

Abstract
Apolipophorin III (apoLp-III) is an insect apolipoprotein (18 kDa) that comprises a single five-helix bundle domain. In contrast, human apolipoprotein A-I (apoA-I) is a 28 kDa two-domain protein: an α-helical N-terminal domain (residues 1-189) and a less structured C-terminal domain (residues 190-243). To better understand the apolipoprotein domain organization, a novel chimeric protein was engineered by attaching residues 179 to 243 of apoA-I to the C-terminal end of apoLp-III. The apoLp-III/apoA-I chimera was successfully expressed and purified in E. coli. Western blot analysis and mass spectrometry confirmed the presence of the C-terminal domain of apoA-I within the chimera. While parent apoLp-III did not self-associate, the chimera formed oligomers similar to apoA-I. The chimera displayed a lower α-helical content, but the stability remained similar compared to apoLp-III, consistent with the addition of a less structured domain. The chimera was able to solubilize phospholipid vesicles at a significantly higher rate compared to apoLp-III, approaching that of apoA-I. The chimera was more effective in protecting phospholipase C-treated low density lipoprotein from aggregation compared to apoLp-III. In addition, binding interaction of the chimera with phosphatidylglycerol vesicles and lipopolysaccharides was considerably improved compared to apoLp-III. Thus, addition of the C-terminal domain of apoA-I to apoLp-III created a two-domain protein, with self-association, lipid and lipopolysaccharide binding properties similar to apoA-I. The apoA-I like behavior of the chimera indicate that these properties are independent from residues residing in the N-terminal domain of apoA-I, and that they can be transferred from apoA-I to apoLp-III.
Keywords: apolipoprotein, lipid binding, apolipophorin III, lipoprotein
Graphical Abstract
1. Introduction
Exchangeable apolipoproteins are proteins that are highly adaptable to different lipid environments found in lipoproteins [1]. They are able to exist in a lipid-free state, but are often associated with the surface of lipoproteins. ApoA-I is found predominantly on high density lipoprotein (HDL), an antiatherogenic lipoprotein that mediates reverse cholesterol transport [2,3]. The 243 amino acid protein is composed of an assembly of amphipathic α-helices, providing the protein with the ability to interact with hydrophobic lipoprotein surfaces. ApoA-I shows characteristics of a two-domain structure: an N-terminal (NT) helix bundle domain, and a much smaller and structurally less defined C-terminal (CT) domain [4,5]. Isolated CT domain of apoA-I forms oligomers, indicating that this domain is responsible for self-association [6]. Removal of the CT domain leaves the overall helix bundle structure intact, but the deletion significantly decreases the ability of apoA-I to interact with lipid surfaces [4,5,7]. While all amphipathic α-helices of apoA-I associate with lipids, it has been proposed that helices of the CT domain initiate apoA-I lipid binding interaction [4]. Insect apolipophorin III (apoLp-III) is a small exchangeable apolipoprotein which has been used as a model for structural and functional analysis of apolipoproteins, and many important insights have been derived of how apolipoproteins interact with lipids [8]. ApoLp-III high-resolution structures, both X-ray and NMR, are available for two unrelated apoLp-IIIs in their lipid-free state [9–11]. These structures show that apoLp-III is a one-domain protein composed of a bundle of five amphipathic α-helices. ApoLp-III is monomeric and does not self-associate even at high protein concentrations [12]. In contrast to apoA-I, apoLp-III is primarily present in a lipid-free form, but associates with lipoprotein surfaces during insect flight when large amounts of neutral lipids are transferred to circulating lipoproteins for transport to flight muscles. Thus, while apoA-I and apoLp-III are both helix bundle apolipoproteins, they differ in respect to their structural organization and functional properties. To better understand the domain organization of these apolipoproteins, a chimera of apoLp-III and the CT domain of apoA-I was engineered. Addition of CT-apoA-I to apoLp-III allowed for the study of CT-apoA-I in a protein context without interference of the NT domain of apoA-I. A disulfide bridge was introduced in a critical position in the apoLp-III helix bundle rendering it inactive in terms of lipid binding. This allows for analysis of the chimera with an inactive apoLp-III, but also a functionally active apoLp-III upon reduction of the disulfide bond. The structural and functional properties of the novel two-domain apoLp-IIIcys/CT-apoA-I chimera were determined, showing that apoLp-III adopts apoA-I like properties.
2. Materials and methods
2.1. Protein constructs and site-directed mutagenesis
Optimized codon sequences for 6xHis-tagged human apoA-I, Locusta migratoria apoLp-III, and the chimera apoLp-IIIcys/apoA-I (179–243) were synthesized and inserted into the pET-20b(+) vector (Eurofins MWG Operon). Two cysteines were introduced into apoLp-III (Thr-20 and Ala-149) using the QuikChange-II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The following primers were used to replace Thr-20 by Cys: 5′-GTCCAGCAGCTCAATCACTGCATCGTGAATGCTGCGC-3′ (forward), and 5′-GCGCAGCATTCACGATGCAGTGATTGAGCTGCTGGAC-3′ (reverse). Ala-149 was replaced by cysteine using forward primer 5′-GACGAAAGAAGCTGCTTGCAATCTGCAGAACAGCATTCAGTCG -3′, and reverse primer 5′-CGACTGAATGCTGTTCTGCAGATTGCAAGCAGCTTCTTTCGTC-3′. Mutations were verified by DNA sequencing (Genewiz, La Jolla, CA).
2.2. Recombinant protein expression
Proteins were expressed in Escherichia coli BL21(DE3) pLysS cells (Agilent Technologies, Santa Clara, CA). Overnight cultures of freshly transformed cells were grown in 50 mL 2xYT broth containing 50 μg/mL ampicillin and chloramphenicol, then used to seed 2 L cultures and grown on a benchtop shaker (Barnstead, Mellrose Park, IL) at 37 °C. Protein overexpression was induced by 0.5 mM isopropyl-β-D-1-thiogalactopyranoside (Gold Biotechnology, St. Louis, MO) when the optical density of E. coli cultures at 600 nm was 0.6. Cells were collected by centrifugation at 8,000 x g, and resuspended in phosphate buffered saline (PBS; 150 mM NaCl, 20 mM sodium phosphate, pH 7.4) and lysed by sonication using a digital sonifier (Branson, Shanghai, China) in five 30 s cycles at 30% amplitude. Sonicated samples were subjected to two rounds of centrifugation at 20,000 x g for 30 min at 4°C to remove cell debris. The supernatant was mixed with an equal volume of sample loading buffer (2x PBS, 6 M guanidine-HCl, pH 7.4) and loaded onto two 5 mL HiTrap™ chelating columns (GE Healthcare, Piscataway, NJ). The column was washed with 20 mL of 40 mM imidazole, 3 M guanidine-HCl in PBS. Proteins were eluted using 20 mL elution buffer (500 mM imidazole in PBS). The protein solution was transferred to 6–8 kDa cut-off dialysis tubes (Spectrum laboratories, Rancho Dominguez, CA) and dialyzed against 4 L of 10 mM ammonium bicarbonate, pH 7.8 containing 1 mM EDTA with 3 additional buffer changes within 48 h. The recombinant proteins were further purified by size-exclusion chromatography using Superdex 200 (XK-26/70 column, GE Healthcare, Pittsburgh, PA). Proteins were eluted in 10 mM ammonium bicarbonate, pH 7.8, 1 mM EDTA, lyophilized and stored at −20 °C. Individual sample fractions were assessed for purity by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE). Molecular weight of the recombinant proteins was measured in an AB SCIEX 4800 MALDI mass spectrometer using sinapinic acid as a matrix at the IIRMES facility at CSU Long Beach. For experimental analysis, proteins were dissolved in 6 M guanidine-HCl followed by dialysis in the appropriate buffer for 2 days with 3 buffer changes at 4 °C. To reduce the cysteine-containing proteins, protein samples were incubated in the presence of a 100-fold molar excess of dithiothreitol (DTT).
2.3. Electrophoresis and Western blot analysis
To separate protein by SDS-PAGE, 20 μg of purified protein was incubated at 70 °C for 10 min with lithium dodecyl sulfate sample loading buffer and NuPAGE sample reducing agent (Life Technologies, Grand Island, NY). Proteins were separated on a 10% Bis-Tris gel (NuPAGE) in MES buffer, at 200 V for 30 min. For non-denaturing electrophoresis, protein (20 μg, 1 μg/μL) was mixed with an equal volume of Novex native Tris-glycine sample buffer, and separated on 4–20% gradient Tris-glycine gels (Life Technologies, Grand Island, NY) at 125 V for 120 min. Gels were fixed and stained in 0.5% (m/v) naphthol blue-black in 10% glacial acetic acid, 45% methanol (v/v).
For Western blot analysis, 0.5 μg protein was subjected to SDS-PAGE and transferred to PVDF membrane using a Trans-Blot SD semi-dry transfer cell (Biorad, Hercules, CA). PVDF membrane was soaked in NuPage transfer buffer and proteins were transferred at 15 V for 35 min. The PVDF membrane was blocked with 5% non-fat dry milk in Tween-PBS (0.2%) and washed with Tween-PBS. The membrane was incubated at 4 °C overnight with goat anti-human apoA-I HRP conjugated antibody diluted 4,000-fold in 1% non-fat dry milk in Tween-PBS (Abcam, Cambridge, MA). The membrane was washed 3x in Tween-PBS prior to addition of 400 μL of Amersham ECL Plus Western Blotting Detection Reagent (GE Healthcare). Chemiluminescence was detected using a FluorChem M system (ProteinSimple, San Jose, California).
2.4. Self-association analysis
To determine the extent of self-association, proteins were cross-linked with dimethylsuberimidate (DMS, ThermoFisher Scientific, Waltham, MA). Proteins (20 μg, 1 μg/μL) were incubated in the presence of 13 mM DMS for 2 h at 24 °C in 63 mM triethanolamine, pH 9.0, and analyzed by SDS-PAGE. In addition, size-exclusion chromatography was used to obtain the elution profile to determine self-association. Protein (0.5 mg, 1 μg/μL) was applied to a Superdex 200 10/300 GL column using an AKTA purifier (GE Healthcare, Pittsburgh, PA). Elution of proteins at a flow rate of 1 mL/min was monitored at 210 nm.
2.5. Circular dichroism
Ellipticity of the protein samples was measured in the far-UV range from 185 to 260 nm in a Jasco 810 polarimeter (Jasco, Japan). Protein samples (0.2 mg/mL) were prepared in 20 mM sodium phosphate (pH 7.4) and transferred to a 1.0 mm pathlength cylindrical cuvet (Hellma Cells, Plainview, NY). At a scan speed of 50 nm/min, 4 scans were recorded at 24 °C. The molar ellipticity ([θ], deg•cm2•dmol−1) at 222 nm was calculated using the equation: [θ] 222 = (MRW • θ)/(l • c) where MRW is mean residue weight, θ is ellipticity at 222 nm (deg), l is the cuvet pathlength (cm) and c is the protein concentration (g/mL). The α-helical content of each protein sample was determined using the ellipticity at 222 nm: % α-helix = (([θ] 222 + 3,000)/39,000) × 100 [13]. To determine the protein stability, proteins (0.2 mg/mL) were incubated in guanidine-HCl (MP Biomedicals, Solon, OH) in the presence or absence of DTT for 16 h at 24 °C after which the ellipticity at 222 nm was measured. The free energy change of unfolding in the absence of guanidine-HCl was calculated (ΔGD) with the following equation: ΔGD= −RT ln KD [14]. The equilibrium constant (KD) between unfolded and native states of the protein was determined with KD = fD/(1−fD), in which the fraction of denatured protein fD was calculated by (θ-θN)/(θD- θN), wherein θ is the ellipticity at 222 nm at a particular guanidine-HCl concentration, θN is the ellipticity in the absence of denaturant, and θD is the ellipticity of unfolded protein determined at 3 M guanidine-HCl.
2.6. Lipid binding analysis
To prepare phospholipid vesicles, 10 mg of dimyristoylphosphatidylcholine (DMPC, Avanti Polar Lipids, Birmingham, AL) was dissolved in 0.5 mL 3:1 chloroform/methanol (v/v) and solvent was removed under a gentle stream of N2; complete dryness of the thin lipid film was achieved under vacuum for 3 h. Lipid films were hydrated in 1 mL PBS at 37 °C and vortexed for 1 min. The suspension was extruded through a 200 nm pore size membrane to prepare large unilamellar vesicles (LUV) using an Avanti Mini-Extruder at 37 °C. The LUV suspension (0.5 mg in 1 mL PBS) was incubated with 0.5 mg apolipoprotein at 24.1 °C in a 1 mL cuvet and the absorbance at 325 nm was monitored in a UV-2401PC spectrophotometer (Shimadzu) equipped with a Peltier unit. Based on the decrease in light scatter intensity, first order rate constants (k) were calculated.
For formation of calcein-containing vesicles, a solution of calcein (Sigma Aldrich, Saint Louis, MO) was prepared in a phosphate-free glass tube by dissolving 62.5 mg in 1 mL of Tris-HCl buffer (20 mM Tris HCl, 150 mM NaCl, 0.5 mM EDTA, pH 7.4) and extruded through 100 nm membrane with 6 mg of L-α-phosphatidyl-DL-glycerol (eggPG, Avanti Polar Lipids, Birmingham, AL) as described above. Gel filtration chromatography (Sephadex G-75, GE Healthcare Life Sciences, Pittsburg, PA) was used to separate LUVs from free calcein. The concentration of vesicles was determined with the Ames phosphate assay [15]. Calcein release was monitored in a LS 55 Fluorescence Spectrometer (Perkin Elmer, Shelton, CT) using 10 μM eggPG and a molar lipid to protein ratio of 1000:1. Excitation and emission wavelength were set at 490 and 520 nm, respectively, using a slit width of 5 nm. After 2 min equilibration, protein was added while release of remaining calcein was obtained by addition of 10 μL of 10 % Triton X-100 detergent (VWR International, Radnor, PA) at the 10 min time interval.
To assess binding to lipoproteins, human low density lipoprotein (LDL, Sigma Aldrich, St. Louis, MO) was treated with phospholipase-C [16]. Apolipoprotein (150 μg) in Tris buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM CaCl2, pH 7.4) was combined with 50 μg of LDL protein in 200 μL in a 96-well plate. The reaction was carried out at 37°C and started by addition of 200 mU of phospholipase-C; the turbidity was measured at 5 min intervals for 2 h at 340 nm using a Varioskan plate reader (Thermo Electron Corp., Milford, MA). The percent LDL protection (P %) was determined at 80 min using the equation: P % = [1−(Ap−Ab/A0−Ab)]*100, wherein Ap is the absorbance of the solution in the presence of protein, Ab is the baseline absorbance at zero min and A0 is the absorbance in the absence of protein.
2.7. LPS binding and neutralization
E. coli LPS serotype O55:B55 (Sigma Aldrich, St. Louis, MO) was purified by size exclusion chromatography (Sepharose CL-6B) and ethanol precipitation to remove contaminating bacterial components as described previously [17]. LPS concentration was determined by the 3-deoxy-D-manno-octulosonic acid assay [18]. Proteins were incubated at 37°C for 1 h in the presence of various amounts of LPS and separated using non-denaturing PAGE (pre-cast 4–20% Tris-Glycine gels, Invitrogen, Carlsbad, CA). Gels were stained with 0.5% naphthol blue black in 45% methanol and 10% acetic acid.
To determine the ability of the proteins to neutralize LPS, macrophages were incubated with LPS and apolipoprotein after which the tumor necrosis factor-α (TNF-α) secretion was measured. Macrophage cells (Raw 264.7; ATCC, Manassas, Virginia) were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal calf serum (Hyclone, Logan, UT) and 100 units/mL penicillin/streptomycin (Invitrogen, Carlsbad, CA). Ten μL of LPS (100 ng/mL) and 10 μL of each apolipoprotein (1000 ng/mL) was pre-incubated for 1 h at 37°C. Five μL of each LPS-protein solution was added to 250 μL of resuspended macrophages in X-vivo15 media (Lonza, Basel, Switzerland) supplemented with 1 % L-glutamine and 1 % penicillin/streptomycin. Cells were incubated for 18 h at 37°C in 5 % CO2. The cell supernatant was harvested and the amount of TNF-α released by the murine macrophages was measured using a murine TNF-α mini TMB ELISA development and buffer kit (PeproTech, Rocky Hill, NJ). Plates were coated with rabbit anti-murine TNF-α, and blocked with 1% BSA in PBS. Diluted supernatant samples and murine TNF-α standard were added to the wells and incubated at room temperature for 2 h. Upon washing, wells were incubated for 2 h with detection antibody (0.25 μg/mL). TMB (100 μL) substrate solution was added to each well, and incubated 20 min, after the color development was halted with 100 μL 1 M HCl. The absorbance was measured at 450 nm (with background subtraction at 620 nm) using a Varioskan multi-plate reader (Thermo Electron Corp., Milford, MA). The amount of TNF-α in each sample was calculated by interpolating from the average absorbance value to TNF-α concentration using the standard curve.
3. Results
3.1. Expression and initial characterization
Apolipophorin III was selected as a carrier for the CT domain of apoA-I (Fig. 1A). A DNA construct was designed comprising a His-tag, L. migratoria apoLp-III (residues 3-164), and CT-apoA-I. The first two residues of apoLp-III (Arg-Pro) were omitted as they lead to an unwanted N-terminal modification when the protein is over-expressed in E. coli; thus the amino acid sequence of apoLp-III resembles the apoLp-IIIb isoform abundantly present in insect hemolymph [19]. In addition, two cysteine residues were introduced in apoLp-III: Thr-20 in helix 1 and Ala-149 in helix 5 represented as apoLp-IIICys. Previous studies have shown that such a cysteine couple locks the helix bundle in a closed conformation under oxidizing conditions, but reduction of the protein will allow helix bundle opening and restore lipid binding if required [20–21]. Based on structure analysis by hydrogen/deuterium exchange, residues 179 to 243 of human apoA-I were selected to represent the CT domain of apoA-I (CT-apoA-I) [22–23]. The chimera construct was inserted into the pET-20b(+) vector and used to transform E. coli expression cells. Induction of protein over-expression resulted in yields of ~25 mg purified protein per liter culture, similar to that obtained when over-expressing wild-type apoLp-III (apoLp-IIIWT).
Figure 1.

Panel A: Schematic representation of the apoLp-IIIcys/CT-apoA-I chimera. Residues 179-243 from apoA-I were attached to apoLp-III from L. migratoria creating a 26 kDa chimeric apolipoprotein. Residues Thr-20 and Ala-149 were substituted with Cys to prevent opening of the apoLp-III helix bundle. Panels B and C: Identification of apoLp-IIIcys/CT-apoA-I by SDS-PAGE (B, 20 μg of protein) and Western blot (C, 0.5 μg of protein using goat anti-apoA-I conjugated HRP). Lane 1: apoA-I; lane 2: apoLp-IIIcys/CT-apoA-I; lane 3: apoLp-III.
To confirm the presence of CT-apoA-I in the chimeric protein, SDS-PAGE, mass spectrometry, and Western blot analysis using anti-apoA-I antibodies were performed. SDS-PAGE analysis showed that apoA-I, the chimera and apoLp-IIIWT had molecular masses of 30, 28, and 20 kDa, respectively (Fig. 1B). This result is close to the expected masses of 29.9, 26.4 and 19.1 kDa, respectively. MALDI-TOF analysis of apoLp-IIIcys/CT-apoA-I provided a molecular mass of 26,362 Da, which is identical to the expected mass of the chimeric protein construct based on the amino acid sequence. An anti-apoA-I antibody reacted with apoA-I and apoLp-IIIcys/CT-apoA-I in the Western blot analysis (Fig. 1C, Lanes 1 and 2, respectively), but was unable to detect apoLp-III (Fig. 1C, Lane 3). These results independently confirmed the proper engineering, expression, and purification of the chimera protein.
3.2. Self-association properties
Self-association properties of the chimera were initially assessed by non-denaturing PAGE. As shown in Fig. 2A, the electrophoretic mobility of apoLp-IIIcys/CT-apoA-I in the native state was much slower than that of apoLp-IIIWT. Even though the chimera is a slightly larger protein (26.4 kDa) compared to apoLp-IIIWT (19.1 kDa), the difference in size may not account for the observed large difference in electrophoretic mobility, particularly since the theoretical pI of the proteins is similar (5.71 for the chimera, 5.62 for apoA-I, and 5.62 for apoLp-IIIWT). Thus, the most likely explanation is that addition of CT-apoA-I to apoLp-III caused the protein to self-associate similar to apoA-I, displaying an almost identical electrophoretic mobility as apoA-I. Additionally, the apoA-I and apoLp-IIIcys/CT-apoA-I samples were heterogeneous while apoLp-IIIWT migrated as a distinct band. This suggests that both apoA-I and apoLp-IIIcys/CT-apoA-I exist in multiple oligomeric states. In addition, proteins were analyzed by FPLC using a Superdex-200 size-exclusion column. While apoLp-IIIWT eluted at 17 mL as a single peak (Fig. 2B), both the chimera and apoA-I eluted at 11 mL, thereby indicating a much larger size of the proteins in their native conformation, while peak broadening suggests the presence of multiple oligomeric forms.
Figure 2.

Panel A: Non-denaturing PAGE analysis showing a much greater mobility of apoLp-III compared to apoA-I and the chimera. Twenty μg of protein was electrophoresed in the absence of SDS on a 4–20% Tris-glycine gel. Lane 1: apoA-I, lane 2: apoLp-IIIcys/CT-apoA-I, lane 3: apoLp-III. Panel B: Size-exclusion chromatographic analysis. Protein (0.5 mg at a 1 mg/mL concentration) was applied to a Superdex-200 column. Elution of the proteins was monitored at 210 nm using a flow rate of 0.5 mL/min. ApoLp-III eluted as a single peak at 17 mL (dash-dotted line), while apoA-I (solid line) and the chimera (dotted line) elute much earlier at 11 mL.
To confirm the oligomeric state of the chimera, proteins were cross-linked with DMS and analyzed by SDS-PAGE. Addition of DMS to apoLp-IIIWT did not lead to intermolecular cross-linking (Fig. 3, Lane 6), confirming the predominantly monomeric state of this protein [12]. In contrast, samples of apoA-I (Lane 2) or apoLp-IIIcys/CT-apoA-I (Lane 4) showed dimers, trimers, tetramers, pentamers and hexamers in the presence of DMS. The extensive cross-linking of the chimera is another indication of self-association.
Figure 3.
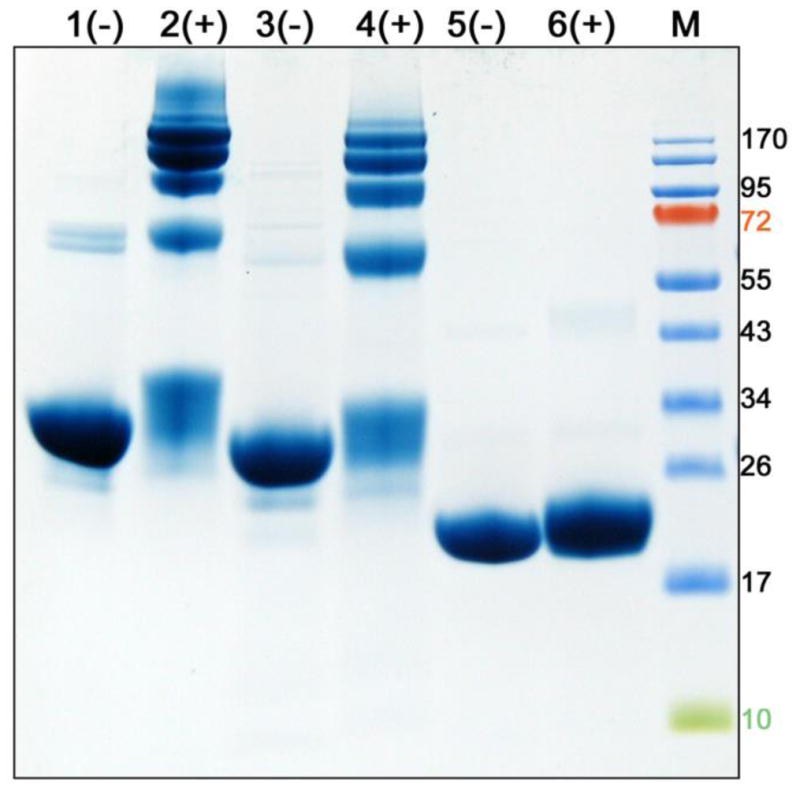
Cross-linking analysis. Twenty μg of reduced protein in the presence (+) or absence (-) of DMS crosslinker was analyzed by SDS-PAGE. Shown are apoA-I (lane 1–2), apoLp-IIIcys/CT-apoA-I (lane 3–4), and apoLp-III (lane 5–6). The molecular mass of marker proteins are shown at the right (M). In the presence of DMS apoA-I and apoLp-IIIcys/CT-apoA-I form oligomers (as evident by multiple protein bands in the range of 50 to 170 kDa) while apoLp-III remains monomeric.
3.3. Secondary structure analysis and protein stability
The secondary structure of the proteins was assessed by far UV circular dichroism. The ellipticity was measured from 260 to 185 nm, showing the typical troughs at 208 and 222 nm for proteins with a predominantly α-helical character (Fig. 4A). The α-helical content of apoLp-III, both apoLp-IIIWT and the double cysteine variant (apoLp-IIICys), was calculated at ~70 %, while the chimera displayed a significant reduction in helical content (~50 %). Thus addition of CT-apoA-I to apoLp-III reduced the overall helical content, consistent with the addition of a less structured domain to the five-helix bundle of apoLp-III. No significant differences were observed when the analysis was carried out under oxidized or reduced conditions (Table 1). To assess the effect of addition of CT-apoA-I to apoLp-III on the overall protein stability, the proteins were denatured with guanidine-HCl. The denaturation plots for the chimera and apoLp-IIICys indicated a much more stable protein compared to apoA-I under oxidizing conditions (Fig 4B). The midpoint of guanidine-HCl denaturation [Gdn-HCl]1/2 of the chimera and apoLp-IIICys was around 1.4 M, while it was 0.98 M for apoA-I (Table 1). However, this could be attributed to the two cysteine residues introduced in the apoLp-III helix bundle. When the proteins were analyzed under reduced conditions (Fig 4C), the [Gdn-HCl]1/2 values dropped to 0.75 M for the chimera and apoLp-IIICys while it remained unchanged for apoA-I. Thus the increased stability is attributed to disulfide bond formation, which tethered the apoLp-III helix bundle in both apoLp-IIICys and the chimera under oxidized conditions. In addition, it was also noticed that the resistance to guanidine-HCl induced denaturation of apoLp-IIICys was slightly higher compared to that of apoLp-IIIWT under reduced conditions, indicating some minor alterations in the apoLp-III helix bundle (Table 1). Thus, addition of CT-apoA-I to apoLp-III did not change the resistance to guanidine-HCl induced denaturation, as plots of apoLp-IIICys and the chimera were overlapping under both oxidizing and reducing conditions. Calculations of the free energy of unfolding showed the same pattern as the [Gdn-HCl]1/2 values: disulfide-bonded proteins had higher ΔGD values compared to the proteins under reducing conditions. Since the chimera free energy change was indistinguishable from apoLp-IIICys (Table 1) the stability of apoLp-IIICys was not affected by addition of CT-apoA-I.
Figure 4.

Secondary structure and protein stability analysis. Panel A: Far-UV spectra of the apolipoproteins at 0.2 mg/mL in 20 mM NaP pH 7.2. The average of 4 scans recorded from 185 to 260 nm at 50 nm/min speed is shown. Shown are apoLp-IIICys (dashed-dotted line), apoLp-IIIcys/CT-apoA-I (dotted line) and apoA-I (solid line). Panels B and C show the denaturation profile in the absence of DTT (B) and in the presence of DTT (C). Protein samples (0.2 mg/mL) were incubated with increasing concentrations of guanidine-HCl. Protein unfolding is plotted as % maximum change in which 100% is a completely unfolded protein. Shown are apoLp-IIIcys/CT-apoA-I (○), apoLp-IIICys (▼), and apoA-I (●), n = 3, ± standard deviation.
Table 1.
Physical and functional properties of the apolipoprotein variants
| Protein (+ or − DTT) | % α-helix | ΔGD (kcal/mol) | [Gdn- HCl]1/2 (M) | % LDL protection | k DMPC solubilization (s−1) |
|---|---|---|---|---|---|
| apoA-I (−) | 61.2 ± 0.9 | 3.85 ± 0.25 | 0.97 ± 0.02 | 98.0 ± 0.5 | 3.26 ± 0.09 × 10−2 |
| apoA-I (+) | 59.3 ± 1.0 | 3.85 ± 0.23 | 0.98 ± 0.02 | 98.9 ± 0.4 | 3.56 ± 0.50 × 10−2 |
| apoLp-IIIcys/CT-apoA-I (−) | 50.2 ± 0.8 | 5.97 ± 0.35 | 1.35 ± 0.06 | 70.5 ± 1.9 | 3.33 ± 0.05 × 10−2 |
| apoLp-IIIcys/CT-apoA-I (+) | 44.3 ± 2.9 | 2.91 ± 0.11 | 0.75 ± 0.01 | 99.2 ± 0.2 | 4.06 ± 0.15 × 10−2 |
| apoLp-III (−) | 70.0 ± 1.7 | 2.69 ± 0.19 | 0.50 ± 0.01 | 77.0 ± 6.3 | 0.67 ± 0.13 × 10−2 |
| apoLp-III (+) | 66.2 ± 2.7 | 2.56 ± 0.05 | 0.51 ± 0.01 | 84.1 ± 5.7 | 0.78 ± 0.25 × 10−2 |
| apoLp-IIICys (−) | 68.6 ± 2.2 | 6.13 ± 0.21 | 1.40 ± 0.01 | 25.3 ± 3.6 | 0.019 ± 0.003 × 10−2 |
| apoLp-IIICys (+) | 71.9 ± 1.4 | 2.96 ± 0.13 | 0.75 ± 0.01 | 85.5 ± 5.6 | 0.36 ± 0.10 ×10−2 |
Values are the average of 3 measurements ± standard deviation.
3.4. Lipid binding
To analyze their lipid binding properties, the apolipoproteins were incubated with LUVs made of pure DMPC. At the phospholipid transition temperature, this leads to the formation of small DMPC-protein discoidal complexes. The disappearance of the LUVs, also known as vesicle solubilization, was monitored by the decrease in sample turbidity as the large vesicles cause extensive light scatter in contrast to the newly formed small discoidal particles (diameter of <15 nm). Rate constants for vesicle solubilization were calculated and used as a measure of lipid binding capability. Addition of oxidized apoLp-IIICys to LUVs resulted in only a small decrease in sample turbidity, and thus this protein was ineffective in converting vesicles into discoidal particles in the time allocated (Fig. 5, curve b). In contrast, the oxidized chimera showed a rapid change in sample turbidity, converting the majority of vesicles into discs in ~ 400 s (curve e). The activity of the chimera was indistinguishable from that of apoA-I. Since the apoLp-III component of the chimera was locked in the closed bundle state, the CT domain of apoA-I in the chimera was responsible for the orders of magnitude increase in solubilization rates, compared to apoLp-IIICys (Table 1). The experiments were also carried out under reducing conditions, wherein no change was observed for the chimera and apoA-I (see Table 1 for rate constants). In contrast, the ability of apoLp-IIICys to solubilize LUVs was restored to a level comparable to that of apoLp-IIIWT. The conversion rate of reduced apoLp-IIICys was still considerably lower than that of apoA-I and the chimera, illustrating that apoA-I is more effective in converting vesicles into discoidal particles compared to apoLp-III.
Figure 5.
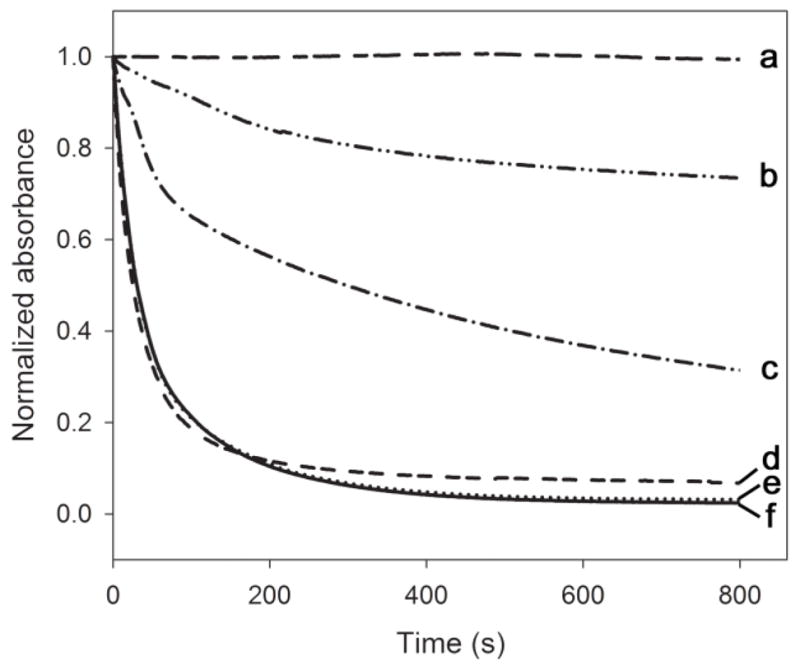
Phospholipid vesicle solubilization. Apolipoprotein was incubated with 0.5 mg/mL DMPC LUVs in a 1:1 mass ratio at 24.1 °C. The conversion of vesicles into the small discoidal complexes was monitored by the decrease in sample turbidity at 325 nm as a function of time. Shown are the average of 3 time scans of: DMPC only (a), apoLp-IIICys (b), apoLp-IIICys with DTT (c), apoLp-IIIcys/CT-apoA-I with DTT (d), apoLp-IIIcys/CT-apoA-I (e), and apoA-I (f).
The lipid binding properties of the apolipoproteins were further assessed using LDL. LDL was incubated with phospholipase-C to create apolipoprotein binding sites on the particle surface. The resultant conversion of phosphatidylcholine into diacylglycerol causes packing defects on the lipoprotein surface, and without further treatment LDL rapidly aggregates. However, inclusion of functional exchangeable apolipoproteins prevents or delays the onset of LDL aggregation [16]. As shown in Fig. 6A (oxidized conditions), apoA-I prevented the aggregation of LDL for the duration of the experiment (2 h). However, the turbidity of LDL steadily increased after 20 min in the presence of apoLp-IIICys, indicating that the tethered protein was not effective in binding to LDL and provide protection. The chimera displayed improved protection, albeit not to the level as observed for apoA-I. When the proteins were analyzed under reduced conditions, a dramatic improvement was observed and all proteins were much more effective preventing the onset of aggregation, although at extended incubation times the turbidity of the sample with apoLp-IIICys increased to a small extent (Fig 6B). Since the chimera was better in protecting LDL aggregation compared to apoLp-III under oxidized conditions, we concluded that the presence of CT-apoA-I in the chimeric protein resulted in a much-improved binding and protection of the LDL surface.
Figure 6.

Inhibition of LDL aggregation. LDL (50 μg protein) was treated with phospholipase-C (160 mU) and simultaneously incubated at 37 °C in the presence of apolipoproteins (150 μg) in the absence of DTT (A) or in the presence of DTT (B). Aggregation of LDL was monitored by the absorbance at 340 nm. Shown are the average of 3 measurements (± standard deviation) of apoLp-IIIcys/CT-apoA-I (○), apoLp-IIIcys(▼), apoA-I(●), and no protein (Δ).
3.5. Association with bacterial membrane components
ApoA-I is able to bind to LPS and PG, which are abundantly found in membranes of gram-negative bacteria, providing antimicrobial properties to the protein [17]. To address the effect of addition of CT-apoA-I to apoLp-III on the interaction with bacterial membranes, binding to PG vesicles and LPS was examined. Binding to negatively charged phospholipid bilayers was measured by encapsulating calcein in PG LUVs through extrusion. Addition of apolipoprotein to the vesicles cause disruption in the phospholipid bilayer packing resulting in release of calcein, which was measured by the increase in fluorescence intensity at 520 nm. Each protein was added to calcein containing PG vesicles at a 1000:1 molar lipid to protein ratio. ApoLp-III was not effective at this ratio, and only 4.3 ± 0.4 % of the available calcein was released (Fig. 7). This improved to 20.2 ± 0.5 % under reduced conditions. However, the chimera was able to release significantly more calcein: 55.7 ± 1.7 % (oxidized) and 62.5 ± 0.6 % (reduced). ApoA-I was most effective in releasing calcein from PG vesicles with 88.8 ± 1.0 %.
Figure 7.

Release of calcein from PG vesicles. Calcein-containing PG vesicles were equilibrated for 2 min, after which apolipoprotein was added triggering release of calcein measured by the increase in fluorescence intensity at 520 nm. At 10 min, detergent was added to release the remaining calcein. Solid line: apoA-I; dotted line: chimera; dash-dotted line: apoLp-III (n=3, ± standard deviation).
LPS association of the apolipoproteins was assessed by comparing the electrophoretic properties of the native protein in the absence or presence of LPS. Incubation of E. coli LPS with apoA-I changed the electrophoretic mobility of apoA-I, due to formation of a complex between LPS and apoA-I, appearing in the upper part of the native PAGE (Fig. 8). Similar results were obtained with the chimeric protein, demonstrating a strong binding interaction with LPS. However, the intensity of the apoLp-III band did not change upon LPS addition, and no protein bands appeared in the top portion of the gel, implying that the protein did not associate with LPS. The results were similar when the incubation was performed under reducing conditions, although binding of apoLp-III to LPS was slightly improved as some LPS/apoLp-III complexes appeared in the top portion of the gel (not shown). Thus, addition of CT-apoA-I to apoLp-III greatly improved binding to LPS.
Figure 8.

Native PAGE of apolipoprotein-LPS incubations. Apolipoproteins (20 μg) were incubated with increasing amounts of LPS for 1 h and analyzed by PAGE under non-denaturing conditions. Shown are apoA-I (lane 1), incubated with 80 μg (lane 2), 130 μg (lane 3) and 180 μg LPS (lane 4). Lanes 5–8 contain chimera only (lane 5) with 80 μg (lane 6), 130 μg (lane 7) and 180 μg LPS (lane 8); lanes 9–12 contain apoLp-III (lane 9) with 80 μg (lane 10), 130 μg (lane 11) and 180 μg LPS (lane 12).
To assess the ability of the apolipoproteins to neutralize LPS, the release of TNF-α by macrophages in response to an LPS challenge was measured. Raw 264.7 murine macrophages were exposed to 100 ng/mL LPS, and mixtures of LPS preincubated with apolipoprotein. Fig. 9 shows that in the absence of LPS, murine macrophages produced 2.3 ± 0.2 ng/mL of TNF-α while following LPS exposure, the release was ~tenfold higher (18.5 ± 0.7 ng/mL). As expected, incubation of LPS with apoA-I prior to macrophages exposure resulted in significantly less TNF-α production (5.9 ± 0.5 ng/mL). The chimera was slightly less effective with 8.3 ± 0.5 ng/mL TNF-α release, but still significantly better compared to apoLp-IIIcys (12.3 ± 0.7 ng/mL).
Figure 9.

TNF-α secretion by macrophages. Apolipoproteins were pre-incubated with LPS (10:1 ratio) after which macrophages were exposed to these mixtures; TNF-α concentrations were measured by ELISA (n = 3, ± standard deviation).
4. Discussion
Insect apoLp-III has served as a valuable model apolipoprotein, leading to novel insights into their structure-function relationship [8]. The protein shares many similarities with vertebrate exchangeable apolipoproteins, but also displays profound differences [24]. The unique helix bundle architecture, with five instead of the typical four amphipathic α-helices, provides the protein with a delicate balance of helix bundle stability coupled with the ability to rearrange its helices when binding to lipids [25]. The biological “trigger” for binding to lipoproteins is the loading of diacylglycerol onto the lipoprotein surface. However, apoLp-III is also capable to transform phospholipid vesicles into discoidal particles in a manner similar to apoA-I and apoE although these particles have not been isolated from insects and their importance for insect physiology remains unknown [26]. The conformation of apoLp-III on the discs is akin to that reported for apoA-I and apoE, with two series of helices oriented side-by-side with the helical axes perpendicular to that of the fatty acyl chains of the bilayer, and shielding them from the aqueous environment [24]. The protein in its lipid-free form is abundantly present in hemolymph, and remains monomeric at high concentrations. High-resolution solution structures of the lipid free protein have been solved, allowing a structure-guided approach to understand intricate details of apolipoprotein function. In earlier studies, apoLp-III was employed to create a chimeric protein harboring the LDL receptor domain of apoE [27]. Helix 5 of apoLp-III from Manduca sexta was replaced with residues 131 to 151 of apoE, and the chimera adopted heparin and LDL receptor binding properties, both of which were absent in apoLp-III. However, the chimeric protein showed indications of helix packing perturbations, likely because the apoE helix did not properly align with parent helices, resulting in increased exposure of hydrophobic residues and fewer helix-helix contacts. To design the apoLp-IIIcys/CT-apoA-I chimera a different approach was adopted. Instead of replacing a helical segment, residues 179-243 of apoA-I were added to the CT end of apoLp-III. This created a protein that potentially resembles the structural organization of apoA-I with an NT helix bundle and a small CT domain. This approach was successful as the structural integrity of the parent protein was maintained, and sufficient quantities of the chimera were produced in a well-established bacterial expression system [28].
The chimeric protein contained less helical structure compared to apoLp-III, consistent with addition of a less structured segment to the highly α-helical parent protein. On the other hand, this addition did not lead to changes in protein stability, as [Gdn-HCl]1/2 values of the disulfide apoLp-III variant and the chimera were indistinguishable, similar as has been observed for full length apoA-I [4]. By joining CT-apoA-I to apoLp-III, the resulting chimeric protein acquired apoA-I like properties that were absent in apoLp-III. The apoLp-IIIcys/CT-apoA-I chimeric protein was oligomeric, similar to apoA-I. The chimera became much more potent in terms of lipid binding as both its ability to solubilize phospholipid vesicles and to bind and protect LDL from aggregation were superior to that of apoLp-III. To block lipid binding of the parent protein, the apoLp-III helix bundle was tethered by a disulfide bond, which formed spontaneously between a newly introduced cysteine pair under oxidizing conditions. For apoLp-IIICys under oxidizing conditions, this led to a large increase in the resistance to guanidine-HCl denaturation while lipid binding was essentially disabled. In addition to helix bundle tethering, the increase in protein stability may have contributed to decreased phospholipid vesicle solubilization rates as this process is inversely correlated with protein stability [29]. Since the chimera harbored the same cysteine pair, lipid binding activity under oxidizing conditions can be attributed to CT-apoA-I alone. While apoLp-IIICys was not effective in prevention of LDL aggregation under oxidizing conditions, the chimera partially protected LDL from aggregation. The level of protection provided by the chimera remained below values obtained for apoA-I, indicating that the NT domain of apoA-I contributes substantially to binding to phospholipase-C-treated LDL. As expected, reduction of the disulfide bond in the chimeric protein resulted in improved protection of lipolyzed LDL as apoLp-III was able to provide additional amphipathic α-helices contributing to lipoprotein association and stability.
The chimeric protein also became more effective in binding to PG vesicles. ApoLp-III was ineffective at a 1000:1 molar ratio of lipid to protein, especially in the oxidized state, while apoA-I was able to release the majority of encapsulated calcein. However, the chimera was much more effective because of addition of CT-apoA-I to apoLp-III. Similar results were obtained when the LPS binding was characterized. Binding of oxidized apoLp-III to LPS was virtually absent, but increased dramatically when CT-apoA-I was present. Thus, LPS binding as well as binding to PG vesicles is mediated by CT-apoA-I, which supports other studies indicating that LPS binding resides in the CT part of apoA-I [17,30]. While the current study implied that binding of L. migratoria apoLp-III to LPS was weak, studies with Galleria mellonella apoLp-III demonstrated substantial changes to the membrane morphology of gram negative bacteria, and that the protein associates better to truncated versions of LPS, indicating a high affinity for lipid A [31,32]. Thus, there are strong similarities in the binding mechanism of apolipoproteins to the diverse hydrophobic compounds used in the present study, which explains why CT-apoA-I greatly enhanced the binding interaction of the chimera to DMPC, PG, lipolyzed LDL and LPS.
In the lipid-free state, apoA-I has characteristics of a two-domain protein, with a helix bundle structure in the NT domain, and a much smaller and less organized CT domain [4,5]. There is general consensus that the CT domain is unstructured, with strong evidence from hydrogen deuterium exchange studies in solution [22,23]. Other studies indicate that it may contain helical segments with some contribution from β-conformers [6,33,34]. Importantly, the helical content of apoA-I increases significantly upon lipid binding when α-helices rearrange into an open conformation, with the CT domain predominantly responsible for the increase in helicity [23]. Deletion of the CT domain (residues 190-243) of apoA-I did not affect helical content or protein stability [4]. However, removal of the CT domain negatively impacts lipid binding of the remaining NT domain. Indeed, various apoA-I CT deletion variants showed impaired ability to solubilize phospholipid vesicles, form lipoprotein complexes and promote cholesterol and phospholipid efflux from macrophages, showed reduced capability of ABCA-I driven lipid efflux in macrophages, reduced lipoprotein association, displayed a preference for smaller HDL and increased plasma clearance [35–37]. Lipoprotein association was partially restored in a chimera substituting CT residues 190-143 with residues 12-77 from apoA-II, which included two hydrophobic helical segments [38–39]. Another chimeric protein composed of the NT domain of mouse apoA-I and the CT domain of human apoA-I showed improved reverse cholesterol transport in mouse macrophages compared to wild type mouse apoA-I [40]. While it is well documented that the CT domain is critical for lipid binding initiation, helical segments in the NT domain make a contribution as destabilization of the NT helix core accelerated lipid binding [41,42], similar as has been seen for apoLp-III [29,43].
It is evident that the CT domain of apoA-I is a critical part of the protein. It is highly hydrophobic, a result of several aromatic amino acids that reside in the non-polar face of the amphipathic helix spanning residues 220-241 [44]. While less hydrophobic and with lower lipid binding affinity, residues 191-220 may modulate ABCA1 dependent formation of HDL. This demonstrates that both helical segments of CT apoA-I are required to initiate effective lipid binding and subsequent processes [45]. Some of the CT apoA-I variants produced showed changes in helical content and stability [46], indicating that the CT domain of apoA-I may interact with the NT domain helix bundle. This is remarkably similar to the structural and functional organization of human apoE [47]. In the case of apoA-I, the details of this domain-domain interaction are not well understood but may involve interactions of NT- and CT-terminal helices [48,49]. Previously, we reported that the S36A mutation in apoA-I caused a significant reduction in self-association [50]. The mutation also caused a decrease in protein stability, and it is plausible that this translated to subsequent changes in other parts of the protein, which may include the CT domain, thereby affecting self-association. Moreover, substitution of all tryptophan residues with phenylalanine, all located in the N-terminal domain, produced a protein with a higher propensity to form oligomers [51]. Self-association has also been observed for N- and C-terminal deletion mutants of apoA-I and apoA-IV, with protein core amino acids participating in dimerization [52,53]. However, this may have been a response to substantial alterations in protein structure upon removal of a sizable apoA-I fragment, and does not necessarily point towards direct involvement of the apoA-I core in self-association in the native protein. Similarly, protein self-association has been observed in response to helix truncation in apoLp-III from G. mellonella [54].
The present study showed that the structural organization and function of apoLp-III dramatically changed upon addition of the apoA-I segment corresponding to the CT domain of apoA-I, resulting in the chimera to adopt apoA-I like properties. This outcome demonstrates that the design of the present chimera in which an unrelated apolipoprotein segment was attached to apoLp-III is a promising approach in pursuit to better understand the domain function of apolipoproteins. The chimera showed that initiation of lipid and LPS binding and self-association, properties that reside in the C-terminal domain of apoA-I, could act independently from its N-terminal domain. The apoLp-IIIcys/CT-apoA-I chimera design demonstrates the feasibility of an in-depth analysis of CT-apoA-I by designing new variants with mutations or deletions in the CT tail to understand the structural and functional properties of this important part of apoA-I.
Highlights.
C-terminal residues of apoA-I were added to apoLp-III
phospholipid vesicle solubilization and lipoprotein binding was much improved
the chimeric protein formed oligomers
apoA-I residues enhanced binding to phosphatidylglycerol and lipopolysaccharides
apoLp-III acquired apoA-I like properties acting as a two-domain protein
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number GM089564 and GM105561. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- apoLp-III
apolipophorin III
- apoA-I
apolipoprotein AI
- CT
C-terminal
- DMPC
dimyristoylphosphatidylcholine
- DTT
dithiothreitol
- ΔGD
free energy change of unfolding
- [Gdn-HCl]1/2
midpoint of guanidine hydrochloride denaturation
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- LUV
large unilamellar vesicles
- NT
N-terminal
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- PG
phosphatidylglycerol
- SDS
sodium dodecyl sulfate
- TNF-α
tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jonas A, Phillips MC. Lipoprotein structure. In: Vance DE, Vance JE, editors. Biochemistry of Lipids, Lipoproteins and Membranes. Elsevier BV; Amsterdam: 2008. pp. 485–506. [Google Scholar]
- 2.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289:24020–24209. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mei X, Atkinson D. Lipid-free Apolipoprotein A-I Structure: Insights into HDL Formation and Atherosclerosis Development. Arch Med Res. 2015;46:351–360. doi: 10.1016/j.arcmed.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson WS, Hazlett T, Mantulin WW, Jonas A. The role of apolipoprotein AI domains in lipid binding. Proc Natl Acad Sci U S A. 1996;93:13605–13610. doi: 10.1073/pnas.93.24.13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Zhu HL, Atkinson D. Conformation and lipid binding of a C-terminal (198–243) peptide of human apolipoprotein A-I. Biochemistry. 2007;46:1624–1634. doi: 10.1021/bi061721z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laccotripe M, Makrides SC, Jonas A, Zannis VI. The carboxyl-terminal hydrophobic residues of apolipoprotein A-I affects its rate of phospholipid binding and its association with high density lipoprotein. J Biol Chem. 1997;272:17511–17522. doi: 10.1074/jbc.272.28.17511. [DOI] [PubMed] [Google Scholar]
- 8.Weers PMM, Ryan RO. Apolipophorin III: role model apolipoprotein. Insect Biochem Mol Biol. 2006;36:231–240. doi: 10.1016/j.ibmb.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Breiter DR, Kanost MR, Benning MM, Wesenberg G, Law JH, Wells MA, Rayment I, Holden HM. Molecular structure of an apolipoprotein determined at 2.5-Å resolution. Biochemistry. 1991;30:603–608. doi: 10.1021/bi00217a002. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Sykes BD, Ryan RO. Structural basis for the conformational adaptability of apolipophorin III, a helix-bundle exchangeable apolipoprotein. Proc Natl Acad Sci USA. 2002;99:1188–1193. doi: 10.1073/pnas.032565999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan D, Zheng Y, Yang D, Wang J. NMR solution structure and dynamics of an exchangeable apolipoprotein, Locusta migratoria apolipophorin III. J Biol Chem. 2003;278:21212–21220. doi: 10.1074/jbc.M208486200. [DOI] [PubMed] [Google Scholar]
- 12.Weers PMM, Kay CM, Oikawa K, Wientzek M, Van der Horst DJ, Ryan RO. Factors affecting the stability and conformation of Locusta migratoria apolipophorin III. Biochemistry. 1994;33:3617–3624. doi: 10.1021/bi00178a019. [DOI] [PubMed] [Google Scholar]
- 13.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 14.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:267–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 15.Ames BN. Assay of inorganic phosphate, total phosphate and phosphatases. Methods Enzymol. 1966;8:115–118. [Google Scholar]
- 16.Liu H, Scraba DG, Ryan RO. Prevention of phospholipase-C induced aggregation of low density lipoprotein by amphipathic apolipoproteins. FEBS Lett. 1993;316:27–33. doi: 10.1016/0014-5793(93)81730-n. [DOI] [PubMed] [Google Scholar]
- 17.Beck WHJ, Adams CP, Biglang-Awa IM, Patel AB, Vincent H, Haas-Stapleton EJ, Weers PMM. Apolipoprotein A-I binding to anionic vesicles and lipopolysaccharides: role for lysine residues in antimicrobial properties. Biochim Biophys Acta. 2013;1828:1503–1510. doi: 10.1016/j.bbamem.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CH, Tsai CM. Quantification of bacterial lipopolysaccharides by the purpald assay: measuring formaldehyde generated from 2-keto-3-deoxyoctonate and heptose at the inner core by periodate oxidation. Anal Biochem. 1999;267:161–168. doi: 10.1006/abio.1998.2961. [DOI] [PubMed] [Google Scholar]
- 19.Van der Horst DJ, Van Doorn JM, Voshol H, Kanost MR, Ziegler R, Beenakkers AMTh. Different isoforms of an apoprotein (apolipophorin III) associate with lipoproteins in Locusta migratoria. Eur J Biochem. 1991;196:509–517. doi: 10.1111/j.1432-1033.1991.tb15843.x. [DOI] [PubMed] [Google Scholar]
- 20.Narayanaswami V, Wang J, Kay CM, Scraba DG, Ryan RO. Disulfide bond engineering to monitor conformational opening of apolipophorin III during lipid binding. J Biol Chem. 1996;271:26855–26862. doi: 10.1074/jbc.271.43.26855. [DOI] [PubMed] [Google Scholar]
- 21.Soulages JL, Arrese EL, Chetty PS, Rodriguez V. Essential role of the conformational flexibility of helices 1 and 5 on the lipid binding activity of apolipophorin-III. J Biol Chem. 2001;276:34162–34166. doi: 10.1074/jbc.M105836200. [DOI] [PubMed] [Google Scholar]
- 22.Chetty PS, Mayne L, Lund-Katz S, Stranz D, Englander SW, Phillips MC. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chetty PS, Mayne L, Kan ZY, Lund-Katz S, Stranz D, Englander SW, Phillips MC. Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2012;109:11687–11692. doi: 10.1073/pnas.1209305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayanaswami V, Kiss RS, Weers PMM. The helix bundle: a reversible lipid binding motif. Comp Biochem Physiol A Mol Integr Physiol. 2010;155:123–133. doi: 10.1016/j.cbpa.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dwivedi P, Rodriguez J, Ibe NU, Weers PMM. Deletion of the N- or C-terminal helix of apolipophorin III to create a four-helix bundle protein. Biochemistry. 2016;55:3607–3615. doi: 10.1021/acs.biochem.6b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan CP, Chiu MH, Wu X, Lee SK, Prenner EJ, Weers PMM. Apolipoprotein-induced conversion of phosphatidylcholine bilayer vesicles into nanodisks. Biochim Biophys Acta. 2011;1808:606–613. doi: 10.1016/j.bbamem.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiss RS, Weers PMM, Narayanaswami V, Cohen J, Kay CM, Ryan RO. Structure-guided protein engineering modulates helix bundle exchangeable apolipoprotein properties. J Biol Chem. 2003;278:21952–21959. doi: 10.1074/jbc.M302676200. [DOI] [PubMed] [Google Scholar]
- 28.Weers PMM, Wang J, Van der Horst DJ, Kay CM, Sykes BD, Ryan RO. Recombinant locust apolipophorin III: characterization and NMR spectroscopy. Biochim Biophys Acta. 1998;1393:99–107. doi: 10.1016/s0005-2760(98)00063-0. [DOI] [PubMed] [Google Scholar]
- 29.Weers PMM, Abdullahi WE, Cabrera JM, Hsu TC. Role of buried polar residues in helix bundle stability and lipid binding of apolipophorin III: destabilization by threonine 31. Biochemistry. 2005;44:8810–8816. doi: 10.1021/bi050502v. [DOI] [PubMed] [Google Scholar]
- 30.Henning MF, Herlax V, Bakas L. Contribution of the C-terminal end of apolipoprotein AI to neutralization of lipopolysaccharide endotoxic effect. Innate Immun. 2011;17:327–337. doi: 10.1177/1753425910370709. [DOI] [PubMed] [Google Scholar]
- 31.Oztug M, Martinon D, Weers PMM. Characterization of the apoLp-III/LPS complex: insight into the mode of binding interaction. Biochemistry. 2012;51:6220–6227. doi: 10.1021/bi300619a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zdybicka-Barabas A, Stączek S, Mak P, Skrzypiec K, Mendyk E, Cytryńska M. Synergistic action of Galleria mellonella apolipophorin III and lysozyme against gram-negative bacteria. Biochim Biophys Acta. 2013;1828:1449–1456. doi: 10.1016/j.bbamem.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL apoA-I: implications for its assembly and function. J Lipid Res. 2008;49:1875–1883. doi: 10.1194/jlr.R800010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oda MN, Forte TM, Ryan RO, Voss JC. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat Struct Biol. 2003;10:455–460. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]
- 35.Burgess JW, Frank PG, Franklin V, Liang P, McManus DC, Desforges M, Rassart E, Marcel YL. Deletion of the C-terminal domain of apolipoprotein A-I impairs cell surface binding and lipid efflux in macrophage. Biochemistry. 1999;38:14524–14533. doi: 10.1021/bi990930z. [DOI] [PubMed] [Google Scholar]
- 36.Favari E, Bernini F, Tarugi P, Guido F, Calabersi L. The C-terminal domain of apolipoprotein A-I is involved in ABCA1-driven phospholipid and cholesterol efflux. Biochem Biophys Res Commun. 2002;299:801–805. doi: 10.1016/s0006-291x(02)02745-6. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt HH, Remaley AT, Stonik JA, Ronan R, Wellmann A, Thomas F, Zech LA, Brewer HB, Jr, Hoeg JM. Carboxyl-terminal domain truncation alters apolipoprotein A-I in vivo catabolism. J Biol Chem. 1995;270:5469–5475. doi: 10.1074/jbc.270.10.5469. [DOI] [PubMed] [Google Scholar]
- 38.Holvoet P, Zhao Z, Deridder E, Dhoest A, Collen D. Effects of deletion of the carboxyl-terminal domain of apoA-I or of its substitution with helices of apoA-II on in vitro and in vivo lipoprotein association. J Biol Chem. 1996;271:19395–19401. doi: 10.1074/jbc.271.32.19395. [DOI] [PubMed] [Google Scholar]
- 39.Holvoet P, Danloy S, Collen D. Role of the carboxy-terminal domain of human apolipoprotein A1 in high-density-lipoprotein metabolism. A study based on deletion and substitution variants in transgenic mice. Eur J Biochem. 1997;245:642–647. doi: 10.1111/j.1432-1033.1997.t01-1-00642.x. [DOI] [PubMed] [Google Scholar]
- 40.Alexander ET, Vedhachalam C, Sankaranarayanan S, de la Llera-moya M, Rothblat GH, Rader DJ, Phillips MC. Influence of apolipoprotein A-I domain structure on macrophage reverse cholesterol transport in mice. Arterioscler Thromb Vasc Biol. 2011;31:320–327. doi: 10.1161/ATVBAHA.110.216226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka M, Dhanasekaran P, Nguyen D, Ohta S, Lund-Katz S, Phillips MC, Saito H. Contributions of the N- and C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry. 2006;45:10351–10358. doi: 10.1021/bi060726t. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka M, Dhanasekaran P, Nguyen D, Nickel M, Takechi Y, Lund-Katz S, Phillips MC, Saito H. Influence of N-terminal helix bundle stability on the lipid-binding properties of human apolipoprotein A-I. Biochim Biophys Acta. 2011;1811:25–30. doi: 10.1016/j.bbalip.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soulages JL, Bendavid OJ. The lipid binding activity of the exchangeable apolipoprotein apolipophorin-III correlates with the formation of a partially folded conformation. Biochemistry. 1998;37:10203–10210. doi: 10.1021/bi980622l. [DOI] [PubMed] [Google Scholar]
- 44.Lyssenko NN, Hata M, Dhanasekaran P, Nickel M, Nguyen D, Chetty PS, Saito H, Lund-Katz S, Phillips MC. Influence of C-terminal α-helix hydrophobicity and aromatic amino acid content on apolipoprotein A-I functionality. Biochim Biophys Acta. 2012;1821:456–463. doi: 10.1016/j.bbalip.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagao K, Hata M, Tanaka K, Takechi Y, Nguyen D, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. The roles of C-terminal helices of human apolipoprotein A-I in formation of high-density lipoprotein particles. Biochim Biophys Acta. 2014;1841:80–87. doi: 10.1016/j.bbalip.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorshkova IN, Liadaki K, Gursky O, Atkinson D, Zannis VI. Probing the lipid-free structure and stability of apolipoprotein A-I by mutation. Biochemistry. 2000;39:15910–15919. doi: 10.1021/bi0014406. [DOI] [PubMed] [Google Scholar]
- 47.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 48.Pollard RD, Fulp B, Samuel MP, Sorci-Thomas MG, Thomas MJ. The conformation of lipid-free human apolipoprotein A-I in solution. Biochemistry. 2013;52:9470–9481. doi: 10.1021/bi401080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melchior JT, Walker RG, Morris J, Jones MK, Segrest JP, Lima DB, Carvalho PC, Gozzo FC, Castleberry M, Thompson TB, Davidson WS. An evaluation of the crystal structure of C-terminal truncated apolipoprotein A-I in solution reveals structural dynamics related to lipid binding. J Biol Chem. 2016;291:5439–5451. doi: 10.1074/jbc.M115.706093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weers PMM, Patel AB, Wan LC, Guigard E, Kay CM, Hafiane A, McPherson R, Marcel YL, Kiss RS. Novel N-terminal mutation of human apolipoprotein A-I reduces self-association and impairs LCAT activation. J Lipid Res. 2011;52:35–44. doi: 10.1194/jlr.M007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jayaraman S, Abe-Dohmae S, Yokoyama S, Cavigiolio G. Impact of self-association on function of apolipoprotein A-I. J Biol Chem. 2011;286:35610–35623. doi: 10.1074/jbc.M111.262485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beckstead JA, Block BL, Bielicki JK, Kay CM, Oda MN, Ryan RO. Combined N- and C-terminal truncation of human apolipoprotein A-I yields a folded, functional central domain. Biochemistry. 2005;44:4591–4599. doi: 10.1021/bi0477135. [DOI] [PubMed] [Google Scholar]
- 53.Deng X, Morris J, Dressmen J, Tubb MR, Tso P, Jerome WG, Davidson WS, Thompson TB. The structure of dimeric apolipoprotein A-IV and its mechanism of self-association. Structure. 2012;20:767–779. doi: 10.1016/j.str.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dettloff M, Niere M, Ryan RO, Luty R, Kay CM, Wiesner A, Weers PMM. Differential lipid binding of truncation mutants of Galleria mellonella apolipophorin III. Biochemistry. 2002;41:9688–9695. doi: 10.1021/bi0200108. [DOI] [PubMed] [Google Scholar]