

Abstract
Mass cytometry is a single cell biology technique that samples >500 cells per second, measures >35 features per cell, and is sensitive across a dynamic range of >104 relative intensity units per feature. This combination of technical assets has powered a series of recent cytomic studies where investigators used mass cytometry to measure protein and phospho-protein expression in millions of cells, characterize rare cell types in healthy and diseased tissues, and reveal novel, unexpected cells. However, these advances largely occurred in studies of blood, lymphoid tissues, and bone marrow, since the cells in these tissues are readily obtained in single cell suspensions. This protocol establishes a primer for single cell analysis of solid tumors and tissues and has been tested with mass cytometry. The cells obtained from this protocol can be fixed for study, cryopreserved for long term storage, or perturbed ex vivo to dissect responses to stimuli and inhibitors.
Keywords: Human tissue, human tumor, dissociation, single cell, cytometry
INTRODUCTION
One key method for understanding a tissue or organ is to dissect and identify the diverse cells which comprise it. Flow cytometry excels at quantifying the abundance and protein expression signatures of hundreds to thousands of cells per second (Chattopadhyay et al., 2014; Roussel et al., 2016) and holds great promise for understanding diseases like cancer, where altered protein expression and signaling activity in rare cell subsets can contribute to oncogenesis and drive treatment resistance (Irish and Doxie, 2014; Irish et al., 2006b). The ability of flow cytometry to quantify proteins on each of millions of cells and reveal signaling in rare, 1-in-10,000 cells, has made it indispensable to modern immunology and clinical hematopathology, where cells in suspension are readily obtained. Mass cytometry is a newly developed form of flow cytometry with the ability to measure 35 or more features at a rate of 500 or more cells per second (Bendall et al., 2011; Chattopadhyay et al., 2014; Greenplate et al., 2016a; Irish, 2014; Newell and Cheng, 2016). This expanded detection capacity is ideal for characterizing the diverse cells present in human tumors, which typically include endothelial cells, epithelial cells, fibroblasts, immune cells, and malignant cells (Leelatian et al., 2016).
Flow cytometry provides outstanding statistical power to detect rare cells and to quantify cellular identity of millions of cells, compared to other techniques that are limited to hundreds or thousands of cells (Chattopadhyay et al., 2014; Greenplate et al., 2016a; Irish and Doxie, 2014; Roussel et al., 2016). Our group and others have implemented this technology in studies of donor and patient cells that are obtained as a suspension, such as blood and bone marrow (Bendall et al., 2011; Hardy et al., 1983; Kordasti et al., 2016; Leelatian et al., 2015; Nicholas et al., 2016; Parks et al., 1984; Qiu et al., 2011; Tung et al., 2004) or that can be disaggregated from lymphoid structures by mechanical force alone (Irish et al., 2006a; Irish et al., 2010; Myklebust et al., 2016; Polikowsky et al., 2015; Wogsland et al., 2016). Clinical diagnoses of blood malignancies use fluorescence flow cytometry characterization of cell surface marker expression, as well as cell subset quantification (Arber et al., 2016; Craig and Foon, 2008; van Dongen et al., 2012; Wood et al., 2007). Additionally, flow cytometry has been used clinically to identify minimal residual disease and to detect disease progression in leukemia (Amir el et al., 2013; Borowitz et al., 2008; van Dongen et al., 2015). Fluorescence flow cytometry has also been applied to studies of solid tissues and tumors for research purposes (Al-Hajj et al., 2003; Chan et al., 2009; Donnenberg et al., 2013; Richards et al., 2012; Singh et al., 2004; Zimmerlin et al., 2011).
In addition to their ability to characterize cell surface markers, flow cytometry technologies allow simultaneous detection and quantification of intracellular targets in individual cells (Irish et al., 2004; Irish et al., 2006b; Krutzik et al., 2004). Commercially available fluorescence flow cytometers generally measure 8–12 targets per cell using target-specific antibodies conjugated to individual fluorophores (Bendall et al., 2012). The number of targets is limited due to the overlap of emission spectra of different fluorophores. Mass cytometry is a newer flow cytometry-based technology that allows detection of more than 35 targets in individual cells. Instead of conjugation to fluorescent dyes, mass cytometry antibodies are conjugated to isotopically pure heavy metals. Specifically, fundamental elements of mass cytometry include 1) the staining of individual cells with isotope-tagged antibodies to detect specific cellular targets and 2) quantification of the isotopic signal via time-of-flight, as in other forms of mass spectrometry, which indicates specific antibody binding (Bandura et al., 2009; Bendall et al., 2011; Spitzer and Nolan, 2016). Therefore, the abundance of a specific metal isotope in each cell corresponds to the abundance of a specific cellular target detected by the antibody. The use of metal isotopes and time-of-flight quantification in mass cytometry results in relatively little spectral overlap between the channels distinguished by isotopes (Maecker et al., 2004; Nicholas et al., 2015; Wang et al., 2012). Additionally, multiple cellular targets of interest can be measured simultaneously, and the numbers are greater than those routinely measured in current fluorescence-based cytometry (Chattopadhyay et al., 2014; Greenplate et al., 2016a; Newell and Cheng, 2016; Spitzer and Nolan, 2016). Mass cytometry has the potential to track evolving cell subsets and to measure features typically associated with one cell type (e.g. mature immune cell or stem cell associated proteins) on all the cells in a sample (Becher et al., 2014; Ferrell et al., 2016a; Ferrell et al., 2016b; Irish, 2014). This type of single cell systems biology has the potential to reveal unexpected, clinically-relevant cell types and measure a wealth of features on cells without the need to return to a sample for repeat measurements (Ferrell et al., 2016a; Gaudilliere et al., 2014; Greenplate et al., 2016b; Kordasti et al., 2016; Levine et al., 2015).
Mass cytometry-based characterization of human bone marrow (Bendall et al., 2011), blood (Newell et al., 2012), and tonsil (Polikowsky et al., 2015) cell subsets has been accomplished in prior studies and described in protocols (Leelatian et al., 2015). However, mass cytometry has just recently been developed and applied in solid tissues and organs (Diggins et al., in press; Leelatian et al., 2016). One of the major limitations for flow cytometry is the need to generate a suspension of viable single cells derived from the tissue of interest. Although fluorescence flow cytometry has been used to study some solid tissues and cancers, the protocols used to derive viable single cells, even from the same organs, can vary significantly between studies (viz. (Codega et al., 2014; Pastrana et al., 2009; Rahman et al., 2015)). The protocol described here has been optimized to yield viable cells and to preserve known cell subsets from a variety of human tissues, including lymph nodes, gliomas, melanomas, and small cell lung cancer (SCLC) patient-derived xenografts (PDXs) (Leelatian et al., 2016). It is thus suitable for preparing single cells for fluorescence cytometry, mass cytometry, and other applications requiring isolated single cells. We also provide a protocol detailing cellular immunostaining for detection of cell-surface and intracellular epitopes in mass cytometry analysis of cells from human tonsils, gliomas, and melanomas, and a support protocol describing computational analysis of multi-dimensional data obtained from mass cytometry based on established approaches (Amir el et al., 2013; Diggins et al., 2015; Qiu et al., 2011).
Basic Protocol 1: PREPARATION OF VIABLE SINGLE CELLS FROM HUMAN TISSUE AND TUMORS
Introduction
This section describes a method for preparing single cell suspensions from human tissues. It has been experimentally tested to preserve cell subsets detected using imaging platforms and maximize cell viability for cells from human tonsils, glioma tumors, melanoma tumors, and small cell lung cancer (SCLC) patient-derived xenografts (PDX) (Leelatian et al., 2016). Human tonsils, glioma tumors, and melanoma tumors were resected from patients and transported directly to the laboratory (within 1 hour after collection for human gliomas and melanomas, and within 4 hours after collection for human tonsils). SCLC PDXs were flank xenografts in immunocompromised mice, generated from patient specimens. When grown as flank tumors, these xenografts form a solid tissue about 1–2 cm in diameter. SCLC PDXs were transported to lab within 1 hour after collection. We expect this protocol to work in other human tissue and cancer types, as well as solid tissues from other species. However, it is important to note that 1) choice of enzymes, and 2) total dissociation time need to be tested before routine use of the protocol in tissues not indicated here.
Materials
Tissue sample: This protocol is for preparation of single cells from human tissues from surgical resections. Samples should be placed in appropriate experimental medium (see below), phosphate buffered saline (PBS), or normal saline solution immediately after surgical resection. The volume of media or normal saline should be enough to immerse the entire sample (Figure 1). Ideally, samples should be transported directly to lab for preparation at room temperature (~23°C).
Transfer medium: PBS (Catalog no. 21040CV, Corning/Mediatech, Corning, NY) at room temperature.
- Experimental media:
- Glioma, DMEM/F12+Glutamax (Catalog no. 10565018, Gibco/Life Technologies, MA) with a defined hormone and salt mix (Reynolds et al., 1992) and 50 μg/mL gentamicin sulfate (Catalog no. 30-005-CR, Corning, NY)
- Melanoma, MEM (Catalog no. 10010CV, Corning/Mediatech, Corning, NY) with 10% FBS (Catalog no. 26140079, Thermo Fisher Scientific, MA) + 100 units/mL Penicillin and 100 μg/mL Streptomycin (Penicillin-Streptomycin solution, catalog no. SV30010, GE Healthcare, Pittsburgh, PA)
- Tonsils, RPMI 1640 (Catalog no. 10040CV, Corning/Mediatech, Corning, NY) with 10% FBS (Catalog no. 26140079, Thermo Fisher Scientific, MA) + 1X 100 units/mL Penicillin and 100 μg/mL Streptomycin (Penicillin-Streptomycin solution, catalog no. SV30010, GE Healthcare, Pittsburgh, PA)
- Note: Experimental medium may vary by cell type, as different cell types may have distinct nutrient and supplement requirements. For this protocol, media were selected based on established cell culture protocols for each cell type. Furthermore, if additional assays, such as a signaling response assay using phospho-specific flow cytometry (Krutzik and Nolan, 2003; Schulz et al., 2012), are to be performed, it is important to test different types of medium for those specific assays. For example, to preserve lymphocyte signaling capability for subsequent detection by phospho-specific flow cytometry, medium containing FBS is superior to serum-free medium (Irish et al., 2006a; Irish et al., 2010; Polikowsky et al., 2015). Conversely, multiple growth factor supplements are added to the neurosphere culture medium to ensure growth of human glioma cells (Azari et al., 2011; Lee et al., 2006).
Scalpels with blade no.10 (Catalog no. 12-460-451, Fisher Scientific, MA)
60 mm petri dish (Catalog no. FB0875713, Fisher Scientific, MA)
15 mL (Catalog no. 430055, Corning, NY) and 50 mL (Catalog no. 430829, Corning, NY) conical tubes
Incubator set at 37 °C, 5% CO2
Nutating platform placed inside incubator set to 18 rpm (Catalog no. 05-450-213, Fisher Scientific, MA)
P1000 tips, trimmed to make a wide opening ~2–3 mm.
70 μm (Catalog no. 431751, Corning, NY) and 40 μm (Catalog no. 431750, Corning, NY) cell strainers sized to fit 50 mL conical tubes
Benchtop centrifuge with swing-out rotor (Model Sorvall ST 16, Thermo Scientific, MA)
Rotor adapters with round buckets that accommodate 5 mL FACS tubes (Catalog no. 75003680, Thermo Fisher Scientific, MA)
Trypan Blue, (Catalog no. SV30084.01, Hyclone, UT, prepared as recommended by manufacturer)
Inverted phase contrast microscopy for cell culture (use 10X objective magnification for quantifying cell viability)
DMSO (Catalog no. BP231-1, Fisher Scientific, MA)
1.8 mL cryogenic tubes with cap (Catalog no. 377267, Thermo Fisher Scientific, MA)
ACK lysing buffer (Catalog no. 10-548E, Lonza, MD)
- 100X DNase I (10,000 Kunitz/mL) (Catalog no. DN25, Sigma-Aldrich, MO)
- DNase is diluted in PBS to yield the indicated concentration for long-term storage at −80 °C
- 20X Collagenase II 2500 CDU/mL (20 mg/mL) (Catalog no. C6885, Sigma-Aldrich, MO)
- Collagenases II, IV, V, and XI displayed equivalent activity on tumor and tissue types tested (Leelatian et al., 2016).
- Collagenase II is diluted in PBS to yield the indicated concentration for long-term storage at −80 °C.
Figure 1. Step-by-step illustration of tissue dissociation protocol.

Surgically resected patient samples were transported in PBS or experimental medium at room temperature. Mechanical dissociation was followed by 1-hour enzymatic dissociation using Collagenase II and DNase I (see text). ACK lysis was used to eliminate red blood cell contamination, prior to cell counting and cryopreservation or experiment.
Steps and Annotations
Mechanical and Enzymatic Dissociation (Figure 1):
Transfer pieces of human tissue from surgery to a cell preparation laboratory while keeping the sample submerged in room temperature PBS (see Time Considerations in Commentary).
Once in lab, transfer tissue pieces and PBS to one or more 50 mL conical tubes, ensure tubes are well balanced, and centrifuge at 100 × g at room temperature for 5 min to pellet cells and tissue pieces.
- Carefully discard supernatant by pipetting and resuspend tissue in 5 mL or more of warm (37°C) experimental medium, as need to cover tissue.
- Note: For larger tissue (larger than 1 cm3), use multiple rounds of mincing as in Step 4 and Step 5.
- Note: Dead cells will not pellet effectively at 100 × g and will be contained in the supernatant with other, non-cellular tissue components and secreted factors.
Transfer tissue and experimental medium into a 60 mm petri dish.
Mince tissue in experimental medium with scalpel to obtain ~1–3 mm3 pieces.
Transfer minced tissue and cells in experimental medium into 15 or 50 mL conical tubes, as dictated by the total volume of the cell and medium suspension.
Centrifuge tissue and cells in experimental medium at 100 × g at room temperature for 5 min.
- Discard supernatant by pipetting and add ~4.7 mL of warm experimental medium.
- Note: This volume of experimental medium leaves room for ~300 μL of enzyme solutions in the next step and is recommended for tissue that was originally ~1 cm3 in size. For larger tissue, the volumes in Step 8 and Step 9 should be increased proportionately to match tissue size. For example, ~9.4 mL of warm experimental medium would be used in Step 8 for tissue that was originally ~2 cm3 in size.
Add 250 μL of 20X Collagenase II and 50 μL of 100X DNase I, and mix with serological pipet. The final concentrations of collagenase II and DNase I should be 1 mg/mL and 100 Kunitz/mL, respectively
Incubate the tube on a nutating platform (18 rpm) in an incubator (37°C, 5% CO2) for 60 min.
Remove tubes from the incubator and carefully triturate (pipette 25–50 times) the cell suspension using a 10 mL plastic serological pipet. When complete, the cell suspension should look homogeneous and have no visible tissue pieces.
Strain with 70 μm cell strainer into a new 50 mL conical tube.
Strain flow-through from Step 12 with 40 μm cell strainer into a new 50 mL conical tube.
Wash 10 mL of warm (37°C) experimental medium through the 40 μm strainer into the same tube.
Centrifuge the collected strained cell suspension at 100 × g at room temperature for 10 min, discard supernatant by pipetting.
If pellet contains red blood cells or platelets, add 5 mL or more of ACK lysis buffer following manufacturer protocols, mix with serological pipet, and leave at room temperature for 60 seconds to allow for hypotonic lysis.
Add 5 mL or more of warm experimental medium (the same volume used in Step 16 for ACK lysis buffer to a final 1:1 proportion), centrifuge at 100 × g at room temperature for 10 min, and discard supernatant.
Resuspend cells in warm experimental medium and count cells to quantify viable cells using Trypan Blue (Figure 2).
Cells are now ready to be prepared for mass cytometry analysis. If mass cytometry analysis is to be performed on a different day, or if the cells need to be preserved for long-term storage, cryopreservation is required. This can be performed per a previously established protocol (Leelatian et al., 2015).
Figure 2. Trypan Blue stain for viable cell quantification.
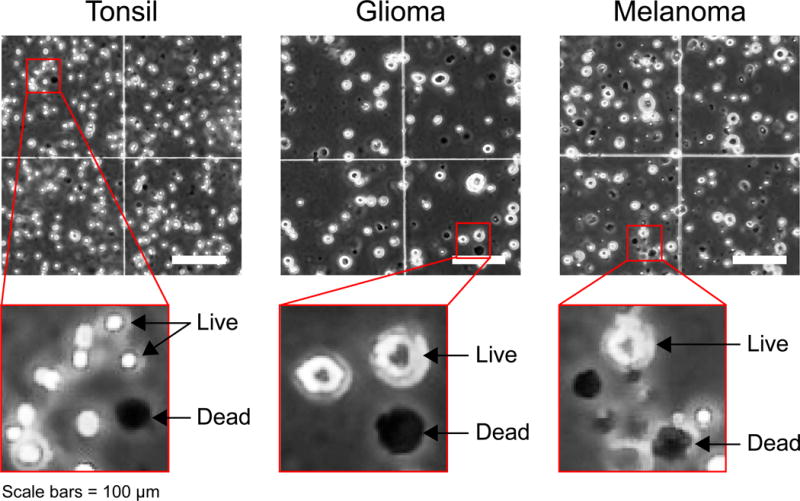
Trypan Blue stain was used to quantify cell viability after mechanical and enzymatic dissociation. Representative images of dissociated human tissues including tonsil, glioma, and melanoma are shown. Red boxes show higher resolution of live (Trypan Blue-negative, white) and dead cells (Trypan Blue-positive, black) of each tissue type. Note that some pigmented cell types, such as melanocytes or neurons of the substantia nigra, can be brown or red and therefore appear dark in monochrome phase contrast images. These cells should be distinguished from dead cells in counting. Scale bars = 100 μm.
Basic Protocol 2: PREPARATION OF CELLS FOR MASS CYTOMETRY
Introduction
This section describes a protocol for immunostaining of single-cell suspensions derived from human tissues and tumors. Tonsils, glioma tumors, and melanoma tumors are used as examples. Using antibodies listed in Table 1, this protocol allows characterization of immune cell subsets (CD45+) in tonsils, as well as infiltrating immune cells in glioma tumors and melanoma tumors. These antibodies allow characterization of immune cells into distinct groups: myeloid lineage (CD11b, CD11c, CD14, CD16, CD64, CD68, HLA-DR), B cell and plasma cell lineage (CD19, CD38, CD27, IgM, IgD, HLA-DR), and T cell lineage (CD3, CD4, CD8, CD8a, CD45RA, CD45RO). Additionally, antibodies that were specifically selected for identifying non-immune cell subsets in glioma (CD31, TUJ1, S100B, PDGFRα, c-MET, SOX2, CD24, Nestin, CD44, GFAP, αSMA, and CD56) and melanoma (CD31, β-catenin, S100B, vimentin, CD49F, cytokeratin, SOX2, Nestin, CD44, αSMA, and CD56) tumors were included. The antibodies described here are isotope-tagged antibodies for mass cytometry analysis. This protocol was adapted from previously established fluorescence flow cytometry protocols (Krutzik et al., 2005; Krutzik et al., 2004; Leelatian et al., 2015; McLaughlin et al., 2008).
Table 1.
Tissue-specific antibody panels
| Metal | Antibody | Clone | Working conc (μg/mL) | Dilution | Volume to use in 100 μL stain (μL) | Sample type | Staining condition | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ton | Glio | Mel | Surf | Sap | MeOH | ||||||
| 141Pr | HLA-ABC | W3-32 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 142Nd | cCasp3 | D3E9 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ✓ | ✓ | ||
|
| |||||||||||
| 144Nd | CD11b | ICRF44 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 145Nd | CD4 | RPA-T4 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
| CD31 | WM59 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ✓ | ||||
|
| |||||||||||
| 146Nd | IgD | IA6-2 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
| CD64 | 10.1 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | |||||
| CD8a | RPA-T8 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | |||||
|
| |||||||||||
| 147Sm | β-catenin | D10A8 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 148Nd | CD16 | 3G8 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 149Sm | CD45RO | UCHL1 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ✓ | ✓ | ||
|
| |||||||||||
| 152Sm | TUJ1 | TUBB3 | 50 | 1:100 | 1 | ✓ | ✓ | ||||
|
| |||||||||||
| 153Eu | CD45RA | HI100 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
| S100B | 19-S100B | 100 | 1:100 | 1 | ✓ | ✓ | ✓ | ||||
|
| |||||||||||
| 154Gd | CD45 | HI30 | Fluidigm | 1:400 | 0.25 | ✓ | ✓ | ✓ | ✓ | ||
|
| |||||||||||
| 155Gd | CD27 | L128 | Fluidigm | 1:100 | 1 | ✓ | ✓ | ||||
|
| |||||||||||
| 156Dy | Vimentin | RV202 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 159Tb | CD11c | Bu15 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
| CD49F | GoH3 | 100 | 1:100 | 1 | ✓ | ✓ | |||||
|
| |||||||||||
| 160Gd | CD14 | M5E2 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 161Dy | CD19 | HIB19 | 100 | 1:100 | 1 | ✓ | ✓ | ||||
| PDGFRα | 16A1 | 200 | 1:100 | 1 | ✓ | ✓ | |||||
| Cytokeratin | C-11 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | |||||
|
| |||||||||||
| 162Dy | c-MET | L6E7 | 100 | 1:100 | 1 | ✓ | ✓ | ||||
|
| |||||||||||
| 163Dy | SOX2 | O30-678 | 100 | 1:100 | 1 | ✓ | ✓ | ✓ | |||
|
| |||||||||||
| 166Er | CD24 | ML5 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 167Er | CD38 | HIT2 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 168Er | CD8 | SK1 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
| Nestin | 10C2 | 100 | 1:100 | 1 | ✓ | ✓ | |||||
|
| |||||||||||
| 169Tm | CD44 | BJ18 | 100 | 1:100 | 1 | ✓ | ✓ | ✓ | |||
|
| |||||||||||
| 170Er | CD3 | SP | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ✓ | ✓ | ||
|
| |||||||||||
| 171Yb | CD68 | Y1/82A | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
| GFAP | 1B4 | 25 | 1:100 | 1 | ✓ | ✓ | |||||
|
| |||||||||||
| 172Yb | IgM | MHM-88 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ||||
|
| |||||||||||
| 173Yb | αSMA | Ab54723 | 50 | 1:100 | 1 | ✓ | ✓ | ✓ | |||
|
| |||||||||||
| 174Yb | HLA-DR | L243 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ✓ | ✓ | ||
|
| |||||||||||
| 175Lu | CD56 | HCD56 | 50 | 1:100 | 1 | ✓ | ✓ | ✓ | ✓ | ||
|
| |||||||||||
| 176Yb | Histone H3 | D1H2 | Fluidigm | 1:200 | 0.5 | ✓ | ✓ | ✓ | ✓ | ||
Conc = concentration; Ton = tonsil; Glio = glioma; Mel = melanoma; Surf = surface; Sap = saponin; MeOH = post-methanol Fluidigm = use antibodies provided by Fluidigm
Materials
Dissociated single cells derived from Basic Protocol 1
Water bath set to 37°C (Catalog no. 15-462-10, Fisher Scientific, MA)
15 mL conical tubes (Catalog no. 430055, Corning, NY)
5 mL round-bottom FACS tubes without cap (Catalog no. 352052, Corning, NY)
5 mL round-bottom FACS tubes with filter caps (Catalog no. 352235, Corning, NY)
PBS (Catalog no. 21040CV, Corning/Mediatech, Corning, NY)
Deionized water
- Metal-conjugated antibodies (see Table 1)
- Note: All new antibodies should be titrated prior to use with appropriate positive control cells that express the target of interest and negative control cells that are known to not express the target of interest. The goal of an antibody titration is to determine the optimal concentration of an antibody that separates the true signal of the positive control cells from any background or non-specific signal observed in the negative control cells. Antibody titration is required for every combination of antibody clone, tissue preparation technique, and antibody conjugation. This extensive validation is because clones can perform differently under different antigen exposure conditions, such as permeabilization of cells by detergent or alcohol, and protocols to conjugate fluorochrome or metal reporter tags to antibodies can change their binding properties. Examples of appropriate validation and titration have been shown (Hulspas et al., 2009; Krutzik et al., 2005; Krutzik and Nolan, 2003; Schulz et al., 2012).
Staining media (1% bovine serum albumin (BSA (Catalog no. BP9703100, Fisher Scientific, MA) in PBS)
1.5 mL Eppendorf tubes (Catalog no. 05-408-129, Fisher Scientific, MA)
16% paraformaldehyde (PFA) (Catalog no. 15710, Electron Microscopy Sciences, PA)
Perm 1: Room-temperature 0.02% saponin (Catalog no. 558255, Calbiochem, MA) (w/v) in PBS
- Perm 2: Ice-cold 100% methanol (kept at −20°C until immediately prior to adding to cells) (Catalog no. A412-4, Fisher Scientific, MA)
- Note: The final concentration of ice-cold methanol after added to cells should be > 95%. Other permeabilization concentrations and reagents might be used after optimization (Krutzik and Nolan, 2003).
- Note: For each cellular target the user aims to detect, it must first be determined if the target is exposed on the cell-surface (i.e. extracellular) or present within the cell (i.e. intracellular). Most extracellular targets are detected with live cell staining (see “Staining of viable cells to detect extracellular targets” below). However, if the target of interest is an intracellular target, it is especially important to optimize the permeabilization technique and reagents (Krutzik and Nolan, 2003). Examples of permeabilization reagents include saponin (Perm 1), methanol (Perm 2), ethanol, Triton-X, among many others. For the protocol described here, saponin permeabilization (Perm 1) (for SOX2 antibody staining) is used prior to methanol permeabilization (Perm 2) (for staining of the remaining intracellular targets) in human glioma and melanoma.
1X Four Elements Calibration Beads (Catalog no. 201078, Fluidigm, CA)
Steps and Annotations
Antibody Preparation:
- Example reagent mixes for healthy human tonsil tissue, glioma tumors, and melanoma tumors are shown in Table 1 and separated according to staining step. Prepare reagent mixes separately for each of three example staining steps: live cell staining (Live), staining in 0.02% saponin (Perm 1), and staining after methanol treatment (Perm 2).
-
cNote: adaptation of this protocol for phospho-flow should detect cell surface proteins following fixation as described in “Live cell staining” even though the cells are no longer viable, as described by (Krutzik and Nolan, 2003; Leelatian et al., 2015; Schulz et al., 2012).
-
dNote: this protocol does not use metal barcoding, but this technique can be useful in addressing potential batch effects from staining and collecting data at different times (Behbehani et al., 2014; Zunder et al., 2015).
-
c
Staining of viable cells to detect extracellular targets:
-
2
If preparing cells from cryopreservation, thaw cryovial in warm water bath (37°C) for 1–2 minutes (until just completely thawed). If preparing cells that were freshly dissociated, skip to Step 5.
-
3
Transfer cells from cryopreservation tubes to a 15 mL conical tube.
-
4Resuspend cells in 10 mL of warm experimental medium.
- Note: The goal is to dilute and remove DMSO as quickly as possible after thawing.
- Note: Addition of DNase may be helpful upon sample thaw to preserve viability. Use the same concentration of DNase as used during tissue dissociation as described in Basic Protocol 1, step 9. Specifically, resuspend the cells in this step in 9.9 mL of warm experimental medium and add 100 μL of 100X DNase to the cell suspension and proceed to the next step.
-
5
Pellet cell suspension at 100 × g for 5 min at room temperature and discard supernatant.
-
6
Resuspend cells in 1 mL of staining medium and transfer cell suspension to a 5 mL FACS tube.
-
7Pellet cell suspension at 100 × g for 5 min at room temperature and discard supernatant by briskly decanting.
- Note: For all centrifuge steps involving a cell pellet, invert and decant only once. After placing the tube upright again, cells typically enter solution and the pellet can detach. Thus, additional decanting significantly lowers viable cell yield.
-
8Resuspend cell pellet in staining medium to achieve the transfer volume.
- Note: The “transfer volume” is the volume transferred in Step 9 and it is calculated by subtracting the summed volume of staining antibodies (the “antibody volume”) from the total volume in which staining will occur in Step 10. For example, consider a protocol where approximately 10 μL of cells in staining medium from Step 8 are to be stained with 1 μL each of 30 antibodies in a total volume of 100 μL in Step 10. In this case, on Step 8, at least 60 μL of staining medium should be added to the 10 μL of cells in staining medium to achieve a transfer volume of 70 μL for Step 9. For Steps 8–10, the volume of antibodies varies and is specified in Table 1 for tonsil, glioma, and melanoma. The total volume in Step 10 is 100 μL.
-
9
Transfer cell suspension in staining medium to a new FACS tube.
-
10Add Live Stain reagent mix to the same FACS tube. Briefly vortex to mix cells with antibodies.
- Note: Total staining volume (cells in staining medium plus antibodies) should be exactly 100 μL.
- Note: Live Stain reagents in Table 1 can be combined in a 1.5 mL Eppendorf tube prior to mixing with cells in FACS tubes.
-
11
Leave cells at room temperature for 30 minutes.
-
12
Add 1 mL of staining medium to the FACS tube and pellet cells at 100 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
13
Repeat Step 12 once.
-
14
Add 1 mL of PBS to FACS tube and resuspend the cells by gentle vortexing until there is no visible cell clumps.
-
15
Add 100 μL of 16% PFA to FACS tube, for a final concentration of 1.6%, and vortex to mix. This step is for cell fixation prior to further intracellular staining steps.
-
16
Leave cells at room temperature for 10 minutes.
-
17
Pellet cells at 800 × g for 5 minutes at room temperature and decant to discard supernatant (See Note in Step 7).
Cells are now ready for intracellular staining.
Staining to detect intracellular targets:
-
18Determine the optimal permeabilization conditions required for each intracellular target. If all intracellular antibodies have been shown to effectively detect target antigens after permeabilization with ice-cold methanol (as is the case for the antibodies that were used to stain tonsils), skip to Step 28.
- Note: Antibodies for mass cytometry are pre-labeled with metal isotopes. Many of these antibodies are commercially available (see Table 1). For antibodies that are not commercially available in isotope-tagged formats, they can be labeled with a metal isotope using a commercial conjugation kits (Leipold et al., 2015). The isotope-labelled antibodies can then be used for immunostaining by following the protocol described below.
-
19
Resuspend cells in 1 mL of Perm 1, pellet at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
20
Repeat Step 19 once.
-
21
Resuspend cell pellet in appropriate volume of Perm 1 (see Note on Step 8).
-
22
Transfer appropriate volume of cell suspension in Perm 1 to a new FACS tube (see Note on Step 8).
- 23
-
24
Leave cells at room temperature for 30 minutes.
-
25
Add 1 mL of Perm 1 to the FACS tube and pellet cells at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
26
Repeat Step 25 once.
-
27
Add 1 mL of PBS to the FACS tube and pellet cells at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
28
Vigorously vortex cell pellet to resuspend cells in void volume.
-
29
Add 1 mL of Perm 2 to FACS tube. Vortex to thoroughly resuspend cells with Perm 2.
-
30Keep cells at −20°C for at least 20 minutes or overnight.
- Note: Alternatively, cells can be stored in Perm 2 at −80°C for days or weeks before proceeding if necessary.
-
31
Remove FACS tubes from −20°C and add 1 mL of PBS to the tubes.
-
32
Pellet cells at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
33
Add 1 mL of staining medium to FACS tubes. Pellet cells at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
34
Repeat step 33 once.
-
35
Resuspend cell pellet in appropriate volume of staining medium (see Note on Step 8).
-
36
Transfer appropriate volume of cell suspension in staining medium to a new FACS tube (see Note on Step 8).
-
37Add Methanol Stain reagent mix to the same FACS tube. Briefly vortex to mix cells with antibodies.
-
38
Incubate cells at room temperature for 30 minutes.
-
39
Add 1 mL of staining medium to the FACS tube and pellet cells at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
40
Repeat Step 39 once.
-
41
Add 1 mL of PBS to the FACS tube and pellet cells at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
42
Add 1 mL of deionized water to the FACS tube and pellet cells at 800 × g for 5 min at room temperature, decant to discard supernatant (See Note in Step 7).
-
43
Resuspend cell pellets in 1X Four Elements Calibration Beads in deionized water prior to mass cytometry analysis. Use ~ 1 mL for every 0.5 × 106 cells.
-
44
Filter cells with FACS tubes with filter caps.
Cells are now ready for mass cytometry analysis.
COMMENTARY
Background Information
Flow cytometry technologies employing fluorescence- and mass-based reporters have been successfully applied to characterize protein phenotype and to quantify the abundance of diverse human cell types. Flow cytometry protocols commonly use reporter-conjugated antibodies to make relative quantitative measurements for tens of features in each of hundreds of thousands of cells in minutes (Chattopadhyay and Roederer, 2012; Leipold et al., 2015; Mahnke and Roederer, 2007; Ornatsky et al., 2010; Perfetto et al., 2004; Robinson, 2005). Mass cytometry, a newer form of flow cytometry based on mass spectrometry, has gained attention for the relative ease with which more than 35 cellular features can be measured (Bandura et al., 2009; Bendall et al., 2011; Bjornson et al., 2013). However, as flow cytometry requires individual cells in suspension, mass cytometry’s application to solid tumors and tissues has previously been modest compared to its rapid adoption in immunology and blood cancer research, where samples of viably cryopreserved cells have been collected and characterized for decades (Bendall et al., 2014; Borowitz et al., 2008; Ferrell et al., 2016a; Hardy et al., 1983; Lee et al., 1999; Parks et al., 1984; Roederer et al., 2015). The key limitation has been the perceived difficulty in preparing cells from solid tissues into single cell suspensions that are viable and representative of different cell types present in the original tissue. Furthermore, mass cytometry antibody sets had not been designed and tested to effectively identify cells outside the immune system. It is only recently that mass cytometry has been tested and applied to solid tissues and tumors (Diggins et al., in press Leelatian et al., 2016). Key to this work was the development of a protocol that preserved the viability and diversity of the tissue cells in a way compatible with detection of cell surface and intracellular features by mass cytometry.
Critical Parameters and Troubleshooting (Table 2)
Table 2.
Troubleshooting and optimizing solid tissue dissociation
| Issues | Potential causes | Troubleshooting |
|---|---|---|
| Few viable cells after dissociation |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
| Red blood cell or platelet contamination |
|
|
|
| ||
| Few viable cells after cryopreservation |
|
|
|
|
|
|
|
|
|
| ||
| Cell subsets of interest were not detected |
|
|
|
|
|
|
|
|
|
|
|
|
| ||
| Non-specific staining |
|
|
Tissue quality and transportation
This protocol is applicable to human tissues extracted by surgery or to animal tissues isolated after dissection. To preserve tissue viability, samples should be transported to the laboratory for further preparation as rapidly as possible. The dissociation protocol presented here was tested on samples that were processed between 30 minutes and 4 hours after surgical resection (Leelatian et al., 2016). Additionally, samples should be transported in sterile PBS, appropriate experimental medium, or other sterile transport medium that has been tested to preserve cell viability and representative cell subsets for specific tissue types. Samples should be entirely submerged in the transport medium in a closed container. Unless specifically optimized and validated using other conditions, samples should be transported at room temperature (~23°C) immediately to the laboratory for further preparation.
Mechanical dissociation
As described in Basic Protocol 1, tissue should be mechanically dissociated into fine pieces (1–3 mm3 pieces) to maximize surface contact with dissociation enzymes in subsequent steps. During mechanical dissociation in the petri dish, tissue pieces should be adequately covered in warm (37°C) experimental medium. For larger samples, the tissue should be divided into batches for mincing and combined prior to addition of dissociation enzymes, collagenase II and DNase I.
Selection of enzymes and duration of dissociation
Dissociation enzymes are incorporated in the protocol to break down the extracellular matrix to yield a single cell suspension. Many types of enzymes are available, such as collagenases, Trypsin, Papain, and HyQTase, among others. These enzymes can be used individually or in combination. The types of dissociation enzymes used can affect the viability of single cells derived from the starting tissue. As described above, a suitable dissociation protocol would maximize cell viability as well as preserve representative cell subsets in the original tissue. Variables to consider when testing the dissociation conditions include the use of a single enzyme or combinations of enzymes, and duration of dissociation (Leelatian et al., 2016). Additionally, inclusion of DNase I in the dissociation protocol described here significantly improved viable cell yield from multiple tissues (Leelatian et al., 2016). Therefore, it is highly recommended to include DNase I in the dissociation solution unless it is specifically demonstrated experimentally that DNase I is not required to improve cell viability. The protocol described here uses a time of 1 hour of enzymatic dissociation, which has been shown to result in highest viable cell yield for various human tissues and tumors (Leelatian et al., 2016). Specifically, for most tissues, shorter dissociation time led to release of fewer cells, whereas longer dissociation led to increased cell death. It is recommended that the type of dissociation enzyme and duration of enzymatic dissociation be tested and optimized for a new tissue type to achieve optimal viable cell yield. This can be quantified by using Trypan blue staining (see Basic Protocol 1). In addition to overall cell viability, it is crucial to determine preservation of known cell types and cells of interest as part of the optimization process. When testing the dissociation on a particular tissue, imaging techniques such as colorimetric immunohistochemistry or immunofluorescent detection of known cellular targets can be used to characterize the presence of cell subsets in the original tissue.
Immunostaining for mass cytometry
It is highly recommended to optimize the immunostaining protocol of each antibody in a panel to ensure target-specific staining and to optimize signal to background (Krutzik and Nolan, 2003). Parameters that need optimization include 1) antibody specificity (which can be tested using positive and negative control cells that are known to express and lack the target of interest), 2) antibody concentration (to allow maximal distinction between positive and negative cells, and minimize non-specific background staining), 3) staining order (extracellular staining or intracellular staining), and 4) compatibility of permeabilization reagents for intracellular targets (saponin, ice-cold methanol, or other reagents) (Krutzik and Nolan, 2003). If multiple permeabilization reagents are required for different intracellular targets, specificity and sensitivity of the antibodies should be tested to ensure that the targets are still detectable after multiple permeabilization steps. For glioma immunostaining presented here, saponin permeabilization was used for SOX2 detection, prior to subsequent permeabilization by ice-cold methanol. It has been previously shown that saponin does not destroy intracellular targets normally detectable after methanol permeabilization and therefore these reagents can be used in the same protocol, with the use of saponin preceding methanol (Behbehani et al., 2014). However, each antibody should be specifically tested and optimized prior to use.
Treatment of cells with reagents for detection of intact cells via mass cytometry
In flow cytometry analyses, an initial step is to identify intact cells and remove cellular debris or enucleated cells from further analyses. Conventional fluorescence flow cytometry relies on measurement of cell size (forward scatter, FSC) and cell granularity (side scatter, SSC) to identify cells. Additionally, cellular debris can be distinguished due to its smaller size (low FSC) and higher granularity (high SSC). In contrast to fluorescence flow cytometry, mass cytometry does not have direct parameters to distinguish intact cells from cellular debris. Therefore, mass cytometry analysis requires measurement of indirect parameters to identify intact cells.
Iridium-conjugated DNA intercalator is commonly used to identify intact cells by mass cytometry analysis (Ornatsky et al., 2008). The per-cell quantity of DNA-intercalated iridium provides information about DNA content, which can be used to define intact cells. However, iridium-conjugated DNA intercalator cannot be detected on fluorescence flow cytometry. In this protocol, we used anti-histone H3 antibody staining for detection of intact nucleated cells (Leelatian et al., 2016). The advantage of using an antibody-based technique is that it is readily applied across different flow cytometry platforms (fluorescence and mass cytometry).
Anticipated Results
This protocol produces viable, single-cell suspensions from solid tumors and tissues and is expected to identify most common cell types, including endothelial cells, immune cells, epithelial cells, neural cells, and fibroblasts. This protocol has been validated for human tonsil tissue, glioma tumors, melanoma tumors, and small cell lung cancer patient-derived xenografts. Maximum viable cell yield per gram of tissue from the dissociation of human tonsils, glioma, and melanoma, using collagenase II plus DNase I, should be achieved after 1 hour incubation (Leelatian et al., 2016). Histone H3 staining should allow highly specific identification of nucleated, intact cells. Additionally, cells derived from this protocol are suitable for quantitative measurement of protein expression in individual cells and cell subset abundance using either fluorescence flow or mass cytometry, among other applications. For other tissue types not mentioned above, tissue-specific optimization of the dissociation protocol that takes into consideration the critical parameters described here is highly encouraged. Specifically, a systematic comparison of different dissociation durations, as well as different enzyme combinations, is required. For every condition, it is crucial to quantify cell viability using techniques such as Trypan blue stain. Additionally, the relative abundance of known cell subsets after different dissociation conditions should be quantified, as has been done above using flow cytometry. These data should be compared to prior knowledge of cell types present in tissue, and possibly with immunohistochemistry stains of the original intact tissue (for more details, refer to (Leelatian et al., 2016)).
Signal normalization using bead standards
During each mass cytometry analysis, the detection sensitivity of the mass cytometer can vary between individual samples. Additionally, signal can vary between different mass cytometry experiments. Specifically, the signal intensity of a given cellular target, which is known to be consistent, can vary between different samples for different mass cytometry analyses. To allow accurate comparison between samples, bead standards are used (Finck et al., 2013). Known metal isotopes with standard signal intensity are embedded within polystyrene beads. These beads are mixed and analyzed simultaneously with each sample to allow monitoring of signaling variation during data acquisition. The change of the isotope signal of the beads proportionately correlates with the variation of each sample, due to detection variability. Therefore, the variation of signal between each sample can be adjusted to allow direct comparison of the signal between samples. Normalization is performed using publicly available MATLAB normalization software prior to further data analysis. (For more details, refer to (Finck et al., 2013)).
Quantification of single cell protein expression by biaxial analysis
Biaxial plots are a mainstay in cytometry and typically are used to compare the abundance of cells with differing relative intensity of two or more quantified cellular targets. A protocol to generate biaxial and other common plots of mass cytometry data was previously established (Leelatian et al., 2015). In a typical cytometry analysis workflow (Diggins et al., 2015; Saeys et al., 2016), cells are filtered or assigned to populations based on expression profiles of cellular targets in a process called gating (Figure 3). Gating can be repeated sequentially on increasingly refined cell subsets, resulting in a nested hierarchy of cell types that traditionally captures a developmental continuum or indicates an increasingly polarized and specific cell identity (Bendall et al., 2011; Diggins et al., in press DuPage and Bluestone, 2016; Saeys et al., 2016). Examples of sequential biaxial gating of cells derived from mass cytometry analysis of healthy tonsil (Figure 3A), a patient glioma (Figure 3B), and a patient melanoma (Figure 3C) are shown here and based on prior studies (Leelatian et al., 2015; Leelatian et al., 2016). For samples with known cell types, such as healthy peripheral blood mononuclear cells (PBMCs), established sets of identity markers can be used to distinguish cell types (Anonymous, 1984; Maecker et al., 2012). However, concepts of cell identity are still under active discussion in established single-cell fields like immunology (DuPage and Bluestone, 2016). Furthermore, methods of defining and identifying cell populations are likely to be refined as the field of single cell biology matures. Key areas of growth include measurement platforms like mass cytometry, analysis tools from machine learning (Amir el et al., 2013; Irish, 2014; Shekhar et al., 2014; Wang et al., 2016), reference knowledge bases of established cell identities (Shen-Orr et al., 2009), annotated repositories of single cell data (Kotecha et al., 2010; Spidlen et al., 2012), and quantitative labels of cell type (Diggins et al., in press).
Figure 3. Biaxial analysis of cells derived from human tissue and tumors.
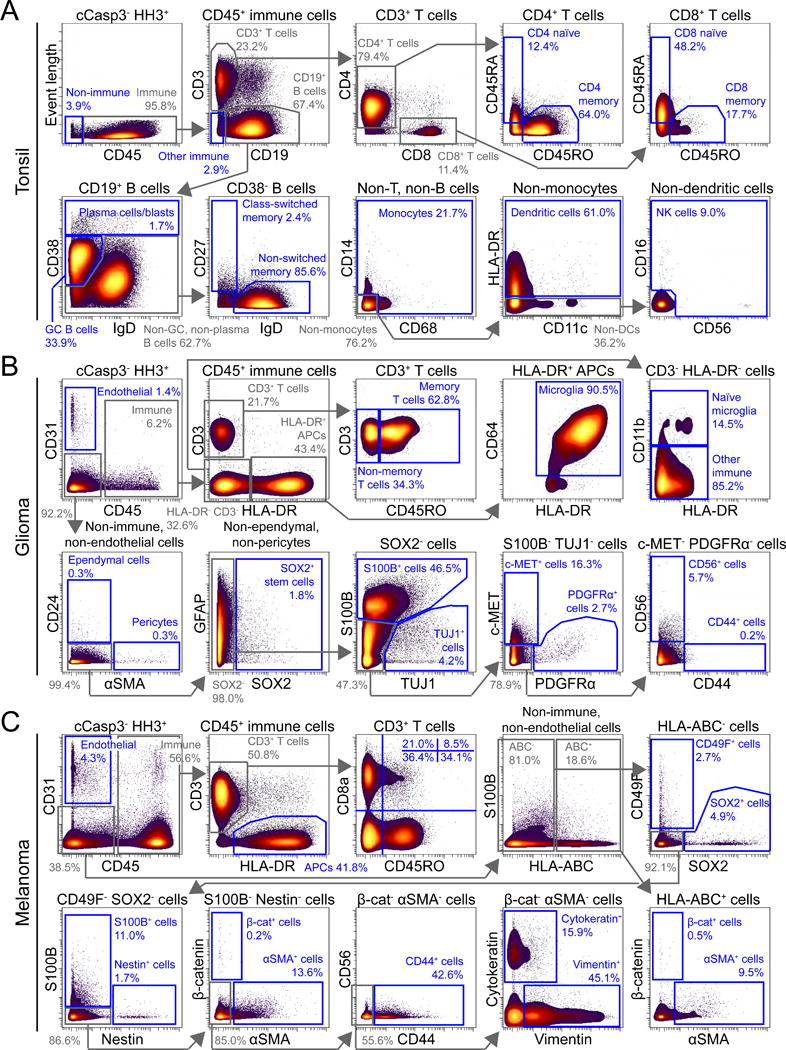
Biaxial plots of non-apoptotic (cCasp3−), nucleated (HH3+) cells from mass cytometry analysis of (A) tonsil, (B) glioma, and (C) melanoma are shown. Intermediate gates are shown in grey, and terminal gates are shown in blue. Cell types or protein identity of cells in each gate are indicated. The percentages of cells in gates are also specified.
Heat plots and viSNE analysis
A new generation of flow cytometry and single cell analysis tools compress multiple dimensions of information into rich two-dimensional views. Examples include mountain plots (Irish et al., 2010), viSNE and related views of t-SNE axes (Amir el et al., 2013; Shekhar et al., 2014), SPADE plots (Qiu et al., 2011), and many other tools that have the potential for machine learning of cell identity (for more details, see (Diggins et al., 2015; Diggins et al., in press Leelatian et al., 2015; Saeys et al., 2016; Wang et al., 2016)). Here, we demonstrate two-dimensional viSNE plots (Amir el et al., 2013). viSNE analysis places cells on a 2-dimensional map that reflects how individual cells are similar to or different from each other when every measured cellular target is simultaneously taken into account. Cell density and expression of individual markers are displayed for a wide range of features using the same t-SNE axes. Data collected from the same patients (tonsil, Figure 4A; glioma, Figure 4B; and melanoma, Figure 4C) as shown in biaxial analyses in Figure 3 were analyzed by viSNE and are shown here. For each sample, a separate pair of t-SNE axes was created (i.e. each sample was analyzed separately in viSNE to create sample-specific viSNE plots, each of which has its own, sample-specific t-SNE axes). All computational analysis was performed using Cytobank software (http://www.cytobank.org) (Kotecha et al., 2010).
Figure 4. High-dimensional analysis of mass cytometry data using viSNE.
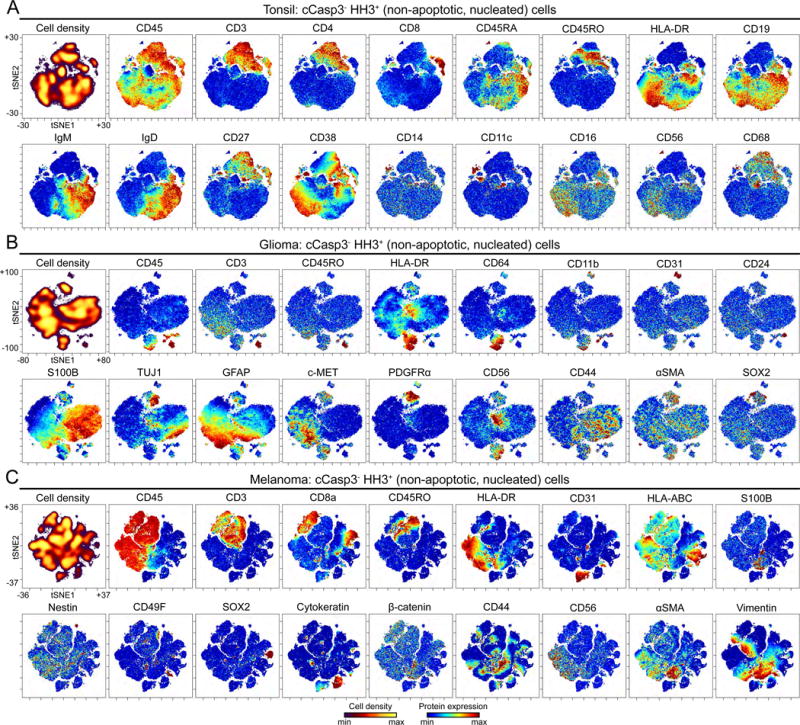
Non-apoptotic (cCasp3−), nucleated (HH3+) cells from mass cytometry analysis of (A) tonsil (plots of 106,568 cells), (B) glioma (plots of 65,834 cells), and (C) melanoma (plots of 94,810 cells), are shown. The first plot of each tissue type depicts cell density of the viSNE map. The remaining plots display expression of indicated protein. viSNE maps of each tissue type were generated separately and the markers shown here for each tissue type were used to generate the maps.
Time Considerations
This protocol was experimentally tested on human gliomas, human melanomas, and SCLC PDXs that were transported to the laboratory within 1 hour after surgical resection. Human tonsils were transported within 4 hours after resection. After tissue is transported to the laboratory, this dissociation protocol can be completed in 2–3 hours, depending on the size of the tissue sample. The size of the tissue sample determines the time that is needed for mechanical dissociation (larger tissue takes more time to be properly minced, whereas smaller tissue takes less time). The time for enzymatic dissociation is not affected by tissue size. The approximate timing of the protocol is: 10–30 minutes for mechanical dissociation, 1 hour for enzymatic dissociation, 15–30 minutes for cell straining, 10–30 minutes for red blood cell lysis and counting, and 10–30 minutes for diluting cells for cryopreservation, if needed (see Basic Protocol 1). Once viable single cells are obtained (either from immediate dissociation or from cryopreservation), live surface immunostaining can be completed in 1 hour, followed by 10–15 minutes of cell fixation. Duration of intracellular staining varies depending on whether permeabilization and staining with saponin (1 hour) is required for the panel of interest. Permeabilization with methanol is usually performed overnight, but can be performed for as little as 10 minutes (Krutzik and Nolan, 2003). Once all cells are permeabilized by ice-cold methanol, an additional 1 hour is required for intracellular immunostaining with isotope-labeled antibodies.
SIGNIFICANCE STATEMENT.
The protocol presented here obtains viable, single cell suspensions from solid tumors and tissues and identifies the vast majority (>99%) of the major cell types obtained, including endothelial cells, immune cells, epithelial cells, neural cells, and fibroblasts. This protocol has been validated for human tonsil tissue, glioma tumors, melanoma tumors, and small cell lung cancer patient-derived xenografts. This protocol enables researchers to begin preparing viable single cells from solid tissues for other aspects of basic and clinical research protocols. In the long term, this protocol is expected to spur new cancer and developmental cell biology research based on quantitative single cell techniques.
Acknowledgments
The authors thank Dr. Jay A. Werkhaven for providing tonsil specimens (Vanderbilt IRB #121328); Dr. Lola B. Chambless, Dr. Kyle D. Weaver, and Dr. Reid C. Thompson for providing glioma specimens (Vanderbilt IRB #131870); Dr. Mary A. Hooks, and Dr. Rondi M. Kauffmann for providing melanoma specimens (Vanderbilt IRB #030220); Dr. Jonathan M. Lehman and Dr. Pierre P. Massion for providing SCLC PDX specimens (with Vanderbilt IACUC approval).
NIH/NCI R00 CA143231 (J.M.I.),
R25 GM062459 (D.B.D.),
T32 CA009592 (D.B.D.),
F31 CA199993 (A.R.G.),
T32 HD007502 (J.S.),
The Vanderbilt-Ingram Cancer Center (VICC, P30 CA68485),
The Vanderbilt International Scholars Program (N.L.),
A Vanderbilt University Discovery Grant (J.M.I. and N.L.),
A VICC Provocative Question award (J.M.I.),
R01 NS096238 (R.A.I.),
DOD W81XWH-16-1-0171 (R.A.I.),
A VICC Discovery Grant (R.A.I.),
VICC Ambassadors awards (J.M.I. and R.A.I.).
Funding sources
Study and researchers were supported by NIH/NCI R00 CA143231 (J.M.I.), R25 GM062459 (D.B.D.), T32 CA009592 (D.B.D.), F31 CA199993 (A.R.G.), F31 HD007502 (J.S.), the Vanderbilt-Ingram Cancer Center (VICC, P30 CA68485), the Vanderbilt International Scholars Program (N.L.), a Vanderbilt University Discovery Grant (J.M.I. and N.L.), a VICC Provocative Question award (M.C.K. and J.M.I.), R01 NS096238, DOD W81XWH-16-1-0171, and a VICC Discovery Grant (R.A.I) and VICC Ambassadors awards (J.M.I. and R.A.I.).
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Literature Cited
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature biotechnology. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. Nomenclature for clusters of differentiation (CD) of antigens defined on human leukocyte populations. IUIS-WHO Nomenclature Subcommittee. Bull World Health Organ. 1984;62:809–815. [PMC free article] [PubMed] [Google Scholar]
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- Azari H, Millette S, Ansari S, Rahman M, Deleyrolle LP, Reynolds BA. Isolation and expansion of human glioblastoma multiforme tumor cells using the neurosphere assay. J Vis Exp. 2011:e3633. doi: 10.3791/3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KW, Low D, Ruedl C, Riccardi-Castagnoli P, Poidinger M, Greter M, Ginhoux F, Newell EW. High-dimensional analysis of the murine myeloid cell system. Nat Immunol. 2014;15:1181–1189. doi: 10.1038/ni.3006. [DOI] [PubMed] [Google Scholar]
- Behbehani GK, Thom C, Zunder ER, Finck R, Gaudilliere B, Fragiadakis GK, Fantl WJ, Nolan GP. Transient partial permeabilization with saponin enables cellular barcoding prior to surface marker staining. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2014;85:1011–1019. doi: 10.1002/cyto.a.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe’er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends in immunology. 2012;33:323–332. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson ZB, Nolan GP, Fantl WJ. Single-cell mass cytometry for analysis of immune system functional states. Curr Opin Immunol. 2013;25:484–494. doi: 10.1016/j.coi.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, Linda S, Martin PL, Pullen DJ, Viswanatha D, Willman CL, Winick N, Camitta BM, Children’s Oncology, G Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111:5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, Gill H, Presti J, Jr, Chang HY, van de Rijn M, Shortliffe L, Weissman IL. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14016–14021. doi: 10.1073/pnas.0906549106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay PK, Gierahn TM, Roederer M, Love JC. Single-cell technologies for monitoring immune systems. Nature immunology. 2014;15:128–135. doi: 10.1038/ni.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay PK, Roederer M. Cytometry: today’s technology and tomorrow’s horizons. Methods. 2012;57:251–258. doi: 10.1016/j.ymeth.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codega P, Silva-Vargas V, Paul A, Maldonado-Soto AR, Deleo AM, Pastrana E, Doetsch F. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron. 2014;82:545–559. doi: 10.1016/j.neuron.2014.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig FE, Foon KA. Flow cytometric immunophenotyping for hematologic neoplasms. Blood. 2008;111:3941–3967. doi: 10.1182/blood-2007-11-120535. [DOI] [PubMed] [Google Scholar]
- Diggins KE, Ferrell PB, Jr, Irish JM. Methods for discovery and characterization of cell subsets in high dimensional mass cytometry data. Methods. 2015;82:55–63. doi: 10.1016/j.ymeth.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggins KE, Leelatian N, Greenplate AR, Wogsland CE, Irish JM. Characterizing cell subsets in heterogeneous tissues using marker enrichment modeling. Nature methods. doi: 10.1038/nmeth.4149. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg VS, Landreneau RJ, Pfeifer ME, Donnenberg AD. Flow cytometric determination of stem/progenitor content in epithelial tissues: an example from nonsmall lung cancer and normal lung. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2013;83:141–149. doi: 10.1002/cyto.a.22156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nature reviews Immunology. 2016;16:149–163. doi: 10.1038/nri.2015.18. [DOI] [PubMed] [Google Scholar]
- Ferrell PB, Jr, Diggins KE, Polikowsky HG, Mohan SR, Seegmiller AC, Irish JM. High-Dimensional Analysis of Acute Myeloid Leukemia Reveals Phenotypic Changes in Persistent Cells during Induction Therapy. PloS one. 2016a;11:e0153207. doi: 10.1371/journal.pone.0153207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell PB, Maier CR, Roussel M, Savona MR, Irish JM. Expression of S100A9 in Bone Marrow Cells Differentiates Refractory Cytopenia with Multilineage Dysplasia (RCMD) from Refractory Anemia with Excess Blasts (RAEB) and Acute Myeloid Leukemia (AML) Blood. 2016b;128:4303–4303. [Google Scholar]
- Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe’er D, Nolan GP, Bendall SC. Normalization of mass cytometry data with bead standards. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Fragiadakis GK, Bruggner RV, Nicolau M, Finck R, Tingle M, Silva J, Ganio EA, Yeh CG, Maloney WJ, Huddleston JI, Goodman SB, Davis MM, Bendall SC, Fantl WJ, Angst MS, Nolan GP. Clinical recovery from surgery correlates with single-cell immune signatures. Science translational medicine. 2014;6:255ra131. doi: 10.1126/scitranslmed.3009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenplate AR, Johnson DB, Ferrell PB, Irish JM. Systems immune monitoring in cancer therapy. European journal of cancer. 2016a;61:77–84. doi: 10.1016/j.ejca.2016.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenplate AR, Johnson DB, Roussel M, Savona MR, Sosman JA, Puzanov I, Ferrell PB, Jr, Irish JM. Myelodysplastic Syndrome Revealed by Systems Immunology in a Melanoma Patient Undergoing Anti-PD-1 Therapy. Cancer immunology research. 2016b;4:474–480. doi: 10.1158/2326-6066.CIR-15-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K, Parks DR, Herzenberg LA. Demonstration of B-cell maturation in X-linked immunodeficient mice by simultaneous three-colour immunofluorescence. Nature. 1983;306:270–272. doi: 10.1038/306270a0. [DOI] [PubMed] [Google Scholar]
- Hulspas R, O’Gorman MR, Wood BL, Gratama JW, Sutherland DR. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry B Clin Cytom. 2009;76:355–364. doi: 10.1002/cyto.b.20485. [DOI] [PubMed] [Google Scholar]
- Irish JM. Beyond the age of cellular discovery. Nature immunology. 2014;15:1095–1097. doi: 10.1038/ni.3034. [DOI] [PubMed] [Google Scholar]
- Irish JM, Czerwinski DK, Nolan GP, Levy R. Altered B-cell receptor signaling kinetics distinguish human follicular lymphoma B cells from tumor-infiltrating nonmalignant B cells. Blood. 2006a;108:3135–3142. doi: 10.1182/blood-2006-02-003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish JM, Doxie DB. High-dimensional single-cell cancer biology. Curr Top Microbiol Immunol. 2014;377:1–21. doi: 10.1007/82_2014_367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–228. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Irish JM, Kotecha N, Nolan GP. Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nature reviews Cancer. 2006b;6:146–155. doi: 10.1038/nrc1804. [DOI] [PubMed] [Google Scholar]
- Irish JM, Myklebust JH, Alizadeh AA, Houot R, Sharman JP, Czerwinski DK, Nolan GP, Levy R. B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12747–12754. doi: 10.1073/pnas.1002057107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasti S, Costantini B, Seidl T, Perez Abellan P, Martinez Llordella M, McLornan D, Diggins KE, Kulasekararaj A, Benfatto C, Feng X, Smith A, Mian SA, Melchiotti R, de Rinaldis E, Ellis R, Petrov N, Povoleri GA, Sook Chung S, Thomas NS, Farzaneh F, Irish JM, Heck S, Young NS, Marsh JC, Mufti GJ. Deep-phenotyping of Tregs identifies an immune signature for idiopathic aplastic anemia and predicts response to treatment. Blood. 2016 doi: 10.1182/blood-2016-03-703702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Current protocols in cytometry/editorial board, J Paul Robinson, managing editor … [et al] 2010;17 doi: 10.1002/0471142956.cy1017s53. Chapter 10:Unit 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzik PO, Clutter MR, Nolan GP. Coordinate analysis of murine immune cell surface markers and intracellular phosphoproteins by flow cytometry. J Immunol. 2005;175:2357–2365. doi: 10.4049/jimmunol.175.4.2357. [DOI] [PubMed] [Google Scholar]
- Krutzik PO, Irish JM, Nolan GP, Perez OD. Analysis of protein phosphorylation and cellular signaling events by flow cytometry: techniques and clinical applications. Clinical immunology. 2004;110:206–221. doi: 10.1016/j.clim.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2003;55:61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nature medicine. 1999;5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- Leelatian N, Diggins KE, Irish JM. Characterizing Phenotypes and Signaling Networks of Single Human Cells by Mass Cytometry. Methods Mol Biol. 2015;1346:99–113. doi: 10.1007/978-1-4939-2987-0_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelatian N, Doxie DB, Greenplate AR, Mobley BC, Lehman JM, Sinnaeve J, Kauffmann RM, Werkhaven JA, Mistry AM, Weaver KD, Thompson RC, Massion PP, Hooks MA, Kelley MC, Chambless LB, Ihrie RA, Irish JM. Single cell analysis of human tissues and solid tumors with mass cytometry. Cytometry B Clin Cytom. 2016 doi: 10.1002/cyto.b.21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold MD, Newell EW, Maecker HT. Multiparameter Phenotyping of Human PBMCs Using Mass Cytometry. Methods Mol Biol. 2015;1343:81–95. doi: 10.1007/978-1-4939-2963-4_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, Finck R, Gedman AL, Radtke I, Downing JR, Pe’er D, Nolan GP. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maecker HT, Frey T, Nomura LE, Trotter J. Selecting fluorochrome conjugates for maximum sensitivity. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2004;62:169–173. doi: 10.1002/cyto.a.20092. [DOI] [PubMed] [Google Scholar]
- Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012;12:191–200. doi: 10.1038/nri3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin Lab Med. 2007;27:469–485. v. doi: 10.1016/j.cll.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin BE, Baumgarth N, Bigos M, Roederer M, De Rosa SC, Altman JD, Nixon DF, Ottinger J, Oxford C, Evans TG, Asmuth DM. Nine-color flow cytometry for accurate measurement of T cell subsets and cytokine responses. Part I: Panel design by an empiric approach. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2008;73:400–410. doi: 10.1002/cyto.a.20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myklebust JH, Brody J, Kohrt HE, Kolstad A, Czerwinski DK, Walchli S, Green MR, Troen G, Liestol K, Beiske K, Houot R, Delabie J, Alizadeh AA, Irish JM, Levy R. Distinct patterns of B-cell receptor signaling in non-Hodgkins’ lymphomas identified by single cell profiling. Blood. 2016 doi: 10.1182/blood-2016-05-718494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Cheng Y. Mass cytometry: blessed with the curse of dimensionality. Nature immunology. 2016;17:890–895. doi: 10.1038/ni.3485. [DOI] [PubMed] [Google Scholar]
- Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas KJ, Greenplate AR, Flaherty DK, Matlock BK, Juan JS, Smith RM, Irish JM, Kalams SA. Multiparameter analysis of stimulated human peripheral blood mononuclear cells: A comparison of mass and fluorescence cytometry. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2015 doi: 10.1002/cyto.a.22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas KJ, Greenplate AR, Flaherty DK, Matlock BK, Juan JS, Smith RM, Irish JM, Kalams SA. Multiparameter analysis of stimulated human peripheral blood mononuclear cells: A comparison of mass and fluorescence cytometry. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2016;89:271–280. doi: 10.1002/cyto.a.22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornatsky O, Bandura D, Baranov V, Nitz M, Winnik MA, Tanner S. Highly multiparametric analysis by mass cytometry. J Immunol Methods. 2010;361:1–20. doi: 10.1016/j.jim.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Ornatsky OI, Lou X, Nitz M, Schafer S, Sheldrick WS, Baranov VI, Bandura DR, Tanner SD. Study of cell antigens and intracellular DNA by identification of element-containing labels and metallointercalators using inductively coupled plasma mass spectrometry. Anal Chem. 2008;80:2539–2547. doi: 10.1021/ac702128m. [DOI] [PubMed] [Google Scholar]
- Parks DR, Hardy RR, Herzenberg LA. Three-color immunofluorescence analysis of mouse B-lymphocyte subpopulations. Cytometry. 1984;5:159–168. doi: 10.1002/cyto.990050210. [DOI] [PubMed] [Google Scholar]
- Pastrana E, Cheng LC, Doetsch F. Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc Natl Acad Sci U S A. 2009;106:6387–6392. doi: 10.1073/pnas.0810407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Polikowsky HG, Wogsland CE, Diggins KE, Huse K, Irish JM. Cutting Edge: Redox Signaling Hypersensitivity Distinguishes Human Germinal Center B Cells. Journal of immunology. 2015;195:1364–1367. doi: 10.4049/jimmunol.1500904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nature biotechnology. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M, Reyner K, Deleyrolle L, Millette S, Azari H, Day BW, Stringer BW, Boyd AW, Johns TG, Blot V, Duggal R, Reynolds BA. Neurosphere and adherent culture conditions are equivalent for malignant glioma stem cell lines. Anat Cell Biol. 2015;48:25–35. doi: 10.5115/acb.2015.48.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JO, Treisman J, Garlie N, Hanson JP, Oaks MK. Flow cytometry assessment of residual melanoma cells in tumor-infiltrating lymphocyte cultures. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2012;81:374–381. doi: 10.1002/cyto.a.22047. [DOI] [PubMed] [Google Scholar]
- Robinson J. Comparative overview of flow and image cytometry. Curr Protoc Cytom. 11:2005. doi: 10.1002/0471142956.cy1201s31. Chapter 12:Unit 12. [DOI] [PubMed] [Google Scholar]
- Roederer M, Quaye L, Mangino M, Beddall MH, Mahnke Y, Chattopadhyay P, Tosi I, Napolitano L, Terranova Barberio M, Menni C, Villanova F, Di Meglio P, Spector TD, Nestle FO. The genetic architecture of the human immune system: a bioresource for autoimmunity and disease pathogenesis. Cell. 2015;161:387–403. doi: 10.1016/j.cell.2015.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel M, Greenplate AR, Irish JM. Experimental Approaches for the Investigation of Innate Immunity. WORLD SCIENTIFIC; 2016. Dissecting Complex Cellular Systems with High Dimensional Single Cell Mass Cytometry; pp. 15–26. [Google Scholar]
- Saeys Y, Gassen SV, Lambrecht BN. Computational flow cytometry: helping to make sense of high-dimensional immunology data. Nature reviews Immunology. 2016;16:449–462. doi: 10.1038/nri.2016.56. [DOI] [PubMed] [Google Scholar]
- Schulz KR, Danna EA, Krutzik PO, Nolan G. Single-cell phospho-protein analysis by flow cytometry. Curr Protoc Immunol. 17:2012. 11–20. doi: 10.1002/0471142735.im0817s96. Chapter 8:Unit 8. [DOI] [PubMed] [Google Scholar]
- Shekhar K, Brodin P, Davis MM, Chakraborty AK. Automatic Classification of Cellular Expression by Nonlinear Stochastic Embedding (ACCENSE) Proceedings of the National Academy of Sciences of the United States of America. 2014;111:202–207. doi: 10.1073/pnas.1321405111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Goldberger O, Garten Y, Rosenberg-Hasson Y, Lovelace PA, Hirschberg DL, Altman RB, Davis MM, Butte AJ. Towards a cytokine-cell interaction knowledgebase of the adaptive immune system. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2009:439–450. [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Spidlen J, Breuer K, Brinkman R. Preparing a Minimum Information about a Flow Cytometry Experiment (MIFlowCyt) compliant manuscript using the International Society for Advancement of Cytometry (ISAC) FCS file repository (FlowRepository.org) Current protocols in cytometry/editorial board, J Paul Robinson, managing editor … [et al] 2012;18 doi: 10.1002/0471142956.cy1018s61. Chapter 10:Unit 10. [DOI] [PubMed] [Google Scholar]
- Spitzer MH, Nolan GP. Mass Cytometry: Single Cells, Many Features. Cell. 2016;165:780–791. doi: 10.1016/j.cell.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung JW, Parks DR, Moore WA, Herzenberg LA, Herzenberg LA. Identification of B-cell subsets: an exposition of 11-color (Hi-D) FACS methods. Methods in molecular biology. 2004;271:37–58. doi: 10.1385/1-59259-796-3:037. [DOI] [PubMed] [Google Scholar]
- van Dongen J, Lhermitte L, Bottcher S, Almeida J, van der Velden VHJ, Flores-Montero J, Rawstron A, Asnafi V, Lecrevisse Q, Lucio P, Mejstrikova E, Szczepański T, Kalina T, de Tute R, Brüggemann M, Sedek L, Cullen M, Langerak AW, Mendonça A, Macintyre E, Martin-Ayuso M, Hrusak O, Vidriales MB, Orfão A, EuroFlow Consortium (EU-FP6, L.-C.-.-. 2012) EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. 1908–1975 doi: 10.1038/leu.2012.120. [*This reference does not have a pre-defined format and therefore uses the “Generic” format] [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen JJ, van der Velden VH, Bruggemann M, Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125:3996–4009. doi: 10.1182/blood-2015-03-580027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhu J, Pierson E, Batzoglou S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. bioRxiv. 2016 doi: 10.1038/nmeth.4207. [DOI] [PubMed] [Google Scholar]
- Wang L, Abbasi F, Ornatsky O, Cole KD, Misakian M, Gaigalas AK, He HJ, Marti GE, Tanner S, Stebbings R. Human CD4+ lymphocytes for antigen quantification: characterization using conventional flow cytometry and mass cytometry. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2012;81:567–575. doi: 10.1002/cyto.a.22060. [DOI] [PubMed] [Google Scholar]
- Wogsland CE, Greenplate AR, Kolstad A, Myklebust JH, Irish JM, Huse K. Mass cytometry of follicular lymphoma tumors reveals intrinsic heterogeneity in proteins including HLA-DR and a deficit in non-malignant plasmablast and germinal center B cell populations. Cytometry B Clin Cytom. 2016 doi: 10.1002/cyto.b.21498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood BL, Arroz M, Barnett D, DiGiuseppe J, Greig B, Kussick SJ, Oldaker T, Shenkin M, Stone E, Wallace P. 2006 Bethesda International Consensus recommendations on the immunophenotypic analysis of hematolymphoid neoplasia by flow cytometry: optimal reagents and reporting for the flow cytometric diagnosis of hematopoietic neoplasia. Cytometry B Clin Cytom. 2007;72(Suppl 1):S14–22. doi: 10.1002/cyto.b.20363. [DOI] [PubMed] [Google Scholar]
- Zimmerlin L, Donnenberg VS, Donnenberg AD. Rare event detection and analysis in flow cytometry: bone marrow mesenchymal stem cells, breast cancer stem/progenitor cells in malignant effusions, and pericytes in disaggregated adipose tissue. Methods Mol Biol. 2011;699:251–273. doi: 10.1007/978-1-61737-950-5_12. [DOI] [PubMed] [Google Scholar]
- Zunder ER, Finck R, Behbehani GK, Amir el AD, Krishnaswamy S, Gonzalez VD, Lorang CG, Bjornson Z, Spitzer MH, Bodenmiller B, Fantl WJ, Pe’er D, Nolan GP. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nature protocols. 2015;10:316–333. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]