

Abstract
Dynamic nuclear polarization is an emerging technique for sensitizing solid-state NMR experiments by transferring polarization from electrons to nuclei. Stable biradicals, the polarization source for the cross effect mechanism, are typically codissolved at millimolar concentrations with proteins of interest. Here we describe the high-affinity biradical tag TMP-T, created by covalently linking trimethoprim, a nanomolar affinity ligand of dihydrofolate reductase (DHFR), to the biradical polarizing agent TOTAPOL. With TMP-T bound to DHFR, large enhancements of the protein spectrum are observed, comparable to when TOTAPOL is codissolved with the protein. In contrast to TOTAPOL, the tight binding TMP-T can be added stoichiometrically at radical concentrations orders of magnitude lower than in previously described preparations. Benefits of the reduced radical concentration include reduced spectral bleaching, reduced chemical perturbation of the sample, and the ability to selectively enhance signals for the protein of interest.
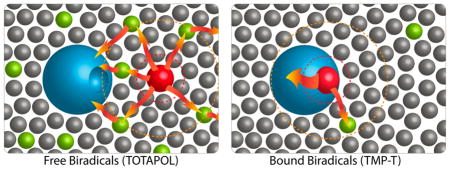
Graphical abstract
INTRODUCTION
The recent past has seen a dramatic increase in the range of methods and applicability for solid state NMR of biological systems.1–3 Nevertheless, for complex systems or systems available in limited quantity, detection sensitivity continues to be an important challenge. Dynamic nuclear polarization (DNP) experiments enhance NMR signals by transferring electron polarization from paramagnetic compounds to nearby nuclei.4 One particularly robust mechanism, the cross effect,5–8 uses nitroxide biradicals such as TOTAPOL cosolubilized with a system of interest,9–11 with measurements performed near 100 K. DNP experiments with biradical polarizing agents have enabled studies of amyloidogenic peptides, bacteriorhodopsin, acetylcholine receptors,12–15 and many other systems.16–19
For DNP studies of heterogeneous biomolecular mixtures, the biradical is typically added at millimolar concentrations and the distance between the radical and the protein of interest is not deliberately controlled. A number of adventitious effects of co-added radicals make it of interest to minimize biradical concentration and, where appropriate, control their spatial location in the sample. Added paramagnetic compounds may lead to NMR signal bleaching, line broadening, and other paramagnetic relaxation effects.20 These effects can be useful structural probes,21,22 but can also lead to signal losses. Moreover, free radicals can perturb cellular function at high concentrations.23,24 Lastly, hydrophobic radicals, such as TOTAPOL, may precipitate, aggregate, or bind proteins,14,25,26 possibly at surface sites, which can provide a nonspecific way to target biradicals. Recent attempts to control radical solubility and location include solubilization in surfactants,27,28 caging in cyclodextrin,29 labeling peptides with biradicals,30 radical tagged lipids,31,32 “gluing” with trehalose,33 use of endogenous paramagnetic cofactors,34,35 labeling with paramagnetic metal chelators,36 and the use of a biradical-tagged peptide that binds the protein of interest.37 Covalent attachment of the biradicals TOTAPOL and AMUPOL via cysteine-specific methanthiosulfonate chemistry has also been reported as a method for matrix-free membrane protein enhancement.22,38
Here, we prepared a high affinity biradical-tagged ligand specifically targeted to a protein of interest and investigated how binding the biradical to the protein affects key parameters in the DNP experiment. We adapted the TMP-DHFR tagging approach,39–41 predicated on the high affinity and selectivity of the inhibitor trimethoprim (TMP) for E. coli dihydrofolate reductase (DHFR) (Kd ~ 15 nM42), whose binding mode is known from crystallography.43 TMP has been derivatized with fluorophores for in vivo imaging of DHFR fusion proteins in mammalian cells, making the DHFR-based system a generalizable biradical tagging approach through the use of fusion proteins. We derivatized trimethoprim with the popular nitroxide biradical TOTAPOL, yielding the tight-binding biradical tag we dubbed TMP-T. We characterized TMP-T and its complex with DHFR using high field DNP, demonstrating the potential of a ligand-based tag to enable novel DNP applications.
MATERIALS AND METHODS
Synthesis of TMP-T (2-(4-((2,4-Diaminopyrimidin-5-yl)methyl)-2,6-dimethoxyphenoxy)-N-(1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl)-N-(2-hydroxy-3-((1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl)oxy)propyl)-acetamide)
TOTAPOL (1-(2,2,6,6-Tetramethyl-1-oxy-4-piperidinyl)oxy-3-(2,2,6,6- tetramethyl-1-oxy-4-piperidinyl)-amino-propan-2-ol [1-(TEMPO-4- oxy)-3-(TEMPO-4-amino)-propan-2-ol]) and TMP-COOH (2-(4-((2,4-diaminopyrimidin-5-yl)methyl)-2,6-dimethoxyphenoxy)acetic acid) were synthesized according to literature procedures (see Scheme 1).10,41 To couple TOTAPOL and the TMP acid, TMP-COOH (20 mg, 0.05 mmol) was dissolved in DMF (2 mL) followed by the addition of EDC·HCl (20 mg, 0.1 mmol). Then TOTAPOL (20 mg, 0.05 mmol), HOAT (2 mg, 0.015 mmol), and diisopropylethyleneamine (187 μL) were added sequentially. The resulting red solution was stirred at room temperature under nitrogen for 48 h. After TLC showed that TOTAPOL was completely consumed, the reaction was quenched by adding ethyl acetate (5 mL). The solvents were removed under vacuum and the resulting solid was subjected to flash column chromatography with methylene chloride and methanol as the mobile phase (first 6:1, then 1:1 v/v). The second fraction was collected as the product (20 mg, yield 56%). The product was characterized by LC-MS (ESI+) and FT-IR. The m/z calculated for C36H57N7O8 (M+H) is 716.43, and the observed mass is 716.56. Supporting Information Figure S1 shows ATR- FTIR analysis of TMP-T.
Scheme 1.

Synthesis of TMP-T from TMP-COOH and TOTAPOL
Protein Expression and Labeling with TMP-T
U–13C,15N DHFR was prepared as in ref 39 with the modification that E. coli cultures were grown in 13C,15N-minimal media (see Figure S3).39 The purified protein was buffer exchanged into 98.5 ± 1.0% 2H-PBS, pH 7.4 and concentrated at 4 °C using an Amicon Ultracel 10k centrifugal concentrator at 4150 g. Protein concentration was measured by UV/vis (ε280= 31,100 for DHFR) and d8-glycerol was promptly added to 30% v/v to prevent precipitation. For non-stoichiometric samples, concentrated DHFR stocks were mixed with biradical stocks of known concentration, also containing 30% v/v glycerol-d8, along with any additional additives (e.g., 13C-glucose, DMSO, TMP). The composition of all samples is available in Table S1.
For samples with 1:1 TMP-T:DHFR stoichiometry, we capitalized on the tight binding of DHFR and TMP-T in the micromolar range. Approximately 0.3 mM DHFR stocks were incubated with an 8-fold excess of TMP-T. Unbound TMP-T was removed by concentrating this mixture using a 10 kDa filter and washing twice with deuterated PBS buffer. The final concentration of free or excess TMP-T was assessed by EPR to be <5 μM, orders of magnitude lower than the DHFR concentration. Kickoff experiments (Results and Discussion Section, Figure 1b) indicated that these samples had a 1:1 stoichiometry of TMP-T:DHFR.
Figure 1.

Room temperature 9.5 GHz EPR spectra of (a) 50 μM TMP-T before (red) and after (black) addition of 100 μM DHFR, with the bound spectra expanded to show broadening. (b) 100 μM 1:1 TMP-T:DHFR complexes before (black) and after (red) addition of 20-fold excess trimethoprim to kick off TMP-T.
For some samples, extra care was taken to achieve high (>98%) deuteration levels (as indicated in “Other” column in Table S1). This entailed minimizing atmosphere exposure to under a few minutes, and using a stock solution of 10× concentrated deuterated PBS to prepare fresh 1× buffer for each sample so that 90% of the solution would be “new” buffer. Solutions were adjusted to a pH of 7.4 using a 12 M NaOH solution in H2O; protons introduced were calculated to be <0.05%.
For frozen solution DNP-SSNMR, approximately 25 μL of sample was centrifuged into a 3.2 mm Bruker DNP sapphire rotor using a custom-built centrifugal packing funnel and a benchtop centrifuge. All samples were stored at −80 °C.
CW X-Band EPR and DNP Experiments
CW-EPR data were acquired at room temperature using a Bruker EMX X-band EPR spectrometer at a microwave frequency of 9.756 GHz, with a center field of 3480 G and sweep width of 100 G. MW power and modulation amplitude were varied to ensure that line shape and intensity were unchanged, and conditions chosen to maximize S/N. Final EPR parameters were a modulation frequency of 100 kHz, modulation amplitude of 1.00 G, conversion time of 164 s, and microwave power of 20.1 mW.
DNP-SSNMR experiments were performed on a 600 MHz (14.1 T) Bruker AVANCE III-DNP system at the New York Structural Biology Center (NYSBC), equipped with a 395 GHz gyrotron, a HCN E-free probe and a 4-channel HCND probe. A 400 MHz (9.4 T) Bruker AVANCE-III-DNP system at Bruker Biospin (Billerica, MA), equipped with 263 GHz gyrotron and 3-channel HCN DNP probe, was used for measurements at a lower magnetic field. MAS rates of 8 kHz were used for all samples except the spectrum acquired at 400 MHz (9.4 T) (see Figure 3), which was acquired at 9 kHz. Typical LTMAS temperatures were 98 K for variable-temperature (VT) gas, ~ 105 K for bearing gas, and ~104 K for drive gas. Prior to experiments, gyrotron parameters were calibrated to yield a smooth power curve/enhancement profile, and TOTAPOL enhancement was checked at the experimental conditions using a U–13C,15N proline sample, yielding an enhancement factor of 23 at 600 MHz. All spectra were acquired with a recycle delay of 3 s. CP experiments were performed with a 10% tangential ramp, a 1H 90° pulse of 2.5 μs (100 kHz), and with proton decoupling field strength of 100 kHz.
Figure 3.

2D 13C–13C DARR spectrum of 0.5 mM 1:1 TMP-T complexes. Spectrum was collected at 400 MHz 1H field with continuous gyrotron irradiation at 263 GHz, with a 9 kHz MAS rate and a sample temperature of 111 K. Spectrum was collected in 17 h with 64 scans per row.
RESULTS AND DISCUSSION
EPR Characterization of TMP-T and Binding to DHFR
Trimethoprim can be extensively derivatized while keeping the Kd in the nanomolar range.40,42 We therefore chose it as a scaffold for TOTAPOL derivatization and synthesized the high-affinity biradical tag TMP-T (see Scheme 1). To characterize TMP-T, we used EPR spectroscopy, which is exquisitely sensitive to the nitroxide’s environment44 and reflects molecular structure as well as interactions with proteins or other macromolecules.45–47 We compared the experimental CW X-band EPR spectra of aqueous TMP-T to that of TOTAPOL (see Figure S5). Relative to TOTAPOL, TMP-T has a slightly increased line width, likely indicating that the trimethoprim derivatization leads to hindered rotation of the nitroxides and intramolecular interactions with the hydrophobic trimethoprim. When bound to DHFR, the EPR spectrum of TMP-T undergoes both intensity and line width changes (see Figure 1a), indicative of a change in the nitroxide’s environment. We used these spectral changes to confirm that binding of TMP-T to DHFR is indeed stoichiometric (i.e., greater than 95%) under our DNP sample preparation conditions by preparing 100 μM 1:1 DHFR:TMP-T complexes that were washed to remove unbound TMP-T (see Materials and Methods Section). We then “kicked-off” the bound TMP-T by adding a 20-fold excess of trimethoprim and recovered the EPR spectrum of free TMP-T (see Figure 1b). The protein concentration, as measured by UV/vis spectroscopy, and the biradical concentration, as measured by EPR using a standard curve, agree within error of the two methods (55 ± 5 μM protein concentration, 63 ± 8 μM biradical concentration), indicating that binding is essentially 1:1.
To further quantify the binding affinity of TMP-T for DHFR, we used a fluorescence polarization competition experiment where a fluorescent analog of trimethoprim, TMP-TAMRA (Figure S6) was competitively displaced with TMP-T, resulting in changes in the measured fluorescence anisotropy (see SI and Figure S7). These experiments showed that TMP-T binds DHFR tightly with a Kd of 165 ± 7 nM.
Enhancements/Dilution
When stoichiometrically bound to TMP-T, a frozen solution of U–13C,15N-DHFR in 30/70 v/v d8-glycerol/D2O was enhanced by DNP using a 395 GHz gyrotron at 14.1 T (600 MHz 1H-field), and detected using CPMAS 13C NMR spectroscopy. We obtained excellent enhancement factors ε (defined as the ratio of carbonyl intensity with gyrotron on to gyrotron off48), comparable to optimal conditions for TOTAPOL experiments (see Table S1). In fact, 4.5 mM 1:1 TMP-T:DHFR complexes had the same enhancement as a sample with 10 mM TOTAPOL and a similar protein concentration (Table S1). To explore how specific binding affects DNP parameters, we prepared samples at a range of TMP-T/DHFR concentrations and ratios and compared them to TOTAPOL samples collected under similar conditions.
The dependence of enhancement on biradical concentration for both TMP-T and TOTAPOL is shown in Figure 2. For TOTAPOL, consistent with prior reports,48,49 optimal enhancements were obtained at a radical concentration of 20 mM, with an enhancement of 28 (Figures 2 and S8 and Table S1). Buildup times (TB, measured according to pulse sequence in Figure S4) were closely dependent on radical concentration, decreasing from 5.0 to 2.3 s as the TOTAPOL concentration increased from 4 to 20 mM (Table S1). When TOTAPOL was diluted, the enhancement degraded dramatically, providing little enhancement at 1 mM TOTAPOL:0.5 mM DHFR, with a significantly lengthened TB of ~27 s (Table S1). Moreover, enhancement for 20–21 mM TOTAPOL was very similar even when protein concentration differed by an order of magnitude (6.5 mM vs 0.5 mM, see Table S1), indicating that, for TOTAPOL, enhancement is dominated by biradical concentration, regardless of analyte concentration.
Figure 2.

Enhancement and bleaching as a function of biradical concentration for TMP-T (top) and TOTAPOL (bottom) (Table S1). For TMP-T, ε is measured at a fixed 1:1 biradical:protein ratio, with the exception of the 2:1 sample indicated by the green star. Bleaching is defined as % integrated intensity relative to an identical sample without biradicals present.
In contrast, for TMP-T, enhancement and polarization buildup times depended on the stoichiometry of TMP-T bound to DHFR. For a stoichiometric and superstoichiometric sample ([TMP-T]/[DHFR] = 4.5 mM/4.5 mM or 7.0 mM/3.0 mM), ε = 23 and 26 respectively. As expected, for a substoichiometric sample ([TMP-T]/[DHFR] = 2.7 mM/5.9 mM), ε was considerably smaller at 14 (Table S1). Notably, as stoichiometric protein:biradical complexes were diluted to the low micromolar regime ([TMP-T]/[DHFR] = 0.05 mM/0.05 mM), samples retained high enhancements (ε>15) and short buildup times (Figures 2 and S8 and Table S1).
The TMP-T data indicates the importance of biradical-protein proximity to achieving enhancement and presents a key advantage of the biradical tagging approach. Namely, the enhancement barely changes between 4.5 mM radical (stoichiometric) and 7 mM radical samples, suggesting that the bulk of the protein polarization comes from the bound, proximal biradical. The stoichiometric data points in the micromolar regime imply that as long as the biradical is bound to the protein, it can effectively provide polarization, regardless of absolute concentration. To support this interpretation, TB remained short at ~4–5s throughout the concentration range, as expected since the biradical-protein geometry was constant. Although there is some concentration dependence to overall enhancement for stoichiometric complexes (Figure 2), which can be attributed to a decrease in overall biradical density, excellent enhancement is retained even at 50-μM absolute biradical concentration since all protein molecules are proximal to at least one biradical.
To further underscore the benefit of biradical proximity to DNP parameters, we measured enhancement for samples in which TMP-T was competitively displaced with TMP. EPR confirmed that TMP-T can be displaced by a large excess of TMP (Figure 1b). Addition of ~10-fold excess TMP to the substoichiometric TMP-T sample (2.3 mM TMP-T, 4 mM DHFR) caused TB to lengthen significantly to 12.5 s, and the DNP enhancement to degrade (Table S1). For TOTAPOL samples, TB is inversely proportional to radical concentration; these data are consistent with the expectation that TMP-T has enhancement properties similar to free TOTAPOL when displaced from DHFR.
The aforementioned enhancement factors were obtained at 600 MHz. DNP enhancements are generally higher at lower field; for comparison, at 400 MHz/263 GHz, ε = 42 for a stoichiometric sample.18 With these enhancements, we collected well-resolved 2D spectra of 1:1 DHFR:TMP-T complexes at 0.5 mM (Figure 3), allowing characterization of DHFR with only ~15 nmol of protein.
Bleaching
We saw little evidence of bulk signal loss (paramagnetic bleaching) due to the bound biradical when TMP-T was used at concentrations up to 7 mM (Figures 2 and S9). In contrast, since TOTAPOL must be used at higher absolute concentrations to achieve optimal enhancements, bleaching was significant at 20 mM, as previously observed.49 Moreover, 2D 13C–13C, 15N–13C, and 3D 15N–13C–13C spectra of 1:1 TMP-T: DHFR showed relatively well-resolved peaks, with half widths of 1–1.5 ppm, comparable to prior encouraging reports,15,50,51 and enabled site-specific NMR assignments (Figure 4). On the other hand, when 10 mM TOTAPOL was present, peaks due to (presumed solvent-exposed) histidine, arginine, and lysine residues were partially bleached (Figures S10 and S11). Our encouraging results are in contrast to recent literature reports that indicate significant bleaching with covalently attached biradicals.22,38 We attribute this to the fact that TMP-T binds DHFR at a solvent-exposed face of the protein (see SI Figure S2) and protrudes out into the solvent, offering enhancement advantages without generalized bleaching.
Figure 4.

Slices from NCoCx (blue) and NCaCx (red) 3D spectra of 4.5 mM U(13C,15N) 1:1 TMP-T:DHFR complexes. (a) shows assignment of T35/N34, (b) shows a slice at I82 resonance, and (c) is a close-up of I82 crosspeaks in (b). Spectra were collected at 600 MHz with MAS frequency of 8 kHz, sample temperature of 106 ± 1 K, and continuous microwave irradiation. NCo/NCa DCP transfers were followed by 25 ms of 13C–13C DARR mixing57 to acquire Cx. 1H decoupling was at 100 kHz, with CW during DCP and SPINAL-64 during acquisition. The NCoCx spectrum was collected in 41 h, with 16 transients per row. The NCaCx spectrum was collected in 42 h with 12 transients per row.
Additionally, we see no evidence for a strong MAS-dependent depolarization effect that others have reported,52 despite the fact that the biradical is tightly tethered to the protein (see Figure S12). We conclude that depolarization is not a significant concern in the concentration range used in this work. Furthermore, this lack of observed depolarization effects allows us to assess the DNP enhancement simply as the ratio of intensities, IμW on/IμW off; since the microwaves off signal is not significantly perturbed by depolarization, there is no need to invoke more complex metrics to assess enhancement factors.
Preferential Enhancement
In relatively dilute samples, DHFR’s NMR peaks can be enhanced preferentially over those of cosolutes, such as glycerol (added for cryoprotection). For example, for 1:1 DHFR:TMP-T complexes at 0.5 mM concentration, enhancement was ~19 for the protein but only 9 for the glycerol (Table S1). The buildup time for the glycerol peak was also longer (TB = 12 s for glycerol but 4 s for the protein, Figure 5c). When the DHFR/TMP-T complexes were further diluted to 0.05 mM, the glycerol resonances were undetected while the protein signals had an enhancement factor of 15 (Figure 5a and Table S1).
Figure 5.

(a) Overlay of 0.5 mM (red) and 0.05 mM (blue) fully deuterated TMP-T samples shows that dilution leads to selective protein enhancement; the glycerol is unenhanced in the dilute limit. (b) Comparison of >98% 2H and 20% 1H solvents shows excellent selectivity for the protein in the former but nonselective enhancements in the latter. (c) Polarization buildup curves for TMP-T bound samples at two protonation levels. When fit to saturation recovery functions, TB= 4.1 s for C′ and 12.1 s for glycerol at >98% 2H; however at 20% 1H, TB= 32 s for C′ and 37 s for glycerol. Spectra were acquired via CP from 1H at 8 kHz MAS frequency with 100 kHz SPINAL decoupling during detection, 106 ± 1 K, and continuous microwave irradiation. Spectra in (a) and (b) were scaled to account for differences in protein concentration and enhancement.
These selective enhancement experiments were carried out using a solvent that was approximately 98% deuterated. When the solvent contained 20% 1H, there was no selectivity, as shown in Figures 5 and S13, and the buildup times were long (>30 s) for both protein and glycerol. TOTAPOL exhibited neither the potential for selectivity nor sensitivity to deuteration levels (Figures S13 and S14), likely due to differences in protein-biradical geometry between the two systems. These data in aggregate support a picture in which ultralow proton content is useful for “containing’” the polarization locally, or defeating spin diffusion on the seconds time scale.
CONCLUSIONS
We report the synthesis and characterization of the biradical tag TMP-T, which, together with its protein target DHFR, presents an excellent alternative to the use of high biradical concentrations in DNP-enhanced SSNMR experiments. TMP-T binds DHFR tightly, as indicated by fluorescence polarization experiments, EPR data, and short DNP polarization buildup times for protein molecules with a bound biradical. When bound stoichiometrically to DHFR, TMP-T provides sizable NMR signal enhancements that enable multidimensional NMR characterization of DHFR. These enhancements do not come at the expense of resolution or global paramagnetic bleaching.
Moreover, with TMP-T stoichiometrically bound to DHFR, we are able to lower protein and radical concentrations to 50 μM and still obtain enhancements of 15. These concentrations correspond to a situation where it is difficult to express the protein of interest in high yield (e.g., mammalian proteins) or the in vivo expression level of many E. coli proteins.53,54 Under these conditions, DHFR is selectively enhanced over cosolutes, favorably implying that a ligand-based strategy can be used to selectively pick out and study a protein of interest in an in vivo setting. Even with a 50 μM sample, the enhancement provided by TMP-T would deliver a 15N–13C–13C 3D spectrum with reasonable signal-to-noise ratio in 4 days, which is shorter than typically required for such a spectrum using conventional SSNMR.
The TMP-T/DHFR approach is broadly applicable through the use of DHFR fusion proteins, which are robust and preserve TMP binding properties.40 By synthesizing other TMP-linked biradicals with higher enhancements, such as AMUPOL,55 specific enhancement can likely be increased to factors of 50 or higher. We anticipate that the TMP-T/DHFR tagging system, as well as other biradical tag-based strategies,56 can be used to enhance proteins in whole cells or whole cell fractions, thereby enabling DNP SSNMR characterization of sparingly expressed proteins in their native environments.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Science Foundation (NSF): MCB1412253 to A.E.M. R.R. was supported by the National Institutes of Health Training Program in Molecular Biophysics T32GM008281. The authors thank Drs. Tracy Wang, Zhixing Chen, and Yao Ze Ng for help with the TMP/DHFR system and fluorescent polarization binding assays, Dr. Steffen Jockusch for help with EPR, and Drs. Mike Goger and Boris Itin of the New York Structural Biology Center for support with NMR instrumentation. The data collected at NYSBC was enabled by a grant from NYSTAR and ORIP/NIH facility improvement grant CO6RR015495. The 600 MHz DNP/NMR spectrometer was purchased with funds from NIH grant S10RR029249. DNP spectrometer time at 400 MHz was kindly provided by Bruker Biospin.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.6b09021.
Details of sample preparation, fluorescence characterization, DNP parameters, and supplementary figures (PDF)
References
- 1.McDermott A. Structure and Dynamics of Membrane Proteins by Magic Angle Spinning Solid-State NMR. Annu Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 2.Sergeyev IV, Day LA, Goldbourt A, McDermott AE. Chemical Shifts for the Unusual DNA Structure in Pf1 Bacteriophage from Dynamic-Nuclear-Polarization-Enhanced Solid-State NMR Spectroscopy. J Am Chem Soc. 2011;133:20208–20217. doi: 10.1021/ja2043062. [DOI] [PubMed] [Google Scholar]
- 3.van der Cruijsen EAW, Nand D, Weingarth M, Prokofyev A, Hornig S, Cukkemane AA, Bonvin AMJJ, Becker S, Hulse RE, Perozo E, et al. Importance of Lipid-Pore Loop Interface for Potassium Channel Structure and Function. Proc Natl Acad Sci U S A. 2013;110:13008–13013. doi: 10.1073/pnas.1305563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes aB, De Paëpe G, van der Wel PCA, Hu KN, Joo CG, Bajaj VS, Mak-Jurkauskas ML, Sirigiri JR, Herzfeld J, Temkin RJ, et al. High-Field Dynamic Nuclear Polarization for Solid and Solution Biological NMR. Appl Magn Reson. 2008;34:237–263. doi: 10.1007/s00723-008-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wollan DS. Dynamic Nuclear Polarization with an Inhomogeneously Broadened ESR Line. I. Theory. Phys Rev B. 1976;13:3671–3685. [Google Scholar]
- 6.Hu KN, Debelouchina GT, Smith Aa, Griffin RG. Quantum Mechanical Theory of Dynamic Nuclear Polarization in Solid Dielectrics. J Chem Phys. 2011;134:125105–19. doi: 10.1063/1.3564920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu K, Yu H, Swager TM, Griffin RG. Dynamic Nuclear Polarization with Biradicals. J Am Chem Soc. 2004;126:10844–10845. doi: 10.1021/ja039749a. [DOI] [PubMed] [Google Scholar]
- 8.Hovav Y, Feintuch A, Vega S. Theoretical Aspects of Dynamic Nuclear Polarization in the Solid State - the Cross Effect. J Magn Reson. 2012;214:29–41. doi: 10.1016/j.jmr.2011.09.047. [DOI] [PubMed] [Google Scholar]
- 9.Matsuki Y, Maly T, Ouari O, Karoui H, Le Moigne F, Rizzato E, Lyubenova S, Herzfeld J, Prisner T, Tordo P, et al. Dynamic Nuclear Polarization with a Rigid Biradical. Angew Chem, Int Ed. 2009;48:4996–5000. doi: 10.1002/anie.200805940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song C, Hu KN, Joo CG, Swager TM, Griffin RG. TOTAPOL: A Biradical Polarizing Agent for Dynamic Nuclear Polarization Experiments in Aqueous Media. J Am Chem Soc. 2006;128:11385–11390. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 11.Siaw TA, Fehr M, Lund A, Latimer A, Walker Sa, Edwards DT, Han S-I. Effect of Electron Spin Dynamics on Solid-State Dynamic Nuclear Polarization Performance. Phys Chem Chem Phys. 2014;16:18694–18706. doi: 10.1039/c4cp02013h. [DOI] [PubMed] [Google Scholar]
- 12.van der Wel PCA, Hu KN, Lewandowski J, Griffin RG. Dynamic Nuclear Polarization of Amyloidogenic Peptide Nanocrystals: GNNQQNY, a Core Segment of the Yeast Prion Protein Sup35p. J Am Chem Soc. 2006;128:10840–10846. doi: 10.1021/ja0626685. [DOI] [PubMed] [Google Scholar]
- 13.Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG. DNP Enhanced Frequency-Selective TEDOR Experiments in Bacteriorhodopsin. J Magn Reson. 2010;202:9–13. doi: 10.1016/j.jmr.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linden AH, Lange S, Franks WT, Akbey U, Specker E, van Rossum BJ, Oschkinat H. Neurotoxin II Bound to Acetylcholine Receptors in Native Membranes Studied by Dynamic Nuclear Polarization NMR. J Am Chem Soc. 2011;133:19266–19269. doi: 10.1021/ja206999c. [DOI] [PubMed] [Google Scholar]
- 15.Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG. Functional and Shunt States of Bacteriorhodopsin Resolved by 250 GHz Dynamic Nuclear Polarization-Enhanced Solid-State NMR. Proc Natl Acad Sci U S A. 2009;106:9244–9249. doi: 10.1073/pnas.0900908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maciejko J, Mehler M, Kaur J, Lieblein T, Morgner N, Ouari O, Tordo P, Becker-Baldus J, Glaubitz C. Visualizing Specific Cross-Protomer Interactions in the Homo-Oligomeric Membrane Protein Proteorhodopsin by DNP-Enhanced Solid-State NMR. J Am Chem Soc. 2015;137:9032–9043. doi: 10.1021/jacs.5b03606. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan M, Cukkemane A, van Zundert GCP, Narasimhan S, Daniëls M, Mance D, Waksman G, Bonvin AMJJ, Fronzes R, Folkers GE, et al. Probing a Cell-Embedded Megadalton Protein Complex by DNP-Supported Solid-State NMR. Nat Methods. 2015;12:649–652. doi: 10.1038/nmeth.3406. [DOI] [PubMed] [Google Scholar]
- 18.Smith AN, Long JR. Dynamic Nuclear Polarization as an Enabling Technology for Solid State Nuclear Magnetic Resonance Spectroscopy. Anal Chem. 2016;88:122–132. doi: 10.1021/acs.analchem.5b04376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mao J, Do N, Scholz F, Reggie L, Mehler M, Lakatos A, Ong Y, Ullrich SJ, Brown LJ, Brown RCD, et al. Structural Basis of the Green – Blue Color Switching in Proteorhodopsin as Determined by NMR Spectroscopy. J Am Chem Soc. 2014;136:17578–17590. doi: 10.1021/ja5097946. [DOI] [PubMed] [Google Scholar]
- 20.Jaroniec CP. Solid-State Nuclear Magnetic Resonance Structural Studies of Proteins Using Paramagnetic Probes. Solid State Nucl Magn Reson. 2012;43–44:1–13. doi: 10.1016/j.ssnmr.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 21.Nadaud PS, Helmus JJ, Höfer N, Jaroniec CP. Long-Range Structural Restraints in Spin-Labeled Proteins Probed by Solid-State Nuclear Magnetic Resonance Spectroscopy. J Am Chem Soc. 2007;129:7502–7503. doi: 10.1021/ja072349t. [DOI] [PubMed] [Google Scholar]
- 22.van der Cruijsen EAW, Sauvée C, Koers EJ, Hulse RE, Weingarth M, Ouari O, Perozo E, Tordo P, Baldus M. Biomolecular DNP-Supported NMR Spectroscopy Using Site-Directed Spin Labeling. Chem - Eur J. 2015;21:12971–12977. doi: 10.1002/chem.201501376. [DOI] [PubMed] [Google Scholar]
- 23.Soule BP, Hyodo F, Matsumoto KI, Simone NL, Cook Ja, Krishna MC, Mitchell JB. The Chemistry and Biology of Nitroxide Compounds. Free Radical Biol Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couet WR, Brasch RC, Sosnovsky G, Tozer TN. Factors Affecting Nitroxide Reduction in Ascorbate Solution and Tissue Homogenates. Magn Reson Imaging. 1985;3:83–88. doi: 10.1016/0730-725x(85)90012-8. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi H, Hediger S, De Paëpe G. Matrix-Free Dynamic Nuclear Polarization Enables Solid-State NMR 13C-13C Correlation Spectroscopy of Proteins at Natural Isotopic Abundance. Chem Commun (Cambridge, U K) 2013;49:9479–9481. doi: 10.1039/c3cc45195j. [DOI] [PubMed] [Google Scholar]
- 26.Ravera E, Corzilius B, Michaelis VK, Luchinat C, Griffin RG, Bertini I. DNP-Enhanced MAS NMR of Bovine Serum Albumin Sediments and Solutions. J Phys Chem B. 2014;118:2957–2965. doi: 10.1021/jp500016f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiesewetter MK, Michaelis VK, Walish JJ, Griffin RG, Swager TM. High Field Dynamic Nuclear Polarization NMR with Surfactant Sheltered Biradicals. J Phys Chem B. 2014;118:1825–1830. doi: 10.1021/jp410387e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lelli M, Rossini AJ, Casano G, Ouari O, Tordo P, Lesage A, Emsley L. Hydrophobic Radicals Embedded in Neutral Surfactants for Dynamic Nuclear Polarization of Aqueous Environments at 9.4 T. Chem Commun (Cambridge, U K) 2014;50:10198–10201. doi: 10.1039/c4cc02152e. [DOI] [PubMed] [Google Scholar]
- 29.Mao J, Akhmetzyanov D, Ouari O, Denysenkov V, Corzilius B, Plackmeyer J, Tordo P, Prisner TF, Glaubitz C. Host-Guest Complexes as Water-Soluble High-Performance DNP Polarizing Agents. J Am Chem Soc. 2013;135:19275–19281. doi: 10.1021/ja409840y. [DOI] [PubMed] [Google Scholar]
- 30.Vitzthum V, Borcard F, Jannin S, Morin M, Miéville P, Caporini Ma, Sienkiewicz A, Gerber-Lemaire S, Bodenhausen G. Fractional Spin-Labeling of Polymers for Enhancing NMR Sensitivity by Solvent-Free Dynamic Nuclear Polarization. ChemPhysChem. 2011;12:2929–2932. doi: 10.1002/cphc.201100630. [DOI] [PubMed] [Google Scholar]
- 31.Smith AN, Caporini Ma, Fanucci GE, Long JR. A Method for Dynamic Nuclear Polarization Enhancement of Membrane Proteins. Angew Chem, Int Ed. 2015;54:1542–1546. doi: 10.1002/anie.201410249. [DOI] [PubMed] [Google Scholar]
- 32.Fernández-de-Alba C, Takahashi H, Richard A, Chenavier Y, Dubois L, Maurel V, Lee D, Hediger S, De Paëpe G. Matrix-Free DNP-Enhanced NMR Spectroscopy of Liposomes Using a Lipid-Anchored Biradical. Chem - Eur J. 2015;21:4512–4517. doi: 10.1002/chem.201404588. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi H, Hediger S, De Paëpe G. Matrix-Free Dynamic Nuclear Polarization Enables Solid-State NMR 13C-13C Correlation Spectroscopy of Proteins at Natural Isotopic Abundance. Chem Commun. 2013;49:9479–9481. doi: 10.1039/c3cc45195j. [DOI] [PubMed] [Google Scholar]
- 34.Maly T, Cui D, Griffin RG, Miller AF. 1H Dynamic Nuclear Polarization Based on an Endogenous Radical. J Phys Chem B. 2012;116:7055–7065. doi: 10.1021/jp300539j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wenk P, Kaushik M, Richter D, Vogel M, Suess B, Corzilius B. Dynamic Nuclear Polarization of Nucleic Acid with Endogenously Bound Manganese. J Biomol NMR. 2015;63:97–109. doi: 10.1007/s10858-015-9972-1. [DOI] [PubMed] [Google Scholar]
- 36.Kaushik M, Bahrenberg T, Can TV, Caporini MA, Silvers R, Heiliger J, Smith AA, Schwalbe H, Griffin RG, Corzilius B. Gd(III) and Mn(II) Complexes for Dynamic Nuclear Polarization: Small Molecular Chelate Polarizing Agents and Applications with Site-Directed Spin Labeling of Proteins. Phys Chem Chem Phys. 2016;18:27205–27218. doi: 10.1039/c6cp04623a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viennet T, Viegas A, Kuepper A, Arens S, Gelev V, Petrov O, Grossmann TN, Heise H, Etzkorn M. Selective Protein Hyperpolarization in Cell Lysates Using Targeted Dynamic Nuclear Polarization. Angew Chem, Int Ed. 2016;55:10526. doi: 10.1002/anie.201603205. [DOI] [PubMed] [Google Scholar]
- 38.Voinov Ma, Good DB, Ward ME, Milikisiyants S, Marek A, Caporini Ma, Rosay M, Munro Ra, Ljumovic M, Brown LS, et al. Cysteine-Specific Labeling of Proteins with a Nitroxide Biradical for Dynamic Nuclear Polarization NMR. J Phys Chem B. 2015;119:10180–10190. doi: 10.1021/acs.jpcb.5b05230. [DOI] [PubMed] [Google Scholar]
- 39.Wang TY, Friedman LJ, Gelles J, Min W, Hoskins AA, Cornish VW. The Covalent Trimethoprim Chemical Tag Facilitates Single Molecule Imaging with Organic Fluorophores. Biophys J. 2014;106:272–278. doi: 10.1016/j.bpj.2013.11.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller LW, Cai Y, Sheetz MP, Cornish VW. In Vivo Protein Labeling with Trimethoprim Conjugates: A Flexible Chemical Tag. Nat Methods. 2005;2:255–257. doi: 10.1038/nmeth749. [DOI] [PubMed] [Google Scholar]
- 41.Calloway NT, Choob M, Sanz A, Sheetz MP, Miller LW, Cornish VW. Optimized Fluorescent Trimethoprim Derivatives for in Vivo Protein Labeling. Chem Bio Chem. 2007;8:767–774. doi: 10.1002/cbic.200600414. [DOI] [PubMed] [Google Scholar]
- 42.Roth B, Aig E, Rauckman S, Strelitz JZ, Phillips AP, Ferone R, Bushby SRM, Sigel CW. 2,4-Diamino-5-Benzylpyrimidines and Analogues as Antibacterial Agents. 5. 3′,5′-Dimethoxy-4′-substituted-Benzyl Analogues of Trimethoprim. J Med Chem. 1981;24:933–941. doi: 10.1021/jm00140a005. [DOI] [PubMed] [Google Scholar]
- 43.Matthews Da, Bolin JT, Burridge JM, Filman DJ, Volz KW, Kaufman BT, Beddell CR, Champness JN, Stammers DK, Kraut J. Refined Crystal Structures of Escherichia Coli and Chicken Liver Dihydrofolate Reductase Containing Bound Trimethoprim. J Biol Chem. 1985;260:381–391. [PubMed] [Google Scholar]
- 44.Nesmelov YE. Protein Structural Dynamics Revealed by Site-Directed Spin Labeling and Multifrequency EPR. Methods Mol Biol. 2014;1084:63–79. doi: 10.1007/978-1-62703-658-0_4. [DOI] [PubMed] [Google Scholar]
- 45.Cocco L, Blakley RL. Synthesis of a Spin-Labeled Analogue of Nicotinamide Adenine Dinucleotide Phosphate and Its Interaction with Dihydrofolate Reductase. Biochemistry. 1979;18:2414–2419. doi: 10.1021/bi00578a042. [DOI] [PubMed] [Google Scholar]
- 46.Porel M, Ottaviani MF, Jockusch S, Jayaraj N, Turro NJ, Ramamurthy V. Suppression of Spin–spin Coupling in Nitroxyl Biradicals by Supramolecular Host–guest Interactions. Chem Commun. 2010;46:7736–7738. doi: 10.1039/c0cc02587a. [DOI] [PubMed] [Google Scholar]
- 47.Ionita P, Wolowska J, Chechik V, Caragheorgheopol A. Ligand Dynamics in Spin-Labeled Au Nanoparticles. J Phys Chem C. 2007;111:16717–16723. [Google Scholar]
- 48.Corzilius B, Andreas LB, Smith AA, Ni QZ, Griffin RG. Paramagnet Induced Signal Quenching in MAS-DNP Experiments in Frozen Homogeneous Solutions. J Magn Reson. 2014;240:113–123. doi: 10.1016/j.jmr.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lange S, Linden AH, Akbey U, Franks WT, Loening NM, van Rossum BJ, Oschkinat H. The Effect of Biradical Concentration on the Performance of DNP-MAS-NMR. J Magn Reson. 2012;216:209–212. doi: 10.1016/j.jmr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Barnes AB, Corzilius B, Mak-Jurkauskas ML, Andreas LB, Bajaj VS, Matsuki Y, Belenky ML, Lugtenburg J, Sirigiri JR, Temkin RJ, et al. Resolution and Polarization Distribution in Cryogenic DNP/MAS Experiments. Phys Chem Chem Phys. 2010;12:5861–5867. doi: 10.1039/c003763j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopez del Amo JM, Schneider D, Loquet A, Lange A, Reif B. Cryogenic Solid State NMR Studies of Fibrils of the Alzheimer’s Disease Amyloid-β Peptide: Perspectives for DNP. J Biomol NMR. 2013;56:359–363. doi: 10.1007/s10858-013-9755-5. [DOI] [PubMed] [Google Scholar]
- 52.Thurber KR, Tycko R. Perturbation of Nuclear Spin Polarizations in Solid State NMR of Nitroxide-Doped Samples by Magic-Angle Spinning without Microwaves. J Chem Phys. 2014;140:184201–11. doi: 10.1063/1.4874341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moran U, Phillips R, Milo R. SnapShot: Key Numbers in Biology. Cell. 2010;141:1262. doi: 10.1016/j.cell.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 54.Ishihama Y, Schmidt T, Rappsilber J, Mann M, Hartl FU, Kerner MJ, Frishman D. Protein Abundance Profiling of the Escherichia Coli Cytosol. BMC Genomics. 2008;9:102. doi: 10.1186/1471-2164-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sauvée C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P. Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency. Angew Chem, Int Ed. 2013;52:10858–10861. doi: 10.1002/anie.201304657. [DOI] [PubMed] [Google Scholar]
- 56.Jing C, Cornish VW. Chemical Tags for Labeling Proteins Inside Living Cells. Acc Chem Res. 2011;44:784–792. doi: 10.1021/ar200099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Igumenova TI, Wand aJ, McDermott AE. Assignment of the Backbone Resonances for Microcrystalline Ubiquitin. J Am Chem Soc. 2004;126:5323–5331. doi: 10.1021/ja030546w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.