

Abstract
Achieving selective C–H activation at a single and strategic site in the presence of multiple C–H bonds can provide a powerful and generally useful retrosynthetic disconnection. In this context, the directing group serves as a compass to guide the transition metal to C–H bonds using distance and geometry as powerful recognition parameters to distinguish between proximal and distal C–H bonds. However, the installation and removal of directing groups is a practical drawback. To improve the utility of this approach, one can seek solutions in three directions. First, simplifying the directing group; Second, use of common functional groups or protecting groups as directing groups; Third, attaching the directing group to substrates via a transient covalent bond to render directing groups catalytic. This review describes the rational development of an extremely simple and yet broadly applicable directing group for Pd(II), Rh(III) and Ru(II) catalysts, namely, the N-methoxy amide (CONHOMe). Through collective efforts in the community, a wide range of C–H activation transformations using this type of simple directing groups has been developed.
Keywords: Directing groups, C–functionalization, Pd catalysis, Rh catalysis, Ru catalysis
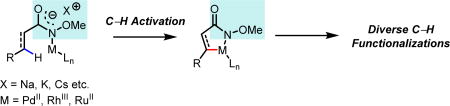
This review describes the development of and emergence of a simple and versatile amide directing group for a wide range of C–H functionalization reactions using Pd, Rh and Ru catalysis. The initial choice of the N-methoxy amide as the directing group for Pd-catalyzed sp2 and sp3 C–H activation is based upon the studies on the alkali carboxylate coordination with Pd catalysts. The application and modification of this simple directing group has also led to the discovery of an array of novel Rh(III)-catalyzed C–H activation reactions. The successful development of enantioselective hydroarylation of olefins based aryl C–H activation is a highlight. Finally, the use of cheaper ruthenium catalysts with this directing group significantly improves the practicality of these directed C–H activation reactions.
1. Introduction
1.1 Regioselectivity
Transition metal catalyzed carbon-hydrogen bond (C–H) activation and functionalization has become a prominent area of research within synthetic organic chemistry and the last decade has been witness to the development of a plethora of transformations that directly functionalize “inert” C–H bonds.[1] The ubiquitous presence of C–H bonds on organic molecules offers overwhelming opportunities for structural modification via C–H activation. On the other hand, achieving site-selectivity can be enormously challenging due to the same reasons that make C–H functionalization so appealing. The fundamental challenge in addressing this issue is to identify parameters that can be recognized by the interactions between the substrates and catalysts. Inspired by enzymatic oxidation chemistry (Cytochrome P450 oxygenase),[2] extensive efforts have been dedicated to the development of biomimetic catalysts for selective C–H oxidation through recognition of intrinsic electronic and steric biases.[3] Inevitably, such approaches are challenging to adopt when substrates contain multiple C–H bonds possessing similar bond strengths and electronic properties. Alternatively, a potentially useful approach is to develop catalytic systems that can distinguish C–H bonds based on their distal and geometric relationship with respect to an existing functional group.[1i] In this context, the interaction between the catalyst and the existing functional group or installed directing group play a pivotal role in both promoting the C–H activation and controlling the selectivity. To advance this approach, it is essential to develop practical and simple directing groups that have the following attributes: easy to install and remove, small in terms of mass, and relatively weak coordination with metal catalysts so that external ligands can coordinate and exert stronger influence on the metal catalysts, thereby controlling the reactivity, stereoselectivity and site selectivity. While proximity to a functional group would appear to limit the number of C–H bonds that can be activated, the development of directing groups and ligands to activate distal C–H bonds could drastically expand the scope of this approach as demonstrated recently.[4]
1.2 Reactivity
Historically, directed C–H activation with transition metal catalysts has predominantly employed strongly coordinating heteroatoms to achieve reactivity through a chelating effect. In this context, pyridines,[5a,5b] oxazolines[5c,5d] and imines[5e] are widely used as directing groups (eq 1–3, Scheme 1). These directing groups have been shown to be highly efficient at promoting transition metal catalyzed C–H cleavage and have found widespread adoption in the literature,[1,5] partly because these cyclometalation intermediates are thermodynamically stable and easy to isolate. However, due to the thermodynamic stability of the metallacycle complexes formed with strongly coordinating directing groups, at times the functionalization step can be difficult due to a lack of reactivity. One could argue that the relatively poor reactivity of the stable metallacycles significantly hampered the discovery of catalytic conditions for achieving wide range of C–H functionalization transformations.[1i] A critical disadvantage of strongly coordinating directing groups also emerges as the field moves forward to develop ligand-controlled and accelerated C–H activation reactions.[6] Reasons for this are two-fold. First, two equivalents of strongly coordinating substrates could bind to Pd(II)X2 at the same time thereby preventing a ligand from binding. Second, Pd(II) coordinated to electron-donating ligand and substrate may not possess the appropriate electronic properties required for the cleavage of C–H bonds. In light of these considerations, our group has systematically developed an alternative approach whereby weakly coordinating directing groups or existing functional groups are used to direct the metalation of C–H bonds. This strategy allows for the formation of less thermodynamically stable metallacycle complexes that can be more readily functionalized than strongly coordinated complexes, thereby greatly accelerating the discovery of a diverse range of C–H activation transformations. Most importantly, a number of ligands have been discovered to match these weakly coordinating directing groups leading to C–H activation reactions in which the reactivity is primarily or purely driven by the ligands instead of being driven by the directing group.[6] The extensive development of the N-methoxy amide directing group is an exemplary example of this approach. Preliminary rationalization of the superior reactivity of this directing group and the simplicity of installing and removing this directing group will be discussed in section 2 (Palladium Catalysis).
Scheme 1.

Evolution of β-C(sp3)–H functionalizations of aliphatic acids.
1.3 Synthetic disconnection
For the purpose of late-stage diversification of advanced intermediates or final products, it is essential to have different catalysts or strategies that can activate C–H bonds at multiple sites selectively in order to achieve maximum diversity for probing chemical space.[7] On the other hand, achieving selective C–H activation at a single and strategic site proximate to an existing functional group in the presence of multiple C–H bonds is sufficient to provide a powerful and generally useful retrosynthetic disconnection. Indeed, the relationship between the existing and newly created functional groups is a corner stone in retrosynthetic analysis.[8] In this context, it is important that the intended directed C–H activation and functionalization will create commonly encountered structural patterns in synthesis so that the directing group is not a burden, but rather an essential part of the new structural pattern that constitutes the next level of complexity. With this in mind, our first research project focused on β-C(sp3)–H functionalizations of aliphatic acids, mirroring the well-established disconnection involving conjugate addition of α,β-unsaturated carboxylic acids (scheme 2). This review will systematically reveal the evolution of the development of directing groups for β-C(sp3)–H functionalizations of aliphatic acids and the advent of a relatively simple and weakly coordinating N-methoxy amide directing group (Scheme 3). The utility of this directing group in directed C–H activation and functionalization with other substrates and a variety of transition metal catalysts is also discussed. Compatibility of this directing group with a broad range of substrates, transformations, various metal catalysts (Pd, Rh and Ru) and mild conditions (room temperature) showcase the efficiency of this weakly coordinating directing group. Most notably, this directing group can cooperate with a ligand to significantly boost the reactivity in C(sp3)–H activation.
Scheme 2.

Different disconnections (conjugate addition vs C–H functionalization) to the synthesis of β-functionalized aliphatic acids.
Scheme 3.

The rational design of the N-methoxy amide directing group from alkali carboxylate directing group.
2. Palladium Catalysis
Our early development of the catalytic cycle for Pd(II)-catalyzed coupling of C–H bonds with organometallic reagents including organotin[9] and organoboron reagents[10,11a] prompted us to extend this catalytic reaction to simple substrates such as carboxylic acids. While the ortho-C(sp2)–H coupling of benzoic acids and phenyl acetic acids afford high yields,[11] the coupling of the β-C(sp3)–H bonds in aliphatic acids with organoboron compounds is inefficient due to the homo-coupling of the organoboron reagents prior to C–H activation.[11a] Considering the utility and availability of aryl and alkyl boronic acid reagents, we set out to overcome this limitation. We reasoned, while the weakly coordinating potassium carboxylate is capable of directing C–H activation, the side reaction of the aryl or alkyl boronic acids with Pd(II) is more facile. Therefore we need to slightly increase the binding strength of the directing group to sequester the Pd(II) more efficiently.
Based on structural information regarding the coordination of alkali carboxylates with Pd(II),[11,12] we proposed that the alkali carboxylate coordinates with Pd(II) as a neutral σ-donor in the transition state (Scheme 3). We therefore envisioned that a more Lewis basic carboxylate mimetic could bind to Pd(II) more efficiently, minimizing homo-coupling background reactions of boronic acid derivatives in Pd(II)/Pd(0) catalytic manifolds. At the same time, it was hoped that the carboxylate mimetics can adopt a similar coordination mode to facilitate the C–H insertion step. We envisioned that an acidic amide moiety could be deprotonated to form an imidate structure similar to the alkali carboxylate. From a brief search of the literature, we noticed that O-methyl hydroxamic acid had been used as a masked ester in synthesis.[13a,13b] Furthermore, a few examples of ortho-lithiation using this chelating group have also been reported[13c,13d] We reasoned that the well-balanced acidity would allow us to generate an imidate structure under mildly basic conditions. Such an imidate is anticipated to have a slightly improved coordinating ability to Pd(II) as compared to an alkali carboxylate. It is also believed that the sp2 hybridization of the coordinating atom is beneficial for minimizing the dihedral angle between Pd(II) and the target C–H bonds in the desired transition state.[14] The coordination via the anionic nitrogen of the amide could also be responsible for this enhanced reactivity as supported by isolated X-ray structure.[18,19] The facile installation and removal of N-methoxy amide directing group as fully demonstrated recently in β-C(sp3)–H arylation is also an important practical advantage.[27]
With this in mind, we tested the N-methoxy amide directing group for the coupling of β-C(sp3)–H bonds with organoboronic acids in 2008 (Scheme 4). To our delight, this directing group allowed for β-C(sp3)–H alkylation of carboxylate derivatives with alkyl boronic acids and for the first time established a method for C(sp3)–H/C(sp3)–B cross coupling.[15] Notably, this coupling reaction allowed the use of pressurized air as the sole oxidant in this Pd(II)/Pd(0) catalytic cycle.
Scheme 4.

The first report of Pd(II)-catalyzed C(sp3)–H cross-coupling with aryl(alkyl) boronic acids via Pd(II)/Pd(0) catalysis using the N-methoxy amide directing group (Yu et al., 2008).[15] BQ = benzoquinone, tetramethylTHF = 2,2,5,5-tetramethyl tetrahydrofuran.
The utility of this directing group was further examined by our group in the same year to affect an intramolecular C(sp2)–H amidation and an intermolecular C(sp3)–H chlorination via Pd(II)/Pd(0) catalysis (Scheme 5).[16] The success of these two reactions hinged on the use of the N-methoxy amide and suggested that this directing group might be generally applicable to both C(sp2)–H and C(sp3)–H activation systems.
Scheme 5.

Pd(II)-catalyzed C(sp2)–H amidation and C(sp3)–H chlorination via Pd(II)/Pd(0) catalysis using the N-methoxy amide directing group (Yu et al., 2008).[16] DCE = 1,2-dichloroethane.
In 2009, Wang and coworkers reported an efficient C(sp2)–H alkoxylation reaction using the N-methoxy amide directing group in the presence of a strong external oxidant. This reaction is proposed to proceed through a Pd(II)/Pd(IV) catalytic cycle due to the requirement of a strong oxidant to promote the catalysis (Scheme 6).[17] Interestingly, when screening oxidants for the direct C(sp2)–H methoxylation of benzoic acid using this directing group, they found that PhI(OAc)2 only gave trace oxidation products. Considering the structural similarity between oximes and the imidate form of the N-methoxy amide, it was surprising to the authors that no product was formed with this oxidant, even though this oxidant is commonly used in oxime directed C–H oxidations.[5e] This result exemplifies the fact that the N-methoxy amide does not behave in the same ways as oxime directing groups and should not necessarily be considered kin to these strongly coordinating directing groups. After further screening, Wang and coworkers achieved the direct alkoxylation of benzoic acid derivatives utilizing the N-methoxy amide directing group in a Pd(II)/Pd(IV) catalytic system with K2S2O8 as the bystanding oxidant.
Scheme 6.

The first example of Pd(II)-catalyzed C(sp2)–H alkoxylation via Pd(II)/Pd(IV) catalysis using the N-methoxy amide directing group (Wang et al., 2010).[17]
In 2011, the Wang group further showcased the robustness of this directing group to a variety of redox cycles by combining a directed Pd(II)/Pd(IV) C(sp2)–H arylation with an intramolecular Pd(II)/Pd(0) C(sp2)–H amidation to provide phenanthridinones from benzoic acid derivatives containing the N-methoxy amide directing group and aryl iodides (Scheme 7)[18a] Notably, a stable five-membered Pd(II) complex was obtained after C–H cleavage in which the N-methoxy amide is an X type directing group. However, the N-methoxy amide may coordinate to Pd(II) catalyst as a neutral (L type) directing group in the form of an imidate in transition state for the C–H activation step as shown in a related acidic amide directing group (Scheme 3).[19] Subsequent isomerization of the directing group from an imidate structure to more stable amide structure could account for the formation of this palladacycle. Very recently, the Bhanage group reported a similar approach by replacing aryl iodides with aryl diazonium salts generated in situ by the diazotization of anilines in the presence of tert-butyl nitrite (t-BuONO).[18b]
Scheme 7.

Pd(II)-catalyzed dual C(sp2)–H activation via Pd(II)/Pd(IV) and Pd(II)/Pd(0) catalysis using the N-methoxy amide directing group (Wang et al., 2011).[18a]
In the same year, the Cheng group developed an alternative strategy to the synthesis of phenanthridinones through the oxidative coupling of N-methoxy benzamide derivatives and arene solvents (Scheme 8).[20] Though the use of arene as a solvent was required for this reaction, the conditions were surprisingly mild providing the desired products after three separate C(sp2)–H activations at room temperature. The efficiency of the N-methoxy amide directing group was demonstrated uniquely in this reaction by enabling these three C–H activations at such a mild temperature. Together, these two methods developed in 2011 provide rapid access to phenanthridinone derivatives.
Scheme 8.

Pd(II)-catalyzed triple C(sp2)–H activation via Pd(II)/Pd(IV) and Pd(II)/Pd(0) catalysis using the N-methoxy amide directing group (Cheng et al., 2011).[20] TFA = trifluoroacetic acid.
Also in 2011, Brooker-Milburn and Lloyd-Jones published a collaborative paper disclosing the tandem C(sp2)–H olefination/Wacker oxidation and C(sp2)–H carbonylation of N-methoxy benzamide substrates (Scheme 9).[21a] Worthy of mention in this particular paper is the observation of a subsequent intramolecular Wacker oxidation after an initial C(sp2)–H olefination reaction. This is in stark contrast to the use of other acidic amide directed olefinations at the time, as cyclization post C(sp2)–H activation typically occurs through conjugate addition. Notably, this is also in contrast to rhodium systems (discussed below) that had been disclosed with this directing group which provides solely the olefination product (Scheme 23). Simultaneously, Wang and coworkers reported the C(sp2)–H olefination of N-methoxy benzamide substrates to afford isoindolinones.[21b]
Scheme 9.

Pd(II)-catalyzed C(sp2)–H olefination and carbonylation via Pd(II)/Pd(0) catalysis using the N-methoxy amide directing group (Lloyd-Jones, Booker-Milburn and Wang et al., 2011).[21a,21b]
Scheme 23.

Rh(III)-catalyzed C(sp2)–H annulation with 1,3-diynes using the N-pivaloyloxy amide directing group (Glorius et al., 2014) [37]
Internal alkynes were also introduced as coupling partners to make isoquinolinones by Huang and coworkers via Pd(II)/Pd(0) catalysis (Scheme 10).[22] Interestingly, the examination of additives showed that alkali metal salts were beneficial to this reaction. Among these salts, NaI·2H2O was the most effective additive. Notably, atomspheric air was the sole oxidant in this reaction to oxidize Pd(0) and regenerate Pd(II) catalyst. Finally, it was determined that this reaction favored the formation of products wherein the large group on an unsymmetrical alkyne is positioned adjacent to the nitrogen atom.
Scheme 10.
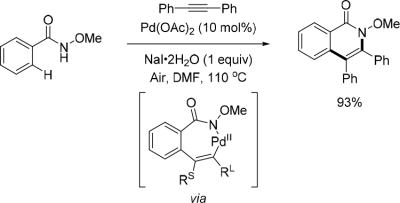
Pd(II)-catalyzed C(sp2)–H annulation with internal alkynes via Pd(II)/Pd(0) catalysis using the N-methoxy amide directing group (Huang et al., 2012).[22] DMF = dimethylformamide, RS = small group, RL = large group.
Another beautiful example of palladium catalysis was demonstrated by Zhao and coworker by achieving the C(sp2)–H acylation of N-methoxy benzamides using aldehydes as coupling partners in 2013 (Scheme 11).[23a] Subsequent cyclization afforded the hydroxyl isoindolones. Both aromatic and aliphatic aldehydes were employed, indicating a broad coupling partner scope. Notably, mechanistic study revealed that a radical process is likely to be involved as adding a radical scavenger shut down the reaction. In 2016, the Zhang group disclosed a similar reaction by using large excess amount of toluene derivatives as corresponding benzaldehyde equivalents in the presence of TBHP.[23b]
Scheme 11.

Pd(II)-catalyzed C(sp2)–H annulation with aldehydes using the N-methoxy amide directing group (Zhao and Huang et al., 2013).[23a] TBHP = tert-butyl hydroperoxide.
Inspired by the annulation reaction of internal alkynes, more recently, the Jeganmohan group[24a] and the Xu group[24b] independently reported a Pd(II)-catalyzed C(sp2)–H activation with arynes by adopting the N-methoxy amide as the directing group affording the phenanthridinone derivatives (Scheme 12).
Scheme 12.

Pd(II)-catalyzed C(sp2)–H annulation with arynes via Pd(II)/Pd(0) catalysis using the N-methoxy amide directing group (Jeganmohan and Xu et al., 2014).[24a,24b] TBAB = tetra-n-butylammonium bromide, DMSO = dimethyl sulfoxide.
A significant challenge for C–H functionalization is achieving robust control of positional selectivity. The use of a weakly coordinating directing group to control the selectivity would seem, at the fundamental level, problematic with the majority of medicinally important heterocyclic substrates as the strongly coordinating heteroatoms can interfere with the catalyst leading to either poisoning or undesired selectivity. However, to overcome this limitation when using this directing group, we envisioned that this acidic amide directing group could serve as an acetate surrogate to assist the aerobic oxidation of Pd(0) to Pd(II) thereby anchoring the catalyst and cleaving the proximal C–H bonds despite the presence of the Lewis basic heterocycles. We anticipated that the key to this approach is to deprive the reaction conditions of any external anions and to use a Pd(0) pre-catalyst. With this idea in mind, in 2014, we developed an aerobic C(sp2)–H annulation with isocyanide using the N-methoxy amide directing group that effectively overcomes catalyst poisoning by heterocycles and overrides the commonly observed positional selectivity dictated by heterocycles. The success of this reaction with pyridine-containing substrates demonstrates that the interference from the heterocycles in C–H functionalization methods can be overcome by choice of directing group and catalytic conditions (Scheme 13).[25]
Scheme 13.

Pd(II)-catalyzed C(sp2)–H annulation with isocyanide using the N-methoxy amide directing group (Dai and Yu et al., 2014).[25] dba = dibenzylideneacetone.
Later, our group found this strategy could be applied to olefinic C–H activation. Different from β-olefinic C–H activation involving the same five-membered cyclometalation intermediates with directed aromatic C–H activation. This time, the palladium catalyst selectively cleaves α-olefinic C–H bonds of α,β-unsaturated olefins for the first time due to the special reaction mechanism (Scheme 14).[26] A range of 4-imino-β-lactams can be readily synthesized through this method. Notably, the β-olefinic C–H activation can also proceed when the α-position of α,β-unsaturated olefins is blocked.
Scheme 14.

Pd(II)-catalyzed C(sp2)–H annulation with isocyanide using the N-methoxy amide directing group (Dai and Yu et al., 2016).[26] dba = dibenzylideneacetone.
While exploitation of the C–H activation reactivity solely driven by the directing group has proven to be productive in developing a variety of transformations, it is crucial for the field to achieve C–H activation reactions in which the reactivity is enabled by ligands. This is of great importance moving forward as both enantioselectivity and regioselectivity can be potentially controlled by catalytic amounts of ligands. As an attempt to address this, in 2014, a ligand-controlled β-C(sp3)–H mono- or di-arylation of alanine derivatives with an acidic amide (CONHArF, ArF = 4-CF3(C6F4)) as the directing group was developed by our group.[6g] However, the directing group utilized with this protocol was rather difficult to remove when appended to sterically bulky substrates, such as the products arising from di-arylation. In order to enhance the practicality of this method, we developed conditions and ligands that would enable us to utilize the N-methoxy amide auxiliary for this transformation as it is far easier to remove (Scheme 15).[27] The facile removal of the N-methoxy amide to provide the corresponding ester or acid proceeds with quantitative yield and complete retention of chirality, demonstrating the usefulness of this directing group. A range of β-(hetero)aryl alanine derivatives were assembled rapidly through C(sp3)–H activation utilizing this methodology. The N-methoxy amide was also capable of activating the secondary benzylic C(sp3)–H bond of the phenylalanine derivatives by a simple switch of the ligand. Using this method a series of β,β’-homo/hetero-diaryl alanines could be obtained in a one-pot fashion. The N-methoxy amide was easily converted to the methyl ester with full retention of the chirality.
Scheme 15.

Ligand-controlled Pd(II)-catalyzed C(sp3)–H arylation of α-amino acids via Pd(II)/Pd(IV) catalysis using the N-methoxy amide directing group (Yu et al., 2015).[27] 2-picoline = 2-methylpyridine, 2,6-lutidine = 2,6-dimethylpyridine, HFIP = hexafluoro-2-propanol.
In 2015, the Kanai group reported the first example of Pd(II)-catalyzed C(sp2)–H functionalization with epoxides under very mild conditions by using the pyridyl directing group. Interestingly, they found the N-methoxy amide directing group can also be used in this reaction (Scheme 16).[28] Several 3-substituted isochroman-1-ones were synthesized through C–H alkylation with epoxides and subsequent intramolecular condensation.
Scheme 16.

Pd(II)-catalyzed C(sp2)–H annulation with epoxides using the N-methoxy amide directing group (Kanai et al., 2015).[28] HFIP = hexafluoro-2-propanol.
It is clear that the N-methoxy amide directing group has found great utility in palladium catalysis with significant contribution to heterocycle synthesis via C(sp2)–H annulation with various coupling partners. Both Pd(II)/Pd(0) and Pd(II)/Pd(IV) catalytic cycles have been shown to be compatible with this directing group, demonstrating the versatility of this auxiliary for a variety of catalysis. As the ligand effects for this directing group begin to become clear, as shown by our group very recently, many new transformations may become possible with palladium catalysis. Given the versatility of this directing group, it is unsurprising that it has also gained popularity in C–H activation reactions catalyzed by other metal catalysts as discussed below.
3. Rhodium Catalysis
3.1 Rh(III)-catalyzed C(sp2)–H activation with alkynes
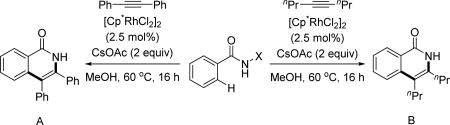
Owing to its simplicity and finely tuned coordination strength, the N-methoxy amide directing group has also been widely adopted in the field of rhodium catalyzed C–H activation. The first Rh(III)-catalyzed C(sp2)–H activation utilizing the N-methoxy amide was disclosed by Fagnou and Guimond for the synthesis of nitrogen-containing heterocycles via an annulation with alkynes in 2010 (Table 1).[29] Their initial choice of conditions was influenced by previously developed methods for Rh(III)-catalyzed C(sp2)–H annulations with alkynes using other directing groups,[30,31,32] but they found that when utilizing the N-methoxy amide directing group a mixture of products were obtained. Initially, the minor product was misassigned as a C–N cyclization product. This side product was later correctly identified by Huang[22] to arise from C–O cyclization with the retention of the N–OMe moiety (Table 1) which has significant mechanistic implication. Intriguingly, the O-cyclized product appears to be formed through a direct reductive elimination from a Rh(III) center forging the C–O bond, which is consistent with an alkali carboxylate-type coordination mode (Scheme 3). It is likely that in the case of C–O bond reductive elimination that Rh(I) is oxidized by Cu(OAc)2·H2O to regenerate the Rh(III) catalyst and close the catalytic cycle. Meanwhile, the desired isoquinolone product was obtained via C–N bond formation, after which the Rh(I) was oxidized to Rh(III) by N–O bond cleavage. Recognizing this, Cu(OAc)2·H2O was replaced with redox innocent CsOAc to improve the selectivity and the isoquinolone was formed cleanly in high yields. This result indicated that the N-methoxy amide directing group could act as an internal oxidant in the absence of an external oxidant. With the optimal conditions in hand, a range of benzoic acid derivatives and internal alkynes were employed to afford isoquinolones. It is interesting to note that, in the presence of an appropriate external oxidant, Pd(II)/Pd(0) catalysis provides the C–N bond forming product without cleaving the N–OMe bond (Scheme 10), whereas the Rh(III)/Rh(I) catalysis yielded the C–O bond forming side product.[22] It is likely Pd(II) favors coordination by the nitrogen atom of the directing group, whereas Rh(III) equilibrates between Rh–O and Rh–N bound isomers (Scheme 17). Notably, this newly established system has proven to be very general in rhodium and ruthenium catalysis.
Table 1.
The first report of Rh(III)-catalyzed C(sp2)–H annulation with internal alkynes using the N-methoxy amide directing group (Guimond and Fagnou et. al., 2010).[29]
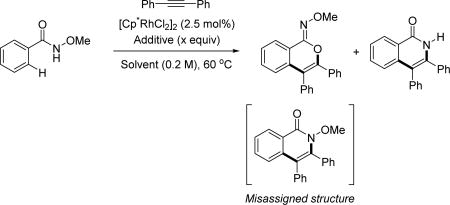
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Solvent | Additive (x equiv) | % Yield[a] (O+N)-CP[c] |
Ratio (O:N)-CP[c] |
| 1 | DMF | Cu(OAc)2·H2O (2) | 89 | 1:1.1 |
| 2 | DMF | CsOAc (2) | 38 | 1:20 |
| 3 | MeOH | CsOAc (2) | 97 (92)[b] | 1:20 |
| 4 | MeOH | CsOAc (0.3) | 97 (90)[b] | 1:20 |
1H NMR yield.
Isolated yield.
CP = Cyclized Product.
Cp* = pentamethylcyclopentadienyl.
Scheme 17.

Different coordination modes in rhodium catalysis with the N-methoxy amide directing group.[29]
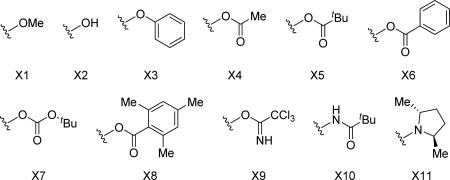
In 2011, Glorius and coworkers explored the reactivity of a related N-pivaloyoxy amide directing group (Scheme 25) and the Fagnou group later applied this directing group to their annulation reaction with internal alkynes.[33] Although the reaction conditions when using the N-methoxy amide were fairly mild to begin with, the authors set out to modify the nature of the directing group to obtain improved conditions which would enable lower catalyst loadings and improved substrate scope. They reasoned that using hydroxamic acids bearing better leaving groups, or groups that could better stabilize intermediates of the catalytic cycle, would be beneficial to the reaction. To ensure that the N-pivaloyoxy amide directing group was optimal, a series of hydroxamic acid derivatives were synthesized and examined under the standard conditions to shed light on the reaction mechanism (Table 2). They found that though most of the directing groups were efficient for coupling with diphenylacetylene, only the O-carboxylate-based directing groups were effective with both internal diarylalkynes and less reactive internal dialkylalkynes. The increased reactivity might arise from the better leaving group ability and the interaction of the carbonyl oxygen lone pair with the rhodium catalyst. With the enhanced N-pivaloyloxy amide directing group, the new system with lower rhodium catalyst loading (as low as 0.5 mol%) at room temperature was established to not only improve the substrate scope of internal alkynes, but also broaden the scope to various terminal alkynes (Scheme 18).
Scheme 25.

Rh(III)-catalyzed C(sp2)–H activation with tertiary propargylic carbonates using the N-methoxy amide directing group (Ma et al., 2015).[39]
Table 2.
The directing group optimization (Guimond and Fagnou et. al., 2011).[33]
| |||||
|---|---|---|---|---|---|
| |||||
|
| |||||
| X | % Yield[a] A |
% Yield[a] B |
X | % Yield[a] A |
% Yield[a] B |
| 1 | 90 | 12 | 7 | 86 | 63 |
| 2 | 52 | n/d | 8 | 78 | 63 |
| 3 | 74 | 14 | 9 | 0 | n/d |
| 4 | 93 | 77 | 10 | 0 | n/d |
| 5 | 98 | 85 | 11 | 0 | n/d |
| 6 | 97 | 85 | |||
1H NMR yield using trimethoxybenzene as an internal standard.
Scheme 18.

Direct comparison between previously reported system and newly designed system (Guimond and Fagnou et al., 2011).[33]
A systematic mechanistic study was carried out to elucidate the reaction mechanism of this annulation reaction. First, they conducted the reaction of the N-methoxy benzamide and diphenylacetylene under the standard reaction conditions in deuterated methanol to partial conversion. The unreacted benzamide and the product were isolated and analyzed by 1H NMR to probe for deuterium incorporation. They found no deuterium incorporation in either of the compounds, which indicated that the C–H activation step was irreversible or that the following step was extremely fast relative to the C–H cleavage. Next, by measuring the initial rate of substrate and deuterated substrate in parallel for the N-methoxy and N-pivaloyoxy amide directing groups, a large KIE value for the N-pivaloyoxy amide directed C–H activation was observed, which is consistent with the conclusion that the C–H activation step was the rate-limiting step. In contrast, there was no kinetic isotope effect for the N-methoxy amide directing group. With these insights and DFT calculation, a stepwise mechanism was proposed to account for the C-N bond formation and N–O bond cleavage by way of a Rh(III)/Rh(I) catalytic cycle (Scheme 19).
Scheme 19.

A stepwise mechanism for the N–O bond cleavage (Guimond and Fagnou et al., 2011).[33]
Subsequently, Park and coworkers tethered the alkyne coupling partner to the substrate so as to construct the indolizidine scaffold via Rh(III)-catalyzed intramolecular C(sp2)–H activation and annulation (Scheme 20) [34] This tethering approach gave entry to the total synthesis of (±)-antofine, (±)-septicine and (±)-tylophorine.
Scheme 20.

Rh(III)-catalyzed intramolecular C(sp2)–H annulation with alkynes and the application in total synthesis (Park et al., 2012).[34] TMS = trimethylsilyl.
In 2012, the Glorius group took the advantage of Rh(III)-catalyzed C(sp2)–H annulation with alkynes and their newly developed N-pivaloyloxy amide directing group to employ an alkynyl MIDA boronate as the coupling partner to make heterocyclic boronic acid derivatives (Scheme 21).[35] Interestingly, the MIDA group was crucial for the reaction to proceed. One limitation for the substrate scope was that internal alkynyl MIDA boronates failed to provide any product. The heterocyclic boronic acid products could be converted to corresponding arylated compounds via a Pd(0)-catalyzed Suzuki cross-coupling reaction.
Scheme 21.

Rh(III)-catalyzed C(sp2)–H annulation with alkyne MIDA boronates using the N-pivaloyloxy amide directing group (Glorius et al., 2012).[35]
Intrigued by Fagnou’s and Park’s work, Lin and coworkers cleverly engineered a 1,6-enyne coupling partner to react with the N-methoxy benzamide or the N-pivaloyloxy benzamide to afford hydrobenzofurans or isoquinolones respectively (Scheme 22).[36] The steric congestion of the cyclohexadienone motif from R2 suppressed the direct Michael addition between the benzamides and the coupling partners. The different reactivity was due to the coordination difference of the N-pivaloyloxy and the N-methoxy directing groups. For the N-pivaloyloxy benzamide, the chelation effect allowed a direct C–N bond reductive elimination followed by an intramolecular conjugate addition of the N–Rh bond to form the product. However, the weaker coordinating ability of the N-methoxy benzamide led to protonation and gave an opened vinyl-Rh(III) intermediate. Subsequently, the C–Rh bond added across to the cyclohexadienone in a Michael addition fashion to afford the corresponding product.
Scheme 22.

Rh(III)-catalyzed C(sp2)–H annulation with 1,6-enynes using the N-methoxy(pivaloyloxy) amide directing group (Tian and Lin et al., 2014).[36]
In 2014, the Glorius group explored the reactivity of 1,3-diynes in Rh(III)-catalyzed C(sp2)–H activation of N-pivaloyloxy benzamides to synthesize diverse bisheterocycles.[37] The challenges encountered by utilizing 1,3-diynes as coupling partners were the control of chemoselectivity, regioselectivity and mono/di selectivity. Gratifyingly, they found the N-pivaloyloxy benzamide could couple with both symmetrical and nonsymmetrical 1,3-diynes with good to excellent selectivities to afford bisisoquinolones. Using a 1:1 ratio of N-pivaloyloxy benzamides to 1,3-diynes under milder conditions, a high degree of mono-selectivity was achieved. Further, the mono-annulated product was subjected to many other substrates under Rh(III)-catalysis and converted to various nonsymmetrical bisheterocycles. This protocol could be adapted to a one-pot procedure to rapidly obtain diverse bisheterocycles (Scheme 23). Based on their observations, it appears the regioselectivity of the migratory insertion was highly affected by the hybridization of the carbon atom and the coordinative ability of the substituents, with the order of preference being alkynyl and isoquinolonyl, phenyl, and finally alkyl groups to remain in the position adjacent to the heteroatom of the heterocycles.
Utilizing N-pivaloyloxy urea as the directing group, the Cui group was able to functionalize the 2-position of indoles and pyrroles with various alkynes by adopting similar conditions to those developed by Fagnou (Scheme 24)[38]
Scheme 24.

Rh(III)-catalyzed C(sp2)–H annulation with alkynes using the N-pivaloyloxy amide directing group (Cui et al., 2014).[38]
In 2015, the Ma group reported an elegant tetra-substituted allene forming method by applying 2-alkynylic carbonates as coupling partners in Rh(III)-catalyzed C(sp2)–H activation of the N-methoxy benzamides. The rational design for the coupling partner is to have a leaving group attached adjacent to the alkynyl moiety might allow the subsequent β-elimination after alkyne insertion to afford the allene. Among several screened leaving groups, the carbonate is found to be the optimal choice presumably due to it is also a suitable directing group to dictate the regioselectivity during alkyne insertion and orientate the Rh(III) center syn to the carbonate leaving group so as to facilitate the syn-β-oxygen elimination. Notably, only tertiary propargylic carbonate can be used in this reaction. As the alkyne insertion and the syn-β-oxygen elimination steps are stereospecific, enantioenriched coupling partners are employed to form chiral tetra-substituted allenes by chirality transfer. (Scheme 25) [39]
3.2 Rh(III)-catalyzed C(sp2)–H activation with alkenes
The Glorius group reported a Rh(III)-catalyzed C(sp2)–H olefination using an N-methoxy amide as the directing group and internal oxidant under a similar set of conditions as those developed for the coupling of alkynes.[40] It is interesting to notice the different reactivity between the N-methoxy amide and the N-pivaloyloxy amide (Scheme 26). With the coordination of the carbonyl group to the rhodium center, the reaction underwent direct C–N bond reductive elimination and subsequent N–O bond cleavage, whereas the β-hydride elimination proceeded without such interaction when using N-methoxy amide. It is likely that, in contrast to the bidentate N-pivaloyloxy amide directing group, the monodentate N-methoxy amide directing group releases Rh(III), allowing C–C rotation and β-hydride elimination. However, the chelating effect from the N-pivaloyoxy amide directing group prevents release of Rh(III), a requisite for β-hydride elimination, thereby promoting the alternate reductive elimination pathway. In both cases, the N–O bond serves as the internal oxidant for this reaction.
Scheme 26.

Rh(III)-catalyzed C(sp2)–H olefination using the N-methoxy(pivaloyloxy) amide directing group (Glorius et al., 2011).[40]
Despite the plethora of Rh(III)-catalyzed C(sp2)–H functionalization reactions developed with many other directing groups in the past few years, asymmetric reactions which are coupled to Rh(III)-catalyzed C(sp2)–H functionalization reactions are rare. This is partly due to the difficulty associated with incorporating an effective chiral ligand in rhodium catalysis. By realizing the strong coordination of cyclopentadienyl (Cp) ligands to rhodium, the Cramer group reasoned that a suitable chiral Cp ligand might effectively control the spatial arrangement of reactants around the rhodium center. Confirming this idea, in 2012, Cramer and coworkers reported the first example of using a class of C2-symmetric Cp ligands that enable the synthesis of chiral dihydroquinolones based on Glorius and Fagnou’s pioneering work on Rh(III)-catalyzed C(sp2)–H annulation with alkenes using the N-methoxy or N-pivaloyloxy amide directing group[41] In designing these chiral rhodium catalysts, several key features of the ligand class are crucial for the origin of the observed enantioselectivity (Scheme 27). For instance, the tethered alkyl groups serve as a back wall that restricts the approach of the reactant, while the locked axial methyl groups serve as side walls that orient substrates to avoid steric clash. Thus, the substrate is oriented in a fixed conformation with the olefin reactant approaching only from one side, resulting in selective coordination of the olefin which provides the subsequent high enantioselectivity.
Scheme 27.

The first example of chiral Cp ligands-enabled asymmetric Rh(III)-catalyzed C(sp2)–H olefination using the N-tert-butoxycarbonyloxy amide directing group (Cramer et al., 2012).[41] DBPO = dibenzoylperoxide.
At the same time, the Rovis group reported a rhodium catalyzed enantioselective coupling of benzamides and alkenes via artificial metalloenzymatic catalysis (Scheme 28).[42] Unlike other chiral ligands, the highly optimized chiral environments within enzymes provide the distinct advantage of increased reactivity and selectivity. However, their structures are usually tailored to target specific biochemical reactions thus limiting its reaction scope. To overcome this limitation, the Rovis group engineered carboxylate residues inside Streptavidin which work in concert with the biotinylated rhodium(III) complex to promote the C(sp2)–H activation step. The installation of a glutamic or aspartic acid group at a specific position inside the chiral cavity of the metalloenzyme provided accelerated reactivity (nearly 100-fold), increased regioselectivity and excellent enantioselectivity (up to 93:7 er).
Scheme 28.

The first example of biotinylated chiral Cp ligand-enabled asymmetric Rh(III)-catalyzed C(sp2)–H olefination using the N-pivaloyoxy amide directing group (Rovis et al., 2012).[42]
In an very interesting example, Molander and coworkers used potassium vinyltrifluoroborate as an olefin partner in Rh(III)-catalyzed C(sp2)–H activation to afford a regioisomerically complementary substitution pattern to other alkenes in related reactions (Scheme 29).[43] The unusual regioselectivity is likely governed by the electronic factors of the potassium vinyltrifluoroborate. The boron substituent could be further converted to hydroxyl group.
Scheme 29.

Rh(III)-catalyzed C(sp2)–H olefination with potassium vinyltrifluoroborate using the N-pivaloyloxy amide directing group (Molander et al., 2013).[43]
The Rh(III)-catalyzed annulation reaction of benzamides is one of the most efficient ways to build five- or six-membered azaheterocycles. However, in 2012, larger rings had not yet been synthesized through this methodology. In 2013, the Glorius group disclosed a rhodium catalyzed method to effectively construct seven-membered azepinones by using α,β-unsaturated aldehydes and ketones as the coupling partners to react with the N-methoxy benzamide via rhodium catalysis (Scheme 30).[44] To gain insight into the mechanism of this reaction, the hemiaminal was independently synthesized under standard reaction conditions at room temperature. This intermediate was treated separately with each individual component from the reaction conditions and it was found only the rhodium catalyst and/or the silver species yielded the dehydration product. In the proposed mechanism, the Rh(III) catalyst undergoes a protonolysis rather than a β-hydride elimination after alkene insertion. The formed Rh–N bond can intramolecularly add across the carbonyl group delivering the cyclorhodated intermediate, which subsequently undergoes protonolysis to afford the seven-membered hemiaminal. Dehydration of the hemiaminal produces the final product. This method was applied to the synthesis of the homoprotoberberine natural product framework.
Scheme 30.

Rh(III)-catalyzed C(sp2)–H annulation with α,β-unsaturated aldehydes and ketones using the N-methoxy amide directing group (Glorius et al., 2013).[44]
In 2013, the Cui group employed highly strained methylenecyclopropanes in Rh(III)-catalyzed C(sp2)–H activation using the N-pivaloyloxy amide directing group to prepare a variety of spiro dihydroisoquinolines (Scheme 31).[45] Interestingly, when N-pivaloyloxy furan-2-carboxamide was used, an unexpected rearrangement occurred to provide the seven-membered azepinone scaffold.
Scheme 31.

Rh(III)-catalyzed C(sp2)–H olefination with methylenecyclopropanes using the N-pivaloyloxy amide directing group (Cui et al., 2013).[45]
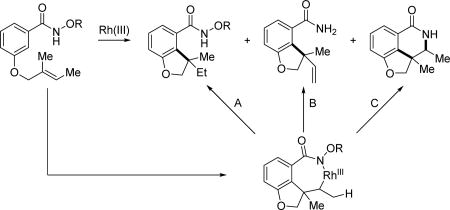
In 2013, the Rovis group systematically studied the directing group effect on Rh(III)-catalyzed intramolecular C(sp2)–H activation with alkenes tethered to the substrate.[46] Three distinct pathways are made possible based on both directing group and conditions. After intramolecular alkene insertion, protonolysis, β-hydride elimination or reductive elimination allow access to different structural scaffolds (Table 3). With the N-methoxy amide directing group, protonolysis and β-hydride elimination are preferred pathways, whereas with the N-pivaloyoxy amide directing group, the reaction pathway favors C-N bond reductive elimination. The rational for the observed selectivity is analogous to that of the simple systems previously discussed (Scheme 26). The Glorius group later also developed a mild and efficient method for the synthesis of fused oligocyclic lactam skeletons via path C.[47a] Very recently, closely related work on aryl spirocycle synthesis via Rh(III)-catalyzed C(sp2)–H activation coupled to an intramolecular Heck-type reaction was published by Chabaud and Guillou (Path B).[47b]
Table 3.
The directing group effect on Rh(III)-catalyzed intramolecular C(sp2)–H olefination (Rovis et al., 2013).[46]
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | R | Condition[a] | Ratio (A:B:C) | % Conv.[b] |
| 1 | Me | A, 17 h | 71:29:0 | 53 |
| 2 | Piv | A, 17 h | 0:0:100 | 61 |
| 3 | Me | B, 2 h | 0:100:0 | 100 |
| 4 | Piv | B, 12 h | 0:22:78 | 100 |
A: [Cp*Rh(MeCN)3](SbF6)2 (1 mol%), PivOH (1 equiv), DCE (0.2 M), 80 °C; B: [Cp*RhCl2]2 (2.5 mol%), CsOAc (2 equiv), MeOH (0.2 M), rt.
Conversion determined by 1H NMR.
Shortly after the report of Rh(III)-catalyzed intramolecular C(sp2)–H olefination by Rovis, the Cramer group further expanded this new chiral Cp ligand class by introducing a sterically tunable biaryl backbone to achieve the reaction in an enantioselective fashion (Scheme 32).[48] Tuning the size of the R group on the biaryl backbone enabled an enantioselective hydroarylation of tethered alkenes, providing enantioenriched dihydrobenzofurans derivatives. For the hydroarylation reaction, it was revealed that the meta-alkoxy group was serving as a secondary directing group, thereby allowing activation of the more hindered ortho-C(sp2)–H bond. The ability to modularly tune the sterics of this Cp ligand should, in principle, allow a plethora of other enantioselective transformations coupled with C–H activation to be realized.
Scheme 32.

Chiral Cp ligand-enabled asymmetric intramolecular Rh(III)-catalyzed C(sp2)–H olefination using the N-methoxy amide directing group (Cramer et al., 2014).[48] Bz = benzoyl, DCM = dichloromethane.
In 2014, the Marsden group studied the behavior of electron-rich alkenes in Rh(III)-catalyzed C(sp2)–H activation.[49] They found vinyl ether was a convenient acetylene equivalent to facilitate the synthesis of 3,4-unsubstituted isoquinolones. Based on their observation, they claimed that electronic factors favored formation of intermediate A by migration of rhodium to the more electron-rich carbon of the enol ethers. However, the regioselectivity was reversed when coordinating groups were present possibly due to directing from OAc group (Scheme 33).
Scheme 33.

Rationale for the regioselectivity of electron-rich alkenes in Rh(III)-catalyzed C(sp2)–H olefination using the N-pivaloyoxy amide directing group (Marsden et al., 2014).[49]
The regioselectivity for the insertion step of styrene derivatives in Rh(III)-catalyzed C(sp2)–H olefination is generally determined by intrinsic substrate control. However, the Cramer group reported a Cp ligand-controlled route enabling regiodivergent synthesis of 3- or 4-aryldihydroisoquinolones (Scheme 34).[50]
Scheme 34.

Cp ligand-controlled regiodivergent Rh(III)-catalyzed C(sp2)–H olefination using the N-tert-butoxycarbonyloxy amide directing group (Cramer et al., 2014)[50]
In 2014, the Wang group utilized a modified terminal olefin to allow a Rh(III)-catalyzed C(sp2)–H allylation of the N-methoxy benzamide substrate (Scheme 35) [51] When R1 = R2 = H or Me, the E/Z ratio for the product was generally greater than 20:1. However, when R1 or R2 was an alkyl or aryl group and the other was H, the E/Z ratio became modest. A tentative mechanism was proposed that implicated a β-oxygen elimination to release CO2 after the alkene insertion.
Scheme 35.

Rh(III)-catalyzed C(sp2)–H allylation with 4-vinyl-1,3-dioxolan-2-ones using the N-methoxy amide directing group (Wang et al., 2014).[51]
By using the N-pivaloyloxy urea directing group, the Cui group was able to functionalize the 2-position of indoles and pyrroles with a variety of alkenes under similar conditions as those used by Fagnou and Glorius (Scheme 36) [38] Impressively, ethylene gas could be used as a coupling partner in this reaction.
Scheme 36.

Rh(III)-catalyzed C(sp2)–H annulation with alkenes using the N-pivaloyloxy amide directing group (Cui et al., 2014).[38]
More recently, the Wang group employed a versatile vinylcyclopropane in the N-methoxy amide directed Rh(III)-catalyzed C(sp2)–H activation to target the allylated product (Scheme 37).[52] Tuning the steric bulk of the ester led to good E/Z selectivity of the product. The reaction pathway likely involves a β-carbon elimination to open the cyclopropane ring.
Scheme 37.

Rh(III)-catalyzed C(sp2)–H allylation with vinylcyclopropanes using the N-methoxy amide directing group (Wang et al., 2015).[52]
Although the alkene insertion into the C–Rh bond derived from C(sp2)–H activation provides numerous useful reactions, when dealing with sterically less demanding terminal alkenes, such as 1-decene, the regioselectivity is commonly problematic. Recently, the Rovis group creatively designed a bulky di-tert-butyl substituted cyclopentadienyl ligand to enable a regioselective synthesis of dihydroisoquinolones.[53] This Cp ligand significantly improved the regioselectivity for migratory insertion that was obtained when compared to the commonly employed Cp* ligand and other Cp ligands (Table 4). Meanwhile, the regioselectivity for the C(sp2)–H activation step decreased when dealing with meta-substituted substrates, which can be explained by the uneven distribution of steric bulk in the Cp3 ligand. Notably, when treating with bulky vinylcyclohexane, alternate regioisomer B was obtained with good selectivity (Scheme 38).
Table 4.
The Cp ligand design for regioselective Rh(III)-catalyzed C(sp2)–H annulation with alkyl substituted terminal alkenes using the N-pivaloyoxy amide directing group (Rovis et. al., 2015).[53]

| |||
|---|---|---|---|
|
| |||
| Entry | Catalyst | % Yield[a] | Regioselectivity |
| 1 | [Cp*RhCl2]2 | 90 | 2.4:1 |
| 2 | [Cp1RhCl2]2 | 85 | 2.4:1 |
| 3 | [Cp2RhCl2]2 | 82 | 12:1 |
| 4 | [Cp3RhCl2]2 | 92 | 15:1 |
1H NMR yield.
Scheme 38.

Rh(III)-catalyzed C(sp2)–H annulation with vinylcyclohexane with different catalysts using the N-pivaloyoxy amide directing group (Rovis et al., 2015).[53]
Based on Glorius and Fagnou’s work, the Xu group extended the alkene coupling partner scope to quinones in order to synthesize dibenzo[b,d]pyran-6-ones (Scheme 39).[54] The N-methoxy amide directing group was crucial for the Rh(III)-catalyzed C(sp2)–H activation. The authors used iodosylbenzene to oxidize hydroquinones to generate quinones in situ.
Scheme 39.

Rh(III)-catalyzed C(sp2)–H annulation with hydroquinones using the N-methoxy amide directing group (Xu et al., 2015).[54]
3.3 Rh(III)-catalyzed C(sp2)–H activation with allenes
Until 2012, only alkynes and alkenes had been shown to be efficient coupling partners in Rh(III)-catalyzed C(sp2)–H activation with both the N-methoxy and N-pivaloyoxy amide directing groups, but their allene cousins had not been demonstrated as effective coupling partners. To address this gap in methodology, the Glorius group reported their exploration into the reactivity of allenes with these two prevalent directing groups and Rh(III) catalysts (Scheme 40).[55] Surprisingly, no desired product was observed when reacting N-methoxy benzamide with cyclohexylallene under standard conditions. Fortunately, the N-pivaloyloxy amide was able to give the allene insertion product. For mono-substituted allenes, the regioselectivity was determined as depicted in scheme 40. However, for sterically more congested allenes, different regioselectivity was obtained.
Scheme 40.

Rh(III)-catalyzed C(sp2)–H annulation with allenes using the N-pivaloyoxy amide directing group and the rationale for regioselectivity of the insertion step (Glorius et al., 2012).[55]
Simultaneously, the Ma group exploited the Rh(III)-catalyzed C(sp2)–H activation with 1,1-disubstituted and trisubstituted allenes using the N-methoxy amide directing group (Scheme 41).[56] A slight modification of the reaction conditions enabled the N-methoxy amide directing group to achieve this reaction and the regioselectivity for migratory insertion was consistent with Glorius’s result for sterically hindered allenes. However, with the N-methoxy amide directing group, protonolysis occurred after allene insertion instead of direct reductive elimination allowing redox neutral catalysis. Notably, this reaction is highly mono selective, enabling a stepwise allylation of benzoic acids.
Scheme 41.

Stepwise Rh(III)-catalyzed C(sp2)–H annulation with allenes using the N-methoxy amide directing group (Ma et al., 2012).[56]
Serendipitously, the Ma group found when simply changing one of the substituents of 1,1-disubstituted allene to a TMS group, the reaction pathway for Rh(III)-catalyzed C(sp2)–H activation with 1,1-disubstituted allenes was completely changed (Scheme 42).[57] Interestingly, this type of allene behaved like a terminal olefin in related reactions. After allene insertion, the Rh(III) intermediate underwent β-H elimination rather than protonolysis or reductive elimination to afford trisubstituted allenylsilanes via Rh(III)/Rh(I) catalysis.
Scheme 42.

Rh(III)-catalyzed C(sp2)–H allenylation with allenylsilanes using the N-methoxy amide directing group (Ma et al., 2013).[57] TMS = trimethylsilyl.
Shortly after Glorius and Ma established Rh(III)-catalyzed C(sp2)–H activation with allenes, the Cramer group set out to find suitable chiral Cp ligands to address the ability to perform this reaction with an enantioselective insertion step (Scheme 43) [58] By tuning the size of the R group on the biaryl backbone of the modified Cp ligand, the enantioselective allylation of benzamides could be achieved.
Scheme 43.

Chiral Cp ligand-enabled asymmetric Rh(III)-catalyzed C(sp2)–H allenylation using the N-methoxy amide directing group (Cramer et al., 2013).[58]
In 2013, the Ma group combined the robustness of Rh(III)-catalyzed C(sp2)–H activation with the N-methoxy amide directing group and the versatile reactivity of allenes disclosing a cascade C(sp2)–H activation with allenols to provide 2,5-dihydrofuran derivatives (Scheme 44).[59]
Scheme 44.

Rh(III)-catalyzed C(sp2)–H activation with allenols using the N-methoxy amide directing group (Ma et al., 2013).[59] Cy = cyclohexyl.
More recently, the Ma group exploited 1,6-allene-enes as coupling partners in Rh(III)-catalyzed C(sp2)–H activation with the N-pivaloyloxy amide directing group to construct eight-membered lactams (Scheme 45).[60]
Scheme 45.

Rh(III)-catalyzed C(sp2)–H activation with 1,6-alleneenes using the N-pivaloyoxy amide directing group (Ma et al., 2015).[60]
3.4 Rh(III)-catalyzed C(sp2)–H cross-coupling with organometallic reagents
Although Pd-catalyzed coupling of C–H bonds with organometallic reagents have been extensively studied since 2005,[9,10,11] analogous transformation using Rh(III) catalysts have not been fully established. In 2012, the Cheng group reported a rare example of such a catalytic transformation through which phenanthridinones could be prepared from the Rh(III)-catalyzed C(sp2)–H coupling with aryl boronic acids using the N-methoxy amide directing group. The reaction proceeds via Rh(III)/Rh(I) catalysis in a similar fashion to that of the Pd(II)/Pd(0) redox catalysis (Scheme 46).[61] Ag2O was used as the external oxidant to enable the dual C(sp2)–H activation. Interestingly, product arising from C–N reductive elimination was the sole product, unlike the original report by Fagnou wherein C–N and C–O reductive elimination proceeded in the presence of an external oxidant. The KIE values were measured by conducting intermolecular KIE experiments for both the substrate and the coupling partner in parallel and used to determine the first C–H activation was the rate-limiting step.
Scheme 46.

Rh(III)-catalyzed C(sp2)–H cross-coupling with aryl boronic acids using the N-methoxy amide directing group (Cheng et al., 2012).[61]
After developing the Rh(III)-catalyzed C(sp2)–H activation and cross-coupling with organoboronic acids, utilizing the N-methoxy benzamides as substrates, the Cheng group further showcased the that aryl silanes served as efficient coupling partners in the same catalytic system to provide a complementary method for phenanthridinones synthesis (Scheme 47).[62] AgF was used to promote the transmetalation of arylsilanes and may serve a secondary role as an external oxidant.
Scheme 47.

Rh(III)-catalyzed C(sp2)–H cross-coupling with arylsilanes using the N-methoxy amide directing group (Cheng et al., 2013).[62]
Based on Cheng’s work on Rh(III)-catalyzed C(sp2)–H cross-coupling with aryl boronic acids utilizing the N-methoxy amide directing group, the Cui group expanded the substrate scope from benzoic acids to indole by attaching the directing group to the indole’s nitrogen atom. By switching the external oxidant, they were able to obtain either 2-arylated product or subsequently cyclized product (Scheme 48).[63]
Scheme 48.

Rh(III)-catalyzed C(sp2)–H cross-coupling with aryl boronic acids using the N-methoxy amide directing group (Cui et al., 2014).[63]
In 2015, the Cheng group expanded the coupling partner scope to organotin compounds by reporting a Rh(III)-catalyzed C(sp2)–H vinylation of the N-methoxy benzamides with vinylstannanes. As the directing group is the internal oxidant, no external oxidant is required in this reaction (Scheme 49).[64]
Scheme 49.

Rh(III)-catalyzed C(sp2)–H vinylation with vinylstannanes using the N-methoxy amide directing group (Cheng et al., 2015).[64]
3.5 Rh(III)-catalyzed C(sp2)–H activation with diazo compounds
As diazo compounds are versatile coupling partners in transition metal catalyzed C(sp2)–H activation, the Rovis group examined the reactivity of donor/acceptor diazo compounds in Rh(III)-catalyzed C(sp2)–H activation with the N-pivaloyoxy amide directing group (Scheme 50).[65] Gratifyingly, a range of γ-lactams could be formed using this method. The mechanism features the generation of a rhodium carbenoid, which can undergo 1,1-migratory insertion to form the six-membered rhodacyclic intermediate. Wing-Yiu Yu and coworkers also reported a similar reaction with an N-acetoxy amide directing group.[66]
Scheme 50.

Rh(III)-catalyzed C(sp2)–H annulation with diazo compounds using the N-pivaloyoxy amide directing group (Rovis et al., 2013).[65]
Following Rovis’s work on Rh(III)-catalyzed C(sp2)–H annulation with diazo compounds, in 2013, the Cui group engineered a cascade reaction for the synthesis of seven-membered azepinones by using vinyl diazo compounds as coupling partners (Scheme 51).[67] The reaction pathway is likely to involve a 1,3-allylic migration after diazo insertion.
Scheme 51.

Rh(III)-catalyzed C(sp2)–H annulation with vinyl diazo compounds using the N-pivaloyoxy amide directing group (Cui et al., 2013).[67]
Interested in further investigating the ability of their chiral Cp ligands to enable enantioselective transformations, the Cramer group re-investigated the annulation reaction using diazo compounds as coupling partners (Scheme 52).[68] Notably, the substitution on the ester group had a significant impact on the enantioselectivity. They also found alkyl substituted diazo compounds performed better than aryl substituted ones in terms of enantioselectivity. After optimization of the ligand structure by tuning the R groups, a series of enantiomerically enriched isoindolones could be obtained utilizing this methodology.
Scheme 52.

Chiral Cp ligand-enabled Rh(III)-catalyzed C(sp2)–H annulation with diazo compounds using the N-pivaloyoxy amide directing group (Cramer et al., 2014).[68] Bz = benzoyl.
After Rovis repoted the Rh(III)-catalyzed C(sp2)–H activation of benzoic acid derivatives with diazo compounds, the Cui group expanded the substrate scope to indoles and pyrroles functionalizing the 2-position with many diazo compounds by attaching the directing group to the indole or pyrrole nitrogen as they have done previously (Scheme 53).[38]
Scheme 53.

Rh(III)-catalyzed C(sp2)–H annulation with diazo compounds using the N-pivaloyoxy amide directing group (Cui et al., 2014).[38]
In an attempt to expand the coupling partner scope for Rh(III)-catalyzed C(sp2)–H activation with diazo compounds, the Cui group found that in situ generation of diazo compounds through the reaction of ketones and hydrazine in the presence of excess MnO2 could expand the scope to donor/donor diazo compounds (Scheme 54).[69] Both aryl alkyl ketones and diaryl ketones were employed to afford isoindolinones.
Scheme 54.

Rh(III)-catalyzed C(sp2)–H annulation with generated in situ diazo compounds using the N-pivaloyoxy amide directing group (Cui et al., 2015).[69]
In 2015, the Yi group discovered the reaction between N-methoxy benzamides and an α-diazotized Meldrum’s acid derivative via Rh(III)-catalyzed C(sp2)–H activation, providing access to N-methoxy isoquinolinediones (Scheme 55).[70]
Scheme 55.

Rh(III)-catalyzed C(sp2)–H annulation with α-diazotized Meldrum’s acid using the N-methoxy amide directing group (Xu and Yi et al., 2015).[70]
3.6 Rh(III)-catalyzed C(sp2)–H amination
In 2012, the Glorius group reported an elegant C–H amination enabled by the N-pivaloyloxy amide directing group via rhodium catalysis under mild conditions (Scheme 56).[71] A series of six-membered heterocyclic amines were employed to give monoaminated benzoic acid derivatives. However, the scope appears to be limited to cyclic amines as only one acyclic amine was reported in low yield. Deuterium incorporation observed in the absence of aminating reagent displayed an efficient and reversible Rh(III)-catalyzed C–H cleavage, whereas no deuterium incorporation was found in both the starting material and the product when conducting the model reaction in deuterated MeOH. This indicated that the C–N bond formation was significantly faster than the C–H activation step. A large KIE value was measured by subjecting a 1:1 ratio of benzamide and the corresponding deuterated benzamide to the standard reaction conditions. A plausible mechanism was proposed to involve an electrophilic amination process after the C–H activation step via a redox neutral catalysis.
Scheme 56.

Rh(III)-catalyzed C(sp2)–H amination using the N-pivaloyloxy amide directing group (Glorius et al., 2012).[71]
The Zhang and Xu group reported a unique hydroxyamidation protocol by using the N-hydroxycarbamate as the amidating reagent under synergistic Rh/Cu catalysis using N-methoxy benzamide substrates (Scheme 57).[72] It was found 10 mol% of CuCl was essential for the reaction to proceed. They speculated that the Cu(I)-catalyzed oxidation of the N-hydroxycarbamate to the nitrosocarbonyl compound which then acted as an electrophile and reacted with the rhodacycle derived from Rh(III)-catalyzed C(sp2)–H activation. The C–Rh bond added across the N=O bond followed by protonolysis and intramolecular condensation furnishing benzo[c]isoxazol-3(1H)-ones in good yields.
Scheme 57.

Rh(III)-catalyzed C(sp2)–H hydroxyamidation using the N-methoxy amide directing group (Xu and Zhang et al., 2015).[72]
3.7 Rh(III)-catalyzed C(sp2)–H activation with other coupling partners
In 2014, the Glorius group first introduced α-halo and pseudohalo ketones as terminal alkyne equivalents to accomplish the synthesis of N-heterocycles via Rh(III)-catalyzed C(sp2)–H activation of N-methoxy benzamide substrates.[73] Among the (pseudo)halides screened (Table 5), Cl, OTs and OMs were effective. The authors proposed a mechanism involving a Rh(III)-catalyzed Grignard-type C–H alkylation, an intramolecular condensation and a dehydration.
Table 5.
Rh(III)-catalyzed C(sp2)–H annulation with α-(pseudo)halo ketones using the N-methoxy amide directing group (Glorius et al., 2014).[73]
Isolated yield.
1H NMR yield.
In 2014, the Loh group first introduced a hypervalent iodine reagent to Rh(III)-catalyzed C(sp2)–H activation with the N-pivaloyloxy amide directing group to achieve an ortho-alkynylation reaction of benzoic acid derivatives (Scheme 58).[74] Notably, the reaction was highly mono selective and compatible with many functional groups on benzamides. However, the scope of alkynes was limited to those containing very sterically hindered groups (TIPS and tBu). It is possible that this hypervalent iodine reagent might behave like an internal alkyne during the alkyne insertion step.
Scheme 58.

Rh(III)-catalyzed C(sp2)–H alkynylation using the N-pivaloyoxy amide directing group (Loh et al., 2014).[74]
In 2015, the Cramer group further showcased the versatility of their chiral Cp ligands by achieving an intramolecular Rh(III)-catalyzed C(sp2)–H activation with tethered aldehydes to afford hydroxychromanes with moderate to good enantioselectivity (Scheme 59).[75]
Scheme 59.

Intramolecular Rh(III)-catalyzed C(sp2)–H annulation with aldehydes using the N-iso-propoxy amide directing group (Cramer et al., 2015).[75] MOM = methoxymethyl.
Since the first demonstration of the simple N-methoxy amide as an efficient directing group for Pd(II)-catalyzed C–H activation reactions, a plethora of Rh(III)-catalyzed C(sp2)–H activation reactions using the N-methoxy or related pivaloyoxy amide directing group have been developed within the last five years. However, to date, this directing group has only been shown to be effective for C(sp2)–H activation with rhodium catalysis. It is likely that future efforts utilizing this directing group will be expanded to the activation of C(sp3)–H bonds as demonstrated in palladium catalysis.[15,16,28]
4. Ruthenium Catalysis
Ru(II) catalysts have also been found to catalyze C–H activation reactions through analogous redox chemistry to that of Pd(II)/Pd(0) and Rh(III)/Rh(I) catalysis. Wang and coworkers reported the first example of using a less-expensive ruthenium catalyst to perform the same annulation with the N-methoxy amide directing group for the synthesis of isoquinolones (Scheme 60).[76] The reaction conditions and substrate scope are similar with the corresponding rhodium catalysis. However, when using meta-substituted benzamide substrates, a noticeable amount of regioisomer generated from hindered C(sp2)–H activation is commonly observed. Especially when the substituent is an electronegative heteroatom, such as oxygen or bromine, the unusual regioisomer becomes dominant. It is proposed the observed selectivity was due to the C–H bond acidity or Ru–C bond stability.
Scheme 60.

The first example of Ru(II)-catalyzed C(sp2)–H annulation with internal alkynes using the N-methoxy amide directing group (Wang et at., 2011).[76] p-cymene = 4-iso-propyltoluene.
Also in 2011, the Ackerman group, following their own extensive studies on Ru(II)-catalyzed C–H activation reactions,[77] further showcased the robustness of the alkyne annulation reaction with H2O as the reaction medium in the presence of a sterically hindered carboxylate salt, KO2CMes (Scheme 61).[78] Using KO2CMes as an additive, the direct use of free hydroxamic acid as the directing group could be achieved. The combination of carboxylate assistance with the N-methoxy amide directing group in ruthenium-catalyzed C(sp2)–H functionalization has enabled mild and environmentally friendly reaction conditions, providing facile access to isoquinolone derivatives. Similar regioselectivity was also observed for meta-substituted substrates, indicating the generality of this trend.
Scheme 61.

Ru(II)-catalyzed C(sp2)–H annulation with internal alkynes using the N-methoxy amide directing group (Ackermann et at., 2011).[78] Mes = mesityl.
By mimicking Rh(III)-catalyzed C(sp2)–H olefination, the Wang group showcased that ruthenium catalysts are also capable of catalyzing the olefination reaction with the N-methoxy amide directing group (Scheme 62).[79] When reacting with acrylate derivatives, similar reactivity with rhodium catalysis was observed. However, cyclized product was formed when styrene or cyclic alkenes were used, indicating that there is indeed a difference in reactivity between rhodium and ruthenium catalysis. The usual regioselectivity for meta-substituted substrates with ruthenium catalysis was also observed.
Scheme 62.

Ru(II)-catalyzed C(sp2)–H activation with alkenes using the N-methoxy amide directing group (Wang et at., 2011).[79]
After extensive study of the Ru(II)-catalyzed C(sp2)–H functionalizations with alkynes and alkenes, allenes were also successfully employed by the Ackermann group in 2015, albeit with relatively limited allene scope when comparing to corresponding rhodium catalysis (Scheme 63).[80] Notably, the reaction becomes sluggish when R are methyl, cyclopropyl, benzyl, or phenyl groups. The bulky TMS group is also crucial for this reaction, replacement with tert-butyl group resulted in lower yield.
Scheme 63.

Ru(II)-catalyzed C(sp2)–H allenylation with allenylsilanes using the N-methoxy amide directing group (Ackermann et at., 2015).[80]
In 2016, the Chegondi group reported a carbonyl-assisted reverse regioselective cascade reaction to build complex molecules via Ru(II)-catalyzed C(sp2)–H annulation of the N-methoxy benzamides with 2-acetylenic ketones (Scheme 64).[81] The intermolecular weak coordination of the carbonyl group to Ru(II) center sets up the regioselectivity for the alkyne insertion. Subsequent reductive elimination and re-oxidation of the catalyst followed by an intramolecular nucleophilic attack from the amide to the carbonyl group delivers the product.
Scheme 64.

Ru(II)-catalyzed C(sp2)–H annulation with 2-acetylenic ketones using the N-methoxy amide directing group (Chegondi et at., 2016).[81]
Though at present Ru(II)-catalyzed reactions using the N-methoxy or N-pivaloyloxy directing group are still under developed, it is reasonable to anticipate further advance in this direction due to its lower cost. Though it may be naive, the question remains how generally can ruthenium, in combination with the N-methoxy or N-pivaloyloxy directing group, replace the more expensive rhodium catalysts that have been used so extensively in the literature. Furthermore, of potentially greater interest will be the discovery of reactivity that diverges from rhodium catalysis.
5. Conclusion
One of the most appealing features of directed C–H activation is that C–H activation can occur highly selectively at a strategically desired position despite the presence of multiple C–H bonds with similar electronic and steric environments. To render this approach broadly useful, the design of simple, effective and broadly applicable directing groups is crucial. This review details the early studies of the coordination mode of alkali carboxylate with Pd(II) catalysts in carboxylate-directed C–H activation and how this understanding provided insight that enabled the development of the N-methoxy amide directing group. A plethora of examples of various C–H activation transformations catalyzed by an array of metal catalysts showcase the extraordinary versatility of the N-methoxy amide and closely related directing groups. It is most exciting that this simple directing group can cooperate with pyridine type ligands to promote C(sp3)–H activation with Pd(II) catalysts. We eagerly anticipate further exploitation of this directing group towards the development of truly practical C–H activation reactions.
Acknowledgments
We gratefully acknowledge TSRI, the NSF (NSF CHE-0615716, the NIH (NIGMS, 1 R01 GM084019). Additional support was provided through the NSF Center for Stereoselective C–H Functionalization (CHE-0943980).
References
- 1.For selected reviews of C–H functionalization, see: Kakiuchi F, Chatani N. Adv. Synth Catal. 2003;345:1077–1101.Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173–1193. doi: 10.1039/b606984n.Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem. 2009;2009;121:5196–5217.Angew. Chem. Int. Ed. 48:5094–5115.Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074–1086. doi: 10.1021/ar9000058.Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e.Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624–655. doi: 10.1021/cr900005n.Yeung CS, Dong VM. Chem. Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d.Wencel-Delord J, Dröge T, Liu F, Glorius F. Chem. Soc. Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a.Engle KM, Mei T-S, Wasa M, Yu J-Q. Acc. Chem. Res. 2012;45:788–802. doi: 10.1021/ar200185g.Engle KM, Yu J-Q. J. Org. Chem. 2013;78:8927–8955. doi: 10.1021/jo400159y.Rouquet G, Chatani N. Angew. Chem. 2013;2013;125:11942–11959. doi: 10.1002/anie.201301451.Angew. Chem. Int. Ed. 52:11726–11743.Ackermann L. Acc. Chem. Res. 2014;47:281–295. doi: 10.1021/ar3002798.
- 2.Ortiz de Montellano PR. Cytochrome P-450. Structure, Mechanism and Biochemistry. 2nd. Plenum Press; New York and London: 1995. [Google Scholar]
- 3.For selected examples of C–H oxidation utilizing biomimetic catalysts, see: Groves JT, Viski P. J. Am. Chem. Soc. 1989;111:8537–8538.Larrow JF, Jacobsen EN. J. Am. Chem. Soc. 1994;116:12129–12130.Kim C, Chen K, Kim J, Que L., Jr J. Am. Chem. Soc. 1997;119:5964–5965.Havranek M, Singh A, Sames D. J. Am. Chem. Soc. 1999;121:8965–8966.Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941–1943. doi: 10.1126/science.1127899.Chen MS, White MC. Science. 2007;318:783–787. doi: 10.1126/science.1148597.
- 4.For selected examples of remote C–H functionalization, see: Leow D, Li G, Mei T-S, Yu J-Q. Nature. 2012;486:518–522. doi: 10.1038/nature11158.Wan L, Dastbaravardeh N, Li G, Yu J-Q. J. Am. Chem. Soc. 2013;135:18056–18059. doi: 10.1021/ja410760f.Tang R-Y, Li G, Yu J-Q. Nature. 2014;507:215–220. doi: 10.1038/nature12963.Li S, Chen G, Feng C-G, Gong W, Yu J-Q. J. Am. Chem. Soc. 2014;136:5267–5270. doi: 10.1021/ja501689j.
- 5.For selected examples of strongly coordinating directing groups utilized in C–H functionalization, for pyridines, see: Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m.Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. For oxazolines, see: Giri R, Chen X, Yu J-Q. Angew. Chem. 2005;117:2150–2153.Angew. Chem. Int. Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884.Giri R, Liang J, Lei J-G, Li J-J, Wang D-H, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q. Angew. Chem. 2004;117:7586–7590.Angew. Chem. Int. Ed. 2004;44:7420–7424. For imines, see: Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c.
- 6.For selected examples of ligand accelerated C–H functionalization, for mono-protected amino acid (MPAA) ligands, see: Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Angew. Chem. 2008;120:4960–4964.Angew. Chem. Int. Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030.Wang D-H, Engle KM, Shi B-F, Yu J-Q. Science. 2010;327:315–319. doi: 10.1126/science.1182512.Engle KM, Wang D-H, Yu J-Q. J. Am. Chem. Soc. 2010;132:14137–14151. doi: 10.1021/ja105044s.Wasa M, Engle KM, Lin DW, Yoo EJ, Yu J-Q. J. Am. Chem. Soc. 2009;133:19598–19601. doi: 10.1021/ja207607s. For pyridine-based ligands, see: Zhang Y-H, Shi B-F, Yu J-Q. J. Am. Chem. Soc. 2009;131:5072–5074. doi: 10.1021/ja900327e.Wasa M, Chan KSL, Zhang X-G, He J, Miura M, Yu J-Q. J. Am. Chem. Soc. 2012;133:18570–18572. doi: 10.1021/ja309325e.He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu J-Q. Science. 2013;343:1216–1220. doi: 10.1126/science.1249198.Wang X-C, Gong W, Fang L-Z, Zhu R-Y, Li S, Engle KM, Yu J-Q. Nature. 2015;519:334–338. doi: 10.1038/nature14214.
- 7.Dai H-X, Stepan AF, Plummer MS, Zhang Y-H, Yu J-Q. J. Am. Chem. Soc. 2011;133:7222–7228. doi: 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- 8.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley-VCH; Weinheim: 1995. [Google Scholar]
- 9.Chen X, Li J-J, Hao X-S, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:78–79. doi: 10.1021/ja0570943. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:12634–12635. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]
- 11.(a) Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J. Am. Chem. Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; (b) Wang D-H, Mei T-S, Yu J-Q. J. Am. Chem. Soc. 2008;130:17676–17677. doi: 10.1021/ja806681z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Thuy-Boun PS, Villa G, Dang D, Richardson P, Su S, Yu J-Q. J. Am. Chem. Soc. 2013;135:17508–17513. doi: 10.1021/ja409014v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:14082–14083. doi: 10.1021/ja8063827. [DOI] [PubMed] [Google Scholar]
- 13.(a) Crawford RJ, Raap R. J. Org. Chem. 1963;28:2419–2424. [Google Scholar]; (b) Cooley JH, Mosher MW, Khan MA. J. Am. Chem. Soc. 1968;90:1867–1871. [Google Scholar]; (c) Misra RN, Botti CM, Haslanger MF, Engebrecht JR, Mahoney EM, Ciosek CP., Jr Bioorg. Med. Chem. Lett. 1991;1:295–298. [Google Scholar]; (d) Fisher LE, Caroon JM, Stabler JSR, Lundberg S, Muchowski JM. J. Org. Chem. 1993;58:3643–3647. [Google Scholar]
- 14.Giri R, Lan Y, Liu P, Houk KN, Yu J-Q. J. Am. Chem. Soc. 2012;134:14118–14126. doi: 10.1021/ja304643e. [DOI] [PubMed] [Google Scholar]
- 15.Wang D-H, Wasa M, Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:7190–7191. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- 16.Wasa M, Yu J-Q. J. Am. Chem. Soc. 2008;130:14058–14059. doi: 10.1021/ja807129e. [DOI] [PubMed] [Google Scholar]
- 17.Wang G-W, Yuan T-T. J. Org. Chem. 2010;75:476–479. doi: 10.1021/jo902139b. [DOI] [PubMed] [Google Scholar]
- 18.(a) Wang G-W, Yuan T-T, Li D-D. Angew. Chem. 2011;2011;123:1416–1419. [Google Scholar]; Angew. Chem. Int. Ed. 50:1380–1383. [Google Scholar]; (b) Yedage SL, Bhanage BM. J. Org. Chem [Google Scholar]
- 19.Zhang X-G, Dai H-X, Wasa M, Yu J-Q. J. Am. Chem. Soc. 2012;134:11948–11951. doi: 10.1021/ja305259n. [DOI] [PubMed] [Google Scholar]
- 20.Karthikeyan J, Cheng C-H. Angew. Chem. 2011;2011;123:10054–10057. [Google Scholar]; Angew. Chem. Int. Ed. 50:9880–9883. [Google Scholar]
- 21.(a) Wrigglesworth JW, Cox B, Lloyd-Jones GC, Booker-Milburn KI. Org. Lett. 2011;13:5326–5329. doi: 10.1021/ol202187h. [DOI] [PubMed] [Google Scholar]; (b) Li D-D, Yuan T-T, Wang G-W. Chem. Commun. 2011;47:12789–12791. doi: 10.1039/c1cc15897j. [DOI] [PubMed] [Google Scholar]
- 22.Zhong H, Yang D, Wang S, Huang J. Chem. Commun. 2012;48:3236–3238. doi: 10.1039/c2cc17859a. [DOI] [PubMed] [Google Scholar]
- 23.(a) Yu Q, Zhang N, Huang J, Lu S, Zhu Y, Yu X, Zhao K. Chem. Eur. J. 2013;19:11184–11188. doi: 10.1002/chem.201302031. [DOI] [PubMed] [Google Scholar]; (b) Yang L, Han L, Xu B, Zhao L, Zhou J, Zhang H. Asian. J. Org. Chem. 2016;5:62–65. [Google Scholar]
- 24.(a) Pimparkar S, Jeganmohan M. Chem. Commun. 2014;50:12116–12119. doi: 10.1039/c4cc05252h. [DOI] [PubMed] [Google Scholar]; (b) Peng X, Wang W, Jiang C, Sun D, Xu Z, Tung C-H. Org. Lett. 2014;16:5354–5357. doi: 10.1021/ol5025426. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y-J, Xu H, Kong W-J, Shang M, Dai H-X, Yu J-Q. Nature. 2014;515:389–393. doi: 10.1038/nature13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong W-J, Liu Y-J, Xu H, Chen Y-Q, Dai H-X, Yu J-Q. J. Am. Chem. Soc. 2016;138:2146–2149. doi: 10.1021/jacs.5b13353. [DOI] [PubMed] [Google Scholar]
- 27.Chen G, Shigenari T, Jain P, Zhang Z, Jin Z, He J, Li S, Mapelli C, Miller MM, Poss MA, Scola PM, Yeung K-S, Yu J-Q. J. Am. Chem. Soc. 2015;137:3338–3351. doi: 10.1021/ja512690x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Kuninobu Y, Kanai M. J. Am. Chem. Soc. 2015;137:6140–6143. doi: 10.1021/jacs.5b02435. [DOI] [PubMed] [Google Scholar]
- 29.Guimond N, Gouliaras C, Fagnou K. J. Am. Chem. Soc. 2010;132:6908–6909. doi: 10.1021/ja102571b. [DOI] [PubMed] [Google Scholar]
- 30.Ueura K, Satoh T, Miura M. J. Org. Chem. 2007;72:5362–5367. doi: 10.1021/jo070735n. [DOI] [PubMed] [Google Scholar]
- 31.Stuart DR, Bertrand-Laperle M, Burgess KMN, Fagnou K. J. Am. Chem. Soc. 2008;130:16474–16475. doi: 10.1021/ja806955s. [DOI] [PubMed] [Google Scholar]
- 32.Guimond N, Fagnou K. J. Am. Chem. Soc. 2009;131:12050–12051. doi: 10.1021/ja904380q. [DOI] [PubMed] [Google Scholar]
- 33.Guimond N, Gorelsky SI, Fagnou K. J. Am. Chem. Soc. 2011;133:6449–6457. doi: 10.1021/ja201143v. [DOI] [PubMed] [Google Scholar]
- 34.X Xu, Liu Y, Park C-M. Angew. Chem. 2012;2012;124:9506–9510. [Google Scholar]; Angew. Chem. Int. Ed. 51:9372–9376. [Google Scholar]
- 35.Wang H, Crohmann C, Nimphius C, Glorius F. J. Am. Chem. Soc. 2012;134:19592–19595. doi: 10.1021/ja310153v. [DOI] [PubMed] [Google Scholar]
- 36.Fukui Y, Liu P, Liu Q, He Z-T, Wu N-Y, Tian P, Lin G-Q. J. Am. Chem. Soc. 2014;136:15607–15614. doi: 10.1021/ja5072702. [DOI] [PubMed] [Google Scholar]
- 37.Yu D-G, de Azambuja F, Gensch T, Daniliuc CG, Glorius F. Angew. Chem. 2014;2014;126:9804–9809. doi: 10.1002/anie.201403782. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 53:9650–9654. [Google Scholar]
- 38.Zhang Y, Zheng J, Cui S. J. Org. Chem. 2014;79:6490–6500. doi: 10.1021/jo500902n. [DOI] [PubMed] [Google Scholar]
- 39.Wu S, Huang X, Wu W, Li P, Fu C, Ma S. Nat. Commun. 2015;6:7946. doi: 10.1038/ncomms8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rakshit S, Grohmann C, Besset T, Glorius F. J. Am. Chem. Soc. 2011;133:2350–2353. doi: 10.1021/ja109676d. [DOI] [PubMed] [Google Scholar]
- 41.Ye B, Cramer N. Science. 2012;338:504–506. doi: 10.1126/science.1226938. [DOI] [PubMed] [Google Scholar]
- 42.Hyster TK, Knörr L, Ward TR, Rovis T. Science. 2012;338:500–503. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Presset M, Oehlrich D, Rombouts F, Molander GA. Org. Lett. 2013;15:1528–1531. doi: 10.1021/ol400307d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi Z, Grohmann C, Glorius F. Angew. Chem. 2013;2013;125:5503–5507. doi: 10.1002/anie.201301426. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 52:5393–5397. [Google Scholar]
- 45.Cui S, Zhang Y, Wu Q. Chem. Sci. 2013;4:3421–3426. [Google Scholar]
- 46.Davis TA, Hyster TK, Rovis T. Angew. Chem. 2013;2013;125:14431–14435. doi: 10.1002/anie.201307631. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 52:14181–14185. [Google Scholar]
- 47.(a) Shi Z, Boultadakis-Arapinis M, Koester DC, Glorius F. Chem. Commun. 2014;50:2650–2652. doi: 10.1039/c4cc00029c. [DOI] [PubMed] [Google Scholar]; (b) Chabaud L, Raynal Q, Barre E, Guillou C. Adv. Synth. Catal. 2015;357:3880–3884. [Google Scholar]
- 48.Ye B, Donets PA, Cramer N. Angew. Chem. 2014;2014;126:517–521. doi: 10.1002/anie.201309207. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 53:507–511. [Google Scholar]
- 49.Webb NJ, Marsden SP, Raw SA. Org. Lett. 2014;16:4718–4721. doi: 10.1021/ol502095z. [DOI] [PubMed] [Google Scholar]
- 50.Wodrich MD, Ye B, Gonthier JF, Corminboeuf C, Cramer N. Chem. Eur. J. 2014;20:15409–15418. doi: 10.1002/chem.201404515. [DOI] [PubMed] [Google Scholar]
- 51.Zhang S-S, Wu J-Q, Lao Y-X, Liu X-G, Liu Y, Lv W-X, Tan D-H, Zeng Y-F, Wang H. Org. Lett. 2014;16:6412–6415. doi: 10.1021/ol503229c. [DOI] [PubMed] [Google Scholar]
- 52.Wu J-Q, Qiu Z-P, Zhang S-S, Liu J-G, Lao Y-X, Gu L-Q, Huang Z-S, Li J, Wang H. Chem. Commun. 2015;51:77–80. doi: 10.1039/c4cc07839j. [DOI] [PubMed] [Google Scholar]
- 53.Hyster TK, Dalton DM, Rovis T. Chem. Sci. 2015;6:254–258. doi: 10.1039/c4sc02590c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang W, Wang S, Zhang Q, Liu Q, Xu X. Chem. Commun. 2015;51:661–664. doi: 10.1039/c4cc08260e. [DOI] [PubMed] [Google Scholar]
- 55.Wang H, Glorius F. Angew. Chem. 2012;2012;124:7430–7434. [Google Scholar]; Angew. Chem. Int. Ed. 51:7318–7322. [Google Scholar]
- 56.Zeng R, Fu C, Ma S. J. Am. Chem. Soc. 2012;134:9597–9600. doi: 10.1021/ja303790s. [DOI] [PubMed] [Google Scholar]
- 57.Zeng R, Wu S, Fu C, Ma S. J. Am. Chem. Soc. 2013;135:18284–18287. doi: 10.1021/ja409861s. [DOI] [PubMed] [Google Scholar]
- 58.Ye B, Cramer N. J. Am. Chem. Soc. 2013;135:636–639. doi: 10.1021/ja311956k. [DOI] [PubMed] [Google Scholar]
- 59.Zeng R, Ye J, Fu C, Ma S. Adv. Synth. Catal. 2013;355:1963–1970. [Google Scholar]
- 60.Wu S, Zeng R, Fu C, Yu Y, Zhang X, Ma S. Chem. Sci. 2015;6:2275–2285. doi: 10.1039/c5sc00092k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karthikeyan J, Haridharan R, Cheng C-H. Angew. Chem. 2012;2012;124:12509–12513. [Google Scholar]; Angew. Chem. Int. Ed. 51:12343–12347. [Google Scholar]
- 62.Senthilkumar N, Parthasarathy K, Gandeepan P, Cheng C-H. Chem. Asian. J. 2013;8:2175–2181. doi: 10.1002/asia.201300356. [DOI] [PubMed] [Google Scholar]
- 63.Zheng J, Zhang Y, Cui S. Org. Lett. 2014;16:3560–3563. doi: 10.1021/ol5014312. [DOI] [PubMed] [Google Scholar]
- 64.Prakash S, Muralirajan K, Cheng C-H. Chem. Commun. 2015;51:13362–13364. doi: 10.1039/c5cc04211a. [DOI] [PubMed] [Google Scholar]
- 65.Hyster TK, Ruhl KE, Rovis T. J. Am. Chem. Soc. 2013;135:5364–5367. doi: 10.1021/ja402274g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lam H-W, Man K-Y, Chan W-W, Zhou Z, Yu W-Y. Org. Biomol. Chem. 2014;12:4112–4116. doi: 10.1039/c4ob00512k. [DOI] [PubMed] [Google Scholar]
- 67.Cui S, Zhang Y, Wang D, Wu Q. Chem. Sci. 2013;4:3912–3916. [Google Scholar]
- 68.Ye B, Cramer N. Angew. Chem. 2014;2014;126:8030–8033. [Google Scholar]; Angew. Chem. Int. Ed. 53:7896–7899. [Google Scholar]
- 69.Zhang Y, Wang D, Cui S. Org. Lett. 2015;17:2494–2497. doi: 10.1021/acs.orglett.5b01016. [DOI] [PubMed] [Google Scholar]
- 70.Shi J, Zhou J, Yan Y, Jia J, Liu X, Song H, Xu HE, Yi W. Chem. Commun. 2015;51:668–671. doi: 10.1039/c4cc08407a. [DOI] [PubMed] [Google Scholar]
- 71.Grohmann C, Wang H, Glorius F. Org. Lett. 2012;14:656–659. doi: 10.1021/ol203353a. [DOI] [PubMed] [Google Scholar]
- 72.Yang W, Sun J, Xu X, Zhang Q, Liu Q. Chem. Commun. 2014;50:4420–4422. doi: 10.1039/c3cc49496a. [DOI] [PubMed] [Google Scholar]
- 73.Yu D-G, de Azambuja F, Glorius F. Angew. Chem. 2013;2013;125:2792–2796. [Google Scholar]; Angew. Chem. Int. Ed. 52:2754–2758. [Google Scholar]
- 74.Feng C, Loh T-P. Angew. Chem. 2014;2014;126:2760–2764. [Google Scholar]; Angew. Chem. Int. Ed. 53:2722–2726. [Google Scholar]
- 75.Ye B, Cramer N. Synlett. 2015;26:1490–1495. [Google Scholar]
- 76.Li B, Feng H, Xu S, Wang B. Chem. Eur. J. 2011;17:12573–12577. doi: 10.1002/chem.201102445. [DOI] [PubMed] [Google Scholar]
- 77.Sarkar SD, Liu W, Kozhushkov SI, Ackermann L. Adv. Synth. Catal. 2014;356:1461–1479. [Google Scholar]
- 78.Ackermann L, Fenner S. Org. Lett. 2011;13:6548–6551. doi: 10.1021/ol202861k. [DOI] [PubMed] [Google Scholar]
- 79.(a) Li B, Ma J, Wang N, Feng H, Xu S, Wang B. Org. Lett. 2012;14:736–739. doi: 10.1021/ol2032575. [DOI] [PubMed] [Google Scholar]; (b) Manikandan R, Madasamy P, Jeganmohan M. ACS Catal. 2016;6:230–234. [Google Scholar]
- 80.Nakanowatari S, Ackermann L. Chem. Eur. J. 2015;21:16246–16251. doi: 10.1002/chem.201502785. [DOI] [PubMed] [Google Scholar]
- 81.Gollapelli KK, Kallepu S, Govindappa N, Nanubolu JB, Chegondi R. Chem. Sci. doi: 10.1039/c6sc01456a. [DOI] [PMC free article] [PubMed] [Google Scholar]