by Tukey’s HSD post-hoc test.
Statistics source data for a, b, c, d, e, f, h can be found in Supplementary Table 2. Uncropped images of blots are shown in Supplementary Fig. 8.
We next delivered adeno-associated virus (AAV) expressing WT and C277A forms of ACAT2 to ACAT2−/− mice. AAV2/8 is a hepatotropic serotype of AAV, and as expected, ACAT2 was selectively expressed in the liver (Supplementary Fig. 6c). In addition, the mRNA level of exogenous ACAT2 was comparable to that of endogenous ACAT2 in WT mice (Supplementary Fig. 6d), suggesting that AAV2/8-mediated delivery mimics the knock-in situation of ACAT2. When fed on HFD for 12 weeks, ACAT2 (C277A)-expressing mice showed much lower FFA levels (14% and 18% lower in the plasma and liver respectively) and much higher CEs content (12% and 16% higher in the plasma and liver respectively) than ACAT2 (WT)-expressing mice (Fig. 7d). In addition, the CEs contents of lipoproteins in ACAT2 (C277A)-expressing mice were higher as revealed by FPLC analysis (Supplementary Fig. 6e).
GTT and ITT showed that HFD-induced insulin resistance in both ACAT2 (WT)- and (C277A)-expressing mice. However, ACAT2 (C277A)-expressing mice displayed higher glucose clearance ability and insulin sensitivity than did ACAT2 (WT)-expressing mice (Fig. 7e and 7f). Consistently, insulin-induced phosphorylation level of AKT in ACAT2 (C277A) mice was higher than ACAT2 (WT) mice when fed on HFD (Fig. 7g and 7h). Together, these data further validated that the stabilization of ACAT2 ameliorates lipotoxicity by catalyzing CEs formation.
According to these findings, we proposed a model to illustrate the protective effects of ACAT2 on lipotoxicity and insulin resistance induced by lipids, as depicted in Supplementary Fig. 6f. When cellular lipid levels are low, the gp78/Insigs complex binds ACAT2 and catalyzes the ubiquitination of C277, thus promoting ACAT2 degradation. When cholesterol and FA levels are elevated, the ROS is induced which oxidizes C277 and prevents ACAT2 from ubiquitination. As a result, increased ACAT2 converts cholesterol and FA to CEs, thereby reducing lipotoxicity. The utilization of a Cys as an unconventional ubiquitination site represents a lipid-induced ROS sensing and protection mechanism.
Discussion
The current study shows that ACAT2 is stabilized by cholesterol and FA. It is ubiquitinated on a Cys rather than a Lys. Lipids including free cholesterol and saturated FAs induce ROS that oxidizes C277 of ACAT2 and protects the protein from degradation. The stabilized ACAT2 further converts cholesterol and FAs to CEs, thereby preventing the lipotoxicity of polar lipids.
gp78 is a membrane-anchored ubiquitin ligase with five transmembrane domains19,20. It binds to Ufd1, VCP and UBE2G2 via the cytosolic domain, and associates with Insig1/2 through the transmembrane domain16, 21, 22, 23. Insigs work as adaptors to recruit substrates including ACAT2 and HMGCR24. Interestingly, although both ACAT2 and HMGCR are gp78 substrates, their ubiquitination is regulated in opposite directions. When cholesterol level rises, HMGCR binds gp78/Insigs and is targeted to degradation16. Meanwhile, high level of cholesterol and FA induces the generation of ROS to oxidize C277 and prevent ACAT2 degradation. These two mechanisms in concert decrease cholesterol biosynthesis and increase the conversion of cholesterol to CE, protecting the cell from high level of toxic free cholesterol. Other important mechanisms controlling cholesterol biosynthesis include cholesterol-regulated degradation of 7-DHC reductase and squalene monooxygenase25, 26, 27. When cellular cholesterol level drops, HMGCR dissociates from gp78/Insigs and becomes stabilized. Also, the availability of C277 confers ACAT2 to be ubiquitinated by gp78/Insigs. By this way, the cellular cholesterol level elevates quickly. In sum, the gp78/Insigs E3 complex plays a central role in cellular cholesterol homeostasis through modulating de novo synthesis and esterification.
The specific sterols and FAs capable of stabilizing ACAT2 have been determined. Cholesterol, sterol intermediates in mevalonate pathway and oxysterols could increase ACAT2 (Supplementary Fig. 1a and 1b). This sterol specificity is especially important because ACAT2 is required for intestinal cholesterol absorption when dietary cholesterol level is high. The stabilization of ACAT2 by cholesterol, therefore, substantiates highly efficient cholesterol absorption. Interestingly, neither ergosterol nor plant sterols could stabilize ACAT2 (Fig. 1e). This might be another mechanism governing the specificity of cholesterol absorption, in addition to selective cholesterol uptake by NPC1L128, 29 and preferential plant sterols export by ABCG5/830, 31. On the other hand, only saturated but not unsaturated FAs increase the stability of ACAT2 (Fig. 1f). As saturated FAs function as strong activators of ROS and insulin resistance32, this specificity grants ACAT2 a protective role against saturated FA-induced lipotoxicity. Interestingly, oleoyl-CoA has been shown to be the predominant intracellular fatty acid substrate for ACAT2 since the CE content dramatically reduces in the liver of SCD1−/− mice33. However, results from in vitro assay identify palmitoyl-CoA as the most preferred fatty acyl CoA for ACAT234. We found that the content of saturated FAs in hepatic CEs, despite in relatively small amounts, is substantially increased in mice fed on HFD (Supplementary Fig. 7d), suggesting that the ROS contributed by saturated FA upon HFD may be partially relieved by ACAT2-mediated CE synthesis. Together, these data suggest ACAT2 stabilization may counteract ROS through multiple angles. We also looked up the SNPs at ACAT2 (±20 kb) in Chinese GWAS35 and DIAGRAM Consortium databases36, 37, but found no significant correlation between SNPs in this region and type 2 diabetes (P ≥ 0.07). Whether ACAT2 SNPs or mutation contributes to metabolic diseases in human population needs further investigation.
Lipid-induced ROS elevation is a common pathophysiological characteristic of metabolic diseases. Our study reveals that ACAT2 acts as a key ROS-responding protein to sense lipid overload and oxidative stress. These results suggest that pharmacologically increasing the stability of ACAT2 could be an effective strategy to treat type II diabetes.
Methods
Reagents
We obtained MG132 from Calbiochem; menadione from Sigma; di-ubiquitins (M1-linked, K6-linked, K11-linked, K29-linked, K33-linked, K48-linked and K63-linked) from Boston Biochem; K27-linked di-ubiquitin from Lifesensor; Myc peptide (3×) was synthesized at ChinaPeptides Co. Ltd; fatty acid-free BSA, lovastatin, mevalonate and fatty acids from Sigma; sterols from Steraloids, Inc; horseradish peroxidase-conjugated donkey anti-mouse and anti-rabbit IgG from Jackson ImmunoResearch Laboratories; 60% high fat diet (D12492) from Research Diets, Inc. The 2% high cholesterol diet was prepared by adding 2% (w/w) cholesterol to regular chow diet. Lipoprotein-deficient serum (LPDS) and delipid-fetal calf serum (D-FCS) was prepared from newborn calf serum as described before38.
Antibodies
Primary antibodies used for immunoblotting were as follows: mouse monoclonal anti-ubiquitin antibody P4D1 (SC-8017, Santa Cruz, 1:500); rabbit polyclonal antibodies against ubiquitin M1 (ABS175, 1:1000), K11 (MABS107, 1:1000) K48 (05-1307, 1:1000) and K63 (05-1308, 1:1000) linkages were from Millipore; mouse monoclonal anti-T7 (69522, Millipore, 1:500); mouse monoclonal anti-HA (clone 16B12) (901503, Biolegend, 1:500); mouse monoclonal anti-β-actin (clone AC-15) (A1978, Sigma, 1:3000); mouse monoclonal anti-Myc (clone 9E10) (2 μg/ml) was prepared from hybridomas (ATCC); rabbit polyclonal antibody against Hrd1 (SC-130889, Santa Cruz, 1:1000); rabbit polyclonal antibody against Sel1L (SC-48081, Santa Cruz, 1:1000); rabbit polyclonal antibody against phosphorylated-AKT (ser473) (#4060, Cell Signaling Technology, 1:1000), rabbit polyclonal antibody against total AKT (#9272, Cell Signaling Technology, 1:1000); rabbit polyclonal antibody against human ACAT1 (SC-130578, Santa Cruz, 1:1000); rabbit polyclonal antibody against human ACAT2 (100027, Cayman, 1:1000). Rabbit polyclonal antibodies against mouse ACAT1 (2 μg/ml) and ACAT2 (2 μg/ml) were generated in our lab39. Rabbit polyclonal antibody against gp78 (2 μg/ml) was prepared as described before21. Peroxidase affinipure donkey anti-mouse IgG secondary antibody (715-035-150, 1:5000) was from Jackson ImmunoResearch Laboratories. Goat anti-rabbit IgG secondary antibody (31460, 1:5000) was from Pierce. All homemade antibodies were affinity-purified with antigens. Anti-Myc-coupled agarose beads (E6654, 40 μl/sample) were from Sigma. Ni-NTA agarose beads (30210, 40 μl/sample) were from Qiagen.
Plasmids
The plasmids of 6×Myc-ACAT2, His6-6×Myc-ACAT2, His6-ACAT2, His6-T7-Insig1, His6-Ub, HA-Ub and HA-gp78 were constructed by standard molecular cloning techniques. Various ubiquitin linkage mutants, ACAT2 truncations or site-directed mutants and gp78 dominant negative mutants were constructed using either the QuikChange site-directed or multisite-directed mutagenesis kits (Stratagene) and confirmed by sequencing.
Media
Medium A contains Dulbecco’s Modified Eagle Medium (DMEM) containing 100 units/mL penicillin and 100 μg/mL streptomycin sulfate. Medium B contains a 1:1 mixture of Ham’s F-12 Medium and DMEM containing 100 units/mL penicillin and 100 μg/mL streptomycin sulfate. Medium C contains Media 199 containing 100 units/mL penicillin and 100 μg/mL streptomycin sulfate. Medium D contains Ham’s F-12 Medium containing 100 units/mL penicillin and 100 μg/mL streptomycin sulfate. Lipid-depleting medium was prepared by adding 5% LPDS or D-FCS, 1 μM lovastatin and 50 μM mevalonate into each medium.
Cell culture
HepG2 (a human hepatocellular liver carcinoma cell line), Huh7 (a human hepatocellular carcinoma cell line), Hepa1-6 (mouse hepatocellular carcinoma cell line), Caco2 (a cell line derived from a human colon adenocarcinoma), SV589 (an immortalized line of human fibroblasts) and CHO-7 (a clone of CHO-K1 cells selected for growth in LPDS) were obtained from ATCC. SRD15 (an Insig-1/Insig-2 deficient cell line) was a generous gift from Dr. Russell Debose-Boyd at UT Southwestern Medical Center. All cells were grown in a monolayer at 37 °C with 5% CO2. HepG2, Huh7, Hepa1-6, SV589 cells were maintained in medium A supplemented with 10% FBS; Caco2 cells were maintained in medium A supplemented with 20% FBS; CHO-7, SRD15 cells were maintained in medium B supplemented with 5% FBS; primary hepatocytes were maintained in medium C supplemented with 5% FBS; AC29 (an ACAT-deficient CHO cell line, a generous gift from Dr. TY Chang at Dartmouth Medical School) cells were maintained in medium D supplemented with 10% FBS. CHO-7 cells stably expressing pCMV-6×Myc-ACAT2 were generated as previously described40 and maintained in medium C supplemented with 5% FBS plus G418. All the cell lines have been tested and they have no mycoplasma contamination. No cell lines used in this study were found in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI Biosample. The cell lines were not authenticated.
Primary hepatocyte culture
Primary hepatocytes were isolated from the livers of male C57/BL6 mice (8-week-old) using collagenase perfusion as described41. Six-well plates were pre-coated with 0.5 mL of 0.75 mg/mL collagen I solution (collagen gel, gelation: ≥1 h at 37°C) for sandwich cultures. Hepatocytes were plated at a density of 1.5 × 106 cells per well in 2 mL medium C supplemented with 10% FBS and 100 nM dexamethasone. Four hours after seeding, cells on collagen gel were washed twice with ice-cold PBS, overlaid with a second layer of collagen I gel and incubated with indicated medium supplemented with 100 nM dexamethasone. Cell viability, as measured by trypan blue exclusion, was always greater than 80%. After incubation for 16 hrs, cells were switched to indicated medium for various experiments.
Transfection
Transfection was performed using Fugene HD (Promega) according to the manufacturer’s instructions. Primary hepatocytes were transfected with indicated plasmids using Effectene transfection reagent (Qiagen).
Generating gp78 knockout CHO-7 cells
CHO-7 cells deficient in gp78 was generated by TALEN technology. Briefly, a pair of TALEN constructs with puromycin resistance for gp78 knockout was prepared using the Talen construction kit from Sidansai (Shanghai, China) following the manufacturer’ instructions and transfected into CHO-7 cells using Fugene HD (Promega). 12 hr later, 6 μg/mL puromycin was added and maintained for 4 days. Surviving clones were isolated and screened for gp78 depletion by Western blotting.
Fatty acids treatment
Stock solutions of various fatty acids were prepared by mixing free fatty acids with fatty acid-free BSA. After delipidation, cells were treated with fatty acids at concentrations and time durations as indicated.
Ubiquitination assays in non-denaturing or denaturing conditions
For ubiquitination assay in the non-denaturing conditions, cells were homogenized in IP buffer (1×PBS, 1% NP-40, 1% Deoxycholate and 5 mM EDTA, 5 mM EGTA, 0.1 mM Leupeptin) supplemented with protease inhibitors (10 μM MG132, 10 mM N-Ethylmaleimide (NEM) and cocktail). Immunoprecipitation was carried out with the anti-Myc agrose beads as previously described23. For ubiquitination assay in the denaturing conditions, cells were homogenized in buffer A (6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4, 5 mM imidazole, 0.01 M Tris-HCl, pH, 8.0) supplemented with protease inhibitors (10 μM MG132, 10 mM NEM and cocktail). After sonication and centrifugation, the supernatants were collected and incubated with Ni-NTA beads overnight. Beads were then subjected to a series of stringent washes using Buffer B (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH, 8.0), Buffer C (0.2 % Triton X-100, 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH, 6.3) and Buffer D (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH, 6.3). Beads were boiled for 10 min or incubated with Myc peptide for 3 hrs at 4°C, and then the supernatants were analyzed by Western blotting with indicated antibodies.
Coimmunoprecipitation
Cells were harvested and lysed in 0.6 mL of IP buffer (1×PBS, 1% digitonin, 5 mM EDTA, 5 mM EGTA and cocktail) followed by centrifugation at 12,000 × g for 10 min at 4 °C. Immunoprecipitation was carried out as described previously16.
RNA Interference
Duplexes of siRNA were synthesized by Genepharma (Shanghai, China). The sequences of the siRNAs targeting gp78, Hrd1, Insig1, Insig2, G6PD were described previously16, 42. The sequence of siRNA targeting against Sel1L is 5′-GGATATTCACCTTGCGAAA-3′. The siRNA against an irrelevant gene, vesicular stomatitis virus glycoprotein (VSV-G), is 5′-GGCUAUUCAAGCAGACGGUTT-3′ and used as a negative control. Transfection of siRNA was carried out as previously described24.
Measurement of intracellular ROS levels
Intracellular ROS level was determined using oxidation-sensitive DCFH-DA fluorescent dyes (Sigma). Cells were washed twice with PBS and labeled on the culture plates with DCFH-DA for 30 min at 37°C in serum-free medium. At the end of incubation, culture plates were trypsinized, resuspended in PBS and analyzed using a FACScan flow cytometer (λex =488 nm and λem 530nm band-pass filter). The mean fluorescence intensity of 10,000 cells was measured in each sample and corrected for autofluorescence from unlabeled cells.
Detection of cysteine sulfenic acid by DCP-Bio1 probe
Cysteine sulfenic acid detection by DCP-Bio1 was performed as previously described with slight modification43, 44. Briefly, treated cells were washed with PBS and homogenized in DCP-Bio1 working lysis buffer (1×PBS, 1% NP40, 1% sodium deoxycholate, 5 mM EDTA, 5 mM EGTA, 1 mM PMSF, 0.1 mM Aprotinin, 0.1 mM Leupeptin, 10 mM NEM, 10 mM iodoacetamide, 100 μM DTPA, and 200 U/mL catalase). Lysates were centrifuged at 12,000 × g for 10 min. Supernatants were collected and incubated with 1 mM DCP-Bio1 probe (Millipore) for 2 hrs on ice. For affinity capture of the labeled proteins, unreacted DCP-Bio1 was removed using a dialysis device (Pierce). Dialyzed Samples were precleared with Sepharose CL-4B beads (Sigma), applied to plugged columns containing high-capacity streptavidin-agarose beads (Pierce) and incubated overnight at 4 °C. Beads were then subjected to a series of stringent washes (at least four column volumes and two washes each) using 1% SDS, 4 M urea, 1 M NaCl, 10 mM DTT, 100 μM ammonium bicarbonate and water. Beads were boiled for 10 min and then the samples were analyzed by Western blotting analysis.
AAV preparation
AAV2/8-ACAT2 (WT) and AAV2/8-ACAT2 (C277A) were prepared by Obio Technology Co., Ltd. (Shang hai, China). Viral titer was 1.06×1013 vg/mL for AAV2/8-ACAT2 (WT) and 1.32×1013 vg/mL for AAV2/8-ACAT2 (C277A).
Animals and treatments
Male C57BL/6 mice (6–8 week) were obtained from SLAC (Shanghai, China). The ACAT2−/− mice were purchased from Jackson Laboratory. The gp78f/f and liver-specific gp78 knockout mouse was constructed as previously reported45. The gp78−/− mice were generated by crossing gp78f/f mouse with EIIA-Cre mouse. To ensure genetic homogeneity, the ACAT2−/− and gp78−/− mice were both backcrossed to C57BL/6 mice for at least 10 generations. Both types of mice were viable, fertile, normal in size and do not display any gross physical or behavioral abnormalities. Male C57BL/6 mice, ACAT2−/−, gp78−/− and liver-specific gp78−/− mice of 8–12 weeks of age were used in all experiments. Mice were fed ad libitum with standard chow diet prior to study, maintained in a pathogen-free environment in single-ventilated cages, and kept on a 12-h light/dark schedule. All animals were used in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Shanghai Institutes for Biological Sciences. Mice were treated as described in the figure legends.
Glucose Tolerance and Insulin Tolerance Test in mice
Mice were fasted overnight (for GTT) or for 6 hrs (for ITT) in advance, Glucose (1 g/kg body weight) (for GTT) or insulin (0.75 U/kg body weight) (for ITT) was injected intraperitoneally into mice followed by blood sampling from the tail vein for glucose measurements. Tail blood glucose levels were measured at 0, 30, 60 and 120 min after challenge using the Onetouch Ultra blood glucose monitoring system (LifeScan).
Blood and liver chemistry
The livers were homogenized and supernatants were harvested for lipid extraction. Total triglyceride level in the plasma and liver (Triglyceride Assay Kit, KINGHAWK, China), free fatty acid (NEFA C Test Kit, Wako, Japan) and free cholesterol and cholesterol ester (CE) (Amplex Red Cholesterol Assay Kit, Invitrogen) were determined following the manufacturers’ instructions.
Fast performance liquid chromatography (FPLC)
120 μL of pooled plasma from five mice per group was diluted in 300 μL of PBS. Total diluted serum was injected onto a Superose 6 10/300 GL column (GE Healthcare Bio-Sciences AB) and eluted at a constant flow rate of 0.2 mL/min PBS. Fractions of 300 μL were collected and assayed for CE content using Amplex Red Cholesterol Assay Kit (Invitrogen).
Measurement of CEs composition by LC/MS
Chemicals: Solvents for sample preparation and mass spectrometry analysis were purchased from Burdick and Jackson. All other chemical reagents were from Sigma. The lipid internal standard was cholesterol nonadecanoate (CE 19:0).
Preparation of cholesterol ester extracts from liver: Each liver sample (20–30 mg) was weighed and homogenized in 500 μL of ice-cold PBS. After determining protein concentrations by BCA assay (Beyotime), CE (19:0) internal standard was added to each homogenate. Then the homogenate was extracted by Hexane/IPA=3:2 in a glass tube. After vortex and centrifugation, the upper organic phase was collected, mixed with hexane and extract again. All organic phase was dried under argon and reconstituted in hexane prior to injection46.
Liquid Chromatography and Mass Spectrometry: Normal phase (NP) Lipid Chromatography (LC) was carried out using Agilent 1260 Quat pump VL and Accela 1250 pump. We performed normal phase LC separation using 2 mm × 250 mm Beckman silica column. Sample was run at a flow rate of 200 μL/min in mobile C phage [0.5% IPA in hexane]. Eluent from all normal-phase chromatographic methods was modified with 10 mM ammonium acetate (NH4OAc) in IPA introduced prior to the ion source at a flow rate of 40 μL/min. Fractions with retention times were 3 min. The MS was performed on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was operated in positive ion, multiple reaction monitoring (MRM) mode. MRM pairs employed in the detection method used the ammoniated CE precursor mass and the cholesterol product fragment with m/z = 369. The cholesterol fragment is common to all CEs47. They were listed in Supplementary Table 1. The FA compositions of chow diet or high fat diet were measured by LC/MS as previously described48, 49.
ACAT enzyme activity assay
On day 0, AC29 cells stably expressing human ACAT2 (WT) or ACAT2 (C277A) were set up at 1.5×106 per 100-mm dish in media D supplemented with 10% FBS. On day 1, cells were switched to media D supplemented with 5% D-FCS, 1 μM lovastatin and 50 μM mevalonate. On day 2, cells were switched to medium D containing 5% delipid-FCS, 1 μM lovastatin, 50 μM mevalonate in the absence or presence of 15 μg/mL cholesterol/cyclodextrin and 100 μM PA. After 16 hrs, cells were harvested, washed with PBS twice and homogenized in ACAT homogenization buffer (0.25 M sucrose, 1 mM EDTA, 0.1M K2HPO4, pH 7.4) supplemented with protease inhibitor mixture (Sigma) by ultrasound sonication. Lysates was centrifuged at 12,000 × g for 20 min at 4°C. Supernatants were then centrifuged at 100,000 × g for 60 min at 4°C and the microsomal pellet was resuspended in ACAT reaction buffer (0.1 M K2HPO4, pH 7.4) supplemented with protease inhibitor mixture (Sigma). The microsomal suspension was stored at −80°C until use. The activity of acyl-CoA: cholesterol acyltransferase in the microsomes was determined by the rate of incorporation of (1-14C) oleate into the cholesteryl ester fraction. This assay included 100 μg of microsomal protein, 0.1 M K2HPO4, pH 7.4, 50 nM cholesterol, 1 mg of fatty acids-free human serum albumin in a total volume of 290 μL. The esterification of cholesterol was initiated by adding 0.1 μCi of 1-14C Oleoyl-CoA (specific activity 5 Ci/mol, Amersham Biosciences) into the reaction system, making the final incubation volume 300 μL. After incubation for 10 min at 37°C, the reaction was terminated by adding 6 mL of chloroform-methanol (2:1, v/v). 1 hr later, 1.2 mL of 0.88% KCl was added and the phases were allowed to separate overnight at room temperature. The upper aqueous phase in each tube was aspirated and the remaining organic phase was evaporated under liquid N2. Lipid was then resuspended in CHCl3 containing 5 mg/mL cholesteryl oleate and applied to a silica gel-60 TLC plate. The separation of the lipids was done in hexane:ethyl ether:acetic acid (70:30:1) and bands on the TLC plate were visualized with I2 vapor. CE bands were scraped and suspended in scintillation fluid for radioactivity measurement in a liquid scintillation spectrometer.
Western blot analysis
Cells were harvested and homogenized in 120 μL of RIPA buffer supplemented with protease inhibitors. Protein concentration of whole cell lysates was determined according to Lowry method (Bio-Rad). Samples were mixed with 4×SDS loading buffer (150 mM Tris-HCl, pH 6.8, 12% SDS, 30% (v/v) glycerol, 0.02% (w/v) bromophenol blue, 6% (v/v) beta-mercaptoethanol) and boiled for 10 min. Proteins were resolved by SDS-PAGE and transferred onto PVDF membranes. Immunoblots were blocked with 5% BSA in TBS containing 0.075% Tween and probed with primary antibodies overnight at 4 °C. After washing in TBST 3 times, blots were incubated with secondary antibodies for 1 hr at room temperature.
RT-qPCR
Total RNA was extracted using Trizol reagents (Invitrogen) and subjected to reverse transcription (RT) with oligo dT followed by qPCR using target-specific primers in the Stratagene Mx30005P Q-PCR Systems. All reactions were prepared in triplicate and the relative amounts of mRNAs were calculated using the comparative CT method. Human or Mouse Cyclophilin was used as controls. Values show the amount of mRNA relative to control sample, which is arbitrarily defined as 1. Primer sequences are used as follows:
Mouse ACAT1: (Forward: 5′-CCGAGACAACTACCCAAGGA-3′, Reverse: 5′-CACACACAGGACCAGGACAC-3′); Mouse ACAT2 (Forward: 5′-ATGTTCTACCGGGACTGGTG-3′, Reverse: 5′-CCCGAAAACAAGGAATAGCA-3′); Human ACAT1: (Forward: 5′-GATGAAGGAAGGCTGGTGC-3′, Reverse: 5′- GGAAGCTGGTGGCAGTGTAT-3′); Human ACAT2: (Forward: 5′-CATGCTGCTGCTCATCTTCT-3′, Reverse:5′-ACTGCGGAGACCAGGAACA-3′). Mouse Cyclophilin: (Forward: 5′-TGGAGAGCACCAAGACAGACA-3′, Reverse: 5′- TGCCGGAGTCGACAATGAT-3′), Human Cyclophilin: (Forward: 5′-GTCTCCTTTGAGCTGTTTGC-3′, Reverse: 5′-CTGGGAACCATTTGTGTTGG-3′).
Statistical analyses and reproducibility
The between-group variances were similar, and the data were normally distributed. All statistical analyses were performed using the SPSS software (version 11.5). Data were expressed as means ± S.D. and analyzed by unpaired two-tailed Student’s t test or two-way ANOVA as indicated. Statistical tests were justified as appropriate for every figure. Statistical significance was set at p < 0.05. Sample sizes, statistical tests and p values for each experiment are depicted in relevant figure legends. Specific samples size was chosen based on similar experiments of previous studies. No statistical method was used to predetermine sample size. All samples/animals were randomly allocated to experimental groups and processed and the investigators were blinded to the group allocation during the experiment and/or when assessing the outcome. All experiments were successfully repeated for three times in the laboratory. Each immunoblot was representative of 3 independent experiments and uncropped blots are shown in Supplementary Fig. 8.
Data Availability
Source data for Fig. 1b,d, 4a–c, 5d, 6a–c,e 7a–f,h and Supplementary Fig.1d, 4a–r, 6d–e, 7a–f has been provided as Supplementary Table 2. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
We thank Dr. Wei Qi for helpful discussion and critical reading of the manuscript, Xin-Ying Yang, Hong-Hua Miao, Lei Qian, Yu-Xiu Qu, Jie Xu and Jie Qin for technical assistance. This work was supported by the grants from NNSF of China (31690102, 31430044, 31470802, 31230020 and 31271377), MOST of China (2016YFA0500100 and 2014DFG32410) and the 111 Project of Ministry of Education of China (B16036). TYC and CCYC are funded by NIH grant HL 60306.
Footnotes
Author Contributions
B.-L.L. and B.-L.S. conceived the project. Y.-J.W., CY.C., T.-Y.C., B.-L.L. and B.-L.S. designed the experiments. Y.-J.W., Y.B., M.L., Y.X. and Q.L. performed the experiments. S.-Y.G., H.-Y.Y. measured the FA compositions. X.L. performed SNP analysis. Y.-J.W., J.L., CY.C., T.-Y.C., B.-L.L. and B.-L.S. organized and analyzed the data. Y.-J.W., J.L., B.-L.L. and B.-L.S. wrote the manuscript with input from all other authors. All authors approved the final version of the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–130. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- 2.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18:139–143. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anavi S, Hahn-Obercyger M, Madar Z, Tirosh O. Mechanism for HIF-1 activation by cholesterol under normoxia: A redox signaling pathway for liver damage. Free Radic Biol Med. 2014;71:61–69. doi: 10.1016/j.freeradbiomed.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Poli G, Biasi F, Leonarduzzi G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013;1:125–130. doi: 10.1016/j.redox.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maloney E, et al. Activation of NF-kappaB by palmitate in endothelial cells: a key role for NADPH oxidase-derived superoxide inresponse to TLR4 activation. Arterioscler Thromb Vasc Biol. 2009;29:1370–1375. doi: 10.1161/ATVBAHA.109.188813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodeur MR, Bouvet C, Barrette M, Moreau P. Palmitic acid increases medial calcification by inducing oxidative stress. J Vasc Res. 2013;50:430–441. doi: 10.1159/000354235. [DOI] [PubMed] [Google Scholar]
- 7.Chang TY, Chang CC, Cheng D. Acyl-coenzyme A:cholesterol acyltransferase. Annu Rev Biochem. 1997;66:613–638. doi: 10.1146/annurev.biochem.66.1.613. [DOI] [PubMed] [Google Scholar]
- 8.Farese RVJ. Acyl-CoA:cholesterol acyltransferase genes and knockout mice. Curr Opin Lipidol. 1998;9:119–124. doi: 10.1097/00041433-199804000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Chang TY, et al. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr Opin Lipidol. 2001;12:289–296. doi: 10.1097/00041433-200106000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Rudel L, Lee R, Cockman T. Structure, function, and regulation of ACAT. Curr Opin Lipidol. 2001;12:121–127. doi: 10.1097/00041433-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Chang CC, Westover EJ, Covey DF, Chang TY. Investigating the allosterism of acyl-CoA:cholesterol acyltransferase (ACAT) by using various sterols: in vitro and intact cell studies. Biochem J. 2005;391:389–397. doi: 10.1042/BJ20050428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, et al. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol. 2007;177:613–624. doi: 10.1083/jcb.200611063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charles RL, et al. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6:1473–1484. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Poole LB, et al. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddie KG, Seo YH, MuseIii WB, Leonard SE, Carroll KS. A chemical approach for detecting sulfenic acid-modified proteins in living cells. Mol Biosyst. 2008;4:521–531. doi: 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song BL, Sever N, Bose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19:829–840. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Lee PC, Sever N, Debose-Boyd RA. Isolation of sterol-resistant Chinese hamster ovary cells with genetic deficiencies in both Insig-1 and Insig-2. J Biol Chem. 2005;280:25242–25249. doi: 10.1074/jbc.M502989200. [DOI] [PubMed] [Google Scholar]
- 18.Das A, Davis MA, Rudel LL. Identification of putative active site residues of ACAT enzymes. J Lipid Res. 2008;49:1770–1781. doi: 10.1194/jlr.M800131-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu K, et al. The autocrine motility factor receptor gene encodes a novel type of seven transmembrane protein. FEBS Lett. 1999;456:295–300. doi: 10.1016/s0014-5793(99)00966-7. [DOI] [PubMed] [Google Scholar]
- 20.Fang S, et al. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao J, et al. Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA Reductase. Cell Metab. 2007;6:115–128. doi: 10.1016/j.cmet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Zhong X, et al. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem. 2004;279:45676–45684. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
- 23.Chen B, et al. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci USA. 2006;103:341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sever N, et al. Insig-dependent ubiquitinationand degradation of mammalian3-hydroxy-3- methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 25.Sharpe LJ, Cook EC, Zelcer N, Brown AJ. The UPS and downs of cholesterol homeostasis. Trends Biochem Sci. 2014;39:527–535. doi: 10.1016/j.tibs.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Prabhu AV, Luu W, Sharpe LJ, Brown AJ. Cholesterol-mediated Degradation of 7-Dehydrocholesterol Reductase Switches the Balance from Cholesterol to Vitamin D Synthesis. J Biol Chem. 2016;291:8363–8373. doi: 10.1074/jbc.M115.699546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gill S, Stevenson J, Kristiana I, Brown AJ. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 2011;13:260–273. doi: 10.1016/j.cmet.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Brown JM, Rudel LL, Yu L. NPC1L1 (Niemann-Pick C1-like 1) mediates sterol-specific unidirectional transport of non-esterified cholesterol in McArdle-RH7777 hepatoma cells. Biochem J. 2007;406:273–283. doi: 10.1042/BJ20070168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge L, et al. The cholesterol absorption inhibitor ezetimibe acts by blocking the sterol-induced internalization of NPC1L1. Cell Metab. 2008;7:508–519. doi: 10.1016/j.cmet.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Yu L, von Bergmann K, Lutjohann D, Hobbs HH, Cohen JC. Selective sterol accumulation in ABCG5/ABCG8-deficient mice. J Lipid Res. 2004;45:301–307. doi: 10.1194/jlr.M300377-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Berge KE, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 32.Martins AR, et al. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis. 2012;11:30. doi: 10.1186/1476-511X-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyazaki M, Kim YC, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J Biol Chem. 2000;275:30132–30138. doi: 10.1074/jbc.M005488200. [DOI] [PubMed] [Google Scholar]
- 34.Cases S, et al. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J Biol Chem. 1998;273:26755–26764. doi: 10.1074/jbc.273.41.26755. [DOI] [PubMed] [Google Scholar]
- 35.Li H, et al. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes. 2013;62:291–298. doi: 10.2337/db12-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahajan A, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldstein JL, Basu SK, Brown MS. Receptor-mediatedendocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 39.Xie C, et al. Ezetimibe blocks the internalization of NPC1L1 and cholesterol in mouse small intestine. J Lipid Res. 2012;53:2092–2101. doi: 10.1194/jlr.M027359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang CC, et al. Recombinant acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. J Biol Chem. 1998;273:35132–35141. doi: 10.1074/jbc.273.52.35132. [DOI] [PubMed] [Google Scholar]
- 41.Klingmuller U, et al. Primary mouse hepatocytes for systems biology approaches: a standardized in vitro system for modelling of signal transduction pathways. Syst Biol (Stevenage) 2006;153:433–447. doi: 10.1049/ip-syb:20050067. [DOI] [PubMed] [Google Scholar]
- 42.Gupte RS, et al. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem. 2010;285:19561–19571. doi: 10.1074/jbc.M109.092916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klomsiri C, et al. Use of dimedone-based chemical probes for sulfenic acid detection: evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol. 2010;473:77–94. doi: 10.1016/S0076-6879(10)73003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson KJ, et al. Use of dimedone-based chemical probes for sulfenic acid detection; methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu TF, et al. Ablation of gp78 in Liver Improves Hyperlipidemia and Insulin Resistance by Inhibiting SREBP to Decrease Lipid Biosynthesis. Cell Metab. 2012;16:213–225. doi: 10.1016/j.cmet.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 46.Kuksis A, Suomela JP, Tarvainen M, Kallio H. Lipidomic analysis of glycerolipid and cholesteryl ester autooxidation products. Mol Biotechnol. 2009;42:224–268. doi: 10.1007/s12033-009-9146-z. [DOI] [PubMed] [Google Scholar]
- 47.Hutchins PM, Moore EE, Murphy RC. Electrospray MS/MS reveals extensive and nonspecific oxidation of cholesterol esters in human peripheral vascular lesions. J Lipid Res. 2011;52:2070–2083. doi: 10.1194/jlr.M019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zong G, et al. Associations of erythrocyte palmitoleic acid with adipokines, inflammatory markers, and the metabolic syndrome in middle-aged and older Chinese. Am J Clin Nutr. 2012;96:970–976. doi: 10.3945/ajcn.112.040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu Y, et al. Genome-wide meta-analyses identify novel loci associated with n-3 and n-6 polyunsaturated fatty acid levels in Chinese and European-ancestry populations. Hum Mol Genet. 2016;25:1215–1224. doi: 10.1093/hmg/ddw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Fig. 1b,d, 4a–c, 5d, 6a–c,e 7a–f,h and Supplementary Fig.1d, 4a–r, 6d–e, 7a–f has been provided as Supplementary Table 2. All other data supporting the findings of this study are available from the corresponding author on reasonable request.