

Abstract
Purpose
Hypoxia is an important characteristic of the solid tumor microenvironment and constitutes a barrier for effective radiotherapy. Here, we studied the effects of darinaparsin (an arsenic cytotoxin) on survival and radiosensitivity of tumor cells in vitro under normoxia and hypoxia and in vivo using xenograft models, compared to effects on normal tissues.
Experimental Design
The cytotoxicity and radiosensitization of darinaparsin were first tested in vitro in a variety of solid tumor cell lines under both normoxia and hypoxia and compared with arsenic trioxide (ATO, an arsenical with reported cytotoxic and radiosensitizing activities on tumor cells). The effects were then tested in mouse models of xenograft tumors derived from tumor cell lines and clinical tumor specimens. The potential mechanisms of darinaparsin effects, including reactive oxygen species (ROS) generation, cellular damage, and changes in global gene expression, were also investigated.
Results
In comparison with ATO, darinaparsin had significantly higher in vitro cytotoxic and radio-sensitizing activities against solid tumor cells under both normoxia and hypoxia. In vivo experiments confirmed these activities at doses that had no systemic toxicities. Importantly, darinaparsin did not radiosensitize normal bone marrow and actually radioprotected normal intestinal crypts. The darinaparsin-mediated antitumor effects under hypoxia were not dependent on ROS generation and oxidative damage, but were associated with inhibition of oncogene (RAS and MYC)-dependent gene expression.
Conclusion
Darinaparsin has significant and preferential cytotoxic and radiosensitizing effects on solid tumors as compared with normal cells. Darinaparsin may therefore increase the therapeutic index of radiation therapy and has near term translational potential.
Introduction
Solid tumors are often relatively chemo- and radiation resistant, due in part to the presence of hypoxia (1). Inorganic arsenic trioxide (ATO) is an established chemotherapy drug for treatment of acute promyelocytic leukemia (APL) that inhibits the APL-specific oncoprotein, AML-RXRa. We and others have previously reported that ATO has cytotoxic and radiosensitizing effects in solid tumor models, at least partially attributable to the induction of oxidative stress (2-5). Unfortunately, ATO was less efficacious in clinical trials of solid tumors than leukemia, and it has dose-related risks of cardiac and hepatic toxicity (6). Recently, a number of organic arsenics with anticancer activities have been developed (7-11). Their mechanisms of action are different from those of ATO, and include effects on tumor angiogenesis (11), metabolism (12), and cell signaling (NF-κB; ref. 9). As a result, unlike ATO, these organic arsenicals do not depend on AML-RARa inhibition for their antileukemic activities (8, 13, 14), and may be useful for treatment of other cancers including solid tumors.
Darinaparsin is an organic arsenical with potent activity against leukemia and multiple myeloma, with approximately 3 to 10 times greater cytotoxicity than ATO in vitro (15). Remarkably, much higher doses of darinaparsin can be safely used in animal models with significantly less systemic toxicity than ATO (16). These favorable toxicity profiles have been confirmed in phases I and II clinical trials (17, 18). Like other organic arsenics, darinaparsin does not induce differentiation of APL cells and does not cause PML-RARα degradation and rearrangement of PML nuclear bodies (19). Neither myeloma nor APL ATO-resistant cell lines are resistant to darinaparsin (20). In addition, darinaparsin has been reported to have in vitro cytotoxic activity against a variety of solid tumor cell lines (19). These findings suggest that darinaparsin may have different mechanisms of action than ATO and may be an effective antisolid tumor agent. Because hypoxia is an important determinant of radioresistance, we assessed the in vitro activities of darinaparsin under both hypoxia and normoxia, alone and in combination with radiation. Further experiments were conducted to test these darinaparsin activities in vivo on xenograft tumors and normal tissues, and to elucidate the underlying mechanisms of action.
Materials and Methods
Cell culture and reagents
All cell lines were obtained from American Type Culture Collection (ATCC) unless otherwise specified. All cells were maintained in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum. In the hypoxic experiments, the cells were placed in hypoxia chambers (0.5% O2) overnight before drug or irradiation treatment. Darinaparsin was provided by ZIOPHARM Oncology. All other reagents were obtained from Sigma-Aldrich unless otherwise specified. A 137Cs y-ray irradiator (dose rate, 250 cGy/ min) and an X-ray irradiator (dose rate, 126 cGy/min) were used to irradiate cells and animal tumors, respectively.
Animal tumor models
Subconfluent hormone-independent (HI)-LAPC-4 (3 × 106 per mouse) and PANC-1 (2.5 × 106 per mouse) cells were suspended in Matrigel (BD Bioscience) and injected subcutaneously on the lower back of immunodeficient (nude) mice. Darinaparsin treatment was started when xenograft tumors reached approximately 100 to 200 mm3 in volume, as calculated by π/6 × length × width2. The tumor volume was normalized with the starting volume (T0) and fitted by Exponential Growth modeling (Prism), which determined the P value and tumor volume doubling time with 95% CI.
The in vivo model of primary prostate cancer using tissue slice grafts (TSG) has been previously described (21). The TSGs maintain the normal and cancer tissue histology as evidenced by immunohistochemical staining and respond to androgen ablation. Briefly, a prostate cancer specimen (Gleason score 4 + 3) obtained from radical prostatectomy was precision cut into TSGs of 5 mm diameter and 300 μm thickness, which were subsequently implanted under the renal capsules of male RAG2-/- γC-/- mice (Fig. 1C). After 1 month of establishment, mice bearing consecutive TSGs were paired and treated with darinaparsin [100 mg/kg, intraperitoneal (i.p.), thrice weekly] or saline as a control. The TSGs were recovered, fixed, serially sectioned, and stained for p63 (marker for normal basal cells) and AMACR (marker for prostate cancer cells; PIN Cocktail; Biocare Medical), and the area of tumor and TSG in each section was quantified with ImagePro (Media Cybernetics).
Figure 1.
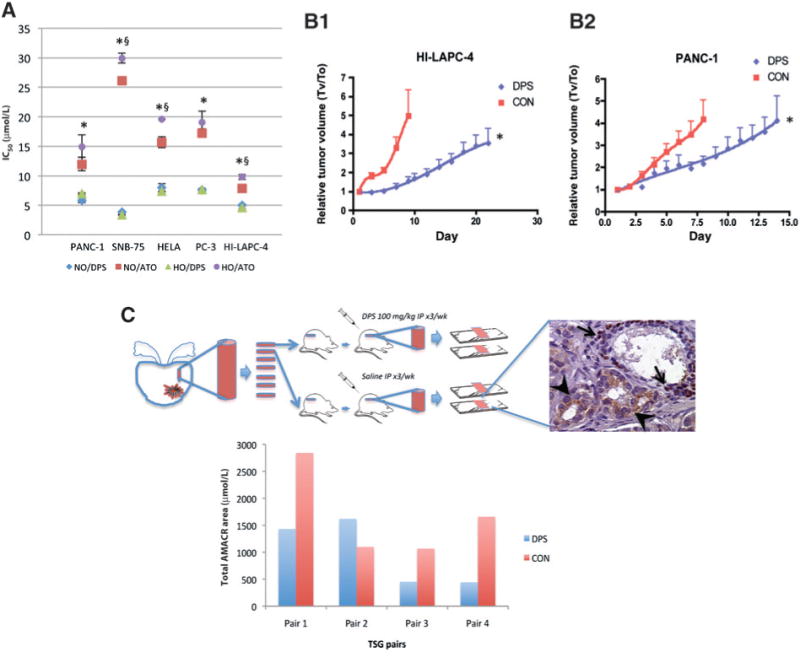
Darinaparsin inhibited tumor cell growth in vitro and in vivo. A, cells were treated with darinaparsin (DPS) for 4 hours under normoxia (NO) or hypoxia (HO), and kept in fresh media for 3 days under NO before MTT assay. Data were analyzed by Sigmoidal dose-response modeling, and expressed as IC50s with SE. *, P < 0.01 for NO/DPS versus NO/ATO and HO/DPS versus HO/ATO; §, P < 0.01 for NO/ATO versus HO/ATO. B, nude mice with subcutaneous HI-LAPC-4 (B1) and PANC-1 (B2) xenograft tumors were given darinaparsin (100 mg/kg, 3 times per week, i.p.) or saline (CON). N = 9 (DPS) and 7 (CON) in B1,andN= 10 (DPS) and 11 (CON) in B2.*,P< 0.0001 for CON versus DPS by exponential growth modeling. C, Rag2−/−γC−/ − mice bearing paired xenograft human prostate cancer TSGs were treated with darinaparsin as in 1B for 4 weeks, and stained for AMACR (cancer, arrowheads) and p63 (normal basal cell, arrows). P = 0.139 by paired t test.
For the study of the darinaparsin effect in combination with radiation on normal intestine, we used an established microcolony assay that measured the intestinal stem cell survival (as the number of crypts/cross section) after radiation (22).
Cytotoxicity, cell death, apoptosis, and clonogenic survival assays
In vitro cytotoxicity was measured using the MTT assay and expressed as the drug concentration causing 50% inhibition of cell growth (IC50). Cell death was measured by trypan blue (0.04%; Invitrogen) exclusion after trypsinization and expressed as the % of positively stained cells divided by total number of cells. Apoptosis was measured by staining with Alexa Fluor 488-conjugated Annexin V (Invitrogen) and propidium iodide (2 μg/mL) followed by flow cytometry. For clonogenic assays, the cells were maintained in fresh medium after treatment for 2 to 3 weeks before staining with 0.25% crystal violet in 95% ethanol. Colonies with 50 cells or more were scored. The surviving fraction was calculated as the colony-forming efficiency (CFE) of treated cells divided by the CFE of untreated cells.
Reactive oxygen species measurement
Cellular reactive oxygen species (ROS) were measured by fluorescent probe CM-H2DCFDA (5 μmol/L for 30 minutes at 37°C; Invitrogen). Mitochondria superoxide (O2·−) was determined using the mitochondria-specific fluorescent probe MitoSox Red (10 μmol/L for 10 minutes at 37°C; Invitrogen). The fluorescence intensity was measured by flow cytometry and expressed as the mean fluorescent unit.
Western blot
After cell lysis, sample proteins were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The membranes were incubated overnight at 4°C with anticleaved caspase-3, anti-γH2AX, or anti-β-actin (1:1,000; Cell Signaling Technology) antibodies, and then with goat anti-rabbit (1:2,000; Cell Signaling) for 1 hour at room temperature.
RNA isolation and microarray analysis
Total RNA was isolated using TRIzol (Invitrogen) and purified using an RNeasy MinElute kit (QIAGEN). The microarray hybridization was conducted by Stanford Functional Genomics Facility using array chips (SurePrint G3 Human GE 8 × 60 k; Agilent Technologies). A Universal Human Reference RNA (Agilent) was used as the control for each sample. The acquired fluorescence intensities for each of the 62,976 features (containing 27,958 Entrez gene RNAs) were obtained with an Agilent G2505C Microarray Scanner System after labeling and available at GenBank (accession no: GSE33001). We selected gene features in which expression levels varied 2.5-fold or more from the untreated controls (CON) in both 4- and 8-hour treatment groups under normoxia; and 1.5-fold or more from CON under hypoxia. The resulting data sets (listed in Supplementary Table S1) were analyzed with Ingenuity Pathway Analysis (Ingenuity Systems), Transcription Factor Target Enrichment Analysis (http://bioinfo.vanderbilt.edu/webgestalt), oncogene-dependent gene expression signature analysis, and Connectivity Map analysis (http://www.broadinsti-tute.org/cmap).
Other assays
Intracellular total glutathione (GSH) concentrations were determined using the GSH reductase recycling assay (23) and normalized with protein concentration. DNA damage was assessed using the neutral comet assay (24). One hundred cells were scored for each sample, and the data were expressed as the mean moment (tail length × intensity of DNA in the tail ± SE). Mitochondrial membrane potential (MMP) was measured by JC-1 (10 μg/mL, 1 hour; Invitrogen) staining and flow cytometry (25). Senescence was measured using senescence-associated (SA)-β-Gal staining (26) and expressed as the ratio of positive stained cells to total number of cells.
Results
Darinaparsin inhibited solid tumor cell growth in vitro and in vivo
Using a panel of cell lines derived from prostate (HI-LAPC-4 and PC-3), pancreas (PANC-1), brain (SNB-75), and cervical (HeLa) tumors, the in vitro cytotoxicity of darinaparsin and ATO was determined using the MTT assay at 3 days after incubation with darinaparsin or ATO for 4 hours under normoxia or hypoxia. Darinaparsin had IC50s of approximately 3 to 8 μmol/L in different cell lines (Fig. 1A). The IC50s were similar under either normoxia or hypoxia, indicating that the effect of darinaparsin on cell proliferation was O2 independent. In comparison, IC50s of ATO were approximately 2- to 6-fold higher than darinaparsin, with hypoxic IC50s generally higher than corresponding normoxic IC50s, indicating that ATO activity against these cell lines was less potent than darinaparsin and O2 dependent to a certain extent. In clonogenic assays which assess the ability of individual cells to proliferate and give rise to colonies, the IC50/4 hours of darinaparsin was approximately 1 to 3 μmol/L for the cell lines studied under both normoxia and hypoxia (Supplementary Table S2).
Next, the effect of darinaparsin on tumor growth in vivo was determined using mouse models with established HI-LAPC-4 and PANC-1 subcutaneous xenograft tumors. The mice were injected with darinaparsin intraperitoneally 3 times per week with a dose of 100 mg/kg for 4 weeks. Toxicology studies, including blood chemistry panels, did not indicate any systemic toxicity with similar doses except for changes in physical activity and body weight (16; Supplementary Fig. S1A and Supplementary Table S3). In pharmacokinetic studies with human subjects (18), this regimen achieved blood arsenic concentrations (Cmax, ∼10 μmol/L) above the IC50s observed in our in vitro studies (Fig. 1A and Supplementary Table S2). In both HI-LAPC-4 and PANC-1 tumor models, darinaparsin significantly inhibited tumor growth (P < 0.0001; Fig. 1B1 and B2), with the average tumor volume doubling time increased from 4 to 13 days in HI-LAPC-4 tumors (P < 0.001), and 4 to 7 days in PANC-1 tumors (P < 0.001).
Finally, the activity of darinaparsin in a novel and clinically relevant tumor tissue slice graft (TSG) model of primary prostate cancer was tested. Mice that received consecutive TSGs (Gleason score 4 + 3) were paired and treated with darinaparsin (100 mg/kg, i.p., 3 times per week for 4 weeks) or saline as a control (Fig. 1C). After recovery, the TSGs were fixed and serially sectioned. The presence of cancer in each section was evaluated by AMACR staining, and AMACR-positive areas were imaged and quantitated. The total areas of the TSG sections from darinaparsin- and saline-treated mice that were evaluated were similar (data not shown). In 3 of the 4 TSG pairs, DPS decreased the total tumor area to less than 50% of the control. Although the difference (P = 0.139 by paired t test) was not statistically significant, possibly due to the small sample size of this pilot study, overall the findings suggest darinaparsin-inducedcytotoxicity in primary prostate cancer as well as advanced cancer as modeled by the cell lines.
Darinaparsin sensitized solid tumor cells to radiation
Because ATO has been reported to sensitize solid tumors to radiation (2–5), we compared the in vitro radiosensitizing effect of darinaparsin to ATO using a panel of relatively radioresistant cell lines (HI-LAPC-4, PC-3, SNB-75, HeLa, and PANC-1). Before irradiation, the cells were incubated with 3 μmol/L darinaparsin or ATO for 4 hours. Figure 2A shows the clonogenic survival of γ-irradiated cells after normalization for the effect of drug alone. As expected, all cell lines were more sensitive to radiation under normoxia as compared with hypoxia. Under hypoxia, ATO had little or no effect on the clonogenic survival of the irradiated cells; in contrast, darinaparsin increased radiation-induced cell killing by approximately one log, which was even greater than the effect of normoxia (oxygen) under normoxic conditions.
Figure 2.
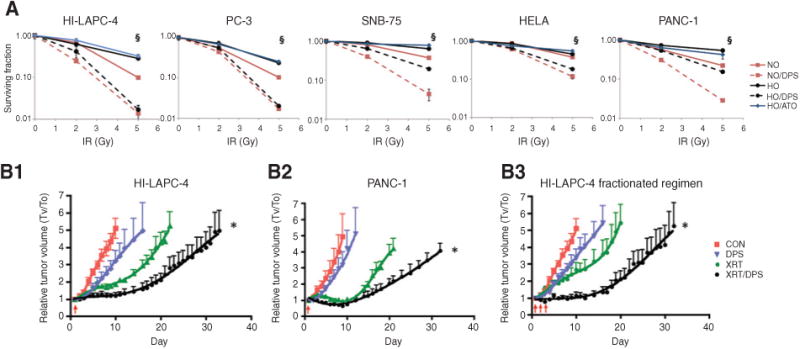
Darinaparsin was a potent radiosensitizer in vitro and in vivo. A, cells were treated with darinaparsin or ATO (3 μmol/L, 4 hours) under NO or HO before irradiation, and the survival assessed by clonogenic assay. §, P < 0.01 of NO versus NO/DPS and HO versus HO/DPS in cells with 5 Gy radiation. B, mice with subcutaneous xenograft tumors were treated with darinaparsin (i.p., 100 mg/kg) and 4 hours later with irradiation (XRT) of 5 Gy (B1 and B2) or 2 Gy (B3) at days indicated by the arrows. *, P < 0.0001 versus all other groups by exponential growth modeling. N = 5 to 7 per group.
Next, the in vivo effect of darinaparsin in combination with local tumor irradiation was evaluated. Nude mice bearing subcutaneous HI-LAPC-4 and PANC-1 xenograft tumors were treated with darinaparsin (100 mg/kg, i.p.) 4 hours before a single dose (5 Gy) of irradiation. In both models, darinaparsin significantly enhanced the radiation-induced tumor growth inhibition, with an increase in the tumor doubling time from 11 to 20 days in HI-LAPC-4 tumors and 16 to 20 days in PANC-1 tumors (Fig. 2B1 and B2, P < 0.0001). The effect of darinaparsin in combination with a clinically relevant fractionated radiation regimen was also tested. In this regimen, darinaparsin (100 mg/kg, i.p.) was administered 4 hours before radiation (2 Gy/day) for 3 consecutive days using the HI-LAPC-4 tumor model. A significant delay of tumor volume doubling time was observed from 7 days with radiation alone and 6 days with darinaparsin alone to 19 days for the combined treatment of radiation and darinaparsin (Fig. 2B3, P < 0.0001).
Darinaparsin did not radiosensitize normal radiosensitive tissues
The body weight loss observed in the radiosensitization regimen, either by darinaparsin alone or in combination with radiation, was minimal (< 5%, Supplementary Fig. S1A). Serial complete blood counts (CBC, indicators of bone marrow function) were obtained following total body radiation (3.5 Gy) with or without darinaparsin treatment (100 mg/kg, 4 hours, i.p.). As expected, the white blood cells (WBC), platelet, red blood cells (RBC), and hemoglobin (HGB) significantly decreased after the radiation and recoveredover time. Darinaparsin did not affect these CBC changes (Fig. 3A). Using the established intestinal microcolony assay, we observed a significant decrease in viable duodenal crypt cells following radiation. However, to our surprise, darinaparsin treatment significantly enhanced the survival of crypt cells following irradiation (Fig. 3B). In comparison with saline-treated controls, the darinaparsin -treated mice had nearly 10 times more viable crypts at 3.5 days after radiation (10–12Gy). Similar results were observed in the jejunum and ileum (data not shown). The data therefore indicate that darinaparsin does not sensitize, and may even protect, normal radiosensitive tissues to radiation.
Figure 3.
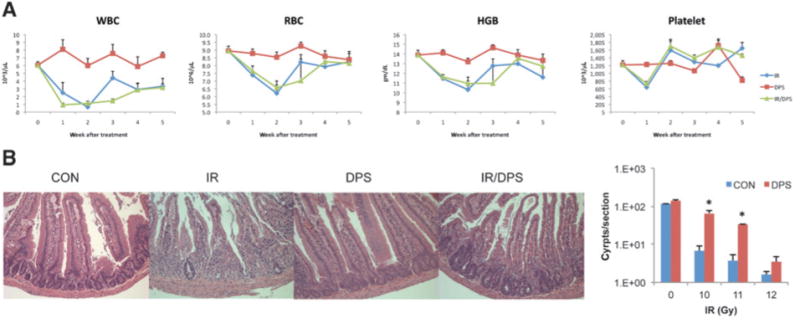
Darinaparsin did not radiosensitize normal radiosensitive tissues. A, Balb/C mice were treated with darinaparsin (100 mg/kg, i.p., 4 hours) alone, total body irradiation (IR) alone, or IR following darinaparsin (IR/DPS). Each group (N = 8) comprised 2 subgroups (N = 4). Each subgroup was retroorbitally bled alternatively (every other week) and counted for WBC, RBC, HGB, and platelets. B, Balb/C mice were divided into 4 groups, including untreated control (CON), darinaparsin (100 mg/kg, i.p.), IR, and IR following 4 hours darinaparsin treatment (IR/DPS). N = 3 per group. The intestinal samples were collected 3.5 days after IR, and viable crypts/cross section were quantified. Data presented are from duodenum samples. *, P < 0.05 versus CON. Data are representative of 3 independent experiments.
Darinaparsin induced apoptosis and senescence but not autophagy in HI-LAPC-4 cells
Darinaparsin caused HI-LAPC-4 cell death in a time (24 and 48 hours)-dependent manner, as shown by trypan blue exclusion assay (Fig. 4A). We therefore assessed the modes of cell death after darinaparsin treatment. After 24 hours of darinaparsin treatment (10 μmol/L, 8 hours), there was a significant increase in caspase-3 cleavage (Fig. 4B) and Annexin V externalization (Fig. 4C) under both normoxia and hypoxia, indicating cell death by apoptosis. Importantly, after 24 hours incubation in hypoxia, darinaparsin was more cytotoxic to HI-LAPC-4 cells than in normoxia, which is in contrast to equivalent effects of darinaparsin in normoxia and hypoxia after only 4 hours incubation (Fig. 1A). In addition, a small proportion of cells that survived darinaparsin treatment had morphologic features of senescence with increased expression of SAβ-galactosidase (Fig. 4D). Finally, morphologic (vacuole formation) and LC-3 activation (an autophagy marker; Supplementary Fig. S2) suggested autophagy was not involved in darinaparsin-induced cell death. A cell-cycle analysis was also conducted and revealed no apparent cell-cycle arrest after 24 hours of darinaparsin treatment (Supplementary Fig. S3).
Figure 4.
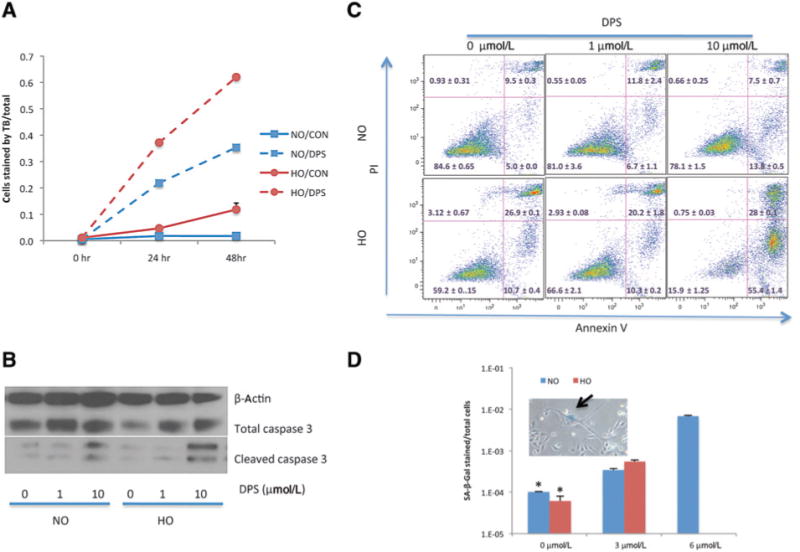
Darinaparsin induced apoptosis and senescence in HI-LAPC-4 cells. A, cells were treated with darinaparsin (10mmol/L) for the time indicated and analyzed with Trypan blue exclusion assay. B and C, cells were treated with darinaparsin (24 hours) under NO and HO before measurements of cleaved caspase-3 (B, Western blot) and Annexin V externalization (C, flow cytometry). D, cells were treated with darinaparsin (24 hours) under NO or HO, and maintained in fresh medium for 24 hours before SA-β-Gal staining. Few cells survived the darinaparsin treatment (6 μmol/L) under HO, and therefore were not counted. P< 0.001 by 1-wayANOVA, and *,P< 0.001 versus 3 and 6 μmol/LbyTukey multiple comparison test. The arrow of the inset indicates a cell with senescence morphology and positive stain of SA-β-Gal.
Darinaparsin cytotoxicity was independent of ROS generation and DNA or mitochondria damage
Because darinaparsin-induced cytotoxicity in hematolog-ic cancer cell lines is mediated by ROS generation, with subsequent mitochondrial damage (19), we determine the effect of darinaparsin on cellular ROS in solid tumor cell lines. Using the fluorescent probe CM-H2DCFDA, we observed a dose-dependent increase of cellular ROS in darinaparsin-treated HI-LAPC-4 cells under normoxia but not hypoxia (Fig. 5A). In addition, darinaparsin depleted cellular antioxidant GSH in normoxia, but not hypoxia (Fig. 5B). Because ROS generation disrupts normal protein folding in the endoplasmic reticulum (ER), and subsequently induces ER stress and unfolded protein response (UPR; ref. 27), the level of ER stress following darinaparsin treatment was assessed using an established luciferase reporter model for UPR-activated transcription factors XBP-1 orATF-4 (28). As expected, darinaparsin caused a dose-dependent activation of XBP-1 and ATF-4 under normoxia, but not under hypoxia (Fig. 5C). Finally, in HI-LAPC-4 cells under normoxia (but not hypoxia), there was a moderate increase of DNA damage (Comet assay; Fig. 5D) and DNA damage response (gH2AX; Fig. 5E) following darinaparsin treatment. However, when cells were pretreated with darinaparsin for 4 hours and then irradiated, there was no synergistic increase in gH2AX at either 0.5 or 12.5 hours after irradiation (Supplementary Fig. S4). In summary, the data above show that darinaparsin did not induce oxidative stress under hypoxia, indicating the existence of ROS-independent mechanisms of darinaparsin cytotoxicity.
Figure 5.
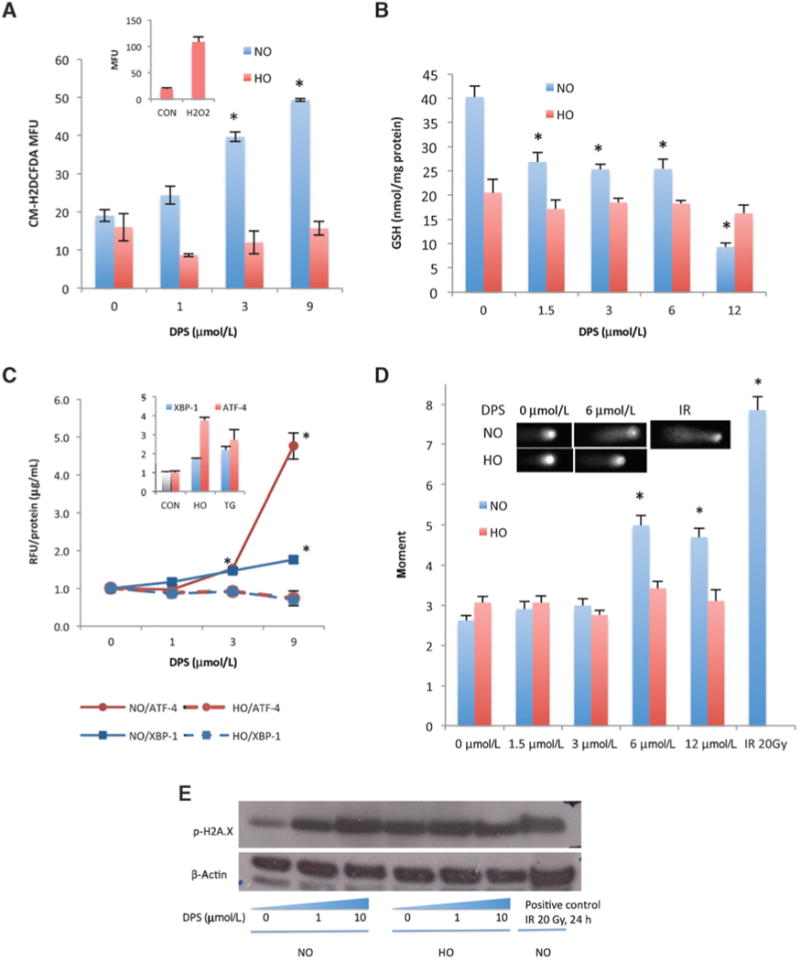
Darinaparsin induced oxidative stress under normoxia but not hypoxia. A, HI-LAPC-4 cells were treated with darinaparsin (8 hours) and assayed with fluorescent probe CM-H2DCFDA. Inset, ROS level of cells with or without H2O2 (positive control) treatment (1 mmol/L, 10 minutes) under HO. B, HI-LAPC-4 cells were treated with darinaparsin (4 hours) before analysis for the total cellular GSH content. C, pancreatic cancer (MIA PaCa-2) cells that stably express a luciferase reporter of ATF-4 or XBP-1 were treated with darinaparsin (8 hours) and thapsigargin (TG, 30 nmol/L, 24 hours, positive control). The luciferin production was expressed as the relative fluorescence intensity (RFU) over untreated control, and normalized by protein concentration. Inset: both HO and TG induced XBP-1 and ATF-4 activity. D and E, HI-LAPC-4 cells were treated with darinaparsin for 4 hours and used for comet assay (D) and γH2AX Western blot (E).IR, irradiation (positive control). Inset, representative comet micrograph. P< 0.0001 for 1-way ANOVA in A-E comparing conditions under NO. *,P< 0.01 for comparison with NO untreated control by the Tukey multiple comparison test.
Although darinaparsin significantly decreased (28%, P < 0.01) MMP in an APL cell line, HL-60 (19, Supplementary Fig. S5A), it did not significantly affect the MMP in the 5 solid tumor cell lines tested, with the exception of HI-LAPC-4undernormoxia (31%,P<0.05).In addition, there was no increase of mitochondrial ROS under either normoxia or hypoxia (Supplementary Fig. S5B), indicating that ROS generation did not cause detectable mitochondrial damage in the majority of the solid tumor cell lines studied, and is not a primary mechanism of darinaparsin cytotoxicity in the these cells.
The antitumor effect of darinaparsin was associated with inhibition of oncogenic signaling pathways
The lack of immediate DNA and mitochondrial damage suggested that darinaparsin-induced cell death was mediated by signaling events. In addition, cycloheximide (CHX, a translation inhibitor) significantly decreased darinaparsin cytotoxicity under both normoxia and hypoxia (P < 0.0001 for the interaction of CHX and darinaparsin by 2-way ANOVA, Supplementary Fig. S6), indicating that darinaparsin activity required de novo protein expression. Next, darinaparsin effects on hypoxia-induced factor Iα and c-jun— NH2—kinase were studied because of their roles as a regulator of cell survival and radiosensitivity under hypoxia (1) and a mediator of darinaparsin cytotoxicity in leukemia cells (15), respectively. The data indicate that neither factor likely plays a significant role in darinaparsin-mediated cytotoxicity in solid tumor cells under hypoxia (Supplementary Figs. S7 and S8).Gene expression profiling was carried out with HI-LAPC-4 cells after 4 and 8 hours incubation with darinaparsin or ATO at the concentrations of IC50/4 hours (Fig. 1A) under normoxia. Among the 16,433 genes analyzed, 343 (up) and 113 (down) genes were regulated by darinaparsin, and 139 (up) and 99 (down) genes were regulated by ATO, with 104 (up) and 57 (down) overlapping genes. Several of the up regulated genes were validated by reverse-transcription and quantitative PCR assay of independently treated samples (Supplementary Table S4A and S4B). An Ingenuity Pathway analysis showed that darinaparsin affected multiple biological processes and pathways that involve cancer, inflammation, and apoptosis (Supplementary Table S4C). Transcription Factor Target Enrichment Analysis identified transcription factors in which target gene expression was significantly affected by darinaparsin (DPS/CON) and ATO (ATO/CON), many of which were associated with once-gene regulated pathways (Supplementary Table S4D).
To further assess the effect of darinaparsin on oncogenic pathways, we interrogated the microarray data against established oncogene-dependent gene expression signatures (29–33), including ontogenesis such as RAS, RAF, MEK, MAPK, AKT, MYC, β-catena, SRC, E2F, androgen receptor (AR), cycling D, and ERBB2. The analysis showed significant overlap of RAS and MYC target genes with darinaparsin-affected genes (Fig. 6). Specifically, darinaparsin significantly decreased the expression of genes that were up regulated by RAS, and increased the expression of genes that were down regulated by MYC. In contrast, ATO only affected AR signaling. Next, we compared RAS and MYC status in HI-LAPC-4 and PC-3 (both prostate cancer) cells with normal fibroblasts, and found increased expression of both proteins in the cancer cell lines (Supplementary Fig. S9). The data are consistent with previous reports of increased MYC and RAS expression in solid tumor cells (34) and suggest a selective effect of darinaparsin on cancer cells via oncogenic pathways.
Figure 6.
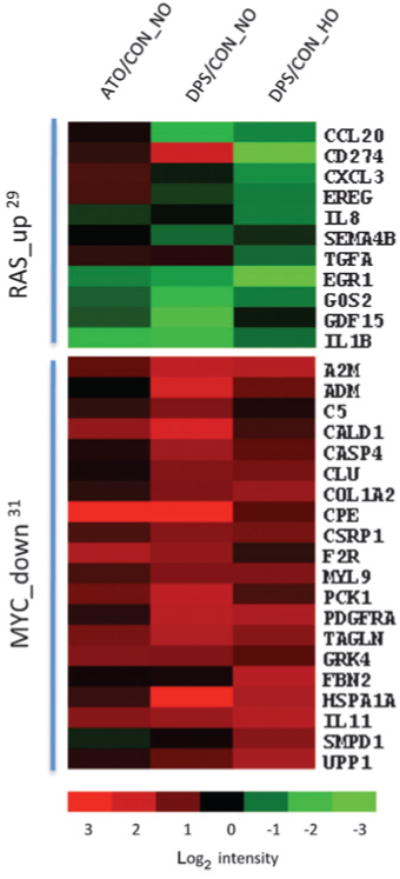
Gene microarray analysis of darinaparsin effect on ontogenesis. HI-LAPC-4 cells were treated with darinaparsin or ATO at IC50 under NO or HO for 4 and 8 hours. The gene expression was assayed by cDNA microarray, and the data analyzed by oncogene-dependent gene expression signature analysis. The heatmap presents the overlaps of darinaparsin-regulated genes and oncogene (RAS and MYC)-dependent signatures under each condition. “up” and “down”, oncogene up- or down regulated signature gene set. The superscripts denote the references of the signature set. P < 0.001 by χ2 test or Yate corrected χ2 test for enrichment of RAS_up genes in DPS/CON_NO and DPS/ CON_HO, and MYC_down genes in DPS/CON_NO.
Connectivity Map analysis (Supplementary Table S4E) was used to compare gene expression changes induced by darinaparsin and ATO to those induced by hundreds of other molecules of known function. Of note, there was no overlap of the top 20 molecules connected with darinaparsin or ATO, again suggesting differential mechanisms of action for these 2 arsenicals. Many of the darinaparsin-connected molecules are known cancer drugs and affect oncogenic pathways, such as HSP90 inhibitors [tanespimycin (35), geldanamycin (36), and alvespimycin (37) ], withaferin a (38), and wortmannin, further supporting the involvement of oncogenic pathways in darinaparsin activity.
Discussion
Resistance of many solid tumors to currently available cancer therapies is an important clinical problem and is determined in part by the presence of hypoxia in the tumor microenvironment. In this study, we report that darinaparsin effectively inhibited the growth of a variety of tumor cell lines in vitro under hypoxia and in vivo in xenograft tumor models. Importantly, darinaparsin significantly radiosensitized tumor cells in vitro under hypoxia and in vivo with a clinically relevant fractionated radiation regimen, but did not enhance the sensitivity of normal radiosensitive intestine and bone marrow to radiation. In fact, darinaparsin actually radioprotected the GI epithelium, suggesting that darinaparsin has the potential to significantly enhance the therapeutic index of radiation therapy. With higher cytotoxic and radiosensitizing potency under hypoxia than ATO, and a more favorable toxicity profile, darinaparsin is more promising for the treatment of solid tumors. Although darinaparsin, a dimethylarsenic conjugated to glutathione, may be generated as an intermediate in the inorganic arsenic metabolic transformation pathway (19, 39), our data suggest that the cellular generation of darinaparsin from ATO is insufficient to fully explain the antitumor effects observed with darinaparsin alone.
Currently, most of the information about the mechanism of action of darinaparsin comes from malignant lympho-hematopoietic cell lines, in which oxidative stress plays an important role. However, under hypoxia (0.5% O2), darinaparsin did not induce oxidative stress and DNA damage in solid tumor cells. We therefore conducted mechanistic studies to elucidate other possible mechanisms of action for darinaparsin. In contrast to ATO, darinaparsin caused significant changes of gene expression that were associated with the ontogenesis RAS and MYC. This targeted effect may help to explain the differential effect of darinaparsin on cancer as compared with normal cells, as well as the decreased systemic toxicity of darinaparsin compared with ATO. In addition, because ontogenesis are important for cancer cell survival after irradiation, and oncogene inhibition can radiosensitize cells (40), it is therefore possible that the selective radiosensitizing effect of darinaparsin is due to inhibition of dysregulated oncogenic pathways in cancer, but not normal, cells.
Of particular interest and potential clinical utility is the observation that darinaparsin enhanced the survival of radiosensitive intestinal cells, while sensitizing tumor cells to radiation. This differential effect may be, in part, due to differences in signaling factors and signal transduction pathways between intestinal and tumor cells. p53, a tumor suppressor that is mutated or silenced in many tumor cells (including the 5 cell lines used in this study; ref. 41–44), has been reported to protect intestinal cells from radiation damage via cell-cycle arrest (45). It is therefore possible that darinaparsin activates p53 and downstream p21, which enhances the radiation-induced cell-cycle arrest in crypt cells. Experiments are ongoing to further elucidate the differential effect of darinaparsin on normal cells as compared to tumor cells.
In summary, the results presented here show that darinaparsin has significant cytotoxic and radiosensitizing activities against normoxic and hypoxic solid tumor cells in vitro and in vivo. Moreover, darinaparsin does not sensitize bone marrow, and protected normal intestinal epithelium from the effects of radiation in vivo, suggesting that darinaparsin has the potential to significantly increase the therapeutic index of radiation therapy. The favorable toxicity profile of darinaparsin and the data presented here are very promising and may have broad clinical applicability for the treatment of solid tumors, in which hypoxia enhances tumor resistance to radiation therapy. Because darinaparsin is already in clinical trials, these results have near term translational potential.
Supplementary Material
Translational Relevance.
Darinaparsin, an organic arsenic compound, is currently in clinical trials as a single agent for the treatment of lymphohematopoietic malignancies and solid tumors. Here, we report that darinaparsin is a more potent cytotoxin and radiosensitizer under hypoxia than arsenic trioxide (ATO). At well-tolerated doses, darinaparsin sensitized tumors, but not normal radiosensitive bone marrow, and protected gastrointestinal epithelium from radiation. These findings therefore suggest that darinaparsin may have the potential to significantly increase the therapeutic index of radiotherapy for the treatment of solid tumors.
Acknowledgments
The authors thank Dr. Albert Koong in the Department of Radiation Oncology at Stanford University for providing the MiaPaca-2 cell line over-expressing markers for endoplasmic reticulum stress
Grant Support: This work was supported by a grant from ZIOPHARM Oncology.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: J. Tian received commercial research grant from ZIOPHARM Oncology. S.J. Knox received commercial research grant from ZIOPHARM Oncology, Inc. and is a consultant and an advisory board member of InterWest Partners. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions: Conception and design: J. Tian, D.M. Peehl, S.J. Knox
Development of methodology: J. Tian, H. Zhao, D.M. Peehl, S.J. Knox
Acquisition of data: J. Tian, H. Zhao, S.W. Reese, S.R. Young, X. Li, D.M, Peehl, S.J. Knox
Analysis and interpretation of data: J. Tian, H. Zhao, D.M. Peehl, S.J. Knox
Writing, review, and/or revision of the manuscript: J. Tian, H. Zhao, S.R., Young, D.M. Peehl, S.J. Knox
Administrative, technical, or material support: J. Tian, R. Nolley, S.W., Reese, X. Li
Study supervision: D.M. Peehl, S.J. Knox
References
- 1.Ruan K, Song G, Ouyang G. Role of hypoxia in the hallmarks of human cancer. J Cell Biochem. 2009;107:1053–62. doi: 10.1002/jcb.22214. [DOI] [PubMed] [Google Scholar]
- 2.Lew YS, Kolozsvary A, Brown SL, Kim JH. Synergistic interaction with arsenic trioxide and fractionated radiation in locally advanced murine tumor. Cancer Res. 2002;62:4202–5. [PubMed] [Google Scholar]
- 3.Ning S, Knox SJ. Optimization of combination therapy of arsenic trioxide and fractionated radiotherapy for malignant glioma. Int J Radiat Oncol Biol Phys. 2006;65:493–8. doi: 10.1016/j.ijrobp.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 4.Griffin RJ, Williams BW, Park HJ, Song CW. Preferential action of arsenic trioxide in solid-tumor microenvironment enhances radiation therapy. Int J Radiat Oncol Biol Phys. 2005;61:1516–22. doi: 10.1016/j.ijrobp.2004.12.058. [DOI] [PubMed] [Google Scholar]
- 5.Chun YJ, Park IC, Park MJ, Woo SH, Hong SI, Chung HY, et al. Enhancement of radiation response in human cervical cancer cells in vitro and in vivo by arsenic trioxide (As2O3) FEBS Lett. 2002;519:195–200. doi: 10.1016/s0014-5793(02)02765-5. [DOI] [PubMed] [Google Scholar]
- 6.Kindler HL, Aklilu M, Nattam S, Vokes EE. Arsenic trioxide in patients with adenocarcinoma of the pancreas refractory to gemcitabine: a phase II trial of the University of Chicago Phase II Consortium. Am J Clin Oncol. 2008;31:553–6. doi: 10.1097/COC.0b013e318178e4cd. [DOI] [PubMed] [Google Scholar]
- 7.Verstovsek S, Giles F, Quintas-Cardama A, Perez N, Ravandi-Kashani F, Beran M, et al. Arsenic derivatives in hematologic malignancies: a role beyond acute promyelocytic leukemia? Hematol Oncol. 2006;24:181–8. doi: 10.1002/hon.787. [DOI] [PubMed] [Google Scholar]
- 8.Golemovic M, Quintas-Cardama A, Manshouri T, Orsolic N, Duzkale H, Johansen M, et al. MER1, a novel organic arsenic derivative, has potent PML-RARalpha-independent cytotoxic activity against leukemia cells. Invest New Drugs. 2010;28:402–12. doi: 10.1007/s10637-009-9267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estrov Z, Manna SK, Harris D, Van Q, Estey EH, Kantarjian HM, et al. Phenylarsine oxide blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, and induces apoptosis of acute myelogenous leukemia cells. Blood. 1999;94:2844–53. [PubMed] [Google Scholar]
- 10.Piatek K, Schwerdtle T, Hartwig A, Bal W. Monomethylarsonous acid destroys a tetrathiolate zinc finger much more efficiently than inorganic arsenite: mechanistic considerations and consequences for DNA repair inhibition. Chem Res Toxicol. 2008;21:600–6. doi: 10.1021/tx7003135. [DOI] [PubMed] [Google Scholar]
- 11.Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, et al. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell. 2003;3:497–509. doi: 10.1016/s1535-6108(03)00109-0. [DOI] [PubMed] [Google Scholar]
- 12.Dilda PJ, Decollogne S, Weerakoon L, Norris MD, Haber M, Allen JD, et al. Optimization of the antitumor efficacy of a synthetic mitochondrial toxin by increasing the residence time in the cytosol. J Med Chem. 2009;52:6209–16. doi: 10.1021/jm9008339. [DOI] [PubMed] [Google Scholar]
- 13.Wang ZG, Rivi R, Delva L, Konig A, Scheinberg DA, Gambacorti-Passerini C, et al. Arsenic trioxide and melarsoprol induce programmed cell death in myeloid leukemia cell lines and function in a PML and PML-RARalpha independent manner. Blood. 1998;92:1497–504. [PubMed] [Google Scholar]
- 14.Chen GQ, Zhou L, Styblo M, Walton F, Jing Y, Weinberg R, et al. Methylated metabolites of arsenic trioxide are more potent than arsenic trioxide as apoptotic but not differentiation inducers in leukemia and lymphoma cells. Cancer Res. 2003;63:1853–9. [PubMed] [Google Scholar]
- 15.Diaz Z, Mann KK, Marcoux S, Kourelis M, Colombo M, Komarnitsky PB, et al. A novel arsenical has antitumor activity toward As2O3-resistant and MRP1/ABCC1-overexpressing cell lines. Leukemia. 2008;22:1853–63. doi: 10.1038/leu.2008.194. [DOI] [PubMed] [Google Scholar]
- 16.Golemovic M, Kantarjian H, Orsolic N, Zingaro R, Gao MZ, Cheng X, et al. Development of an organic arsenic derivative as a therapy for leukemia. Blood. 2003;102:252b. [Google Scholar]
- 17.Wu J, Henderson C, Feun L, Van Veldhuizen P, Gold P, Zheng H, et al. Phase II study of darinaparsin in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2010;28:670–6. doi: 10.1007/s10637-009-9286-9. [DOI] [PubMed] [Google Scholar]
- 18.Tsimberidou AM, Camacho LH, Verstovsek S, Ng C, Hong DS, Uehara CK, et al. A phase I clinical trial of darinaparsin in patients with refractory solid tumors. Clin Cancer Res. 2009;15:4769–76. doi: 10.1158/1078-0432.CCR-08-2984. [DOI] [PubMed] [Google Scholar]
- 19.Mann KK, Wallner B, Lossos IS, Miller WH., Jr Darinaparsin: a novel organic arsenical with promising anticancer activity. Expert Opin Investig Drugs. 2009;18:1727–34. doi: 10.1517/13543780903282759. [DOI] [PubMed] [Google Scholar]
- 20.Matulis SM, Morales AA, Yehiayan L, Croutch C, Gutman D, Cai Y, et al. Darinaparsin induces a unique cellular response and is active in an arsenic trioxide-resistant myeloma cell line. Mol Cancer Ther. 2009;8:1197–206. doi: 10.1158/1535-7163.MCT-08-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao H, Nolley R, Chen Z, Peehl DM. Tissue slice grafts: an in vivo model of human prostate androgen signaling. Am J Pathol. 2010;177:229–39. doi: 10.2353/ajpath.2010.090821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Withers HR, Elkind MM. Microcolony survival assay for cells of mouse intestinal mucosa exposed to radiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1970;17:261–7. doi: 10.1080/09553007014550291. [DOI] [PubMed] [Google Scholar]
- 23.Vandeputte C, Guizon I, Genestie-Denis I, Vannier B, Lorenzon G. A microtiter plate assay for total glutathione and glutathione disulfide contents in cultured/isolated cells: performance study of a new miniaturized protocol. Cell Biol Toxicol. 1994;10:415–21. doi: 10.1007/BF00755791. [DOI] [PubMed] [Google Scholar]
- 24.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–9. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 25.Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) Biochem Biophys Res Commun. 1993;197:40–5. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- 26.Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21–31. doi: 10.1007/978-1-59745-361-5_3. [DOI] [PubMed] [Google Scholar]
- 27.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–93. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 28.Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005;3:597–605. doi: 10.1158/1541-7786.MCR-05-0221. [DOI] [PubMed] [Google Scholar]
- 29.Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics. 2010;3:26. doi: 10.1186/1755-8794-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DePrimo SE, Diehn M, Nelson JB, Reiter RE, Matese J, Fero M, et al. Transcriptional programs activated by exposure of human prostate cancer cells to androgen. Genome Biol. 2002;3:RESEARCH0032. doi: 10.1186/gb-2002-3-7-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260–5. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Creighton CJ. Multiple oncogenic pathway signatures show coordinate expression patterns in human prostate tumors. PLoS One. 2008;3:e1816. doi: 10.1371/journal.pone.0001816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 34.Field JK, Spandidos DA. The role of ras and myc ontogenesis in human solid tumours and their relevance in diagnosis and prognosis (review) Anticancer Res. 1990;10:1–22. [PubMed] [Google Scholar]
- 35.Russell JS, Burgan W, Oswald KA, Camphausen K, Tofilon PJ. Enhanced cell killing induced by the combination of radiation and the heat shock protein 90 inhibitor 17-allylamino-17- demethoxygeldana-mycin: a multitarget approach to radiosensitization. Clin Cancer Res. 2003;9:3749–55. [PubMed] [Google Scholar]
- 36.Fukuyo Y, Hunt CR, Horikoshi N. Geldanamycin and its anti-cancer activities. Cancer Lett. 2010;290:24–35. doi: 10.1016/j.canlet.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 37.Bull EE, Dote H, Brady KJ, Burgan WE, Carter DJ, Cerra MA, et al. Enhanced tumor cell radiosensitivity and abrogation of G2 and S phase arrest by the Hsp90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin. Clin Cancer Res. 2004;10:8077–84. doi: 10.1158/1078-0432.CCR-04-1212. [DOI] [PubMed] [Google Scholar]
- 38.Oh JH, Lee TJ, Kim SH, Choi YH, Lee SH, Lee JM, et al. Induction of apoptosis by withaferin A in human leukemia U937 cells through down-regulation of Akt phosphorylation. Apoptosis. 2008;13:1494–504. doi: 10.1007/s10495-008-0273-y. [DOI] [PubMed] [Google Scholar]
- 39.Hayakawa T, Kobayashi Y, Cui X, Hirano S. A new metabolic pathway of arsenite: arsenic-glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch Toxicol. 2005;79:183–91. doi: 10.1007/s00204-004-0620-x. [DOI] [PubMed] [Google Scholar]
- 40.McKenna WG, Muschel RJ, Gupta AK, Hahn SM, Bernhard EJ. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene. 2003;22:5866–75. doi: 10.1038/sj.onc.1206699. [DOI] [PubMed] [Google Scholar]
- 41.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, et al. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205–25. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 42.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–12. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoppe-Seyler F, Butz K. Repression of endogenous p53 transactiva-tion function in HeLa cervical carcinoma cells by human papillomavirus type 16 E6, human mdm-2, and mutant p53. J Virol. 1993;67:3111–7. doi: 10.1128/jvi.67.6.3111-3117.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–35. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, et al. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science. 2010;327:593–6. doi: 10.1126/science.1166202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.