

Abstract
Objective
Dietary protein dilution (PD) has been associated with metabolic advantages such as improved glucose homeostasis and increased energy expenditure. This phenotype involves liver-induced release of FGF21 in response to amino acid insufficiency; however, it has remained unclear whether dietary dilution of specific amino acids (AAs) is also required. Circulating branched chain amino acids (BCAAs) are sensitive to protein intake, elevated in the serum of obese humans and mice and thought to promote insulin resistance. We tested whether replenishment of dietary BCAAs to an AA-diluted (AAD) diet is sufficient to reverse the glucoregulatory benefits of dietary PD.
Methods
We conducted AA profiling of serum from healthy humans and lean and high fat-fed or New Zealand obese (NZO) mice following dietary PD. We fed wildtype and NZO mice one of three amino acid defined diets: control, total AAD, or the same diet with complete levels of BCAAs (AAD + BCAA). We quantified serum AAs and characterized mice in terms of metabolic efficiency, body composition, glucose homeostasis, serum FGF21, and tissue markers of the integrated stress response (ISR) and mTORC1 signaling.
Results
Serum BCAAs, while elevated in serum from hyperphagic NZO, were consistently reduced by dietary PD in humans and murine models. Repletion of dietary BCAAs modestly attenuated insulin sensitivity and metabolic efficiency in wildtype mice but did not restore hyperglycemia in NZO mice. While hepatic markers of the ISR such as P-eIF2α and FGF21 were unabated by dietary BCAA repletion, hepatic and peripheral mTORC1 signaling were fully or partially restored, independent of changes in circulating glucose or insulin.
Conclusions
Repletion of BCAAs in dietary PD is sufficient to oppose changes in somatic mTORC1 signaling but does not reverse the hepatic ISR nor induce insulin resistance in type 2 diabetes during dietary PD.
Keywords: BCAA, Dietary protein, FGF21, mTORC1, Diabetes
Abbreviations: PD, protein dilution; AA, amino acid; BCAA, branched chain amino acid; AAD, amino acid diluted; mTORC1, mammalian target of rapamycin complex 1; ISR, integrated stress response; HF, high fat; NZB, New Zealand black; NZO, New Zealand obese; FGF21, fibroblast growth factor 21; T2D, type 2 diabetes
Graphical abstract
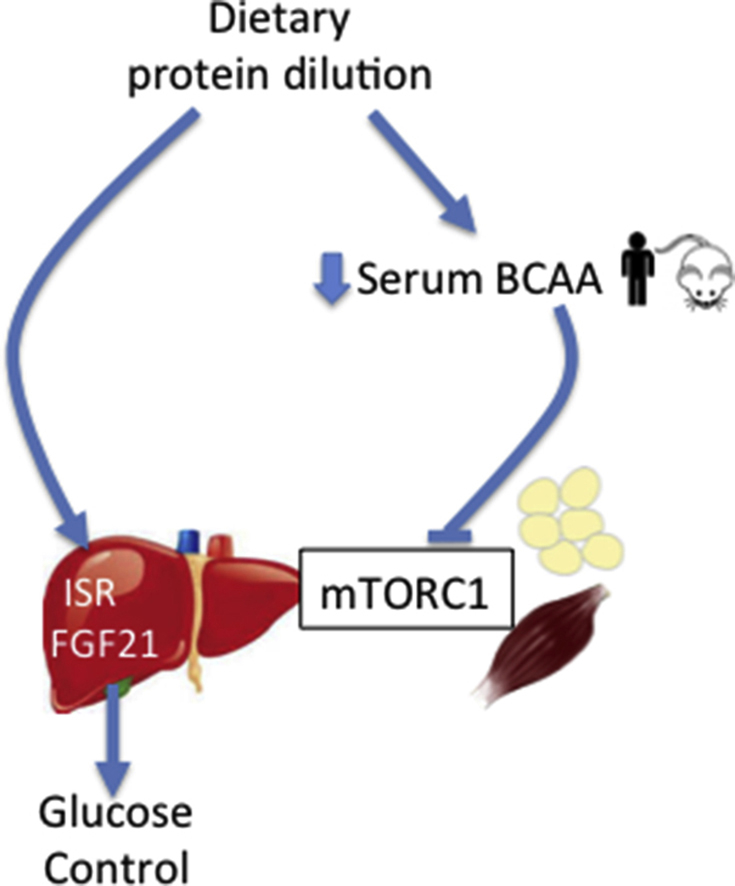
Highlights
-
•
Dietary PD reduces serum BCAAs in humans and mice.
-
•
Repletion of dietary BCAAs reverses somatic mTORC1 but not hepatic ISR signaling.
-
•
Glucose control during dietary PD is unperturbed by BCAA repletion in diabetic mice.
1. Introduction
Dietary protein dilution (PD), in which dietary protein intake is reduced and replaced by calories from carbohydrate and/or fat, has emerged as an influential environmental variable affecting aging and age-related disease such as type 2 diabetes (T2D). Indeed, several studies have demonstrated that DPD promotes longevity in rodents [1], [2], [3] and can retard age-related disease such as cancer [4] and T2D [5]. Furthermore, protein intake rates have been positively associated with increased risk for T2D and all-cause mortality in humans [6], [7].
While the precise mechanisms by which dietary PD can promote health-span are likely to be diverse, we recently demonstrated that DPD can retard the development of insulin resistance and T2D in mouse models, via the induction of the liver-derived hormone fibroblast growth factor 21 (FGF21) [5]. While FGF21 was originally discovered as a liver-enriched FGF [8], it was subsequently shown to be a non-mitogenic metabolic regulator with therapeutic potential for obesity-related-metabolic dysfunction [9], [10], [11], [12], [13], [14], even in humans [15], [16]. More recently, the physiological context of FGF21 has been increasingly explored [17], [18], [19], with several studies from diverse laboratories now having shown that it is an endocrine signal of dietary PD in rodents [5], [20], [21], [22], [23], [24], [25] and in humans [5], [21], [26], [27]. While the downstream mechanisms by which FGF21 improves metabolic health with dietary PD are yet to be fully resolved, it is required for increased energy expenditure, at least in mouse models [5], [21], [22], and heightened energy expenditure is thought to be a promising strategy to combat obesity-related-metabolic disease [28]. Furthermore, although a reduced total dietary amino acid (AA) supply was sufficient to mimic the effects of dietary PD [5], the precise AAs involved has remained unresolved. Evidence to date has suggested that candidates might include neutral AAs [29], a class including branched chain amino acids (BCAAs), which we show here to be consistently affected by dietary PD. Despite normalization of circulating BCAAs and mTORC1 signaling, we demonstrate that dietary BCAA-replacement to an AA-diluted (AAD) diet did not reverse FGF21 induction or restore hyperglycaemia in experimental T2D.
2. Methods
2.1. Human study design
Five healthy, lean male volunteers, age 25.6 ± 0.4 years, body weight 75.9 ± 5.3 kg (mean ± SEM), participated in the human diet study. Subjects consumed a controlled diet low in protein for 7 days. The low protein diet comprised 9 E% protein, 71 E% carbohydrates and 20 E% fat. The protein content was significantly lower in DPR compared to the habitual, normal (“N”) diet which amounted to 20 E% protein (Figure 1A). Diet composition and food intake information were described previously [5]. A meal test was conducted before and at d7 of the experiment, whereby subjects ate an isocaloric meal (i.e. 60 kJ/kg BM containing 15% or 9% protein for N vs. PD diet respectively) with blood samples taken before and at selected times after meal consumption.
Figure 1.

A specific reduction of fed serum BCAAs in humans following dietary PD. Serum urea (A), and BCAAs leucine (Leu), Isoleucine (Ile), and Valine (Val) (B–D) were measured in a meal test from serum collected from healthy men before (normal diet, N) or following 7 days of a protein-diluted (PD) diet regimen. Subjects ate an isocaloric meal (i.e. 60 kJ/kg BM) containing 15% (N group) or 9% protein (PD group). n = 5 per group. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 for significant effect of diet.
2.2. Mouse experiments
Male mice, aged 7 weeks upon arrival, were acclimated to the housing facility (12-12 h light–dark cycle, 22–24 °C) and fed their respective low fat experimental control diet for at least one week prior to experimentation. Mice used for experiments were C57Bl/6NCrl mice (#027, Charles River Laboratories), as well as New Zealand Black (#000993, NZB/BlNJ, Jackson Laboratories) and New Zealand obese (#002105, NZO/HlLtJ, Jackson Laboratories) strains. We tested the effects of dietary PD in the setting of murine obesity-driven insulin resistance and T2D using nutritional and polygenic experimental mouse models: C57Bl6/NCrl mice fed a high fat (HF) diet and the NZO mouse strain. While both models share obesity and insulin resistance in common, male NZO mice display hyperphagia and random fed hyperglycemia with reduced levels of physical activity and energy expenditure [30].
2.3. Studies involving dietary protein or dietary AA dilution
For studies of dietary PD in the setting of HF diet-induced obesity, diet compositions are outlined in Supplementary Table 1 and Figure 2A. Following acclimation, C57Bl/6NCrl mice were switched to a diet containing low protein or a HF diet containing either normal or low protein for 16 weeks [5]. Mice were killed in the random fed state by cervical dislocation, with subsequent trunk blood collection and serum preparation. For studies involving NZO vs. lean NZB mice, half of the mice from each genotype were switched to a low protein diet (Figure 2C) for 10 weeks followed by serum preparation as described above. The same strategy was used for AA-containing diets (Supplementary Table 2 and Figure 3A). For metabolic characterization using AA diets in wildtype mice, an intraperitoneal glucose tolerance test (IPGTT) and insulin tolerance test (ITT) was conducted after 6 weeks and 8 weeks of experimental diet exposure, respectively, and mice were killed in the random fed state for tissue collection after ∼14 weeks. For metabolic characterization using AA diet studies in NZO mice, mice were killed in the random fed state for tissue collection after ∼7 weeks. Feed efficiency was determined over the indicated time periods by dividing the delta body mass by the food energy intake over the same period. Food was weighed during cage changes, on a weekly basis, and bedding was carefully examined for fallen food pellets and accounted for. Tissue mTORC1/ISR signaling was assessed in separate cohorts of wildtype/NZO mice as described in Section 2.5.
Figure 2.

Serum BCAAs are elevated in diabetes and reduced by dietary PD in mice. Total serum AAs (A), BCAAs only (B) or individual BCAAs (C–E) were measured in serum collected from random fed C57Bl/6N mice fed either control diet (“C”) containing 20% caloric energy from protein or a protein-diluted (“PD”) diet containing 5% caloric energy from protein, diluted by added carbohydrate, with either 10% (“LF”) or 60% (“HF”) calories from fat. n = 6–8/group. Total serum AAs (F), BCAAs only (G) or individual BCAAs (H–J) were measured in serum from New Zealand black (“NZB”) or New Zealand obese (“NZO”) mice fed C or PD diets. n = 6–8/group. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 for significant effect of PD. ##p < 0.01, ###p < 0.001 for significant effect of mouse strain.
Figure 3.

Repletion of BCAA in the setting of dietary amino acid dilution attenuates improvements in metabolic health and restores mTORC1 signaling in wildtype mice. Serum BCAA (A), body mass accrual (B) and endpoint tissue weights (C) were measured in random fed mice fed control (“C”), amino acid diluted (“AAD”), or BCAA replete AAD (“AAD + BCAA”) diets. Food intake (D), used to calculate feed efficiency (E), was assessed in the same mice as in (A–C) over experiment days 57–70. An intraperitoneal glucose tolerance test (“IPGTT”, F) was conducted and insulin sensitivity assessed using insulin sensitivity index (G) and an insulin tolerance test (H). Endpoint (day ∼ 100) serum (I) and hepatic transcript (J) levels of FGF21 were measured. n = 6–8/group. Signaling downstream of mTORC1 was assessed by western blot in liver (K–L), abdominal white adipose tissue (epididymal “aWAT”), subcutaneous WAT (inguinal “scWAT”), brown adipose tissue (“BAT”) and gastrocnemius (“GC”) muscle (M–P). Western band intensities expressed relative to respective loading controls are shown below each blot, except 4EBP1 which was expressed using the indicated bands on the total blot as hypophosphorylated: total. n = 3–4/group. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. C. #p < 0.05 ##p < 0.01, ###p < 0.001 vs. AAD.
2.4. In vivo metabolic characterization of mice
Blood glucose measurements for all tests were determined with an Accu-Chek Performa glucometer (Roche). Glucose and insulin tolerance tests were conducted according to established guidelines [31] following a 5–6 h fast. For tolerance tests, glucose was given at 1 g/kg and insulin (Huminsulin Normal, Lilly) at 0.6 U/kg. A fasting ISI was calculated using the equation ISI = 1000/(blood glucose [mM] × plasma insulin [pM]) from 5- to 6-hour-fasted mice. Body composition was determined using a magnetic resonance imaging (MRI) body composition analyzer (Echo Medical Systems, Houston, USA).
2.5. Examination of in vivo tissue signaling pathways
Widtype or NZO mice were fed respective AA-containing diets for ∼3 weeks prior to experimentation (see Figures S3H and S4I). Mice were overnight fasted and refed the same diet with periodic blood collection into EDTA-coated tubes (Sarstedt) for plasma preparation and hormone measurement. After 1–2 weeks of recovery, mice were killed by cervical dislocation following overnight fasting and 45 min of re-feeding. Tissues were rapidly frozen in liquid nitrogen and stored at −80 °C until lysate preparation for western blot analysis.
2.6. Serum/plasma metabolite measurements
Kits were used to measure insulin (Alpco #80-INSMS-E01-AL or Mercodia #10-1247-01) and FGF21 (R&D Systems #MF2100). Serum amino acid profiling was conducted using LC–MS/MS as described previously in detail [29].
2.7. RNA/protein extraction
RNA was extracted from tissues using TRIzol Reagent (Life Technologies) and cDNA was synthesized using the Quantitect Reverse Transcription Kit (Qiagen). qPCR was conducted using Taqman master mix and Taqman primer-probe assays (Life Technologies). Fgf21 transcripts were detected using primer-probe Mm00840165_g1 and Ct values normalized to levels of Tbp (Mm01277042_m1). Tissue protein extraction and immunoblotting were performed using standard methods with antibodies against VCP (valosin containing protein, #ab11433, Abcam) and the following were from Cell Signaling Technologies: S51P-eIF2α (#9721), eIF2α (#9722), S473P-Akt (#9271), T389P-p70 S6K (#9234), p70 S6K (#9202), T37/46P-4EBP1 (#2855), and 4EBP1 (#9452). Western band intensities were quantified using Image Lab software (Bio-Rad Laboratories).
2.8. Statistics
For 1-factorial designs, 2-tailed t tests were used, with paired analysis when comparing the same subjects. For multiple comparisons, statistical analyses were performed using a 1-way or 2-way ANOVA, with repeated measures when applicable, followed by Bonferonni post-hoc tests when significant differences were calculated (Microsoft Excel and GraphPad Prism 5). The significance level was set to P < 0.05.
2.9. Study approval
Animal experiments were conducted according to local, national, and EU (Directive 2010/63) ethical guidelines (Regierungspräsidium Karlsruhe) and adhered to ARRIVE guidelines. For the human study, all subjects gave informed consent prior to their participation. The study was approved by the Copenhagen Ethics Committee (H-3-2012-129) and was executed in accordance with the code of ethics of the World Medical Association (Helsinki II declaration).
3. Results
3.1. Consumption of a protein-diluted diet generates a reduced serum BCAA signature in human and mouse
Although it has been shown that reduced protein intake can result in favorable effects on biomarkers of metabolic health such as plasma glucose and insulin levels in mice and humans [5], we initially asked whether this was also correlated with a serum amino acid signature. Interestingly, humans subjected to one week of dietary PD (Figure 1A) displayed reduced serum levels of urea (Figure 1A), indicative of reduced protein oxidation, as well as lower levels of lysine, threonine and the aromatic amino acids phenylalanine, and tyrosine (Figure 1B). Remarkable, however, were the effects of dietary PD to reduce the meal-related profile of all three BCAAs (Figure 1B–D and Figure S1B). We next assessed whether this diet-induced BCAA signature is conserved in experimental obesity and/or T2D. To this end, we used nutritional and polygenic experimental models: high fat (HF) diet-fed C57Bl6/N (Figure 2A), and hyperphagic New Zealand obese (NZO) mice, respectively [30].
High fat feeding did not affect total serum AAs (Figure 2A) but resulted in slightly lower levels of select amino acids, namely threonine, proline, and aspartate (Figure 2B). Feeding a PD diet, reduced some serum amino acids, often in a dietary fat-dependent manner (Figure 2B). Perhaps most noteworthy was that irrespective of dietary fat content, dilution of dietary protein resulted in lower levels of total serum BCAA (Figure 2B) as well as individual BCAA species (Figure 2C–E). In contrast with the HF-induced obesity model, hyperphagic NZO mice on a control diet had higher levels of total serum AAs (Figure 2F), BCAAs (Figure 2G), as well as the majority of individual amino acids (Figure S2C and D). In congruence with results observed in HF-feeding, dietary PD resulted in lower total and individual BCAAs in NZO mice (Figure 2G–J).
3.2. Repletion of BCAAs in the setting of dietary amino acid dilution attenuates improvements in metabolic health and restores mTORC1 signaling in wildtype mice
The consistent association of reduced serum BCAAs with dietary PD regimens in humans and mice prompted us to test whether reduced dietary BCAA supply per se is necessary for the improved metabolic profile under these dietary conditions. As such, we next utilized purified AA-containing diets (Supplemental Table 2) mirroring the AA supply that would come from casein, the source of protein in our previous experiments (see Figure 2, Figure S2 and [5]). We compared the metabolic effects of an amino acid diluted (AAD) diet to one with all amino acids diluted except BCAAs (denoted as AAD + BCAA) (Figure S3A and B). Like our PD diet feeding experiments, feeding an AAD diet reduced serum levels of BCAAs, while these remained unchanged in mice fed AAD + BCAA (Figure 3A and Figure S3C). Interestingly, the effect of AAD to curb weight gain was attenuated in AAD mice receiving a full complement of BCAAs (Figure 3B). Assessment by MRI of changes in body composition revealed that BCAA-replacement attenuated whole body loss of fat but not lean mass (Figure S3D and E), although this was not reflected in changes in select abdominal and subcutaneous adipose tissue masses (Figure 3C). Despite the finding that replacement of BCAAs attenuated the weight-neutral effect of AAD diet feeding (Figure S3F), caloric intake was equal in all diet groups during this period (Figure 3D). Feed efficiency, which reflects the ratio of body mass accrual to energy intake, was starkly reduced by AAD diet feeding, an effect which was lost when BCAAs were replaced (Figure 3E).
Reversal of metabolic inefficiency by BCAA-replacement, together with previous reports that metabolic inefficiency, without extreme weight loss, is often coupled to metabolic health [5], [28], prompted us to test whether differences would arise between diet groups in glucose homeostasis. Intraperitoneal glucose tolerance was improved following feeding of an AAD diet, an effect attenuated when BCAAs were replaced (Figure 3F). This was likely attributed to an effect of BCAA-replacement to reverse the hypo-insulinemic (Figure S3G) and thus insulin sensitizing effect of AAD diet feeding (Figure 3G) and could be corroborated by insulin tolerance testing (Figure 3H).
We and others have shown that major features of metabolic health following dietary PD require the hepatic hormone FGF21 [5], [21], the expression and secretion of which is induced as a part of a liver integrated stress response (ISR). Notably, although AAD diet re-feeding (Figure S3H) induced markers of the hepatic ISR, namely phosphorylated eIF2α (Figure S3I) as well as serum FGF21 (Figure 3I) and its liver transcript (Figure 3J), these parameters remained unabated by BCAA repletion.
Along with arginine and glutamine, BCAAs are known to promote activation of mTORC1 [32], [33]. Thus, we next assessed whether components of this signaling pathway were perhaps modulated by dietary BCAA supply. Western blot analysis of liver, a major transit site for ingested AAs, illustrated significantly reduced mTORC1 activity, as suggested from hypo-phosphorylation and faster migration of both p70 S6 kinase and 4EBP1 in response to total AAD (Figure 3K–L), while this was completely reversed by dietary BCAA repletion. Levels of hepatic AKT phosphorylation were unchanged by dietary AA manipulation (Figure 3J), consistent with evidence that reduced AAs signal to mTORC1 downstream of PI3-K/AKT/TSC [33]. Interestingly, similar changes in markers of mTORC1 activity were observed in peripheral tissues, namely abdominal and subcutaneous WAT, BAT, and gastrocnemius skeletal muscle (Figure 3M–P). We assessed hepatic and peripheral signaling after 45 min of re-feeding, a time point when diets had no effect on circulating glucose, insulin or FGF21 (Figure S3K–M), suggesting that circulating diet-derived BCAAs per se may be potent determinants of mTORC1 activity in vivo.
3.3. Repletion of BCAA to AAD diet feeding restores mTORC1 signaling in diabetic mice but does not accelerate diabetes onset
We previously showed that dietary PD can effectively attenuate diabetes onset and reverse insulin resistance in mice [5]. Our experiments using wildtype mice showed that markers of metabolic health following dietary AAD can be attenuated or reversed by restoration of BCAA supply (Figure 3). This prompted us to test whether dietary, and thus serum, BCAA repletion (Figure 4A and Figure S4A, B) would affect the salutary metabolic health benefits of dietary AAD in NZO mice. Although body mass accrual was not significantly affected by dietary interventions (Figure 4B), some tissue mass effects of dietary AAD were prevented by repletion of dietary BCAAs, namely subcutaneous white and brown adipose tissue, and skeletal muscle (Figure S4C). Feed efficiency (Figure 4C), reflecting the ratio of delta body mass to energy intake (Figure S4D and E) was unaffected by changes in dietary amino acid supply. Moreover, dietary AAD, irrespective of BCAA repletion, prevented the onset of random fed (Figure 4D) and fasting (Figure S4F) hyperglycemia in NZO mice, although neither dietary intervention significantly affected fasting insulin levels or insulin sensitivity (Figure S4G and H). In congruence with observed effects on glucose homeostasis, dietary BCAA repletion did not curb the induction of the hepatic ISR (Figure 4I and J) and Fgf21 mRNA (Figure 4E). Conversely, while dietary AA supply did not influence energy intake, circulating glucose, insulin or FGF21 levels after 45 min re-feeding (Figure 4K–N), hepatic and peripheral signaling markers of mTORC1 activity were dramatically reduced by total dietary AAD, and fully or partially restored upon BCAA repletion (Figure 4F–K). Hence, while not required for the hepatic ISR-triggered salutary metabolic health profile pursuant to dietary AAD in the diabetic setting, repletion of dietary BCAA supply is sufficient to substantially rescue somatic signaling events downstream of mTORC1.
Figure 4.

Dietary repletion of BCAAs opposes somatic mTORC1 signaling, but not improvements in glucose homeostasis in NZO mice. Endpoint random fed serum BCAAs (A), body mass accrual (B), feed efficiency (C), random fed blood glucose (D) and hepatic FGF21 transcript (E) were assessed in New Zealand obese (NZO) mice fed control (“C”), amino acid diluted (“AAD”), or AAD with BCAA replete (“AAD + BCAA”) diets. n = 6/group. Signaling downstream of mTORC1 was assessed in liver (F and G), white adipose tissue from abdominal (epididymal “aWAT”) and subcutaneous (inguinal “scWAT”) depots, brown adipose tissue (“BAT”) and gastrocnemius (“GC”) muscle (H–K) were assessed by western blot in a separate cohort of mice adapted to diets for 3 weeks, fasted overnight and re-fed for 45 min. Western band intensities expressed relative to respective loading controls are shown below each blot, except 4EBP1 which was expressed using the indicated bands on the total blot as hypophosphorylated: total. n = 3–4. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. C. #p < 0.05 vs. AAD.
4. Discussion
In this study, we recapitulate existing findings that serum levels of BCAAs are clearly reduced following dietary PD in both healthy humans and rodents [3], [27], [34] and show that this effect holds true in the setting of experimental insulin resistance and T2D. We and others have shown that a PD diet is associated with metabolic fitness, improved insulin action and glucose metabolism, and higher levels of FGF21 in both rodents and humans [3], [5], [21]; thus, we hypothesized that the reduced BCAA may be causally related to this metabolic response. Although experimental evidence to date has interrogated the metabolic consequences of adding or subtracting specifically BCAAs from an otherwise complete AA-based diet [27], [35], [36], we have aimed to better understand the concept of general dietary PD, and to address whether BCAA repletion is sufficient to attenuate metabolic health. We believe that this is an important question as, from a dietary perspective, restriction of total protein intake is more feasible than dietary manipulation of selected amino acids. From this derives a need to then understand which particular AA(s) are necessary to confer the effects of PD. While in our experimental design we affect total amino-nitrogen supply by BCAA add-back (see Figure 3A), we believe that this is a rational approach as i) we have previously demonstrated that the in vitro hepatocyte response to AAD is insensitive to total amino-nitrogen supply [5] and ii) using isonitrogenous diets would require simultaneous changes to both BCAAs and other AAs and thus confound the interpretation of results.
Serum metabolite screening of serum from 74 diabetes-free obese and 67 lean individuals identified a metabolite signature that included dysregulated levels of BCAAs and their acylcarnitine metabolites [35]. While the etiology behind this serum signature remains elusive, we show that insulin resistance (induced by HF-feeding) per se is not sufficient to elevate BCAAs on a standard protein diet. Moreover, serum BCAAs were elevated selectively in NZO mice but not in HF-fed mice, despite comparable degrees of insulin sensitivity [5]. It is noteworthy that abnormalities in physical activity, energy expenditure, glycemia, and hyperphagia are less severe or absent in HF-fed wildtype mice in comparison with the NZO strain [30]. Nonetheless, it seems more likely that protein hyperphagia and/or attenuated BCAA metabolism, and not hyperglycemia per se, elevates serum BCAAs [35], [36], [37]. In support of this, higher serum BCAAs were apparent in hyperphagic rats and mice [36], [37], irrespective of diabetes incidence. Indeed, serum BCAAs closely relate to protein intake [27] and higher levels of serum BCAAs as well as aromatic AAs were predictive of future diabetes incidence [38], even after adjustment for serum lipids and HOMA-IR. Notably, we also observed lower BCAAs and aromatic AAs in humans during dietary PD (Figure 1 and Figure S1), and since dietary protein intake correlates with T2D incidence in humans [6], this may imply that dietary protein intake links the AA profile signature with future diabetes risk.
A noteworthy result was the lack of effect of dietary BCAA add-back on glycemia in obese/diabetic NZO mice, despite partial effects in C57Bl/6 mice. Interestingly, BCAA metabolism in white adipose tissue correlates with metabolic health [39], [40], and BCAA supplementation during dietary PD in piglets affects adiposity [41]. Differential effects on adipose tissue mass in C57Bl/6 vs. NZO mice during BCAA repletion (Figure 3, Figure 4) may explain at least part of the paralleled discordant effects on glucose homeostasis between the two strains. Nonetheless, although AAs such as leucine can act as a satiety signal in the brain, we observed that BCAA repletion did not curb food intake in neither wildtype nor NZO mice, an observation consistent with experiments done in rats [42]. On the other hand, BCAA supply may more closely relate with muscle function, as BCAA repletion restored GC muscle mass in both strains (Figure 3, Figure 4), and restriction of dietary BCAAs in an isonitrogenous dietary setting reduced skeletal muscle insulin resistance in Zucker fatty rats [36].
It is interesting that total dietary AA dilution [5], [21], [22] as well as complete dietary deprivation of a single BCAA [43], [44] is sufficient to upregulate hepatic FGF21. We and others have shown that this response during dietary PD involves activation of the ISR within the liver [5], [22], possibly resulting from intracellular accumulation of uncharged tRNAs, and GCN2-dependent phosphorylation of eIF2α [45]. Conversely selective dietary BCAA dilution did not induce [27] and BCAA repletion did not inhibit (Figure 3, Figure 4) FGF21 induction, suggesting that BCAAs are likely not a limiting nutrient in these dietary settings. Collectively, these results illustrate that dietary dilution of BCAAs is neither required, nor sufficient to induce an FGF21 response. This is in agreement with studies showing that repletion of non-essential AAs (such as glutamine), but not leucine alone, can curb FGF21 induction when general protein supply is limited [5], [46], [47]. Nonetheless, changes in dietary BCAA supply affect metabolic efficiency and glucose excursion in lean mice (Figure 3 and [27]), suggesting that an FGF21-independent mechanism is involved that warrants further investigation.
In contrast to the hepatic ISR and FGF21 levels, hepatic and peripheral mTORC1 signaling was strikingly sensitive to dietary BCAA repletion in both wildtype and NZO mice. While it is known that hormones such as insulin [48] and FGF21 [49], [50] can affect cellular mTORC1 activity, it appears that dietary BCAAs are a predominant mode of regulation in vivo, as we could observe a clear congruence between plasma BCAA and tissue mTORC1 activity, whereas plasma levels of insulin and FGF21 did not align (Figure 3, Figure 4 and Figures S3, S4). Even so, in our experimental setting we concede that the divergence between hormonal changes and tissue mTORC1 signaling may be related to relatively quicker regulation by metabolites versus these peptide hormones. While mTORC1 activation is associated with feedback inhibition of insulin signaling and insulin resistance [35], heightened BCAA supply and consequent reactivation of mTORC1 signaling did not reverse glucose control during dietary AAD in NZO mice. Dietary BCAA repletion was sufficient however to restore skeletal muscle but not liver mass in wildtype and NZO mice, suggesting that persistent ISR signaling in the liver likely overrides mTORC1-mediated effects on protein translation, cell growth, and insulin resistance.
In summary, dietary PD in humans and mice results in a reduced serum BCAA profile that, while not required for improved metabolism in experimental diabetes, has implications for somatic mTORC1 activity.
Acknowledgments
We gratefully acknowledge Anja Pfenninger (Sanofi-Aventis), Jonas Schumacher (DKFZ), Andrea Takacs (IDC), Daniela Strzoda (DKFZ), and Dr. Anja Krones-Herzig (IDC) for experimental and/or administrative support. AM was supported by fellowships from the Canadian Institutes of Health Research (CIHR) and the DKFZ. JSKC was supported by a graduate student fellowship from the Hoffmann-Berling International Graduate School. The human diet study was supported by the grant “Physical Activity and Nutrition for Improvement of Health” from the University of Copenhagen Excellence Program for Interdisciplinary Research (2016) to BK. KAS was supported by a postdoctoral research grant from the Danish Council for Independent Research Medical Sciences (grant number 4092-00309). These studies were supported by the Helmholtz Programs “ICEMED” and “Metabolic Dysfunction” as well as the Deutsche Forschungsgemeinschaft (He3260/8-1) to SH and by a project grant from the EFSD/Lilly European Diabetes Research Programme to AJR.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molmet.2017.06.009.
Contributor Information
Adriano Maida, Email: adriano.maida@helmholtz-muenchen.de.
Adam J. Rose, Email: a.rose@dkfz.de.
Conflict of interest statement
D. Schmoll is an employee of Sanofi-Aventis Deutschland, a pharmaceutical company. All other authors declare no conflict of interest.
Appendix A. Supplementary data
The following are the supplementary data related to this article:

Supplementary Figure 1Area under the curve (AUC) for serum AAs following a meal test reveals a BCAA-related profile following dietary PD. Healthy men were switched from their normal diet (“N”) to PD diet (A) for 7 days. Serum AAs were measured before and over 250 min following a meal test. Area under the 250 min AA concentration curve was calculated for each subject using the x-axis as the baseline (B). Subjects ate an isocaloric meal (i.e. 60 kJ/kg BM) containing 15% (N group) or 9% protein (PD group). n = 5 per group. Data are mean ± SEM. *p < 0.05, **p < 0.01, for significant effect of diet.

Supplementary Figure 2Dietary PD influences serum AA profiles in diet-induced or polygenic obesity/diabetes. Wildtype C57Bl/6N mice were fed either control diet (“C”) or a protein-diluted (“PD”) diet, diluted by added carbohydrate, with either low fat (“LF”) or high fat (“HF”) (A). After 16 weeks on diets, individual AAs were measured in serum from random fed mice (B). n = 6–8/group. New Zealand black (“NZB”) or New Zealand obese (“NZO”) mice were fed C or PD diets for 10 weeks (C) followed by serum AA assessment in the random fed state (D). n = 6–8/group. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 for significant effect of PD. #p < 0.05 ##p < 0.01, ###p < 0.001 for significant effect of dietary fat/strain.

Supplementary Figure 3Metabolic and signaling parameters in wildtype mice following repletion of BCAA in the setting of dietary amino acid dilution. Wildtype mice fed control (“C”), amino acid diluted (“AAD”), or AAD with BCAA replete (“AAD + BCAA”) diets (A) underwent metabolic characterization (B), which included the indicated tolerance tests as well as feed efficiency (“FE”) assessment. Random fed serum amino acids (C) and MRI quantification of lean mass (D) and fat mass (E) were assessed in the same mice as in Figure 3A–J. Delta body mass for days 57–70 (F) was used together with food intake (see Figure 3D) to calculate feed efficiency in Figure 3E. Fasting plasma insulin (G), together with corresponding fasting glucose (see Figure 3F) was used to calculate insulin sensitivity index (see Figure 3G). n = 6–8/group. The hormonal and signaling response to these diets was studied in a separate cohort of re-fed mice (H). Phosphorylation eIF2α (I) and AKT (J) were assessed in liver tissue by western and band intensities expressed relative to respective loading controls are shown below each blot. Levels of blood glucose (K), plasma insulin (L) and FGF21 (M) were quantified. n = 3–4/group. Data are mean ± SEM. p < 0.05, **p < 0.01, ***p < 0.001 vs. C. #p < 0.05 vs. AAD.

Supplementary Figure 4Metabolic and signaling parameters in NZO mice following repletion of BCAA in the setting of dietary amino acid dilution. NZO mice fed control (“C”), amino acid diluted (“AAD”), or AAD with BCAA replete (“AAD + BCAA”) diets underwent metabolic characterization (A), which included feed efficiency (“FE”) assessment. Random fed serum amino acids (B) and endpoints tissue weights (C) were assessed in the same NZO mice as in Figure 4A–E. Food intake (D) together with changes in body mass (E) were used to determine feed efficiency in Figure 4C. Fasted blood glucose (F) and plasma insulin (G) were measured on day 45 and used to calculate insulin sensitivity index (H). n = 6/group. The hormonal and signaling response to these diets was studied in a separate cohort of re-fed NZO mice (I). Liver eIF2α phosphorylation (J) was determined and western band intensities expressed relative to respective loading controls are shown below each blot. Levels of energy intake (K), blood glucose (L), plasma insulin (M) and FGF21 (N) were quantified. n = 3–4/group. Data are mean ± SEM. p < 0.05, **p < 0.01, ***p < 0.001 vs. C. #p < 0.05 vs. AAD.
References
- 1.ROSS M.H. Length of life and nutrition in the rat. The Journal of Nutrition. 1961;75:197–210. doi: 10.1093/jn/75.2.197. [DOI] [PubMed] [Google Scholar]
- 2.Miller D.S., Payne P.R. Longevity and protein intake. Experimental Gerontology. 1968;3(3):231–234. doi: 10.1016/0531-5565(68)90006-5. [DOI] [PubMed] [Google Scholar]
- 3.Solon-Biet S.M., McMahon A.C., Ballard J.W.O., Ruohonen K., Wu L.E., Cogger V.C. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metabolism. 2014;19(3):418–430. doi: 10.1016/j.cmet.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine M.E., Suarez J.A., Brandhorst S., Balasubramanian P., Cheng C.-W., Madia F. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metabolism. 2014;19(3):407–417. doi: 10.1016/j.cmet.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maida A., Zota A., Sjøberg K.A., Schumacher J., Sijmonsma T.P., Pfenninger A. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. The Journal of Clinical Investigation. 2016;126(9):3263–3278. doi: 10.1172/JCI85946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Nielen M., Feskens E.J.M., Mensink M., Sluijs I., Molina E., Amiano P. Dietary protein intake and incidence of type 2 diabetes in Europe: the EPIC-INTERACT case-cohort study. Diabetes Care. 2014 doi: 10.2337/dc13-2627. [DOI] [PubMed] [Google Scholar]
- 7.Song M., Fung T.T., Hu F.B., Willett W.C., Longo V.D., Chan A.T. Association of animal and plant protein intake with all-cause and cause-specific mortality. JAMA Internal Medicine. 2016;176(10):1453–1463. doi: 10.1001/jamainternmed.2016.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura T., Nakatake Y., Konishi M., Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochimica Et Biophysica Acta. 2000;1492(1):203–206. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 9.Kharitonenkov A., Shiyanova T.L., Koester A., Ford A.M., Micanovic R., Galbreath E.J. FGF-21 as a novel metabolic regulator. Journal of Clinical Investigation. 2005;115(6):1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metabolism. 2007;5(6):415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metabolism. 2007;5(6):426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Hotta Y., Nakamura H., Konishi M., Murata Y., Takagi H., Matsumura S. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology. 2009;150(10):4625–4633. doi: 10.1210/en.2009-0119. [DOI] [PubMed] [Google Scholar]
- 13.Coskun T., Bina H.A., Schneider M.A., Dunbar J.D., Hu C.C., Chen Y. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149(12):6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 14.Xu J., Lloyd D.J., Hale C., Stanislaus S., Chen M., Sivits G. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58(1):250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Talukdar S., Zhou Y., Li D., Rossulek M., Dong J., Somayaji V. A long-acting FGF21 molecule, PF-05231023, decreases body weight and improves lipid profile in non-human primates and type 2 diabetic subjects. Cell Metabolism. 2016;23(3):427–440. doi: 10.1016/j.cmet.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Gaich G., Chien J.Y., Fu H., Glass L.C., Deeg M.A., Holland W.L. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metabolism. 2013;18(3):333–340. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Kharitonenkov A., DiMarchi R. Fibroblast growth factor 21 night watch: advances and uncertainties in the field. Journal of Internal Medicine. 2017;281(3):233–246. doi: 10.1111/joim.12580. [DOI] [PubMed] [Google Scholar]
- 18.Fisher F.M., Maratos-Flier E. Understanding the physiology of FGF21. Annual Review of Physiology. 2016;78(1):223–241. doi: 10.1146/annurev-physiol-021115-105339. [DOI] [PubMed] [Google Scholar]
- 19.Potthoff M.J., Kliewer S.A., Mangelsdorf D.J. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes & Development. 2012;26(4):312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solon-Biet S.M., Cogger V.C., Pulpitel T., Heblinski M., Wahl D., McMahon A.C. Defining the nutritional and metabolic context of FGF21 using the geometric framework. Cell Metabolism. 2016:1–12. doi: 10.1016/j.cmet.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Laeger T., Henagan T.M., lbarado D.C., Redman L.M., Bray G.A., Noland R.C. FGF21 is an endocrine signal of protein restriction. The Journal of Clinical Investigation. 2014 doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laeger T., Albarado D.C., Burke S.J., Trosclair L., Hedgepeth J.W., Berthoud H.-R. Metabolic responses to dietary protein restriction require an increase in FGF21 that is delayed by the absence of GCN2. Cell Reports. 2016;16(3):707–716. doi: 10.1016/j.celrep.2016.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Marti A., Garcia-Guasch M., Tresserra-Rimbau A., Carrilho-Do-Rosário A., Estruch R., Salas-Salvadó J. A low-protein diet induces body weight loss and browning of subcutaneous white adipose tissue through enhanced expression of hepatic fibroblast growth factor 21 (FGF21) Molecular Nutrition & Food Research. 2017;10:1600725. doi: 10.1002/mnfr.201600725. [DOI] [PubMed] [Google Scholar]
- 24.Ozaki Y., Saito K., Nakazawa K., Konishi M., Itoh N., Hakuno F. Rapid increase in fibroblast growth factor 21 in protein malnutrition and its impact on growth and lipid metabolism. The British Journal of Nutrition. 2015;114(9):1410–1418. doi: 10.1017/S0007114515002846. [DOI] [PubMed] [Google Scholar]
- 25.Pezeshki A., Zapata R.C., Singh A., Yee N.J., Chelikani P.K. Low protein diets produce divergent effects on energy balance. Scientific Reports. 2016;6(1):25145. doi: 10.1038/srep25145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gosby A.K., Lau N.S., Tam C.S., Iglesias M.A., Morrison C.D., Caterson I.D. Raised FGF-21 and triglycerides accompany increased energy intake driven by protein leverage in lean, healthy individuals: a randomised trial. PloS One. 2016;11(8) doi: 10.1371/journal.pone.0161003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fontana L., Cummings N.E., Arriola Apelo S.I., Neuman J.C., Kasza I., Schmidt B.A. Decreased consumption of branched-chain amino acids improves metabolic health. Cell Reports. 2016;16(2):520–530. doi: 10.1016/j.celrep.2016.05.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tseng Y.-H., Cypess A.M., Kahn C.R. Cellular bioenergetics as a target for obesity therapy. Nature Reviews Drug Discovery. 2010;9(6):465–482. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y., Rose A.J., Sijmonsma T.P., Bröer A., Pfenninger A., Herzig S. Mice lacking neutral amino acid transporter B(0)AT1 (Slc6a19) have elevated levels of FGF21 and GLP-1 and improved glycaemic control. Molecular Metabolism. 2015;4(5):406–417. doi: 10.1016/j.molmet.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanasaki K., Koya D. Biology of obesity: lessons from animal models of obesity. Journal of Biomedicine & Biotechnology. 2011;2011(4):197636–197711. doi: 10.1155/2011/197636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayala J.E., Samuel V.T., Morton G.J., Obici S., Croniger C.M., Shulman G.I. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Disease Models & Mechanisms. 2010;3(9–10):525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimobayashi M., Hall M.N. Multiple amino acid sensing inputs to mTORC1. Cell Research. 2016;26(1):7–20. doi: 10.1038/cr.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bar-Peled L., Sabatini D.M. Regulation of mTORC1 by amino acids. Trends in Cell Biology. 2014;24(7):400–406. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fafournoux P., Remesy C., Demigne C. Fluxes and membrane transport of amino acids in rat liver under different protein diets. The American Journal of Physiology. 1990;259(5 Pt 1):E614–E625. doi: 10.1152/ajpendo.1990.259.5.E614. [DOI] [PubMed] [Google Scholar]
- 35.Newgard C.B., An J., Bain J.R., Muehlbauer M.J., Stevens R.D., Lien L.F. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metabolism. 2009;9(4):311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White P.J., Lapworth A.L., An J., Wang L., McGarrah R.W., Stevens R.D. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Molecular Metabolism. 2016;5(7):538–551. doi: 10.1016/j.molmet.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giesbertz P., Padberg I., Rein D., Ecker J., Höfle A.S., Spanier B. Metabolite profiling in plasma and tissues of ob/ob and db/db mice identifies novel markers of obesity and type 2 diabetes. Diabetologia. 2015;58(9):2133–2143. doi: 10.1007/s00125-015-3656-y. [DOI] [PubMed] [Google Scholar]
- 38.Wang T.J., Larson M.G., Vasan R.S., Cheng S., Rhee E.P., McCabe E. Metabolite profiles and the risk of developing diabetes. Nature Medicine. 2011;17(4):448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lackey D.E., Lynch C.J., Olson K.C., Mostaedi R., Ali M., Smith W.H. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. American Journal of Physiology Endocrinology and Metabolism. 2013;304(11):E1175–E1187. doi: 10.1152/ajpendo.00630.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reis F.C.G., Branquinho J.L.O., Brandão B.B., Guerra B.A., Silva I.D., Frontini A. Fat-specific Dicer deficiency accelerates aging and mitigates several effects of dietary restriction in mice. Aging. 2016;8(6):1201–1222. doi: 10.18632/aging.100970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y., Wei H., Li F., Duan Y., Guo Q., Yin Y. Effects of low-protein diets supplemented with branched-chain amino acid on lipid metabolism in white adipose tissue of piglets. Journal of Agricultural and Food Chemistry. 2017;65(13):2839–2848. doi: 10.1021/acs.jafc.7b00488. [DOI] [PubMed] [Google Scholar]
- 42.Laeger T., Reed S.D., Henagan T.M., Fernandez D.H., Taghavi M., Addington A. Leucine acts in the brain to suppress food intake but does not function as a physiological signal of low dietary protein. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2014;307(3):R310–R320. doi: 10.1152/ajpregu.00116.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wanders D., Stone K.P., Dille K., Simon J., Pierse A., Gettys T.W. Metabolic responses to dietary leucine restriction involve remodeling of adipose tissue and enhanced hepatic insulin signaling. BioFactors (Oxford, England) 2015;41(6):391–402. doi: 10.1002/biof.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Sousa-Coelho A.L., Relat J., Hondares E., Perez-Marti A., Ribas F., Villarroya F. FGF21 mediates the lipid metabolism response to amino acid starvation. The Journal of Lipid Research. 2013;54(7):1786–1797. doi: 10.1194/jlr.M033415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anthony T.G., Morrison C.D., Gettys T.W. Remodeling of lipid metabolism by dietary restriction of essential amino acids. Diabetes. 2013;62(8):2635–2644. doi: 10.2337/db12-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cornu M., Oppliger W., Albert V., Robitaille A.M., Trapani F., Quagliata L. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proceedings of the National Academy of Sciences. 2014 doi: 10.1073/pnas.1412047111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris L.-A.L.S., Smith G.I., Patterson B.W., Ramaswamy R.S., Okunade A., Kelly S.C. Alterations in 3-hydroxyisobutyrate and FGF21 metabolism are associated with protein ingestion-induced insulin resistance. Diabetes. 2017 doi: 10.2337/db16-1475. db161475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menon S., Dibble C.C., Talbott G., Hoxhaj G., Valvezan A.J., Takahashi H. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156(4):771–785. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minard A.Y., Tan S.-X., Yang P., Fazakerley D.J., Domanova W., Parker B.L. mTORC1 is a major regulatory node in the FGF21 signaling network in adipocytes. Cell Reports. 2016;17(1):29–36. doi: 10.1016/j.celrep.2016.08.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moyers J.S., Shiyanova T.L., Mehrbod F., Dunbar J.D., Noblitt T.W., Otto K.A. Molecular determinants of FGF-21 activity-synergy and cross-talk with PPARgamma signaling. Journal of Cellular Physiology. 2007;210(1):1–6. doi: 10.1002/jcp.20847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.