

Abstract
Apolipophorin III (apoLp-III) is an exchangeable apolipoprotein found in insects and plays an important function in lipid transport. The protein has an unusual five-helix bundle architecture, deviating from the common four-helix bundle motif. To understand the role of the additional helix in apoLp-III, the N-terminal or C-terminal helix was deleted to create a putative four-helix bundle protein. While the protein lacking helix-1 could be expressed in bacteria albeit at reduced yields, apoLp-III lacking helix-5 could not be produced. Mutational analysis by truncating helix-5 showed that a minimum segment of approximately one third of the C-terminal helix is required for protein expression. The variant lacking helix-5 was produced by inserting a methionine residue between helix-4 and -5; subsequent cyanogenbromide cleavage generated the four-helix variant. Both N- and C-terminal helix deletion variants displayed significantly reduced helical content, protein stability, and tertiary structure. Despite the significantly altered structure, the variants were still fully functional. The rate of dimyristoylphosphatidylcholine vesicle solubilization was enhanced 4- to 5-fold compared to the wild-type protein, and the deletion variants were effective in binding to lipolyzed low density lipoprotein thereby preventing lipoprotein aggregation. These results show that the additional helix of apoLp-III is not essential for lipid binding, but is required for proper folding to keep the protein into a stable conformation.
Graphical abstract
Deletion of the N- or C-terminal helix of apolipophorin III to create a four-helix bundle protein Pankaj Dwivedi, Johana Rodriguez, Nnejiuwa U. Ibe, and Paul M.M. Weers

Apolipophorin III (apoLp-III) is a well-studied exchangeable apolipoprotein found in insects, and known for its role in lipoprotein mediated lipid transport. 1 The protein exists in lipid-bound and a lipid-free states, for the latter X-ray and NMR solution structures have been resolved for apoLp-III from Locusta migratoria and Manduca sexta. 2-4 Recently, backbone assignments have been completed for Galleria mellonella apoLp-III towards elucidating the NMR structure of this apoLp-III, which allows for structure-guided studies to better understand the function in innate immunity. 5 Lipid-free apoLp-III is composed of a bundle of five amphipathic α-helices folded in an up-and-down topology. The protein interior is predominantly made of hydrophobic residues, which are sequestered in the amphipathic α-helices forming a continuous non-polar face directed towards the protein interior. When apoLp-III associates with lipoproteins, the non-polar face of the amphipathic α-helices is presented to the hydrophobic lipoprotein surface. This requires reorientation of the amphipathic α-helices and it has been hypothesized that opening of the helix bundle exposes the protein interior. In vivo, lipoprotein association is triggered by massive diacylglycerol (DG) loading on existing lipoprotein particles in response to flight stimuli. 6-7 This creates suitable binding sites for apoLp-III, and in L. migratoria up to 14 apoLp-IIIs bind to the lipoprotein surface. 8 The diameter of the lipoprotein increases two-fold due to the increased amount of DG and protein, creating a lipoprotein with a significantly lower density: low density lipophorin. The term lipophorin, or apolipophorin for the protein part, is used to distinguish insect from vertebrate lipoproteins. 9 Despite some differences, animal (apo)lipoproteins such as human apolipoprotein A-I and apolipoprotein E share remarkable similarities in structure and function with their insect counterparts. 10
ApoLp-III is a relatively unstable protein, and loosely packed helices presumably facilitate opening of the helix bundle, allowing the switch from a lipid-free to a lipid-bound state.11 While no high-resolution structure of lipid-bound apoLp-III has been resolved to date, there is ample evidence for a distinct conformation compared to the lipid-free protein. A combination of circular dichroism, fluorescence spectroscopy, disulfide-bond engineering and FTIR analysis of discoidal lipoprotein particles made of apoLp-III and phosphatidylcholine suggest an extended helical conformation, supporting the helix bundle opening hypothesis. 12-16
The five-helix bundle motif of apoLp-III is rarely found in other proteins; a few other examples include vinculin tail and a fragment of Talin, although their helix topology is clearly distinct from apoLp-III. 17 Several other protein domains contain five-helix bundles made of short α-helices that form a more compact globular fold such as FK506 binding proteins and HectD1, and aciniform spidroin 1. 18-19 In contrast, the elongated four-helix bundle motif is found in many proteins with a wide variety of functions. 20-22 To gain insight in the unique character of the five-helix bundle, complete helical segments of the protein termini were removed from L. migratoria apoLp-III by site-directed mutagenesis to create a putative four-helix bundle. These and several other truncation variants were analyzed for protein structure and their ability to bind to lipid and lipoprotein surfaces.
Materials and Methods
Generation of truncated apoLp-III variants
All constructs were generated from wild-type L. migratoria apoLp-III. The apoLp-III H2-H5 variant was produced by amplifying the corresponding region by PCR and included the MscI site at the 3′ end, and the EcoRI site at the 5′ end to facilitate ligating the PCR product into the pET-22b (+) vector. Forward and reverse primers were 5′-CCGGCCTG CCCACC CCCGACGAG-3′ and 5′-GCTCGAATTCAGTTGGCGGGCTTCTG-3′. Fifty μg template DNA plus 125 ng of each primer, dNTP mix, and 2.5 U of PfuUltra HF DNA polymerase. The PCR product and vector were digested with EcoRI and MscI and purified using agarose electrophoresis using the Qiagen gel-extraction clean-up kit. One unit of T4 ligase was used to ligate the digested PCR product into the linearized vector. The other mutations were generated by introducing a stop codon in the apoLp-III coding region at the desired location using the QuikChange site-directed mutagenesis kit II (Agilent Technologies, Santa Clara, CA). The following primers were synthesized by MWG Biotech (High Point, NC, complementary primers not shown): apoLp-III H1-H4 5′-GGGCTCCGGTGTAGAGCGCGCTG-3′; apoLp-III Δ136-162 5′-AGAGCGCGCTGTAGGAGGCCGCC-3′; apoLp-III Δ140-162 5′-GCAGGAGGCCGCCTAGAAGACCAAGGA-3′; apoLp-III Δ144-162 5′-CCGAGAAGACCAAGTAGGCAGCGGCCAAC-3′; apoLp-III Δ150-162 5′-CAGCGGCCAACCTGTAGAACTCCATCCAG-3′; apoLp-III Δ154-162 5′-TGCAGAACTCCATCTAGTCGGCCGTCCAG-3′; apoLp-III V131M 5′-CGTGGGCTCCGATGCAGAGCGCG-3′. Template DNA (25 ng) and 125 μg of forward and reverse primers were used to generate the mutation in a thermocycler (PTC 200 Thermocycler, MJ Research, Inc.). The PCR product was incubated with 1 μL of DpnI restriction enzyme (10 units/μL) for 1 h at 37°C to cleave parental DNA, which was then used to transform XL 1-Blue supercompetent cells. Plasmid DNA was extracted using the QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA). To verify that the correct constructs of apoLp-III were obtained, plasmids were analyzed by DNA sequencing at the CSUPERB Microchemical Core Facility DNA Lab at California State University, San Diego.
Protein expression and purification
BL 21 (DE3) E. coli cells were transformed with wild-type and mutant L. migratoria apoLp-III constructs. The wild-type protein and helix-5 deletion mutants lack the N-terminal Arg-Pro segment, and thus resembles the apoLp-IIIb isoform (162 residues total) and amino acid numbering starts with the N-terminal aspartate. In addition, while the native protein is glycosylated, the recombinant protein lacks carbohydrate but this has no effect on the structure of the protein. A 25 mL overnight culture (with 50 μg/mL ampicillin) was prepared and used to seed 2 L minimal medium (47.75 mM Na2PO4, 22 mM KH2PO4, 8.65 mM NaCl, 18.7 mM NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, and 13.3 mM glucose at pH 7.4) with 50 μg/mL ampicillin. The culture was grown for 2-3 h at 37°C in a shaking incubator at 220 rpm until the optical density at 600 nm reached a value of 0.6 (measured in a Shimadzu UV-2401PC UV-Vis spectrophotometer) after which over-expression was induced by 2 mM isopropyl-β-D-thiogalactopyranoside. The culture was grown for an additional 4 to 5 h, and cells were removed by centrifugation at 6,000 rpm for 15 min at 4°C using a Sorvall RC 5C Plus centrifuge with a FiberLite F14 rotor. Since the protein escapes the bacteria during over-expression, the supernatant was further processed and concentrated to 10 mL using a concentrator with a 10k membrane (Pall Corp.). The concentrated protein solution was purified by size-exclusion chromatography using a Sephadex G-75 column equilibrated in phosphate buffer saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, pH 7.4). The flow rate was set at 1 mL/min and elution was monitored by the absorbance at 280 nm. The apoLp-III peak was collected and further purified by reversed-phase HPLC (System Gold, Beckman Coulter) using a semi-preparative column (C8 Zorbax, Agilent, Santa Clara, CA). Proteins were eluted with a linear gradient of water and acetonitrile with 0.05% trifluoroacetic acid. The apoLp-III peak was collected and dried for 2 days using a Labconco freeze dryer, and stored at -20°C until further use. The purity of the protein preparation was verified with sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) using Novex Tris-glycine 4-20% gradient gels (Invitrogen Co., Carlsbad, CA). For experimentation, proteins were dissolved in the appropriate buffer and the concentration determined by the bicinchoninic acid assay (Pierce Co., Rockland, IL) or the absorbance at 280 nm. The mass of the apoLp-III truncation variants was determined by matrix assisted laser desorption/ionization-time of flight spectroscopy (MALDI-TOF) using a sinapinic matrix (IIRMES core facility at CSU Long Beach).
Chemical modification of apoLp-III
Cyanogen bromide (CNBr, Sigma-Aldrich) was used to cleave V131M-apoLp-III into two fragments. Ten mg of the methionine apoLp-III variant was dissolved in 500 μL of 70 % formic acid (Fluka Analytical Inc.), after which 500 μL formic acid containing 11 mg of CNBr was added (100-fold molar excess). 23 The reaction was carried out for 16 h at 24°C and terminated by addition of 5 mL of water. The sample was freeze-dried, and SDS-PAGE was used to verify completion of the CNBr digestion. Cleaved protein was dissolved in 20 mM sodium phosphate buffer (pH 7.4) and the apoLp-III variant comprised of helix-1 to -4 was isolated by reversed-phase HPLC as described earlier.
ApoLp-III was cross-linked with dimethyl suberimidate (DMS, Pierce Co, Rockland, IL) to assess self-association. Eight μL of DMS (20 mg/mL) in 1 M triethanolamine (pH 9.7) was added to 15 μg of apoLp-III and incubated at room temperature for 3 h. 10 The reaction was terminated by addition of 25 μL SDS loading buffer, and subjected to 4-20 % SDS-PAGE.
Secondary structure analysis and fluorescence
For circular dichroism analysis, protein samples were prepared at concentration of 1 mg/mL in 20 mM sodium phosphate, pH 7.2. The protein concentration was determined by the absorbance at 280 nm. Scans were carried out in a JascoJ-810 polarimeter with circular cuvettes with a path length of 0.1 or 0.02 cm. Spectra were recorded between 185 and 260 nm at room temperature. The molar ellipticity [θ] was determined by the following equation: [θ] = (MRW • θ)/(10•d•c), in which θ = ellipticity (in degrees), d = pathlength (cm), c = protein concentration in g/L, and MRW = the mean residue weight. 24 The helical content was estimated using the molar ellipticity at 222 nm: (-[θ]222 nm + 3,000)/ (39,000) 25. Guanidine-HCl denaturation analysis was carried by incubating protein samples of 0.20 mg/mL in 20 mM sodium phosphate, pH 7.2 with varying guanidine-HCl concentration from 0 to 1.5 M. After 16 h to reach full equilibrium, the ellipticity was measured at 222 nm. Denaturation plots were generated and the concentration of guanidine-HCl to cause 50% protein unfolding was determined by sigmoidal curve fitting using Sigma Plot graphing software.
For tryptophan fluorescence emission analysis, apoLp-III (20 μg/mL, in PBS) was excited at 280 nm in a fluorescence spectrophotometer (Perkin Elmer LS 55) using a quartz fluorometer cuvette cell (10 mm pathlength, Starna Cell, Inc). Emission scans were obtained over the range of 290 to 440 nm with a scan speed of 50 nm/s.
Phospholipid solubilization
To measure the rate of phospholipid vesicle solubilization, large unilamellar vesicles (LUV) were prepared by extrusion. 26 Five mg of dimyristoylphosphatidylcholine (DMPC, Avanti Polar Lipids, Birmingham, AL) was dissolved in 1 mL of 3:1 CHCl3/ CH3OH (v/v), and dried under a stream of N2 and placed under vacuum for at least 4 h. The lipid film was rehydrated in 1 mL of PBS, pH 7.0 and incubated for 1 min at 37°C following by vigorous vortexing for 1 min to form multilamellar vesicles. LUVs were made by passing the multilamellar vesciles 15 times through a 200 nm Whatman Nucleopore Track-Etch membranes in a mini-extruder (Avanti Polar Lipids, Alabaster, AL) equilibrated at 30 °C. To measure the rate of vesicle solubilization, protein and DMPC LUVs were mixed and incubated in a 1 mL cuvette at 24°C in a Shimadzu 2401 spectrophotometer. The temperature was maintained in a Peltier temperature-controlled cuvette holder (TCC-240A). The change in light scatter of the protein-lipid suspension was measured by following the absorbance at 325 nm. The first-order rate constants (k) were calculated using the equation: At = A0 e-kt, where At = absorbance at time t, A0 = absorbance at t=0.
Lipoprotein Binding
Human low density lipoprotein (LDL, 100 μg protein, Intracel, Frederick, MD) was incubated with 160 mU of phospholipase-C from Bacillus cereus (Sigma Co., St. Louis, MO) in the presence of 0, 15, or 100 μg of apoLp-III in 50 mM Tris-HCl, 150 mM NaCl, 2 mM CaCl2 at pH 7.5. 27 The reaction was carried out at 37°C for 65 min in a 96 microwell plate with a total volume of 200 μL. LDL aggregation was monitored by measuring the absorbance at 340 nm in Varioscan plate reader (Thermo Electron Corporation, Milford, MA). The % LDL protection was calculated using the following equation: % LDL protection = (A340 in the presence of apoLp-III /A340 no apoLp-III) × 100.
Results
Design and production of truncation variants
Two terminal helix deletion variants were designed by deleting the N- or C- terminal helix in separate mutant proteins, see Fig 1A. To create the N-terminal deletion variant apoLp-IIIH2-H5, the DNA coding region of helix-2 to -5 (amino acid residues 30-162) was amplified by PCR and inserted into the pET-22b(+) expression vector. Similarly, the apoLp-III H1-H4 construct was created by amplifying the DNA coding region of amino acid residues 1-131. DNA sequencing of the constructs showed that both helix deletion mutants were successfully engineered. Attempts were made to over-express the deletion mutants in a well-established bacterial expression system, in which the protein escapes the bacteria and accumulates into the culture medium. 28 SDS-PAGE analysis shown in Fig. 2 demonstrated that apoLp-III H2-H5 was produced in E. coli cultures with a yield of 10 to 12 mg/L, which is significantly below the typical yield of the wild-type protein (apoLp-III WT, ∼30 mg/mL). The apoLp-III H1-H4 construct failed to produce detectable amounts of protein neither in the culture medium nor in the cell pellet, thus it was necessary to employ an alternative strategy to obtain the desired variant. Therefore, a methionine residue was inserted into the loop between helix-4 and -5 in place of valine 131. Since apoLp-III WT is devoid of methionine, this provides a convenient target site for proteolytic cleavage. The apoLp-III V131M variant was expressed and purified with a yield comparable to apoLp-III WT (∼25 mg/L). CNBr treatment of apoLp-III V131M resulted into two fragments, apoLp-III H1-H4 and a small fragment comprised of helix-5 residues. The reaction was ∼ 90 % complete, and apoLp-III H1-H4 was isolated by reversed-phase HPLC (Fig. 2). MALDI-TOF analysis showed that the masses of the purified truncation variants were within 25 Da of the theoretical mass based on the amino acid sequence and thus the desired truncation apolipoproteins were successfully produced.
Figure 1.

Panel A: X-ray structure of L. migratoria apoLp-III. The coding region of helix 1 (light blue) was removed to engineer apoLp-III H2-H5, while removal of helix 5 (blue) of the intact protein created apoLp-III H1-H4. Panel B: Close-up of helix-5 showing amino acid residues targeted for translation termination by introducing stops in the DNA coding region generating truncation variants with varying length to study the effect of helix-5 truncation.
Figure 2.

SDS-PAGE of the isolation and purification of the helix deletion variants. Shown are apoLp-III H2-H5, and apoLp-III H1-H4 which was generated by expressing and purification of apoLp-III V131M followed by CNBr cleavage and HPLC purification.
The lack of apoLp-III H1-H4 in the culture medium indicated that the protein structure has been compromised because of helix-5 removal. To identify the critical residues in helix 5, five truncation variants were engineered by inserting stop codons at various positions in the DNA coding sequence of helix 5. All of the residues selected for stop codon replacement were hydrophilic (glutamine or glutamate) and facing outwards to minimize perturbation of the hydrophobic helix interior (Fig. 1B). The following deletion variants were generated: apoLp-III Δ136-162, Δ140-162, Δ144-162, Δ150-162, and Δ154-162. No measurable amounts of protein could be purified from expression cultures of apoLp-III Δ136-162 and apoLp-III Δ140-162. However, apoLp-III with shorter deletions could be successfully produced (apoLp-III Δ144-162, Δ150-162, Δ154-162) with yields of ∼10 mg/L. Mass spectrometry verified the design and production of the correct variants.
Secondary structure analysis
The circular dichroism spectrum of apoLp-III is characteristic for α-helical proteins, with two prominent troughs at 208 and 222 nm and a positive peak at 192 nm. The circular dichroism plots of the helix truncation mutants apoLp-III H1-H4 and apoLp-III H2-H5 showed the proteins contained α-helical character but the decreased signal intensity compared to apoLp-III WT indicated a significant loss of secondary structure (Fig 3). Calculations based on the ellipticity at 222 nm showed that the helical content for apoLp-III H1-H4 and apoLp-III H2-H5 was 32% and 44%, respectively. This is considerably lower than the 70% calculated for apoLp-III WT. Helical contents of the variants with partial deletions of helix five showed decreased helical contents: 32% for apoLp-III Δ144-162, 36% for apoLp-III Δ150-162 and 41% for apoLp-III Δ154-162. As expected, the longer the helical segment of helix-5, the more helical character the protein retained. However, truncation at position 153 did not restore the helicity of the protein to values found in full-length apoLp-III. Thus, removal of 10 amino acids, most of them lying outside the helix 5 boundary, still caused a 30% reduction in α-helical content.
Figure 3.

Secondary structure analysis of apoLp-III deletion variants. Ellipticity of apoLp-III was measured between 185 and 260 nm. Upon deletion of one of the terminal helices, a significant decrease in helical content was observed as shown in the Far-UV circular dichroism spectra of apoLp-III WT (solid line), apoLp-III H1-H4 (dashed line) and apoLp-III H2-H5 (dotted line).
Guanidine-HCl denaturation
The proteins were denaturated with guanidine-HCl to measure the effect of the truncations on protein stability. Protein samples were incubated with increasing denaturant concentrations after which the ellipticity at 222 nm was measured to reveal the denaturant-induced decrease in α-helical structure. Plots of the change in secondary structure versus the guanidine-HCl concentration were generated, from which the midpoint of denaturation [Gdn-HCl]1/2 was determined. This value indicates the resistance to denaturation, and provides a measure for protein stability. A significant decrease in the resistance to denaturation was observed for the two helix deletion variants, with [Gdn-HCl]1/2 values of 0.29 and 0.19 M for apoLp-III H1-H4 and apoLp-III H2-H5, respectively. This is significantly lower than the [Gdn-HCl]1/2 of 0.50 M observed for apoLp-III WT (Fig. 4). As expected, the partial C-terminal helix deletion variants also displayed less resistance to guanidine-induced denaturation, with [Gdn-HCl]1/2 values in the range of 0.30 M (Table 1).
Figure 4.

Protein denaturation by guanidine-HCl. ApoLp-III deletion variants were incubated in the presence of increasing concentrations of guanidine-HCl followed by measurement of the ellipticity at 222 nm. The % change in ellipticity was plotted against the denaturant concentration. The denaturation profile of apoLp-III WT (filled circles), apoLp-III H1-H4 (filled triangles) and apoLp-III H2-H5 (open circles) demonstrate that removal of one of the terminal helices significantly decreases the stability of the protein as it is less resistant to denaturant induced unfolding. Error bars represent the standard deviation (n=3).
Table 1. Summary of biophysical and functional parameters of apoLp-III.
| ApoLp-III | [GdnHCl]1/2 (M) | α-helix (%) | Vesicle solubilization rate (10-3 s-1) | % LDL protection |
|---|---|---|---|---|
| WT | 0.50 ± 0.00 | 70.1 ± 3.8 | 0.55 ± 0.12 | 64.5 ± 9.9 |
| H1-H4 | 0.29 ± 0.04 | 32.3 ± 5.5 | 3.14 ± 0.60 | 57.6 ± 8.3 |
| H2-H5 | 0.19 ± 0.02 | 44.3 ± 1.1 | 2.85 ± 0.04 | 32.3 ± 4.2 |
| Δ144-162 | 0.26 ± 0.08 | 32.1 ± 2.7 | 2.23 ± 0.08 | 53.8 ± 7.5 |
| Δ150-162 | 0.30 ± 0.06 | 36.4 ± 2.7 | 1.38 ± 0.07 | 62.5 ± 9.4 |
| Δ154-162 | 0.31 ± 0.03 | 41.2 ± 8.7 | 2.28 ± 0.51 | 76.6 ± 14.8 |
For determining the vesicle solubilization rate a protein to lipid ratio of 1:10 (w/w) was used, while 15 μg protein was used for the % LDL protection.
Tertiary structure and oligomerization
Tryptophan emission scans of the apoLp-III variants were obtained to determine the wavelength of emission maximum (λmax), which provides information of tryptophan exposure to the aqueous environment. Two tryptophan residues reside in apoLp-III: one buried in helix 4 and a second more exposed residue in the loop linking helix 4 to 5 and is partially exposed. 29 Both tryptophan residues were retained upon generation of the truncation variants. Deletion of helix-1 led to a large redshift from 334 nm (apoLp-III WT) to 349 nm, while the redshift was more pronounced upon removal of helix-5 (355 nm) (Fig 5). The λmax from the partial helix deletion mutants depended on the extent of deletion, and ranged from 351 (apoLp-IIIΔ154-162) to 354 nm (apoLp-IIIΔ144-162). Thus, in all deletion mutants, tryptophan residues were increasingly more exposed to water, implying decreased tertiary contacts. Since the redshift was more pronounced in the helix-5 deletion variants, up to 21 nm, helix perturbation may have reached a critical threshold and prevented over-expression of apoLp-IIIH1-H4.
Figure 5.

Trp fluorescence emission spectra. The protein samples were excited at 280 nm and emission monitored between 290 and 440 nm, showing apoLp-III WT (solid line), apoLp-III H1-H4 (dashed line), and apoLp-III H2-H5 (dotted line), showing different λmax values for the deletion variants, implying increased exposure of tryptophan to the aqueous environment.
Previous studies of apoLp-III from Galleria mellonella in which two helices were removed to generate three different “three-helix” proteins, resulted in variants with decreased helical content and protein stability. 30-31 These protein constructs displayed a high degree of dimers and trimers of this otherwise monomeric protein. Therefore, self-association of the L. migratoria apoLp-III variants was assessed using DMS cross-linker. However, only a marginal increase in the appearance of dimers was observed for the deletion variants (Fig. 6).
Figure 6.

Crosslinking of apoLp-III. Proteins were cross-linked with DMS and separated by SDS-PAGE to determine the presence of oligomers. The wild-type protein showed a small amount of dimers, which increased slightly in the helix deletion variants.
Phospholipid vesicle solubilization
The ability of apoLp-III to solubilize DMPC bilayer vesicles forming small discoidal particles was used as a measure for lipid binding. This transformation from vesicles into discs occurs mainly at the phosphatidylcholine gel/liquid/crystalline transition temperature (TM), and practically limits this assay to myristylated phosphatidylcholine with a TM of 24°C. The starting vesicle dispersion had a cloudy appearance because of the heavy light scatter caused by LUVs. Induced by addition of apolipoprotein, LUVs were converted into discoidal particles with a diameter of ∼15 nm and thus light scatter intensity was greatly reduced, allowing to monitor the transformation using absorbance spectroscopy. Using a 1:1 ratio of apoLp-III and DMPC (w/w) the rate of transformation was fast for the helix deletion mutants clearing the vesicle dispersion in ∼200 s, while apoLp-III WT light scatter was reduced by only 20% at this time point (Fig. 7). Similar results were obtained for the partial C-terminal helix truncated variants. Since the transformation rate was too fast to detect potential differences among the variants, the amount of protein was reduced to a protein to lipid ratio of 1:10 (w/w) at which the rate of conversion remained sufficiently high to accurately measure the transformation rate. ApoLp-III H1-H4 and apoLp-III H2-H5 displayed faster transformation rates, while the partial helix 5 truncation variants showed a somewhat decreased ability to transform the vesicles albeit still significantly better than the wild-type protein (see Table 1 for rate constants).
Figure 7.

Apolipoprotein-induced vesicle solubilization. ApoLp-III was incubated with DMPC LUVs using a 1:1 lipid to protein weight ratio. The solubilization of vesicles thereby forming small discoidal complexes was followed by changes in the light scatter intensity measured by the absorbance at 325 nm. Shown are apoLp-III WT (solid line), apoLp-III H1-H4 (dashed line) and apoLp-III H2-H5 (dotted line), revealing a much higher rate of discoidal complex formation for the helix deletion variants compared to wild type protein.
Lipoprotein binding
ApoLp-III does not normally bind to human LDL. However, when the LDL surface is incubated with phospholipase-C, surface phosphatidylcholine is converted to DG. The appearance of DG on LDL causes packing defects leading to lipoprotein aggregation. However, surface DG attracts apoLp-III, which main function is to bind and stabilize DG-enriched lipophorins. 6 Upon apoLp-III binding, the LDL surface is protected and aggregation either prevented or delayed. The ability to prevent LDL aggregation was used to measure lipoprotein binding activity of apoLp-III. 27 As shown in Fig. 8, PL-C treated LDL rapidly aggregated evident by a steady increase in sample turbidity, which was monitored by the absorbance at 340 nm for 60 min. To test the ability of the helix deletion variants to delay or prevent the onset of LDL aggregation, LDL and phospholipase-C were simultaneously incubated with 100 μg of the apoLp-III variants. This showed that all proteins were able to provide full protection for the first 45 min, after which the absorbance started to increase. The absorbance values of all variants were similar, although apoLp-III H2-H5 appeared to have a somewhat lower activity. Since it was difficult to differentiate between the activities of the various mutant proteins, a limited amount of protein (15 μg) was used. The absorbance values were then used to calculate the % of protection at the 50 min time point. This showed that most variant proteins and apoLp-III WT provided partial protection between 54 and 74 %. The protection of apoLp-III H2-H5 was lower compared to the wild-type protein (Table 1), confirming our initial results with 100 μg apoLp-III.
Figure 8.

ApoLp-III lipoprotein binding. LDL was treated with phospholipase-C to create apoLp-III binding sites on the LDL surface. Incubations in the absence of apoLp-III led to rapid aggregation, as seen by the increase in sample turbidity (closed squares). Inclusion of the apoLp-III deletion variants delayed the onset of aggregation to 50 min: apoLp-III WT (filled circles), apoLp-III H1-H4 (filled triangles) and apoLp-III H2-H5 (open circles). Error bars represent the standard deviation (n=3).
Discussion
All truncation variants engineered in the current study displayed significant loss of structure. Upon removal of the complete N- or C-terminal helix, the α-helical content decreased from 70% for apoLp-III WT, to 44% for apoLp-III H2-H5 and 32% for apoLp-III H1-H4. In addition, the stability of these deletion variants reduced significantly, as indicated by the smaller amount of denaturant needed to unfold the protein. ApoLp-III is a marginally stable protein with a free energy of unfolding of ∼2 kJ/mol, substantially less than most 4-helix bundle proteins, and may be quite sensitive to amino acid changes in particular when residues are involved in helix packing.3, 32-34 Fluorescence emission scans showed increased exposure of both tryptophans that reside in the protein, and the largest red-shift was observed when helix-5 was removed. This result suggests decreased helical contacts for both deletion variants, in particular for apoLp-III H1-H4. Thus, while the stability of apoLp-III H1-H4 may be slightly higher, apoLp-III H2-H5 has a higher helical content and tertiary structure. While differences in biophysical characteristics between the two variants were observed, it is evident that removal of either helix caused major alterations in protein structure. When designing the helix deletion variants, it was anticipated that the remaining helices would reorient themselves to allow formation of a stable 4-helix bundle. The five α-helices have similar hydrophobic moments, mean hydrophobicity, and the previously published helical wheel diagram indicate the feasibility to remove one terminal helix 35. However, helical content decreased dramatically and stability was significantly reduced, probably because of helix packing defects, suggesting that the protein did not adopt the classic 4-helix bundle fold.
Changes in secondary and tertiary structure likely affected the expression yields. While the apoLp-III H2-H5 variant was produced in reduced but adequate quantities, the apoLp-III H1-H4 variant could not be isolated from media of E. coli cultures. This variant was ultimately generated by introducing a unique methionine between helix-4 and-5, and upon expression, the purified protein was treated with CNBr to release the desired 4-helix protein. While the helix-5 truncation variants apoLp-III Δ136-162, Δ140-162 could not be produced either, other variants with smaller truncations starting from amino acid position 144 were successfully expressed and purified from E. coli cultures. The lack of protein expression of apoLp-III H1-H4 indicates that helix-5 is critical for maintaining the structural integrity of the protein, and does not allow for much manipulation. In another study, variants lacking two successive N- or C-terminal helices of G. mellonella apoLp-III were produced in high quantities in an identical bacterial expression system. 30-31 While these variants also displayed a much lower helical content and protein stability, abundant formation of dimers and trimers may have compensated for the decrease in structure by sequestering hydrophobic residues. Therefore, self-association properties of the deletion variants in L. migratoria were analyzed, but showing no evidence of a substantial increase in oligomerization. Thus, exposure of unstructured sections of the L. migratoria apoLp-III variants produced in the current study may have contributed to lower expression yields or even in failure to produce any detectable amounts of recombinant protein.
Despite the decrease in secondary and tertiary structure, all truncation variant proteins were still able to maintain functionality. The variants were able to bind to the lipolyzed LDL surface and prevent lipoprotein aggregation. This indicates that the intrinsic ability to bind to the lipoprotein surface by the remaining amphipathic α-helical segments was not affected. Even more, removal of the N- or C-terminal helices resulted in a significant increase in binding to phospholipid vesicles. Rates of transformation of vesicles into discoidal particles were increased five-fold for apoLp-IIIH1-H4, H2-H5, and about four-fold for the helix-5 truncation variants. Similar results were reported for the two-helix truncation variants of G. mellonella apoLp-III. 30-31 The increase in phospholipid binding activity is in good agreement with previous studies where it was shown that helix bundle stability inversely correlates with phospholipid vesicle solubilization. 36-40 Conceivably, the equilibrium between open and closed helix bundle states may have shifted to a more open conformation, leading to increased exposure of hydrophobic regions of the amphipathic α-helices, thereby promoting lipid binding. The helical content of the variants decreased substantially which should have led to some degree of disruption of the non-polar faces of the amphipathic α-helices. Nevertheless, other studies have shown that such unstructured regions still maintain the ability to adopt an α-helical conformation in the proper environment. 35
It was intriguing to observe that the protein without helix-5 (ending at amino acid residue 131) could not be expressed in bacterial cell cultures. Many apoLp-III mutants have been engineered in our laboratory and by others, however, this was the first time we were not able to produce an apoLp-III variant. Partial truncation mutant analysis showed that helix-5 could be truncated until residue 144, however, the product was an unstable protein with properties similar to apoLp-III H1-H4. This implies that the protein was able to tolerate removal of residues 144-162 with the remaining helix-5 residues providing sufficient structure for helix-helix contacts for protein folding. Nevertheless, helical content decreased and λmax increased for removal of each consecutive helical segment until a critical value was reached after which the protein may have become too unstable and could not be produced.
Helix-2 and -5 are considered class A amphipathic α-helices 35, which are known for their high propensity to bind lipids. 41-42 Removal of helix-5 did not result in decreased lipid binding, on the contrary, LDL binding remained similar to apoLp-III WT while phospholipid vesicle solubilization increased. Helix-2 is another class A amphipathic α-helix which may have compensated for any lipid binding activity lost in the absence of helix-5. Helix-1 displayed characteristics of class G* α-helix: a high hydrophobic moment and hydrophobicity, a random distribution of charged residues, and lower or no lipid binding properties. 35 Thus, loss of this α-helix may not affect the lipid binding capabilities of apoLp-III, explaining, the results of the lipid binding analysis. While the characteristics of both terminal α-helices were different, neither of them are critical for lipid binding activity of apoLp-III.
When L. migratoria apoLp-III was cleaved into two approximately equal halves by proteolysis, both fragments were devoid of secondary structure. 35 The fragments lacked LDL binding and were not able to spontaneously transform phospholipid vesicles into discoidal particles, although co-sonication could force the protein into a lipid-bound conformation. In addition, the random-coiled protein could be converted into a highly α-helical protein when incubated in the presence trifluorethanol. This implied the protein needs some level of secondary structure to form intramolecular helical contacts to be able to associate with lipid surfaces. In our current study, the deletion variants may have sufficient secondary and tertiary structure to preserve the ability to interact with lipid and lipoprotein surfaces. Since vesicle solubilization was much faster when one of the terminal helices was removed, this provides further support for the proposed critical role these terminal helices play in lipid binding. 43, 44 Furthermore, amino acid changes in helix-1 in G. mellonella apoLp-III resulted in increased protein stability and lowered the ability to bind to lipid vesicles, in agreement with the critical role of helix-1 in initiation of lipid binding.45 Other studies with apoLp-III from three different species in which the protein was tethered by introduction of a disulfide bond between helix-1 and -5 rendered the protein ineffective for phospholipid solubilization. 12, 44, 46 Thus there is ample evidence that helix -1 and -5 are critical to keep the protein in a lipid-free closed conformation. The relatively weaker inter-helical contacts between helix-1 and -5 compared to other helical contacts facilitate separation of the terminal helices as a first step towards the open, lipid-bound conformation. 43, 44 In the absence of one of the terminal helices, the protein is in a more loosely folded “active” conformation. This resembles the pH-induced molten globule-like state of apoLp-III, which displayed a much increased lipid binding activity. 36, 47 The loosely folded state of the helix deletion variants facilitate the more dramatic conformational change in the presence of a suitable lipid surface, see Fig 9. This involves major helical rearrangements, in which the protein switches to a fully open conformation allowing buried hydrophobic faces of all amphipathic α-helices making contact with the lipid surface. 34, 48 This mechanism also provides a functional explanation of why removal of one of the terminal helices did not result in a stable 4-helix bundle protein as this conformation may not promote opening of the helix bundle to allow lipid binding. Thus, the terminal α-helices facilitate the conformational switch, promoting helix separation to allow reorientation to the new lipid surface. Similarly, N-and C-terminal truncation variants of human apoA-I adopt an extended helical conformation, presumably resembling a lipid bound state. 49, 50
Figure 9.
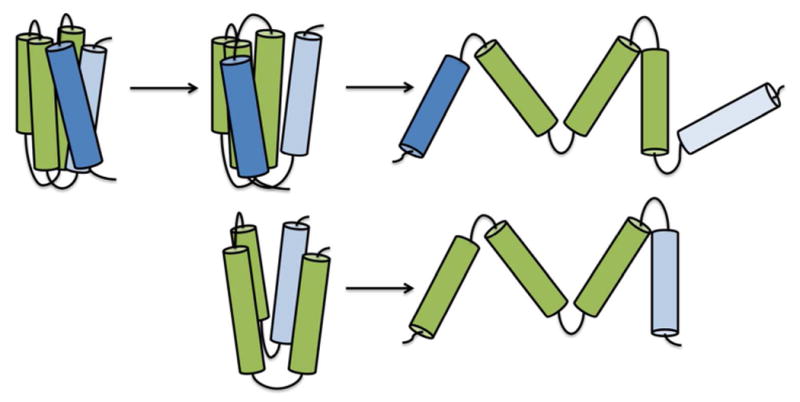
Model of apoLp-III helix bundle opening when binding to lipids. When helix-1 and -5 separate, increased exposure of the helix bundle interior allow for lipid-protein interactions, resulting in a major rearrangement of the amphipathic α-helices. Removal of one of the terminal helices creates a more loosely folded protein, facilitating lipid binding.
Just Accepted.
“Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a free service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are accessible to all readers and citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
Acknowledgments
Research reported in this publication was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number T34GM008074 and GM089564. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health”
Abbreviations
- apoLp-III
apolipophorin III
- CNBr
cyanogen bromide
- DG
diacylglycerol
- DMS
dimethyl suberimidate
- DMPC
dimyristoylphosphatidylcholine
- [Gdn-HCl]1/2
midpoint of guanidine-HCl denaturation
- λmax
wavelength of emission maximum
- LDL
low density lipoprotein
- LUV
large unilamellar vesicles
- MALDI-TOF
Matrix assisted laser desorption/ionization-time of flight spectroscopy
- PBS
phosphate buffered saline
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TM
transition temperature
References
- 1.Weers PMM, Ryan RO. Apolipophorin III: role model apolipoprotein. Insect Biochem Mol Biol. 2006;36:231–240. doi: 10.1016/j.ibmb.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Breiter DR, Kanost MR, Benning MM, Wesenberg G, Law JH, Wells MA, Rayment I, Holden HM. Molecular structure of an apolipoprotein determined at 2.5-Å resolution. Biochemistry. 1991;30:603–608. doi: 10.1021/bi00217a002. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Sykes BD, Ryan RO. Structural basis for the conformational adaptability of apolipophorin III, a helix-bundle exchangeable apolipoprotein. Proc Natl Acad Sci USA. 2002;99:1188–1193. doi: 10.1073/pnas.032565999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan D, Zheng Y, Yang D, Wang J. NMR solution structure and dynamics of an exchangeable apolipoprotein, Locusta migratoria apolipophorin III. J Biol Chem. 2003;278:21212–21220. doi: 10.1074/jbc.M208486200. [DOI] [PubMed] [Google Scholar]
- 5.Crowhurst KA, Horn JV, Weers PMM. Backbone and side chain chemical shift assignments of apolipophorin III from Galleria mellonella. Biomol NMR Assign. 2016;10:143–147. doi: 10.1007/s12104-015-9654-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van der Horst DJ, Van Hoof D, Van Marrewijk WJA, Rodenburg KW. Alternative lipid mobilization: the insect shuttle system. Mol Cell Biochem. 2002;239:113–119. [PubMed] [Google Scholar]
- 7.Van der Horst DJ, Rodenburg KW. Locust flight activity as a model for hormonal regulation of lipid mobilization and transport. J Insect Physiol. 2010;56:844–853. doi: 10.1016/j.jinsphys.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Van der Horst DJ, Van Doorn JM, Voshol H, Kanost MR, Ziegler R, Beenakkers AMTh. Different isoforms of an apoprotein (apolipophorin III) associate with lipoproteins in Locusta migratoria. Eur J Biochem. 1991;196:509–517. doi: 10.1111/j.1432-1033.1991.tb15843.x. [DOI] [PubMed] [Google Scholar]
- 9.Beenakkers AMTh, Chino H, Law JH. Lipophorin nomenclature. Insect Biochem. 1988;18:1–2. [Google Scholar]
- 10.Narayanaswami V, Kiss RS, Weers PMM. The helix bundle: a reversible lipid binding motif. Comp Biochem Physiol A Mol Integr Physiol. 2009;155:123–133. doi: 10.1016/j.cbpa.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weers PMM, Ryan RO. Apolipophorin III: a lipid-triggered molecular switch. Insect Biochem Mol Biol. 2003;33:1249–1260. doi: 10.1016/j.ibmb.2003.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Wientzek M, Kay CM, Oikawa K, Ryan RO. Binding of insect apolipophorin III to dimyristoylphosphatidylcholine. J Biol Chem. 1994;269:4605–4612. [PubMed] [Google Scholar]
- 13.Raussens V, Narayanaswami V, Goormaghtigh E, Ryan RO, Ruysschaert JM. Alignment of the apolipophorin III α-helices in complex with dimyristoylphosphatidylcholine. J Biol Chem. 1995;270:12542–12547. doi: 10.1074/jbc.270.21.12542. [DOI] [PubMed] [Google Scholar]
- 14.Narayanaswami V, Wang J, Kay CM, Scraba DG, Ryan RO. Disulfide bond engineering to monitor conformational opening of apolipophorin III during lipid binding. J Biol Chem. 1996;271:26855–26862. doi: 10.1074/jbc.271.43.26855. [DOI] [PubMed] [Google Scholar]
- 15.Garda HA, Arrese EL, Soulages JL. Structure of apolipophorin III in discoidal lipoproteins. J Biol Chem. 2002;277:19773–19782. doi: 10.1074/jbc.M110089200. [DOI] [PubMed] [Google Scholar]
- 16.Sahoo D, Weers PMM, Ryan RO, Narayanaswami V. Lipid-triggered conformational switch of apolipophorin III helix bundle to an extended helix organization. J Mol Biol. 2002;321:201–214. doi: 10.1016/s0022-2836(02)00618-6. [DOI] [PubMed] [Google Scholar]
- 17.Gingras AR, Bate N, Goult BT, Patel B, Kopp PM, Emsley J, Barsukov IL, Roberts GC, Critchley DR. Central region of talin has a unique fold that binds vinculin and actin. J Biol Chem. 2010;285:29577–29587. doi: 10.1074/jbc.M109.095455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helander S, Montecchio M, Lemak A, Farès C, Almlöf J, Li Y, Yee A, Arrowsmith CH, Dhe-Paganon S, Sunnerhagen M. Basic tilted helix bundle - a new protein fold in human FKBP25/FKBP3 and HectD1. Biochem Biophys Res Commun. 2014;447:26–31. doi: 10.1016/j.bbrc.2014.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tremblay ML, Xu L, Lefèvre T, Sarker M, Orrell KE, Leclerc J, Meng Q, Pézolet M, Auger M, Liu XQ, Rainey JK. Spider wrapping silk fibre architecture arising from its modular soluble protein precursor. Sci Rep. 2015;5:11502. doi: 10.1038/srep11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohn WD, Mant TC, Hodges RS. α-Helical protein assembly motifs. J Biol Chem. 1997;272:2583–2586. doi: 10.1074/jbc.272.5.2583. [DOI] [PubMed] [Google Scholar]
- 21.Harris NL, Presnell SR, Cohen FE. Four helix bundle diversity in globular proteins. J Mol Biol. 1994;236:1356–1368. doi: 10.1016/0022-2836(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 22.Kamtekar S, Hecht MH. The four-helix bundle: what determines a fold? FASEB J. 1995;9:1013–1022. doi: 10.1096/fasebj.9.11.7649401. [DOI] [PubMed] [Google Scholar]
- 23.Narayanaswami V, Kay CM, Oikawa K, Ryan RO. Structural and binding characteristics of the carboxyl terminal fragment of apolipophorin III from Manduca sexta. Biochemistry. 1994;33:13312–13320. doi: 10.1021/bi00249a018. [DOI] [PubMed] [Google Scholar]
- 24.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophy Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 26.Wan CPL, Chiu MH, Wu X, Lee SK, Prenner EJ, Weers PMM. Apolipoprotein-induced conversion of phosphatidylcholine bilayer vesicles into nanodisks. Biochim Biophys Acta. 2011;1808:606–613. doi: 10.1016/j.bbamem.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu H, Scraba DG, Ryan RO. Prevention of phospholipase-C induced aggregation of low density lipoprotein by amphipathic apolipoproteins. FEBS Lett. 1993;316:27–33. doi: 10.1016/0014-5793(93)81730-n. [DOI] [PubMed] [Google Scholar]
- 28.Weers PMM, Wang J, Van der Horst DJ, Kay CM, Sykes BD, Ryan RO. Recombinant locust apolipophorin III: characterization and NMR spectroscopy. Biochim Biophys Acta. 1998;1393:99–107. doi: 10.1016/s0005-2760(98)00063-0. [DOI] [PubMed] [Google Scholar]
- 29.Weers PMM, Prenner EJ, Kay CM, Ryan RO. Lipid binding of the exchangeable apolipoprotein, apolipophorin III induces major changes in fluorescence properties of Trp 115 and Trp 130. Biochemistry. 2000;39:6874–6880. doi: 10.1021/bi992891x. [DOI] [PubMed] [Google Scholar]
- 30.Dettloff M, Weers PMM, Niere M, Kay CM, Ryan RO, Wiesner A. An N-terminal three-helix fragment of the exchangeable insect apolipoprotein apolipophorin III conserves the lipid binding properties of wild-type protein. Biochemistry. 2001;40:3150–3157. doi: 10.1021/bi0013804. [DOI] [PubMed] [Google Scholar]
- 31.Dettloff M, Niere M, Ryan RO, Luty R, Kay CM, Wiesner A, Weers PMM. Differential lipid binding of truncation mutants of Galleria mellonella apolipophorin III. Biochemistry. 2002;41:9688–9695. doi: 10.1021/bi0200108. [DOI] [PubMed] [Google Scholar]
- 32.Ryan RO, Oikawa K, Kay CM. Conformational, thermodynamic, and stability properties of Manduca sexta apolipophorin III. J Biol Chem. 1993;268:1525–1530. [PubMed] [Google Scholar]
- 33.Weers PMM, Kay CM, Oikawa K, Wientzek M, Van der Horst DJ, Ryan RO. Factors affecting the stability and conformation of Locusta migratoria apolipophorin III. Biochemistry. 1994;29:3617–3624. doi: 10.1021/bi00178a019. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Gagné SM, Sykes BD, Ryan RO. Insight into lipid surface recognition and reversible conformational adaptations of an exchangeable apolipoprotein by multidimensional heteronuclear NMR techniques. J Biol Chem. 1997;272:17912–17920. doi: 10.1074/jbc.272.29.17912. [DOI] [PubMed] [Google Scholar]
- 35.Narayanaswami V, Weers PMM, Bogerd J, Kooiman FP, Kay CM, Scraba DG, Van der Horst DJ, Ryan RO. Spectroscopic and lipid binding studies on the amino and carboxy terminal fragments of Locusta migratoria apolipophorin III. Biochemistry. 1995;34:11822–11830. doi: 10.1021/bi00037a021. [DOI] [PubMed] [Google Scholar]
- 36.Soulages JL, Bendavid OJ. The lipid binding activity of the exchangeable apolipoprotein apolipophorin-III correlates with the formation of a partially folded conformation. Biochemistry. 1998;37:10203–10210. doi: 10.1021/bi980622l. [DOI] [PubMed] [Google Scholar]
- 37.Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J Biol Chem. 2002;277:50380–55038. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 38.Segall ML, Dhanasekaran P, Baldwin F, Anantharamaiah GM, Weisgraber KH, Phillips MC, Lund-Katz S. Influence of apoE domain structure and polymorphism on the kinetics of phospholipid vesicle solubilization. J Lipid Res. 2002;43:1688–1700. doi: 10.1194/jlr.m200157-jlr200. [DOI] [PubMed] [Google Scholar]
- 39.Weers PMM, Narayanaswami V, Choy N, Luty R, Hicks L, Kay CM, Ryan RO. Lipid binding ability of human apolipoprotein E N terminal domain isoforms: correlation with protein stability? Biophys Chem. 2003;100:481–492. doi: 10.1016/s0301-4622(02)00300-9. [DOI] [PubMed] [Google Scholar]
- 40.Weers PMM, Abdullahi WE, Cabrera JM, Hsu T. Role of buried polar residues in helix bundle stability and lipid binding of apolipophorin III: destabilization by threonine 31. Biochemistry. 2005;44:8810–8816. doi: 10.1021/bi050502v. [DOI] [PubMed] [Google Scholar]
- 41.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–167. [PubMed] [Google Scholar]
- 42.Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic α-helix: a multifunctional structural motif in plasma apolipoproteins. Adv Prot Chem. 1994;45:302–369. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- 43.Soulages JL, Arrese EL. Dynamics and hydration of the α-helices of apolipophorin III. J Biol Chem. 2000;275:17501–17509. doi: 10.1074/jbc.M909661199. [DOI] [PubMed] [Google Scholar]
- 44.Soulages JL, Arrese EL, Chetty PS, Rodriguez V. Essential role of the conformational flexibility of helices 1 and 5 on the lipid binding activity of apolipophorin III. J Biol Chem. 2001;276:34162–34166. doi: 10.1074/jbc.M105836200. [DOI] [PubMed] [Google Scholar]
- 45.Thistle J, Martinon D, Weers PMM. Helix 1 tryptophan variants in Galleria mellonella apolipophorin III. Chem Phys Lipids. 2015;193:18–23. doi: 10.1016/j.chemphyslip.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leon LJ, Idangodage H, Wan CPL, Weers PMM. Apolipophorin III: lipopolysaccharide binding requires helix bundle opening. Biochem Biophys Res Commun. 2006;348:1328–1322. doi: 10.1016/j.bbrc.2006.07.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weers PMM, Kay CM, Ryan RO. Conformational changes of an exchangeable apolipoprotein, apolipophorin III from Locusta migratoria, at low pH: correlation with lipid binding. Biochemistry. 2001;40:7754–7760. doi: 10.1021/bi010410f. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Narayanaswami V, Sykes BD, Ryan RO. Interhelical Contacts are required for the helix bundle fold of apolipophorin III and its ability to interact with lipoproteins. Prot Sci. 1998;7:336–341. doi: 10.1002/pro.5560070213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286:38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]