

Abstract
Biomaterial scaffolds that enrich and modulate immune cells in situ can form the basis for potent immunotherapies to elicit immunity or reëstablish tolerance. Here, we explore the potential of an injectable, porous hydrogel to induce a regulatory T cell (Treg) response by delivering a peptide antigen to dendritic cells (DCs) in a non-inflammatory context. Two methods are described for delivering the BDC peptide from pore-forming gels in the NOD (non-obese diabetic) mouse model of type 1 diabetes: encapsulation in poly(lactide-co-glycolide) (PLG) microparticles, or direct conjugation to the alginate polymer. While particle-based delivery leads to antigen-specific T cells responses in vivo, PLG particles alter the phenotype of the cells infiltrating the gels. Following gel-based peptide delivery, transient expansion of endogenous antigen-specific T cells is observed in the draining lymph nodes. Antigen-specific T cells accumulate in the gels, and, strikingly, ~60% of the antigen-specific CD4+ T cells in the gels are Tregs. Antigen-specific T cells are also enriched in the pancreatic islets, and administration of peptide-loaded gels does not accelerate diabetes. This work demonstrates that a non-inflammatory biomaterial system can generate antigen-specific Tregs in vivo, which may enable the development of new therapies for the treatment of transplant rejection or autoimmune diseases.
Keywords: alginate hydrogels, dendritic cells, regulatory T cells, antigen-specific tolerance, immunotherapy
1. Introduction
The development of effective, antigen-specific tolerogenic therapies would represent a significant leap in the treatment of autoimmunity, allergy, and transplant rejection. The feasibility of this concept is supported by the finding that even healthy individuals harbor auto-reactive T cells that escape central tolerance,[1] but these cells are kept in check through mechanisms of peripheral tolerance. In addition, the fact that not all individuals who are genetically susceptible to autoimmune diseases actually progress to a disease state suggests that it may be possible to restore tolerance in individuals that do develop disease. Peripheral tolerance can be enforced through inactivation or deletion of self-reactive effector T cells and/or through suppression by regulatory T cells (Tregs). Induction or expansion of antigen-specific Tregs is a potentially powerful approach for inducing tolerance, since Tregs can traffic to target tissues[2,3] and exert dominant suppression over many different types of immune cells, including effector T cells and DCs.[4] Several studies have demonstrated that antigen specificity is important for Treg function and that it is possible for Tregs with a single antigen specificity to suppress disease in NOD (non-obese diabetic) mice.[5–7]
The therapies currently used to promote tolerance are not antigen specific; although broad immunosuppressive agents are effective at dampening unwanted immune responses, patients are at greater risk for infections and cancer development.[8,9] A preferable approach would be to dampen aberrant immune responses towards self-antigens, while maintaining the full capacity of the immune system to mount responses against foreign antigens and pathogens. A number of clinical trials have attempted to treat patients with autoimmune diseases in an antigen-specific manner by delivering disease-associated proteins either orally or nasally to target tolerogenic mucosal DCs.[10] Despite the effectiveness of these treatment strategies in pre-clinical models, trials in humans have not been successful to date.[10] One key difference is that these antigen-specific therapies have mostly been used prophylactically in pre-clinical models, while clinical studies focused on treating patients with recent-onset disease, primarily for ethical reasons.[10] Other possible parameters that may require optimization to treat humans include antigen dose and frequency of administration. While research in this area is still ongoing, concerns have been raised regarding the safety of administering autoimmune disease-associated antigens through systemic routes, which include intravenous, oral, and nasal administration. In pre-clinical models of multiple sclerosis (MS), delivery of soluble disease-associated peptides led to fatal anaphylaxis in some mouse models, as well as disease exacerbation in a non-human primate study.[11] The ability to deliver protein or peptide antigens in a localized and controlled manner could be key to developing safe and effective antigen-specific tolerogenic therapies.
Biomaterial delivery systems could enhance our ability to induce antigen-specific tolerogenic responses by allowing greater control over the spatiotemporal presentation of self-antigens, either alone or in combination with tolerogenic factors. Recently, several promising nanoparticle-based tolerogenic therapies have been reported.[12–17] These technologies are based on nanoparticles that are loaded with antigen alone[12–14] or with tolerogenic factors[15–17] and administered intravenously (i.v.), following which they are taken up by splenic antigen presenting cells (APCs) and processed through tolerogenic pathways. An alternative approach involves biomaterial scaffolds that allow infiltration of the target immune cells into the material. One potential advantage of this approach is that the localization of antigen can be more tightly controlled, limiting its dissemination to sites of inflammation and disease. In addition, subcutaneously administered scaffolds are easily accessible and could be monitored through minimally invasive biopsies. The strategy of recruiting and programming immune cells in a porous biomaterial-based scaffold was shown to be effective in the context of cancer vaccination.[18,19] A polymeric scaffold delivering granulocyte-macrophage colony stimulating factor (GM-CSF), tumor lysate, and CpG oligonucleotides was shown to elicit potent anti-tumor responses in pre-clinical models of melanoma[18,19] and is currently being tested in a phase I clinical trial.[20]
Here, we explore the potential of a biomaterial scaffold system to induce tolerogenic responses in vivo. CD11c+ dendritic cells are targeted with this system, as they continually integrate signals from the environment to regulate the direction and magnitude of immune responses, and are critical for maintaining self-tolerance in steady state conditions.[21,22] In these studies, we specifically utilize alginate-based, pore-forming gels delivering GM-CSF. We previously demonstrated that the porous structure of the gels, as well as the sustained release of GM-CSF from gold nanoparticles (AuNPs) entrapped within the polymer network, allow the gels to mediate infiltration of a large number and high percentage of DCs.[23] The DCs in this system exhibited a non-inflammatory or immature cell surface marker phenotype that is similar to that of bone marrow derived dendritic cells (BMDCs).[23] The impact of this scaffold approach to induce tolerance was explored in a mouse model of type 1 diabetes using NOD mice, which develop spontaneously occurring diabetes as a result of T cell mediated destruction of insulin-producing beta cells in the pancreatic islets. BDC2.5 CD4+ T cells, which were established from pancreatic islet infiltrates of NOD mice,[24,25] recognize an antigen derived from the Chromogranin A protein[26] expressed by pancreatic beta cells. Chromogranin A is one of several proteins, including insulin and glutamic acid decarboxylase (GAD65), that are strongly implicated in diabetes initiation and development; indeed, NOD mice that are deficient in Chromogranin A do not develop diabetes[27]. Interestingly, BDC2.5 T cells have the capacity both to act as diabetogenic effector T cells that initiate beta cell destruction[28,29] as well as to develop into functional Tregs that control or prevent diabetes.[5–7] Additionally, BDC2.5 T cells may have relevance to human disease, since they recognize peptides in the context of the MHC class II I-Ag7, whose peptide binding pocket bears structural similarities with class II human leukocyte antigen (HLA) variants that are associated with susceptibility to type 1 diabetes in patients.[30] Here, we administer pore-forming gels delivering GM-CSF and BDC peptide,[31] a peptide antigen mimotope recognized by BDC2.5 T cells, in NOD mice with the aim of modulating the BDC antigen-specific Treg population. Subcutaneous administration of these gels leads to localized antigen delivery and antigen-specific T cell expansion in the lymph nodes (LN) draining the gels. Over time, antigen-specific CD4+ T cells are enriched at sites where their cognate antigen is found, i.e. in the gels and pancreatic islets. A high percentage of these cells express markers of Tregs in the gels, and administration of 3 gel doses did not accelerate disease progression in NOD mice.
2. Results
2.1. Profile of cells infiltrating pore-forming gels containing peptide-loaded PLG particles
Incorporation of peptide-loaded PLG particles into pore-forming gels was first explored as a method for delivering a peptide antigen in a localized manner. The in vitro release and in vivo biodistribution of peptide were first examined using porous scaffolds fabricated by gas foaming peptide-loaded PLG microparticles. Peptide was released in a gradual and sustained manner in vitro from PLG scaffolds (Figure S1A), as well as from PLG particles incorporated into alginate gels (Figure S1B). In order to analyze the in vivo biodistribution of the peptide following delivery in the scaffolds, an assay designed to detect full-length, intact peptide was used to analyze tissue lysates. Intact, unbound peptide was highly localized in vivo, as it was detected only in the tissue within the scaffold, and not in the adjacent muscle tissue or in the draining lymph nodes (dLNs) (Figure S1C,D). Peptide-loaded PLG microparticles were incorporated into the bulk phase of pore-forming alginate gels (Figure 1A), and the phenotype of cells infiltrating the gels with and without PLG particles was characterized. Gels delivering GM-CSF, either with or without peptide-loaded PLG microparticles, were injected subcutaneously into the flanks of C57BL/6J mice and explanted at various timepoints for analysis. In both conditions, greater than ~95% of infiltrating cells expressed the myeloid cell marker CD11b at all timepoints examined (Figure 1B). However, the fractions of cells expressing CD11c (a marker of conventional DCs), F4/80 (a marker of macrophages and some DC subsets), and MHCII (expressed on DCs and other professional antigen-presenting cells) were significantly reduced in gels containing PLG particles (Figure 1C–E). In gels delivering only GM-CSF, the percentages of cells expressing CD11c, F4/80, and MHCII were initially low at day 1, but increased substantially at days 3 and 5. In contrast, in gels containing peptide-loaded PLG particles, the expression of these three markers did not increase significantly over time. The greatest differences between the two conditions were observed at day 5, at which point gels utilizing PLG to deliver peptide contained 3.5-fold, 28-fold, and 12-fold lower fractions of CD11c+, F4/80+, and MHCII+ cells, respectively. In the case of Gr-1, a marker of monocytes and granulocytes, the opposite trend was observed (Figure 1F). In both conditions, the majority of infiltrating cells expressed Gr-1 at day 1. The expression of Gr-1 greatly decreased over time in gels delivering only GM-CSF, whereas over 90% of the cells remained Gr-1+ at all timepoints in gels delivering peptide-loaded PLG particles and GM-CSF (Supporting Figure S2).
Figure 1.
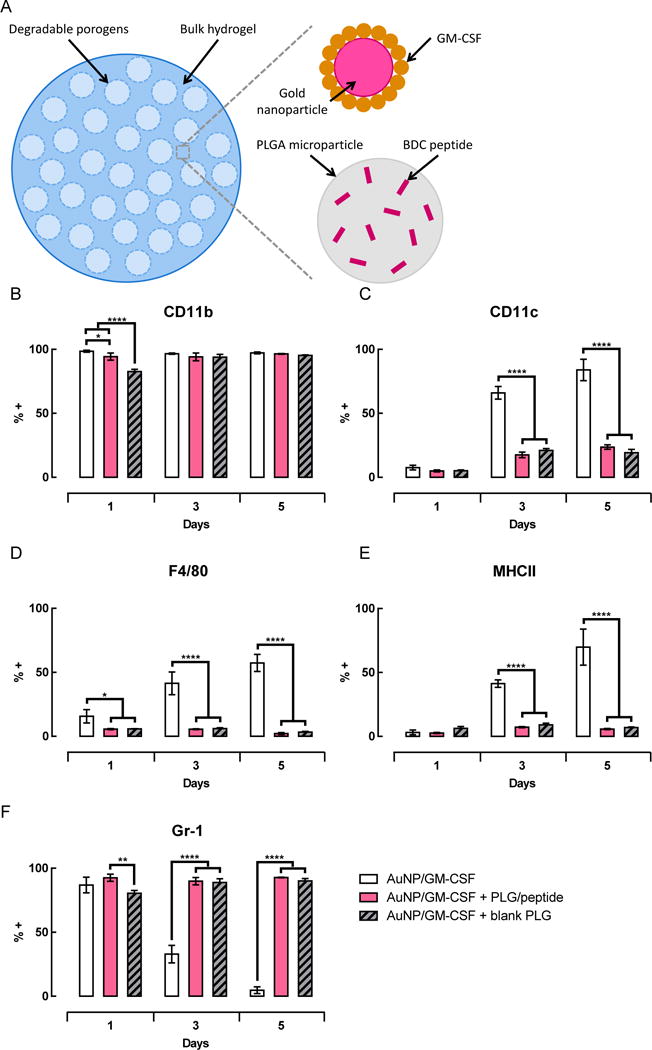
Incorporation of PLG particles in the gels significantly altered the phenotype of cells recruited in response to GM-CSF. (A) Schematic illustrating the different components of pore-forming gels delivering GM-CSF and peptide-loaded PLG particles. (B–F) Gels loaded with AuNP/GM-CSF, AuNP/GM-CSF + peptide-loaded PLG particles, or AuNP/GM-CSF + blank particles were injected subcutaneously into the flanks of C57BL/6J mice. At specified timepoints, gels were isolated and dissociated to analyze infiltrating cells by flow cytometry. Percentage of cells expressing CD11b+ (B), CD11c+ (C), F4/80+ (D), MHCII+ (E), and Gr-1+ (F). (n = 3; mean ± s.d. shown; * p < 0.05; **** p < 0.0001). FACS plots representing CD11c and Gr-1 expression on day 5 shown in Figure S2 (Supporting Information).
To complement this cell surface marker analysis, the expression of inflammatory cytokines in the gels was also examined, both at the mRNA and protein levels. For the analysis of gene expression, cells were sorted into CD11b+ CD11c+ “DC” and CD11b+ CD11c− “myeloid” populations and assayed separately (Supporting Figure S4A). The small population of cells that did not fit into those populations, i.e. lymphocytes (CD3e+ and DX5+ cells) and CD11b− cells, were pooled together and excluded to focus the analysis on DCs and myeloid cells. CD11b+ CD11c+ splenic DCs sorted from naïve mice served as a control representing immature DCs, and all other samples were normalized to this condition. As compared to the CD11b+ CD11c+ splenocyte controls, cells isolated from gels delivering GM-CSF expressed similar or lower levels of all the transcripts examined (Figure 2A,B). A few of the transcripts, such as Il12b, which encodes the IL12p40 subunit of the inflammatory cytokines IL-12 and IL-23, were several orders of magnitude less abundant than in the splenic DC control. The genes assayed did not differ substantially in their expression between gels with and without peptide-loaded PLG particles, with the exception of Il1a, whose expression was significantly increased when peptide-loaded PLG particles were present in the gels (Figure 2A,B). For each of the gel conditions, comparing the CD11b+ CD11c− “myeloid” and CD11b+ CD11c+ “DC” subsets did not reveal any significant differences in gene expression for the transcripts examined here. As a positive control for inflammatory DCs, the CD11b+ CD11c+ splenic DCs were stimulated with LPS for 4h. As expected, stimulation of splenic DCs with LPS induced significant increases in the expression of transcripts encoding inflammatory cytokines, such as Il23a, Il27, Il33, and Il6 (Supporting Figure S4B). Expression of Il12b and Il1a also increased following LPS stimulation, but the differences were not statistically significant at the 4 hour timepoint (Supporting Figure S4B).
Figure 2.
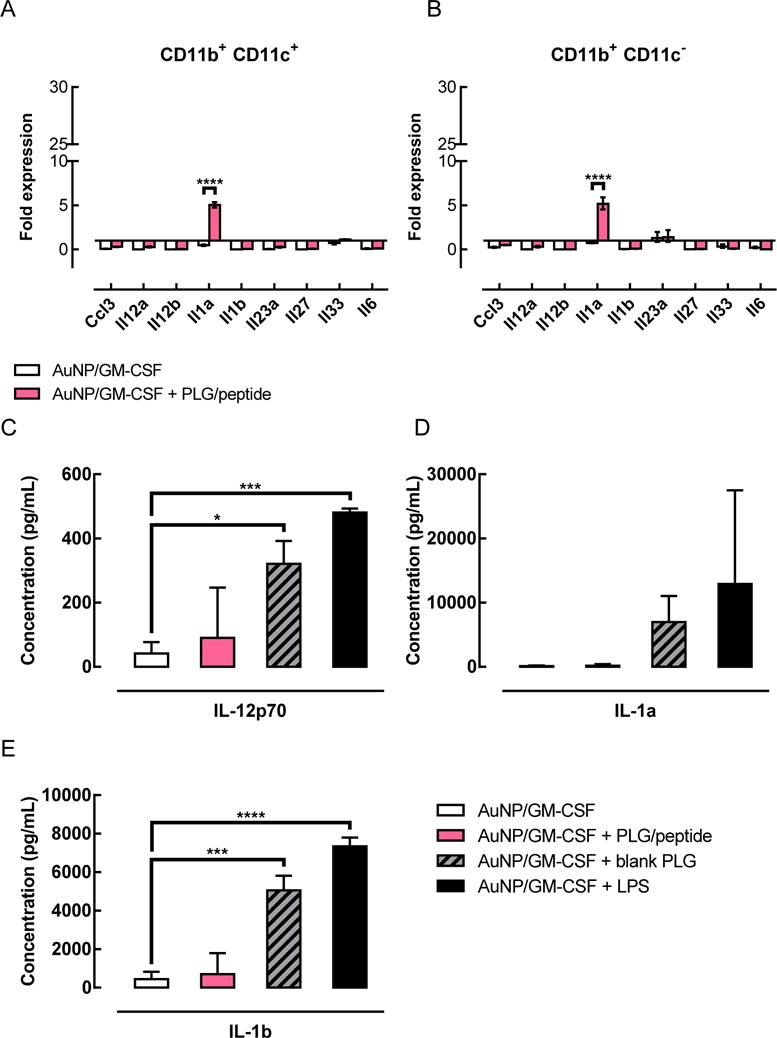
Gels containing blank PLG particles, but not peptide-loaded PLG particles, expressed higher levels of inflammatory cytokines than gels without PLG particles. (A–B) Comparison of gene expression in cells isolated from gels either with or without peptide-loaded PLG particles. Gene expression represented as a fold difference, normalized to splenic DC controls. Gels were injected subcutaneously into the flanks of C57BL/6J mice; after 3 days, gels were isolated, cells were sorted, and gene expression was analyzed by qPCR. The CD11b+ CD11c− “myeloid” (B) and CD11b+ CD11c+ “DC” (A) subsets were analyzed separately. (n = 3; mean ± s.e.m. shown; **** p < 0.0001). (C–E) Concentrations of the inflammatory cytokines IL-12p70 (C), IL-1a (D), and IL-1b (E) in gels delivering either AuNP/GM-CSF alone, or AuNP/GM-CSF together with blank PLG particles or peptide-loaded PLG particles. Gels were injected subcutaneously in the flanks of C57BL/6J mice; at day 3, gels were isolated and subjected to protein extraction for cytokine analysis. Gels delivering AuNP/GM-CSF + 3ug LPS were used as a positive control for inflammatory conditions. (n = 3; mean ± s.d. shown; * p < 0.05; *** p < 0.001; **** p < 0.0001; AuNP/GM-CSF condition was compared to all other conditions.)
Inflammatory cytokines were also analyzed at the protein level by digesting the gels and performing multiplex ELISA assays. Gels delivering GM-CSF alone contained low levels of inflammatory cytokines (Figure 2C–E). When peptide-loaded PLG particles were incorporated into the gels, the level of inflammatory cytokines did not increase significantly compared to gels delivering GM-CSF alone. Unexpectedly, gels containing blank PLG particles exhibited significantly higher levels of IL-12p70 (Figure 2C) and IL-1b (Figure 2E), as well as a trend towards higher levels of IL-1a (Figure 2D). The concentrations of inflammatory cytokines found in these gels approached the concentrations found in gels delivering GM-CSF + LPS, which served as a positive control for inflammatory conditions.
Overall, these data indicate that incorporation of peptide-loaded PLG particles in pore-forming gels is associated with significant differences in the surface marker expression of infiltrating cells, but more subtle differences in cytokine expression.
2.2. Antigen-specific T cell responses to peptide-loaded PLG particles in pore-forming gels
The effects of peptide delivery on antigen-specific T cells in vivo were next examined in a model of type 1 diabetes. Pore-forming gels containing peptide-loaded PLG particles were used to deliver the BDC peptide in NOD mice. BDC2.5 CD4+ T cells, which recognize the BDC peptide in the context of I-Ag7, were labeled with a cell tracker and adoptively transferred prior to gel administration (Figure 3A). As expected, these cells were detectable, but did not proliferate, in naïve mice that did not receive a gel injection or in mice that received gels loaded with a control peptide (OVA). T cells isolated from the draining (dLN), irrelevant (iLN), mesenteric (mLN), and pancreatic (pLN) lymph nodes, as well as in the spleen (spl) were analyzed. Only when their cognate peptide was delivered in the gels did the BDC2.5 T cells proliferate in an antigen-specific manner. The T cells in the lymph nodes draining the gels underwent the greatest extent of proliferation (Figure 3A). In agreement with these data, the frequency of tetramer+ antigen-specific T cells was increased in the same condition where proliferation of the adoptively transferred BDC2.5 T cells was observed. At days 5 (Figure S5) and 10 (Figure 3B), there was a significantly higher percentage of antigen-specific CD4+ T cells found in lymph nodes draining gels delivering BDC peptide, as compared to control peptide. However, by day 20 (Figure 3C), the percentage of antigen-specific T cells returned to baseline levels.
Figure 3.
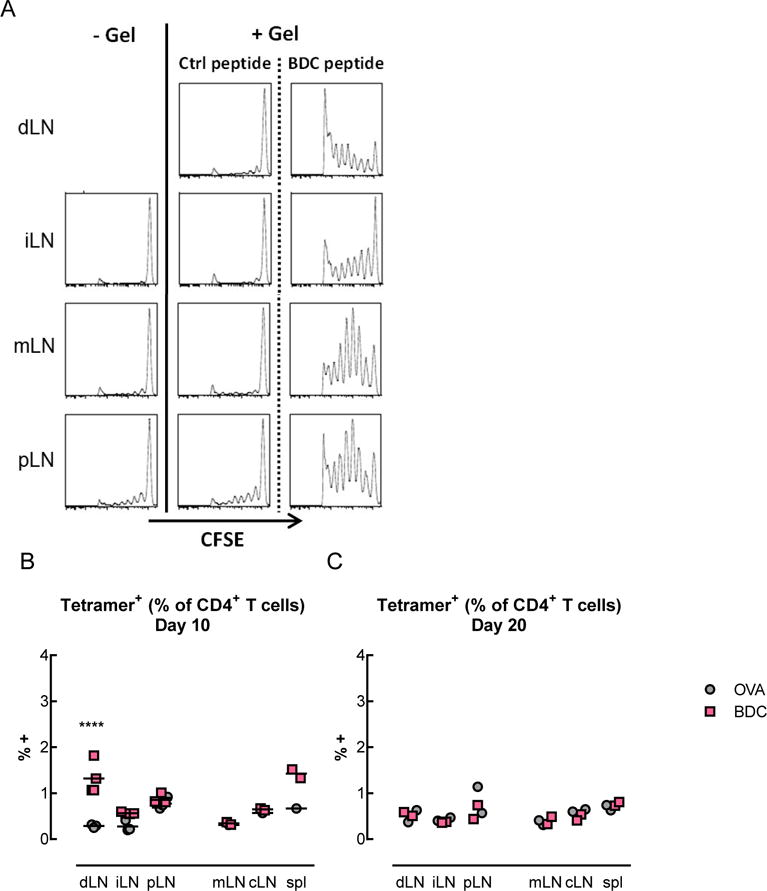
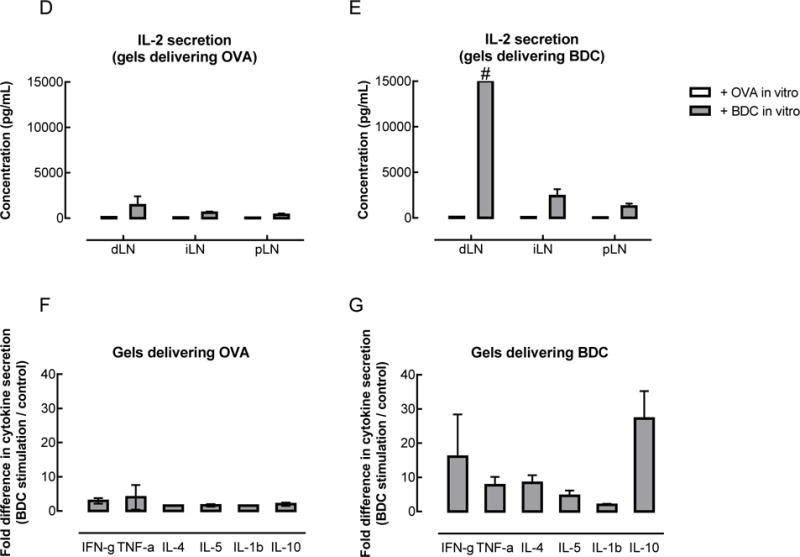
T cells proliferated and secreted cytokines in an antigen-specific manner in vivo following delivery of BDC peptide-loaded PLG particles and GM-CSF in pore-forming gels. (A) Proliferation of adoptively transferred BDC2.5 T cells following administration of gels delivering AuNP/GM-CSF and peptide-loaded PLG particles. BDC2.5 CD4+ T cells were labeled with the cell tracking dye carboxyfluorescein succinimidyl ester (CFSE) and analyzed by flow cytometry to detect cell proliferation (indicated by the progressive halving of CFSE fluorescence intensity). (B–C) Percentage of tetramer+ antigen-specific T cells, which include both adoptively transferred and endogenous cells, in the draining LNs (dLN), irrelevant (iLN), mesenteric (mLN), and pancreatic (pLN) LNs, as well as in the spleen (spl), at days 10 (B), and 20 (C). (n = 2 – 4 per condition per timepoint; individual data points and mean shown; **** p < 0.0001; statistical tests comparing BDC to control were only performed for the conditions where n >= 3). (D–G) Antigen-specific cytokine secretion by T cells. 5 days after administration of gels delivering AuNP/GM-CSF and peptide-loaded PLG particles, T cells were isolated from the draining (dLN), irrelevant (iLN), and pancreatic (pLN) lymph nodes. T cells were restimulated in vitro by BMDCs pulsed with either BDC peptide or control peptide (OVA). Coculture supernatants were analyzed to determine the cytokine secretion profile of the T cells. (D–E) IL-2 secretion by T cells isolated from mice that received control OVA peptide-loaded gels (D) or BDC peptide-loaded gels (E) in vivo. (F–G) Secretion of cytokines associated with Th1, Th2, Th17, and Treg phenotypes by T cells isolated from the draining LNs of mice that received OVA (F) or BDC (G) in gels in vivo, represented as a fold difference in secretion between BDC restimulation and OVA restimulation. (n = 3; mean ± s.d. shown; # data out of range of Bioplex high standard).
The cytokines secreted by these adoptively transferred BDC2.5 T cells were also characterized. CD4+ T cells were isolated from mice that received gels delivering either BDC or control (OVA) peptides; for each of these conditions, the T cells were restimulated in vitro with BMDCs presenting either BDC or OVA peptides. In vitro restimulation with OVA peptide did not lead to substantial cytokine secretion; in contrast, restimulation with BDC peptide triggered cytokine secretion in an antigen-specific manner (Figure 3E,G). T cells isolated from the draining LNs of mice that received BDC-loaded gels secreted substantial amounts of IL-2, a cytokine that induces T cell proliferation, upon restimulation with BDC peptide (Figure 3E). T cells isolated from the irrelevant LNs or the pancreatic LNs of the same mice also secreted IL-2 in response to in vitro restimulation with BDC peptide, but at much lower levels (Figure 3E). Several other cytokines, characteristic of different T helper subsets, were also analyzed. As in the case of IL-2, these cytokines were secreted in an antigen-specific manner by T cells isolated from mice that had received gels delivering BDC in vivo (Fig. 3 F,G). IL-10, a key anti-inflammatory cytokine, exhibited the greatest increase in antigen-specific secretion; 27-fold more IL-10 was released by T cells that were restimulated with BDC peptide as compared to OVA (control) peptide (Figure 3G). The second highest increase in secretion was observed for IFN-γ, a pro-inflammatory anti-viral cytokine, with a 16-fold change relative to the control. The other cytokines assayed, which are associated with different T helper subsets, showed more moderate 2- to 8-fold increases in secretion. T cells isolated from mice that received OVA-loaded gels did not exhibit significant cytokine secretion upon restimulation (Figure 3F).
2.3. Covalent coupling of an MMP-cleavable peptide antigen to alginate
Due to the potential inflammatory response to PLG microparticles, an alternative method was next developed to deliver peptide antigens from pore-forming hydrogels (Figure 4). This approach involved covalent coupling of peptide to the alginate polymer prior to fabrication of the pore-forming gels (Figure 4A,B). Since uptake and presentation of the peptide on MHCII by DCs is critical, a degradable linker was incorporated to allow cell-triggered release of the peptide from the polymer. To determine whether this approach was feasible, MMP activity in the gels was assayed using in vivo imaging with a fluorogenic MMP substrate (Figure 4C,D). MMP activity was found in both gels containing GM-CSF and blank gels, but GM-CSF release led to ~1.7-fold higher MMP activity (Figure 4D). This result indicates that an MMP degradable linker could allow for subsequent peptide release after gel injection. A short spacer sequence and an MMP degradable linker were incorporated on the N-terminal side of the BDC peptide, and a C-terminal Rhodamine B label was used for detection (Figure 4B). Despite the incorporation of aspartic acid residues in the spacer region, the peptide was only soluble in water to a concentration of 2.8 mg/mL (Supporting Figure S6A). To increase the solubility of the peptide and facilitate chemical coupling to alginate in aqueous media, the peptide was first coupled to a 7.5 kDa heterobifunctional polyethylene glycol (PEG) chain via a cysteine residue. The peptide-PEG conjugate, which exhibited increased solubility in water (>= 5 mg/mL; Supporting Figure S6A), was coupled to alginate with a 60% coupling efficiency (Supporting Figure S6B).
Figure 4.
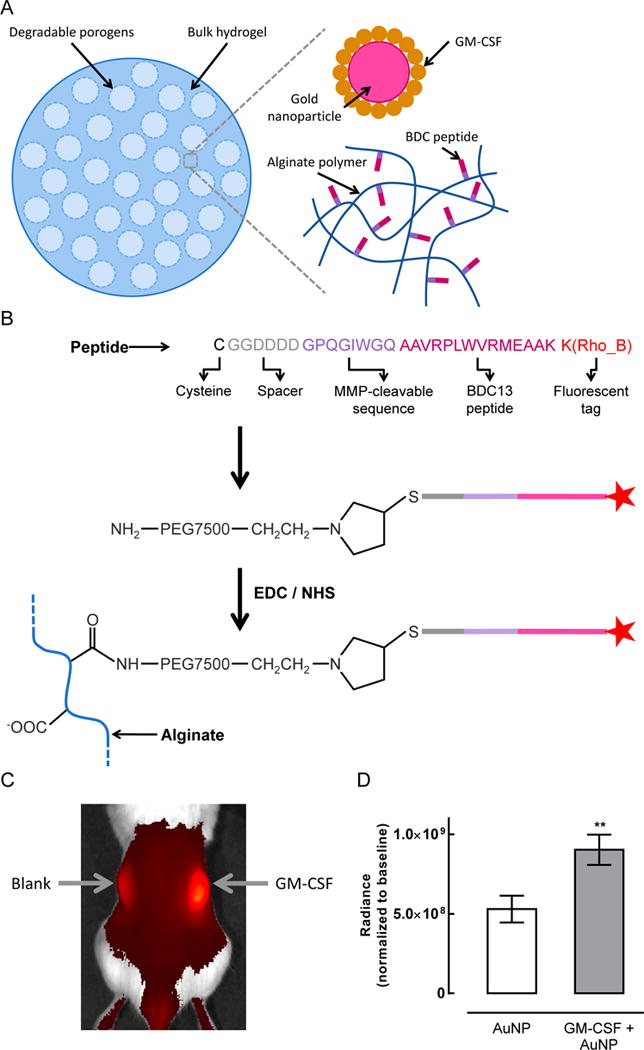
Conjugation of an MMP-cleavable BDC peptide to alginate polymer. (A) Schematic illustrating the different components of pore-forming gels delivering GM-CSF and BDC peptide conjugated directly to the alginate polymer. (B) Strategy for coupling BDC peptide to alginate. Peptide is color-coded to delineate the cysteine (black) used for thiol-maleimide conjugation, a short spacer sequence (grey), the MMP-cleavable sequence (purple), the BDC peptide antigen (pink), and the rhodamine B fluorescent tag (red). (C–D) Pore-forming alginate gels delivering GM-CSF exhibited higher MMP activity than control gels. AuNP control gels and gels delivering GM-CSF + AuNPs were injected subcutaneously into the left and right flanks, respectively, of NOD/ShiLtJ mice. (C) 4 days after administration of the gels, a fluorogenic MMP substrate was injected directly into the gels and 3 replicate mice were imaged. (D) Quantification of the image shown in (C). Regions of interest (ROIs) of the same size were centered over each gel for analysis of the fluorescent signal. (n = 3; mean ± s.d. shown; ** p < 0.01.)
2.4. Antigen-specific T cell responses elicited by peptide-coupled alginate in vitro and in vivo
The ability of the peptide-conjugated alginate to elicit antigen-specific T cells responses was evaluated first in vitro, then in vivo (Figure 5). BDC2.5 CD4+ T cells were co-cultured in vitro with BMDCs in the presence of free BDC peptide or the equivalent molar amount of BDC peptide conjugated to alginate (Figure 5A). The T cells proliferated in the presence of free peptide and peptide-conjugated alginate, but not in the control conditions (vehicle or blank alginate). The peptide-conjugated alginate led to less proliferation than free peptide at the 10−2 and 10−1 μM peptide concentrations; however, increasing doses of free peptide also led to less proliferation under the conditions tested.
Figure 5.
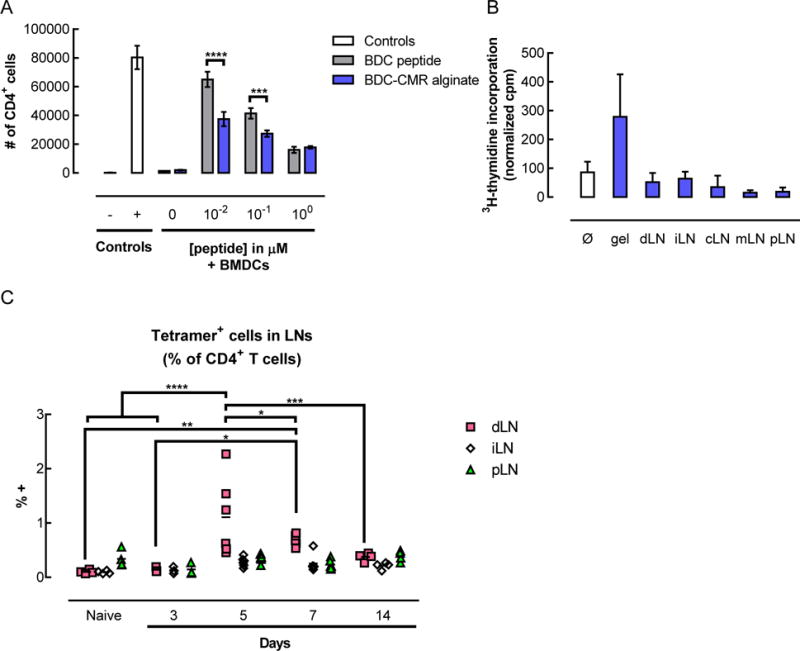
Delivery of peptide-conjugated alginate resulted in functional peptide presentation and in vivo expansion of tetramer+ CD4+ T cells. (A) In vitro proliferation of BDC2.5 T cells in response to soluble BDC peptide or BDC-CMR conjugated alginate, after 3 days of coculture with BMDCs from NOD/ShiLtJ mice. Controls consisted of T cells alone (− control) or T cells cultured with activation beads (+ control for proliferation). (n = 3; mean ± s.d. shown; *** p < 0.001; **** p < 0.0001.) (B–C) In vivo peptide presentation (B) and antigen-specific T cell expansion (C) following subcutaneous administration of pore-forming gels delivering AuNP/GM-CSF and peptide conjugated to alginate in NOD/ShiLtJ mice. (B) In vitro proliferation of BDC2.5 T cells in response to peptide presentation by APCs isolated from various tissues 5 days after injection of gels delivering AuNP/GM-CSF + peptide-conjugated alginate. APCs were isolated from the gels (gel) as well as from the draining (dLN), irrelevant (iLN), cervical (cLN), mesenteric (mLN), and pancreatic (pLN) lymph nodes, then irradiated and co-cultured together with BDC2.5 T cells. No additional BDC peptide was added to the co-cultures in vitro. (n = 3 – 4; mean ± s.d. shown; ** p < 0.01.) (C) Percentage of endogenous antigen-specific CD4+ T cells in the draining (dLN), irrelevant (iLN), and pancreatic (pLN) lymph nodes. At specified timepoints, lymph nodes were isolated and dissociated to analyze lymphocytes by flow cytometry. (n = 3 – 6; individual data points and mean shown; * p < 0.05; **** p < 0.0001; for each condition, the different timepoints were compared to each other.)
To determine whether the peptide could be taken up by DCs in vivo and functionally presented to T cells, peptide-conjugated alginate was used to fabricate pore-forming gels that were injected subcutaneously into NOD mice. Cells were isolated from the gels and various lymph nodes and evaluated for the ability of the APCs found in those tissues to induce proliferation of BDC2.5 CD4+ T cells in an in vitro co-culture. In those in vitro co-cultures, the only source of BDC peptide was peptide that had been taken up and bound to MHCII on APCs in vivo, prior to the cell isolation. The responder BDC2.5 T cells proliferated when co-cultured with cells isolated from the gels, indicating that the DCs within the gel had previously taken up peptide and were able to present it in a functional manner (Figure 5B). On the other hand, APCs isolated from various LNs did not elicit responder T cell proliferation above background levels.
Next, the in vivo effect of gel-based peptide delivery on endogenous antigen-specific T cells was examined (Figure 5C). In the lymph nodes draining the gels, the percentage of antigen-specific CD4+ T cells increased significantly at day 5, reaching greater than 1% of the CD4+ T cells. At day 7, the percentage of antigen-specific cells was lower than at day 5, but still remained elevated relative to baseline. The percentage of antigen-specific T cells continued to decrease with time, returning to levels indistinguishable from baseline at day 14. No significant changes in the percentage of antigen-specific T cells was observed in the other lymph nodes examined (irrelevant and pancreatic). When control gels were administered that delivered either AuNP/GM-CSF alone or AuNP/GM-CSF with an MMP insensitive BDC peptide conjugated to alginate, the percentage of antigen-specific T cells in the LN at day 5 did not vary significantly (Figure S7).
Together, these results show than peptide-conjugated pore-forming gels can deliver a peptide antigen to DCs in vivo, leading to functional presentation of the peptide and antigen-specific expansion of endogenous T cells.
2.5. Enrichment of antigen-specific FoxP3+ cells in gels delivering peptide and in pancreatic islets
In light of the transient antigen-specific T cell expansion observed in the lymph nodes with gel delivery of peptide, antigen-specific T cells were also analyzed in the other relevant tissues, namely, the gels, which contained the BDC peptide mimotope of Chromogranin A, and the pancreatic islets, where the endogenous antigen is found. In the gels, the percentage of CD4+ T cells remained between 2–4% at days 3, 5, and 7, and only increased slightly to ~7% at day 14 (Figure 6A). However, the fraction of those CD4+ T cells that were antigen-specific increased significantly over time (Figure 6B). Initially, at day 3, the tetramer+ population represented less than 1% of the CD4+ T cells in the gels, but subsequently grew to 2.7% and 7% at days 5 and 7, respectively. At day 14, the percentage of antigen-specific cells reached nearly 30%. Notably, a very large fraction of the CD4+ T cells in the gels expressed FoxP3, the master regulator of Tregs. At day 14, greater than 60% of the overall population of CD4+ T cells expressed FoxP3 (Figure 6C). Among the antigen-specific CD4+ T cells, the percentage of FoxP3+ cells was also over 60% in the gels, which was significantly higher than in any of the LNs (Figure 6D). When adjusted for total cell number, this represented an average of ~7,000 antigen-specific FoxP3+ CD4+ T cells in each gel (Figure 6E), which is 1–2 orders of magnitude higher than the number of those cells found in the LNs of naïve mice (i.e. ~90–110 tetramer+ FoxP3+ CD4+ T cells per LN on average). These antigen-specific FoxP3+ cells expressed cell surface receptors that are characteristic of Tregs, such as CD25 (Figure 6F) and CTLA-4 (Figure 6G) at higher levels than the antigen-specific FoxP3− cells.
Figure 6.
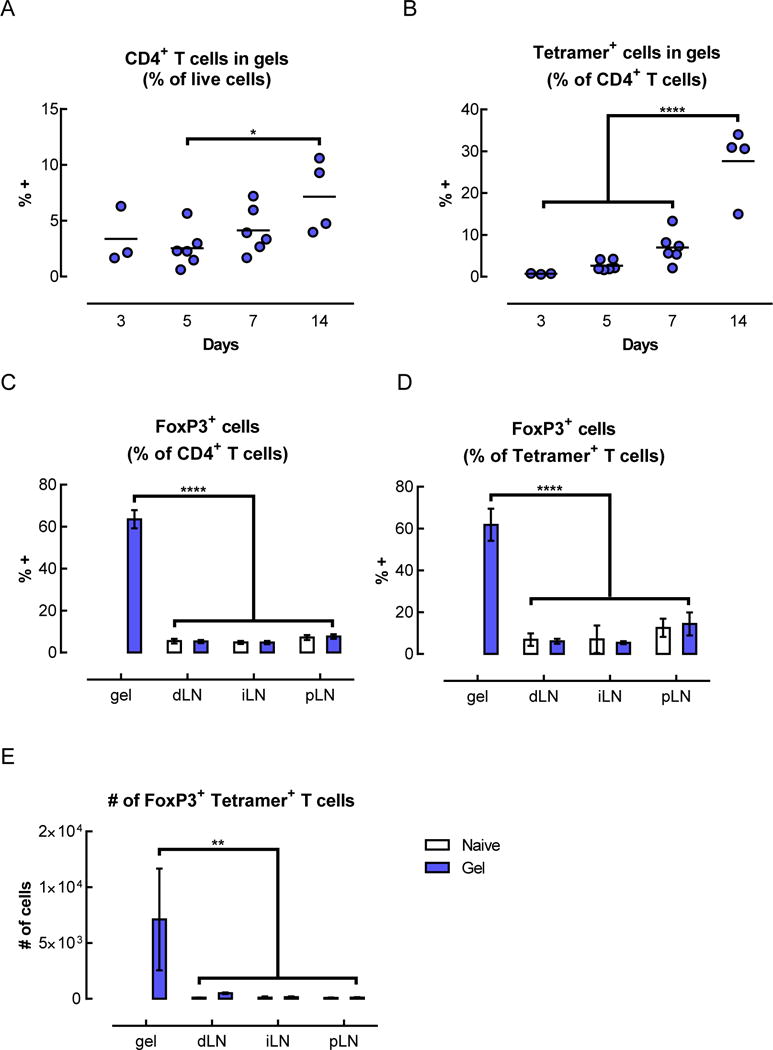
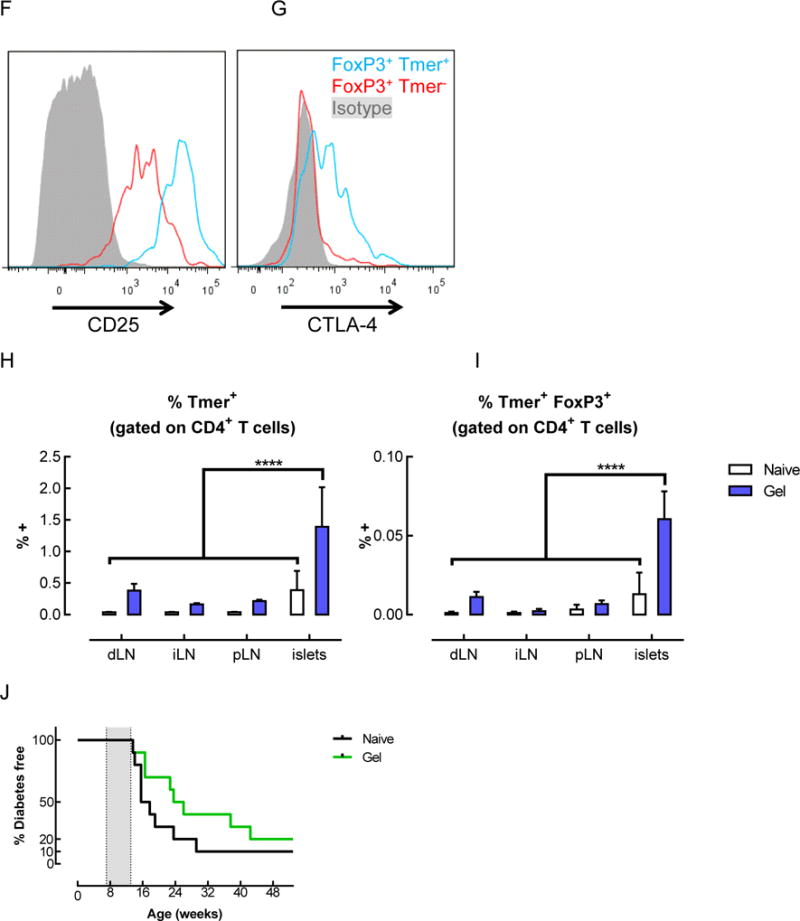
Tetramer+ FoxP3+ CD4+ T cells were enriched in the gels and pancreatic islets of mice that received BDC peptide-conjugated pore-forming gels. (A–G) Flow cytometric analysis of T cells isolated from gels 14 days after subcutaneous injection of gels delivering AuNP/GM-CSF and BDC peptide-conjugated alginate. The percentage of CD4+ cells (A) and tetramer+ CD4+ cells (B) were analyzed. (n = 2 – 6; individual data points and mean shown; * p < 0.05; **** p < 0.0001; all conditions were compared to each other; statistical tests were only performed for the conditions where n >= 3.) (C–E) Analysis of the CD4+ cells in the gels at day 14, showing the fraction of CD4+ cells that were FoxP3+ (C), the fraction of tetramer+ cells that were FoxP3+ (D), and the number of FoxP3+ tetramer+ CD4+ T cells in the gels (E), as calculated by adjusting for total cell number. (n = 3; mean ± s.d. shown; ** p < 0.01; **** p < 0.0001; all conditions were compared to each other.) (F–G) Expression of CD25 (F) and CTLA-4 (G) on tetramer+ CD4+ T cells isolated from peptide-conjugated gels after 14 days. (H–I) Flow cytometric analysis of T cells in the lymph nodes and pancreatic islets of naïve mice or mice that received BDC peptide-conjugated alginate gels. Percentage of tetramer+ cells (H) and tetramer+ FoxP3+ cells (I) among CD4+ T cells at day 14. (J) Spontaneously occurring diabetes study in NOD mice. 3 doses of gels delivering AuNP/GM-CSF and peptide conjugated to alginate were administered between 7–13 weeks (grey shaded region), and mice were monitored for the development of diabetes. (n = 10; p = 0.1259.)
In the pancreatic islets, a site far removed from the subcutaneous gels, the percentage of antigen-specific CD4+ T cells was also altered. The percentage of antigen-specific CD4+ T cells found in the islets was significantly higher for mice that received peptide-conjugated gels (1.4%) as compared to naïve mice (0.4%) (Figure 6H). In addition, the percentage of T cells that were both antigen-specific and FoxP3+ was 4.7-fold higher in mice that received gels delivering the peptide antigen (Figure 6I).
In order to verify that the observed expansion of antigen-specific T cells did not accelerate disease progression in NOD mice, a long-term study of spontaneously occurring diabetes was conducted. Three doses of peptide-conjugated gels were administered to NOD mice between 7 and 13 weeks of age, when islet infiltration is known to have already begun. This treatment did not accelerate disease; on the contrary, disease progression appeared to be delayed, although not in a statistically significant manner (p=0.1259; Figure 6J).
3. Discussion
In this study, a material system that mediates infiltration of a highly enriched population of CD11c+ DCs[23] was used to deliver a defined peptide antigen, and antigen-specific T cell responses were examined. Pore-forming gels, which form the basis for this material system, are injectable and allow in situ formation of large pores suitable for cell infiltration, while retaining the physical and mechanical properties of a nanoporous hydrogel. One potential advantage of pore-forming gels over other types of injectable gels with pre-formed pores, such as cryogels,[32,33] is that their micro-scale stiffness can be more readily tuned to match biological tissues[34]. Previously, the CD11c+ DCs infiltrating pore-forming gels delivering GM-CSF were characterized and found to exhibit a phenotype consistent with immature DCs.[23] Here, their potential to elicit antigen-specific tolerance was explored. Initially, PLG microparticles were used as a vehicle to deliver a peptide antigen in a sustained and spatially localized manner. Intact, unbound peptide was not detected in tissues neighboring the material delivery system, suggesting that the peptide was either degraded or bound to MHC II before reaching detectable concentrations in those tissues. Analysis revealed that incorporation of peptide-loaded PLG particles into the gels significantly altered the phenotype of the cells infiltrating the material. In gels containing peptide-loaded PLG particles, the majority of the cells were Gr-1+, and the percentage of CD11c+, F4/80+, and MHCII+ cells was significantly reduced. In addition to the changes in cell surface marker phenotype, the incorporation of PLG particles also led to more subtle differences in the expression of inflammatory cytokines. At the mRNA level, cells isolated from gels delivering GM-CSF alone expressed similar or lower levels of many genes encoding inflammatory cytokines as compared to immature CD11b+ CD11c+ splenic DCs, supporting the notion that these cells were not mature or inflammatory. When peptide-loaded PLG particles were delivered in addition to GM-CSF, the expression of Il1a, which encodes an inflammatory cytokine that can promote T cell activation, was significantly increased. At the protein level, however, the amount of IL-1a was not statistically significantly different between gels with or without peptide-loaded PLG particles. Expression of the other inflammatory cytokines examined was not increased in a statistically significant manner in the presence of peptide-loaded PLG particles, either at the mRNA or protein levels. Despite clear differences in cell surface marker expression between the sorted CD11c− “myeloid” and CD11c+ “DC” populations, their expression of genes encoding inflammatory cytokines was similar within each gel condition, at least for the transcripts examined here. In contrast, vehicle-only, blank PLG particles led to higher concentrations of inflammatory cytokines in the gels as compared to peptide-loaded PLG particles. This finding was unexpected, considering that the phenotype of cells infiltrating gels loaded either with blank PLG particles or with peptide-loaded PLG particles was very similar, based on the cell surface markers examined here. One possible explanation for the observed difference in cytokine production could be that loading with peptide may alter the non-specific adsorption of proteins at the surface of the particles, which could in turn lead to differential binding or uptake by cells and thus a different inflammatory response.
Delivery of peptide-loaded PLG particles in gels in NOD mice led to antigen-specific T cell responses. Adoptively transferred TCR-transgenic BDC2.5 T cells proliferated following administration of gels delivering their cognate peptide, but not a control peptide. Although antigen-specific T cells were identified in all the lymph nodes that were examined, the extent of proliferation was markedly higher in the lymph nodes draining the gel. This suggests that these T cells were proliferating in the dLN and subsequently trafficking systemically. The frequency of antigen-specific T cells was significantly increased in the dLN of mice that received gels delivering BDC peptide, but not in the other LNs, which is consistent with the fact that the most proliferation was observed in the dLN. The T cells also secreted cytokines in an antigen-specific manner when isolated and restimulated in vitro. Significant amounts of IL-2 were produced upon restimulation by T cells isolated from the draining LNs, and to a lesser extent by T cells isolated from other LNs, which is also consistent with the greatest extent of T cell proliferation having been observed in the dLN. Secretion of cytokines associated with different T helper subsets was also examined, and IL-10, which is indicative of a Treg or Th2 response, exhibited the greatest fold change in antigen-specific secretion. However, cytokines associated with other T helper subtypes, such as Th1 (IFN-g and TNF-a) and Th2 (IL-4 and IL-5), were also secreted upon restimulation, suggesting that polarization towards regulatory subsets was not the clear dominant response. Interestingly, although PLG is highly biocompatible, it can lead to inflammatory responses,[35,36] which is consistent with some of the results seen here. Despite this, several recent studies have demonstrated that PLG nanoparticles could be successfully used to induce tolerance in models of autoimmunity and allergy[12–14,37,16,17]. One strategy involves administering ~500nm particles loaded with antigen (either surface conjugated or encapsulated) by an intravenous (i.v.) route, following which the particles localize to the liver and spleen where they are processed through tolerogenic pathways that normally serve to clear apoptotic cell fragments[12–14]. Interestingly, this system does not require co-delivery of tolerogenic or immunosuppressive factors to be effective, but rather appears to depend on particle uptake by cells expressing the macrophage receptor with collagenous structure (MARCO) scavenger receptor, such as splenic marginal zone macrophages or MARCO+ inflammatory monocytes.[13] In contrast, a different strategy using PLG nanoparticles fundamentally relies on encapsulating the immunosuppressive small molecule rapamycin in the particles,[16,17] but does not require antigen to be co-encapsulated within the particles, since tolerance can be induced following i.v. co-administration of soluble antigen along with rapamycin containing PLG particles[17]. In those studies, both the size and delivery route of the particles differed from the particles used here, which were significantly larger (~30 μm in diameter) and were intended to serve as a depot of antigen that would be retained within subcutaneous gels and release antigen locally.
Due to the observed effect of PLG particles on the phenotype and inflammatory cytokine production of cells infiltrating the gels, an alternate approach was developed to deliver a peptide antigen from pore-forming alginate gels. The BDC peptide was covalently conjugated to the alginate polymer that constitutes the bulk phase of the gels, with an intermediate MMP degradable linker that allowed cell-triggered release and presentation of the peptide by DCs. Pore-forming gels delivering GM-CSF exhibited increased MMP activity in vivo relative to blank gels, suggesting that the cells infiltrating the gels could serve as a source of MMPs to trigger peptide release. The MMP-cleavable linker used here was 8 amino acids long, with MMP cleavage occurring in the middle of the sequence. The 4 amino acids remaining on the N-terminus of the BDC peptide following MMP cleavage were not expected to be an issue for peptide presentation, since the structure of MHCII allows for overhangs on either side of the binding pocket, and, as a result, the length of MHCII epitopes is not tightly restricted. Peptide-conjugated alginate stimulated antigen-specific T cell expansion in vitro, demonstrating that released peptide could indeed be presented by DCs on MHCII and recognized by T cells. As compared to free BDC peptide, the peptide-conjugated alginate at an equivalent peptide concentration yielded a lower number of T cells at the end of the co-culture. This may be a reflection of the fact that the concentration range used was higher than optimal and resulted in T cell overstimulation, since increasing doses of peptide led to a lower final cell count, both for the free peptide and peptide-conjugated alginate conditions. Accordingly, the peptide-conjugated alginate may actually lead to more effective peptide presentation by DCs in vitro as compared to an equivalent dose of free peptide.
In vivo, gels fabricated using peptide-coupled alginate led to localized peptide delivery and to antigen-specific responses. DCs isolated from the gels, but not from other more distant lymph nodes, were able to functionally present peptide in vitro to responder T cells, indicating that peptide was delivered in a localized manner and did not spread systemically. Further, expansion of endogenous antigen-specific T cells was observed specifically in the LNs draining the gels. This expansion was transient, with the highest percentage of antigen-specific cells observed at day 5. In addition, this antigen-specific T cell expansion was dependent on delivery of the BDC peptide as well as on MMP sensitive release of the peptide, since administration of control gels did not lead to a significant changes in this cell population. Over time, antigen-specific T cells accumulated in locations where antigen was present. In the gels, the overall percentage of T cells did not change significantly, but antigen-specific T cells constituted an increasing percentage of CD4+ T cells over time. Strikingly, a very high percentage of CD4+ T cells in the gels expressed FoxP3, a master regulator of Tregs.[3,4] Whereas natural Tregs normally comprise only 5–10% of CD4+ T cells in naïve mice[4,38,39], >~60% of the CD4+ T cells in the gels were FoxP3+. Among antigen-specific CD4+ T cells, the proportion of FoxP3+ cells was also ~60% in the gels, which was several fold higher than in the LNs. The tetramer+ FoxP3+ cells in the gels also expressed CD25 and CTLA-4, which are other characteristic markers of Tregs. When adjusted for total cell numbers, ~7,000 cells in the gels were both tetramer+ and FoxP3+, which is significantly higher than the number found in all the other LNs, but ~20-fold lower than the number of BDC2.5 Tregs that were shown to prevent diabetes in 100% of prediabetic NOD mice upon adoptive transfer.[6] In addition to accumulating in the gels, antigen-specific T cells were also identified at a higher frequency in the pancreatic islets, where the endogenous ChrA derived antigen is found, 14 days after gel administration. The pancreatic lymph nodes were also analyzed, but no differences in the frequency antigen-specific T cells were observed at the timepoints examined. In the islets of mice that received peptide-conjugated gels, both the overall percentage of antigen-specific T cells, as well as the percentage of FoxP3+ antigen-specific T cells, were increased. The enrichment of antigen-specific CD4+ T cells in the gels and islets occurred over the same timeframe as the contraction of that same cell population in the dLN, which suggests that the T cells may have been leaving the draining LNs and trafficking to peripheral tissues.
Finally, a long-term study was carried out in NOD mice to evaluate the effect of peptide-loaded gels on the progression of spontaneously occurring autoimmune diabetes. It is known that BDC2.5 CD4+ T cells have the potential to differentiate into multiple T helper subsets and act either as protective Tregs[5–7] or as pathogenic Th1 effector cells[28,29]. While we observed an expansion of BDC antigen-specific FoxP3+ Tregs following gel administration, the remainder of the antigen-specific T cells that were FoxP3− might have had the potential to act as effector T cells. In addition, the stability of regulatory T cells, even those expressing FoxP3, has been debated in the field,[40,41] leaving open the possibility that some of the antigen-specific Tregs might exhibit plasticity under inflammatory conditions and contribute to disease pathogenesis. In order to address these concerns, three doses of gel were administered between 7 to 13 weeks, at an age when islet infiltration and inflammation become more severe.[42,43] This treatment did not accelerate disease; on the contrary, disease progression was slightly delayed in the mice that received the gels, albeit not in a statistically significant manner. In the future, to improve upon these results, a number of parameters can be optimized, including: the choice of antigen, the antigen dose and release kinetics, and the timing of gel administration. The choice of one or more antigens is an important factor in designing antigen-specific therapies to control disease. It has been shown that Tregs of a single antigen specificity could effectively treat diabetes, but the number of Tregs that needed to be adoptively transferred were relatively high (1.5 × 105 Tregs to prevent diabetes in prediabetic NOD mice or 1.5 × 106 cells to reverse disease in 50% of mice with recent onset diabetes).[6] In addition, it is known that epitope spreading occurs over time as autoimmune disease progresses, so it may be beneficial to deliver a combination of antigens and generate Tregs of more than one specificity to control disease, particularly at later stages of progression. In addition, the antigen dose and release kinetics can impact the T cell response. In in vitro DC : T cell co-cultures, low doses of peptide antigen were shown to be more effective than high doses at inducing and expanding suppressive Foxp3+ Tregs in mouse[44] and human[45] T cells. In vivo, administering low doses of a high affinity antigen favored induction of FoxP3+ Tregs[46,47]; in contrast, delivering high doses of a low affinity antigen led to deletion of antigen-specific cells after an initial proliferation[47]. The doses considered to be “low” depended on the affinity of the peptide mimotopes and varied by 3–4 orders of magnitude[44]. Here, 0.1nmol of peptide were delivered in each gel, and a total of 3 gels were administered to mice for the diabetes disease study. By way of comparison, other studies delivered 0.7pmol to 7nmol per day of HA peptide for 10–14 days[46] or 4.1 nmol per day of insulin peptide for 14 days.[48] In addition to the overall dose of peptide delivered, the kinetics of release could also impact T cell responses. Cell-triggered release is appealing, since it enables peptide release when cells are present to take up the antigen. However, slow release kinetics can potentially result in a longer lasting depot of peptide at the site of the material, which may favor trafficking of T cells into the gels rather than to the site of disease, such as the pancreatic islets. The MMP degradable sequence could easily be varied to modulate release kinetics, since various MMP sequences have been characterized for their MMP specificity and degradation kinetics.[49,50] Beyond optimizing various parameters related to antigen delivery, another promising avenue to explore in future work is the co-delivery of tolerogenic or immunosuppressive factors.
4. Conclusions
An injectable, non-inflammatory material delivery system, which was previously characterized for its ability to locally enrich CD11c+ DCs, was used here to deliver a peptide antigen for the purpose of inducing antigen-specific tolerance. Using two different methods to deliver the BDC peptide mimotope in NOD mice, it was shown that delivery of the peptide antigen was localized, and T cells responded in an antigen-specific manner. Following gel-based peptide delivery, antigen-specific T cells were enriched in locations where the antigen was found, namely in the gels and in the pancreatic islets. In the future, this material system can be further developed by optimizing the dose and kinetics of peptide delivery, as well as exploring the co-delivery of tolerogenic factors. In addition, the high percentage of Tregs in gels raises the possibility of using this material system for applications where a local accumulation of Tregs is beneficial, such as in settings of transplant tolerance.
5. Experimental Section
Animals
All work involving C57BL/6J mice (female, aged 6–10 weeks; Jackson Laboratories), NOD/ShiLtJ mice (female, aged 6–10 weeks; Jackson Laboratories), or BDC2.5 mice (female, aged 6–10 weeks; Jackson Laboratories) was performed in compliance with National Institutes of Health and institutional guidelines.
Peptides
BDC-13 peptide (AAVRPLWVRMEAA), BDC peptide with biotin and DNP tags (Biotin-AAVRPLWVRMEAAK(DNP)), and BDC-CMR (Cysteine, MMP cleavable sequence, Rhodamine B) peptide (CGGDDDDGPQGIWGQAAVRPLWVRMEAAKK(Rho_B)) were obtained from Peptide 2.0 or 21st Century Biochemicals.
Alginate Oxidation and Reduction
Medical grade, high guluronic acid content, high molecular weight alginate (MVG) (FMC Biopolymers) was subjected to 7.5% oxidation and reduction, as described previously.[23]
Alginate Peptide Conjugation
BDC-CMR peptide was reacted with heterobifunctional maleimide PEG amine (7.5kDa, JenKem Technology USA) in the presence of tris(2-carboxyethyl)phosphine (TCEP) in DMSO (1:1:1 molar ratio). The DMSO solvent was removed by lyophilization and the peptide-PEG conjugate was resuspended in H2O. The amine on the PEG was coupled to alginate using EDC/Sulfo-NHS as described previously.[34] Following dialysis, peptide coupling was assayed by measuring the absorbance of the rhodamine B label using a BioTek plate reader.
Alginate Porogen Fabrication
Porogens containing 2% w/v of 7.5% oxidized and reduced alginate together with 0.25% unmodified MVG alginate were fabricated as described previously.[23] Briefly, a solution of alginate in DMEM was dispensed through a glass nebulizer while applying a coaxial flow of N2 gas (5.2 SLPM). Porogen beads were collected in a cross-linking solution of 100 × 10−3 M CaCl2 and 100 × 10−3 M HEPES (pH 7.2). Beads were cross-linked for 5 min and washed thoroughly in HBSS (Sigma Aldrich) by centrifugation at 1800 g.
AuNP Synthesis and Characterization
AuNPs were synthesized via a citrate-reduction method and characterized as described previously.[23] Briefly, a solution of 0.01% w/v gold chloride (III) chloride hydrate (Sigma Aldrich # 254169) in H2O (100 mL) was brought to a boil in a 250 mL Erlenmeyer flask placed on a heating and stirring plate set to 400 °C and 500 rpm. A 1% w/v sodium citrate tribasic dihydrate (Sigma Aldrich # 4641) solution (3 mL) was rapidly added, and the solution was removed from the stirring/heating plate once its color turned to red. The size of the synthesized AuNPs was approximately 13 nm (after subtracting 3 nm to account for the citrate ions and hydration shell), as determined by dynamic light scattering (DLS) on a Malvern Zen 3600 Zetasizer.
AuNP Conjugation
GM-CSF was conjugated to AuNPs as described previously.[23] Briefly, a sterile-filtered AuNP solution (525 μL, 45 μg mL−1) was concentrated 157.5-fold by centrifuging at 20 000 g for 20 min and removing supernatant. Particles were pipetted and sonicated for 10 min in a water bath sonicator to resuspend. Concentrated AuNPs (3.33 μL) were mixed with recombinant murine GM-CSF (3 μg, 1 mg mL−1 in dH2O; Peprotech # 315-03) and incubated at 37 °C for 1 h to allow formation of gold–sulfur bonds.
PLG Microsphere Fabrication
PLG microspheres were fabricated by a standard emulsion technique.[18] Briefly, a 100μL solution of peptide in DMSO or DMSO only (blank control) was added to 1mL of a 5% solution of PLG (85:15, 120 kD copolymer of D, L-lactide and glycolide, Alkermes) in ethyl acetate in a silanized glass test tube. The solution was sonicated at 60% at a continuous setting for 10sec using a Vibracell probe sonicator (Sonics & Materials). 1mL of 1% PVA / 7% Ethyl Acetate in H2O was immediately added and the mixture was vortexed for 10sec. The emulsion was added to 200mL of 0.3% PVA / 7% ethyl acetate in H2O and stirred for 3h to allow solvent evaporation. Microspheres were collected and washed thoroughly in H2O.
In Vitro Peptide Release
Peptide-loaded PLG particles were gas-foamed to form porous scaffolds as described previously[18]. The resulting peptide-loaded scaffolds were incubated at 37°C in 1mL of release buffer (HBSS containing 1% BSA and 1% penicillin/streptomycin). At the indicated timepoints, the release buffer was collected and replaced with 1mL of fresh buffer. The amount of released peptide was quantified by peptide ELISA, as described below.
In Vivo Peptide Biodistribution
Peptide-loaded scaffolds were implanted subcutaneously into the left flanks of C57BL/6J mice, as described previously[18]. At the indicated timepoints, scaffolds and tissue samples were isolated. Samples were taken from muscle tissue directly underlying the material, as well as 4mm, 8mm, and >3cm (i.e. on the right flank) away from the implanted material. The left inguinal and axillary LNs were also isolated. The scaffold was cut into small (~1mm) pieces, and 500μL of T-Per reagent (Pierce) was used to extract proteins from the tissue infiltrating the scaffold, following the manufacturer’s instructions. The pieces of polymer, which were insoluble, were separated by centrifugation. Proteins were extracted from the other tissue samples using 500μL of T-Per reagent, as for the scaffolds. All samples were analyzed by peptide ELISA, as described below.
Peptide ELISA
To enable detection of full length, non-degraded peptide, biotin and DNP tags were incorporated at the N- and C-termini of the peptide, respectively. Peptide was detected using a modified sandwish ELISA assay. StreptaWell (High Bind, Roche) 96 well plates were blocked in assay buffer (1% BSA in PBS) at room temperature for 1h. 50μL of fresh assay buffer + 50μL of standards or samples (in T-Per reagent) were added to the wells and incubated for 1h at room temperature. The DNP tag on the peptide was detected using 100μL of anti-DNP antibody (10μg/mL), followed by 100μL of anti-rat IgG-HRP secondary antibody (10μg/mL). Detection steps were performed by diluting reagents in assay buffer, incubating at room temperature for 1h, and washing wells 5 times in wash buffer (0.05% Tween-20 in PBS) in between each step. 100μL of chromogenic substrate (R&D Systems or BioLegend) were allowed to develop for 20–30min before the addition of 100μL 1N sulfuric acid stop solution. Plates were read for absorbance at 450nm within 30 min, and absorbance at 540nm was used for wavelength correction.
Alginate Hydrogel Fabrication
Unmodified alginate was reconstituted in DMEM without L-cystine (Sigma Aldrich) while stirring at room temperature overnight to obtain a 3% w/v alginate solution. The alginate was mixed with additional cystine-free DMEM containing GM-CSF-loaded AuNPs, yielding a final concentration of 2% w/v unmodified alginate. When specified, PLG microparticles were added to the alginate solution. This mixture, which constituted the bulk phase of the gels, was then combined with 50% v/v of pre-formed porogen beads. Finally, the bulk gel was cross-linked by mixing with a sterile calcium sulfate slurry (1.2 M CaSO4·2H2O in H2O, 4% v/v relative to the bulk alginate). Mixing steps were performed using Luer-Lock syringes (1 mL) joined with Luer-Lock connectors, and a needle (18 G) was used to allow subcutaneous injection immediately after cross-linking the bulk phase of the gel (within ~30–60 s).
In Vivo Cell Characterization
Gels were injected subcutaneously into one flank of mice. At various timepoints, gels were removed and dissociated in EDTA (40 × 10−3 M in PBS) on ice for 10 min, with periodic vortexing. Cells were filtered through 40 μm cell strainers, and live cells were counted using a Countess automated cell counter (Life Technologies). Flow Cytometry : Cells were blocked with Fc Block (BioLegend) for 15 min and stained with antibodies (BioLegend, eBioscience, or BD Biosciences) for 30 min. The antibodies used were specific for CD11b (clone M1/70), CD11c (clone N418), F4/80 (clone BM8), Gr-1 (clone RB6-8C5), MHC II (I-A/I-E) (clone M5/114.15.2), CD3e (145-2C11), CD4 (RM4-5 or GK1.5), FoxP3 (FJK-16s), CD25 (PC61), and CTLA-4 (UC10-4B9). I-Ag7 tetramers loaded with BDC15 peptide (AAAAVRPLWVRMEAA) and chicken hen egg lysozyme (HEL) 11–25 peptide (AMKRHGLDNYRGYSL; used as a staining control) were obtained from the NIH Tetramer Core Facility. 7-AAD (BioLegend) or Fixable Live/Dead dyes (Life Technologies) were used for live and dead cell discrimination. All blocking and antibody staining steps were performed in flow cytometry staining buffer consisting of PBS with 0.5% BSA and 2 × 10−3 M EDTA at 4 °C (except for tetramer staining, which was performed at room temperature). Data were collected on either LSRII or LSRFortessa flow cytometers (BD Biosciences) and analyzed using FlowJo software (TreeStar). Appropriate fluorescence minus one (FMO) controls (using isotype matched antibodies and control tetramer) were run for each experiment to ensure that gating and analysis of the low frequency BDC tetramer+ CD4+ T cells was accurate.
In vivo MMP activity assay
Gels were injected subcutaneously in the flanks of NOD/ShiLtJ mice. 4 days later, 10μL of MMPSense 750 FAST (Perkin Elmer) were injected into the gels. As recommended by the manufacturer, baseline images were taken before injection of the MMPSense reagent, and mice were imaged again 4h after injection. Fluorescent in vivo imaging was performed using an IVIS Spectrum system (Perkin Elmer), and images were analyzed using Living Image software (Perkin Elmer). T Cell Culture : Primary CD4+ T cells were isolated from the spleen and lymph nodes of BDC2.5 mice, sorted using an untouched CD4+ MACS sorting kit (Miltenyi), and cultured in RPMI-1640 medium (with L-glutamine; Sigma Aldrich) supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, 10mM HEPES, and 50μM β-mercaptoethanol. This T cell medium was also used for co-cultures of T cells and DCs. BMDC Culture : Bone marrow cells were isolated using a standard procedure. Briefly, the femur and tibia bones were isolated and flushed with HBSS using a needle (25G). Cells were dissociated and filtered through a 40um cell strainer. For NOD BMDCs, 5 × 106 cells were plated in 10mL of medium in bacteriological Petri dishes. The medium used for BMDC differentiation consisted of RPMI-1640 medium (with L-glutamine; Sigma Aldrich) supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, 50μM β-mercaptoethanol, and 20ng/mL GM-CSF. At day 3, 10mL of fresh medium were added to each dish, and at day 6, 10mL of medium were removed from each dish and replaced with 10mL of fresh medium.
In Vitro Peptide Presentation
Bone marrow cells from NOD mice were isolated and differentiated as described above. On day 6 of the BMDC culture, 50ng/mL LPS was added to mature the BMDCs for 24h. On day 7, cocultures were set up by plating 105 BDC2.5 CD4+ T cells and 2.5 104 BMDCs per well in 96 well plates. The indicated concentrations of BDC peptide or peptide-conjugated alginate were added in a total volume of 250μL/well. The proliferation of the BDC2.5 T cells was analyzed after 3 days. As a positive control for proliferation, T cells were cultured at a 1:4 ratio with mouse anti-CD3/CD28 activation beads (Dynabeads, Life Technologies).
In Vivo Peptide Presentation
Gels were injected subcutaneously into the flanks of NOD/ShiLtJ mice. At the indicated timepoints, gels and LNs were isolated and dissociated, as described for the in vivo cell characterization studies. The cells were resuspended in medium and irradiated with 5krad to inhibit their proliferation. Separately, CD4+ T cells were isolated from the spleen and lymph nodes of BDC2.5 mice and sorted using an untouched CD4+ MACS sorting kit (Miltenyi). Each of the irradiated cell samples was plated into 6 separate wells of a 96 well plate. 3 wells per sample were used for background subtraction, and 105 BDC2.5 CD4+ T Cells were added to the other 3 wells. After 48h, 1μCi of 3H-thymidine was added to each well. After an 18h pulse, the samples were harvested on a Tomtec cell harvester and 3H counts were read using a Microbeta 1450 scintillation counter.
In Vivo T Cell Proliferation
BDC2.5 T cells were labeled with a cell proliferation dye (CFSE or eFluor 670, eBioscience) and 1–2 × 106 cells were adoptively transferred into NOD recipients i.v. by tail vein injection. One day later, gels delivering peptide were injected. LNs were isolated at various timepoints, and the dilution of the cell proliferation dye was analyzed by flow cytometry.
In vivo T cell cytokine secretion
Gels were injected subcutaneously into the flanks of NOD/ShiLtJ mice. At the indicated timepoints, LNs were isolated and dissociated as described for the in vivo cell characterization studies. The samples were enriched for CD4+ T cells using an untouched CD4+ MACS sorting kit (Miltenyi). The enriched T cells were cocultured with 7.5 × 104 NOD BMDCs, and 1μM peptide was added for restimulation. A BMDC culture was started 7 days in advance of the coculture according to the procedure described above, and 24h before the start of the coculture, 50ng/mL LPS was added to mature the BMDCs. Supernatants from the DC : T cell cocultures were collected after 24h and stored at −20°C until cytokine analysis was performed, as described below.
qPCR
Cells were stained for CD11b, CD11c, CD3e, DX5. Calcein blue (eBioscience) was added to a final concentration of 10 μM to discriminate live cells and sorted on a Beckman Coulter MoFlo Astrios. RNA was extracted using TRIzol reagent (Life Technologies) according to the protocol recommended by the Immunological Genome Project.[51] cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The RT-PCR reaction was carried out using TaqMan gene expression assays with the TaqMan Fast Advanced Master Mix on an ABI 7900 HT Real-Time PCR System (Applied Biosystems).
Cytokine analysis
Gels or cell culture supernatants were analyzed by performing BioPlex assays, using either the mouse cytokine 23-plex or 8-plex (Bio-Rad) according to the manufacturer’s instructions. The samples were run on a BioPlex 3D System (Bio-Rad). To extract proteins from gels for cytokine analysis, 500μL of T-Per reagent (Pierce) were added to 100μL gels, and protein extraction was performed according to the manufacturer’s instructions.
Statistical Analysis
Statistical analyses were performed using Prism 6 (GraphPad). When comparing two groups, a two-tailed Student’s t-test was used. When comparing multiple groups, a one-way or two-way analysis of variance (ANOVA) was performed with corrections for multiple comparisons.
Supplementary Material
Figure S1. Sustained delivery of peptide antigen in vivo led to localized antigen presentation in the gel and draining lymph nodes. (A) In vitro release of OVA peptide from PLG particles that were foamed into porous scaffolds. (n = 3 per condition per timepoint; mean ± s.d. shown). (B) In vitro release of BDC peptide from PLG particles incorporated into alginate gels. (n = 4 per condition per timepoint; mean ± s.d. shown). (C–D) In vivo peptide biodistribution. (C) OVA peptide-loaded PLG scaffolds (S) were implanted subcutaneously into the flanks of C57BL/6J mice. (D) Intact OVA peptide was quantified by ELISA at the indicated timepoints in lysates from various tissues, including the muscle underlying the gel (L1), as well as 4mm (L2), 8mm (L3), and >3cm (R) away from the material (n = 4 per condition per timepoint; mean ± s.d. shown).
Figure S2. (A–B) Flow cytometry plots showing expression of CD11c and Gr-1 by cells isolated from gels delivering only AuNP/GM-CSF (A) or AuNP/GM-CSF with peptide-loaded PLG particles (B) at 5 days.
Figure S3. Overview of 23 different cytokines measured in gels delivering GM-CSF conjugated to AuNPs.
Figure S4. (A) Cell sorting strategy to separate CD11b+ CD11c− “myeloid” cells from CD11b+ CD11c+ “DCs” for analysis. (B) Gene expression of control CD11b+ CD11c+ splenocytes before or after 4h LPS stimulation, expressed as fold differences relative to unstimulated controls.
Figure S5. Expansion of antigen specific T cells 5 days after administration of gels delivering peptide-loaded PLG particles. Percentage of tetramer+ antigen-specific T cells, which include both adoptively transferred and endogenous cells, in the draining LNs (dLN), irrelevant (iLN), mesenteric (mLN), and pancreatic (pLN) LNs, as well as in the spleen (spl), at day 5.
Figure S6. Characterization of peptide conjugated alginate. (A) Dissolution of the BDC-CMR peptide in water before and after conjugation to PEG. Dashed line indicates expected concentration (5mg/mL) assuming complete dissolution of the peptide, while bars indicate actual measured peptide in solution. (B) Efficiency of peptide coupling to alginate was determined by measuring the amount of peptide removed during dialysis versus what was measured to be conjugated to the alginate.
Figure S7. In vivo expansion of antigen-specific T cells following administration of pore-forming gels delivering AuNP/GM-CSF either alone or in combination with MMP sensitive or insensitive BDC peptide conjugated to alginate. Gels were injected subcutaneously into the flanks of NOD/ShiLtJ mice; at specified timepoints, lymph nodes were isolated and dissociated to analyze lymphocytes by flow cytometry. Percentage of endogenous antigen-specific CD4+ T cells in the draining (dLN), irrelevant (iLN), and pancreatic (pLN) lymph nodes. (n = 4 – 6; individual data points and mean shown; ** p < 0.01; **** p < 0.0001; for each type of lymph node, the different conditions were compared to each other.)
Acknowledgments
This work was supported by the JDRF (5-2011-434) and the National Institutes of Health (NIH) (1R01DE019917). We thank the NIH tetramer core facility for providing mouse MHC Class II tetramers.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Catia S. Verbeke, School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA.
Dr. Susana Gordo, Dana Farber Cancer Institute, Boston, MA 02215, USA
Dr. David Schubert, Dana Farber Cancer Institute, Boston, MA 02215, USA
Sarah A. Lewin, Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA
Dr. Rajiv M. Desai, School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA.
Dr. Jessica Dobbins, Dana Farber Cancer Institute, Boston, MA 02215, USA
Prof. Kai Wucherpfennig, Dana Farber Cancer Institute, Boston, MA 02215, USA
Prof. David J. Mooney, School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA.
References
- 1.Yu W, Jiang N, Ebert PJR, Kidd BA, Müller S, Lund PJ, Juang J, Adachi K, Tse T, Birnbaum ME, Newell EW, Wilson DM, Grotenbreg GM, Valitutti S, Quake SR, Davis MM. Immunity. 2015;42:929. doi: 10.1016/j.immuni.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li MO, Rudensky AY. Nat Rev Immunol. 2016;16:220. doi: 10.1038/nri.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell DJ. J Immunol. 2015;195:2507. doi: 10.4049/jimmunol.1500801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Immunological Reviews. 2006;212:8. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 5.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. J Exp Med. 2004;199:1467. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, Yang H, Suthanthiran M, Mojsov S, Steinman RM. J Exp Med. 2007;204:191. doi: 10.1084/jem.20061631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaeckel E, von Boehmer H, Manns MP. Diabetes. 2005;54:306. doi: 10.2337/diabetes.54.2.306. [DOI] [PubMed] [Google Scholar]
- 8.Robbins HA, Clarke CA, Arron ST, Tatalovich Z, Kahn AR, Hernandez BY, Paddock L, Yanik EL, Lynch CF, Kasiske BL, Snyder J, Engels EA. J Invest Dermatol. 2015;135:2657. doi: 10.1038/jid.2015.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doycheva I, Amer S, Watt KD. Med Clin North Am. 2016;100:551. doi: 10.1016/j.mcna.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Coppieters KT, Harrison LC, von Herrath MG. Clinical Immunology. 2013;149:345. doi: 10.1016/j.clim.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller SD, Turley DM, Podojil JR. Nat Rev Immunol. 2007;7:665. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- 12.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD. Nat Biotech. 2012;30:1217. doi: 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Getts DR, Terry RL, Getts MT, Deffrasnes C, Müller M, Vreden C van, Ashhurst TM, Chami B, McCarthy D, Wu H, Ma J, Martin A, Shae LD, Witting P, Kansas GS, Kühn J, Hafezi W, Campbell IL, Reilly D, Say J, Brown L, White MY, Cordwell SJ, Chadban SJ, Thorp EB, Bao S, Miller SD, King NJC. Sci Transl Med. 2014;6:219ra7. doi: 10.1126/scitranslmed.3007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smarr CB, Yap WT, Neef TP, Pearson RM, Hunter ZN, Ifergan I, Getts DR, Bryce PJ, Shea LD, Miller SD. PNAS. 2016;113:5059. doi: 10.1073/pnas.1505782113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Proc Natl Acad Sci USA. 2012;109:11270. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maldonado RA, LaMothe RA, Ferrari JD, Zhang A-H, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, Andrian UH von, Kishimoto TK. PNAS. 2015;112:E156. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishimoto TK, Ferrari JD, LaMothe RA, Kolte PN, Griset AP, O’Neil C, Chan V, Browning E, Chalishazar A, Kuhlman W, Fu F, Viseux N, Altreuter DH, Johnston L, Maldonado RA. Nat Nano. 2016 doi: 10.1038/nnano.2016.135. advance online publication. [DOI] [PubMed] [Google Scholar]
- 18.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Nat Mater. 2009;8:151. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali OA, Emerich D, Dranoff G, Mooney DJ. Science Translational Medicine. 2009;1:8ra19. doi: 10.1126/scitranslmed.3000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dendritic Cell Activating Scaffold in Melanoma - ClinicalTrials.gov. can be found under http://www.clinicaltrials.gov/ct2/show/NCT01753089, n.d.
- 21.Ohnmacht C, Pullner A, King SBS, Drexler I, Meier S, Brocker T, Voehringer D. J Exp Med. 2009;206:549. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, von Stebut E, Probst HC, van den Broek M, Riethmacher D, Birnberg T, Blank T, Reizis B, Korn T, Wiendl H, Jung S, Prinz M, Kurschus FC, Waisman A. Immunity. 2012;37:264. doi: 10.1016/j.immuni.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 23.Verbeke CS, Mooney DJ. Adv Healthcare Mater. 2015 Epub ahead of print. [Google Scholar]
- 24.Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Proc Natl Acad Sci U S A. 1989;86:8000. doi: 10.1073/pnas.86.20.8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Cell. 1993;74:1089. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 26.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, Marrack P, Mahata SK, Kappler JW, Haskins K. Nat Immunol. 2010;11:225. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker RL, Bradley B, Wiles TA, Lindsay RS, Barbour G, Delong T, Friedman RS, Haskins K. J Immunol. 2016;196:39. doi: 10.4049/jimmunol.1501190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haskins K, McDuffie M. Science. 1990;249:1433. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 29.Mueller R, Bradley LM, Krahl T, Sarvetnick N. Immunity. 1997;7:411. doi: 10.1016/s1074-7613(00)80362-3. [DOI] [PubMed] [Google Scholar]
- 30.Muixí L, Gay M, Muñoz-Torres PM, Guitart C, Cedano J, Abian J, Alvarez I, Jaraquemada D. Genes Immun. 2011;12:504. doi: 10.1038/gene.2011.26. [DOI] [PubMed] [Google Scholar]
- 31.Jang MH, Seth NP, Wucherpfennig KW. J Immunol. 2003;171:4175. doi: 10.4049/jimmunol.171.8.4175. [DOI] [PubMed] [Google Scholar]
- 32.Bencherif SA, Sands RW, Bhatta D, Arany P, Verbeke CS, Edwards DA, Mooney DJ. PNAS. 2012;109:19590. doi: 10.1073/pnas.1211516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koshy ST, Ferrante TC, Lewin SA, Mooney DJ. Biomaterials. 2014;35:2477. doi: 10.1016/j.biomaterials.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huebsch N, Lippens E, Lee K, Mehta M, Koshy S, Darnell M, Desai R, Madl CM, Xu M, Zhao X, Chaudhuri O, Verbeke CS, Kim WS, Alim K, Mammoto A, Ingber DE, Duda GN, Mooney DJ. Nature Materials. 2015 doi: 10.1038/nmat4407. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson JM, Shive MS. Advanced Drug Delivery Reviews. 1997;28:5. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 36.Athanasiou KA, Niederauer GG, Agrawal CM. Biomaterials. 1996;17:93. doi: 10.1016/0142-9612(96)85754-1. [DOI] [PubMed] [Google Scholar]
- 37.Prasad S, Xu D, Miller SD. Rev Diabet Stud. 2012;9:319. doi: 10.1900/RDS.2012.9.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. Immunity. 2000;12:431. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 39.Fehérvari Z, Sakaguchi S. Journal of Clinical Investigation. 2004;114:1209. doi: 10.1172/JCI23395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overacre AE, Vignali DA. Current Opinion in Immunology. 2016;39:39. doi: 10.1016/j.coi.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey-Bucktrout SL, Bluestone JA. Trends in Immunology. 2011;32:301. doi: 10.1016/j.it.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green EA, Eynon EE, Flavell RA. Immunity. 1998;9:733. doi: 10.1016/s1074-7613(00)80670-6. [DOI] [PubMed] [Google Scholar]
- 43.Veld PI. Semin Immunopathol. 2014;36:569. doi: 10.1007/s00281-014-0438-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner MS, Kane LP, Morel PA. J Immunol. 2009;183:4895. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long SA, Rieck M, Tatum M, Bollyky PL, Wu RP, Muller I, Ho JC, Shilling HG, Buckner JH. J Immunol. 2011;187:3511. doi: 10.4049/jimmunol.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Apostolou I, von Boehmer H. J Exp Med. 2004;199:1401. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gottschalk RA, Corse E, Allison JP. J Exp Med. 2010;207:1701. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daniel C, Weigmann B, Bronson R, von Boehmer H. J Exp Med. 2011;208:1501. doi: 10.1084/jem.20110574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patterson J, Hubbell JA. Biomaterials. 2010;31:7836. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 50.Turk BE, Huang LL, Piro ET, Cantley LC. Nat Biotech. 2001;19:661. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 51.Heng TSP, Painter MW, Elpek K, Lukacs-Kornek V, Mauermann N, Turley SJ, Koller D, Kim FS, Wagers AJ, Asinovski N, Davis S, Fassett M, Feuerer M, Gray DHD, Haxhinasto S, Hill JA, Hyatt G, Laplace C, Leatherbee K, Mathis D, Benoist C, Jianu R, Laidlaw DH, Best JA, Knell J, Goldrath AW, Jarjoura J, Sun JC, Zhu Y, Lanier LL, Ergun A, Li Z, Collins JJ, Shinton SA, Hardy RR, Friedline R, Sylvia K, Kang J. Nat Immunol. 2008;9:1091. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sustained delivery of peptide antigen in vivo led to localized antigen presentation in the gel and draining lymph nodes. (A) In vitro release of OVA peptide from PLG particles that were foamed into porous scaffolds. (n = 3 per condition per timepoint; mean ± s.d. shown). (B) In vitro release of BDC peptide from PLG particles incorporated into alginate gels. (n = 4 per condition per timepoint; mean ± s.d. shown). (C–D) In vivo peptide biodistribution. (C) OVA peptide-loaded PLG scaffolds (S) were implanted subcutaneously into the flanks of C57BL/6J mice. (D) Intact OVA peptide was quantified by ELISA at the indicated timepoints in lysates from various tissues, including the muscle underlying the gel (L1), as well as 4mm (L2), 8mm (L3), and >3cm (R) away from the material (n = 4 per condition per timepoint; mean ± s.d. shown).
Figure S2. (A–B) Flow cytometry plots showing expression of CD11c and Gr-1 by cells isolated from gels delivering only AuNP/GM-CSF (A) or AuNP/GM-CSF with peptide-loaded PLG particles (B) at 5 days.
Figure S3. Overview of 23 different cytokines measured in gels delivering GM-CSF conjugated to AuNPs.
Figure S4. (A) Cell sorting strategy to separate CD11b+ CD11c− “myeloid” cells from CD11b+ CD11c+ “DCs” for analysis. (B) Gene expression of control CD11b+ CD11c+ splenocytes before or after 4h LPS stimulation, expressed as fold differences relative to unstimulated controls.
Figure S5. Expansion of antigen specific T cells 5 days after administration of gels delivering peptide-loaded PLG particles. Percentage of tetramer+ antigen-specific T cells, which include both adoptively transferred and endogenous cells, in the draining LNs (dLN), irrelevant (iLN), mesenteric (mLN), and pancreatic (pLN) LNs, as well as in the spleen (spl), at day 5.
Figure S6. Characterization of peptide conjugated alginate. (A) Dissolution of the BDC-CMR peptide in water before and after conjugation to PEG. Dashed line indicates expected concentration (5mg/mL) assuming complete dissolution of the peptide, while bars indicate actual measured peptide in solution. (B) Efficiency of peptide coupling to alginate was determined by measuring the amount of peptide removed during dialysis versus what was measured to be conjugated to the alginate.
Figure S7. In vivo expansion of antigen-specific T cells following administration of pore-forming gels delivering AuNP/GM-CSF either alone or in combination with MMP sensitive or insensitive BDC peptide conjugated to alginate. Gels were injected subcutaneously into the flanks of NOD/ShiLtJ mice; at specified timepoints, lymph nodes were isolated and dissociated to analyze lymphocytes by flow cytometry. Percentage of endogenous antigen-specific CD4+ T cells in the draining (dLN), irrelevant (iLN), and pancreatic (pLN) lymph nodes. (n = 4 – 6; individual data points and mean shown; ** p < 0.01; **** p < 0.0001; for each type of lymph node, the different conditions were compared to each other.)