

ABSTRACT
The cellular response to DNA double-strand breaks is orchestrated by the protein kinase ATM, which phosphorylates key actors in the DNA repair network. WRAP53β is a multifunctional protein that controls trafficking of factors to Cajal bodies, telomeres and DNA double-strand breaks but what regulates the involvement of WRAP53β in these separate processes remains unclear. Here, we show that in response to various types of DNA damage, including IR and UV, WRAP53β is phosphorylated on serine residue 64 by ATM with a time-course that parallels its accumulation at DNA lesions. Interestingly, recruitment of phosphorylated WRAP53β (pWRAP53βS64) to sites of such DNA damage promotes its interaction with γH2AX at these locations. Moreover, pWRAP53βS64 stimulates the accumulation of the repair factor 53BP1 at DNA double-strand breaks and enhances repair of this type of damage via homologous recombination and non-homologous end joining. At the same time, phosphorylation of WRAP53β is dispensable for its localization to Cajal bodies, where it accumulates even in unstressed cells. These findings not only reveal ATM to be an upstream regulator of WRAP53β, but also indicates that phosphorylation of WRAP53β at serine 64 controls its involvement in the DNA damage response and may also restrict its other functions.
KEYWORDS: γH2AX, ATM, DNA damage response, MDC1, phosphorylation, RNF8, TCAB1, WDR79, WRAP53β, WRAP53
Abbreviations
- 4-OHT
4-Hydroxytamoxifen
- 53BP1
p53-binding protein 1
- ATM
Ataxia Telangiectasia Mutated
- ATR
Ataxia Telangiectasia and Rad3-related protein
- BRCA1
Breast Cancer 1
- BrdU
5-Bromo-2´-deoxyuridine
- BSA
Bovine serum albumin
- CSK
Cytoskeleton
- DAPI
4′,6-diamidino-2-phenylindole
- DNA-PK
DNA-dependent protein kinase
- GFP
Green fluorescent protein
- Gy
Gray
- HR
Homologous recombination
- J
Joule
- LacO
Lac operator
- LacI
Lac repressor
- MDC1
Mediator of DNA damage checkpoint 1
- NHEJ
Non-homologous end joining
- NP40
Nonidet P-40
- UV
Ultraviolet
- IP
Immunoprecipitation
- IR
Ionizing radiation
- PBS
Phosphate buffered saline
- PIC
Protease inhibitor cocktail
- PI3K
Phosphoinositide 3-kinase
- RAD51
Radiation sensitive 51
- RNF8
Ring finger protein 8
- RNF168
Ring finger protein 168
- RPA
Replication protein A
- S64
Serine residue 64
- scaRNAs
Small Cajal body-specific RNAs
- SMN
Survival of motor neuron protein
- SNPs
Single nucleotide polymorphisms
- TCAB1
Telomerase Cajal body protein-1
- U2OS
U-2 osteosarcoma
- WRAP53
WD40-encoding RNA antisense to p53
- WT
Wild Type
- WDR79
WD repeat-containing protein-79
Introduction
The WRAP53 (WD40 encoding RNA Antisense to p53) gene, originally identified in our laboratory as an antisense gene of the p53 tumor suppressor,1 encodes a WD40 protein WRAP53β (also known as WRAP53, WDR79, TCAB1) involved in multiple cellular processes. First, this protein plays a central role in the maintenance of the nuclear organelles known as Cajal bodies, recruiting factors such as the SMN (survival of motor neuron) protein, scaRNAs (small Cajal body-specific RNAs) and telomerase to these bodies.2-4 Upon loss of WRAP53β these organelles collapse and cannot reform, resulting in mislocalization of associated factors.2 Second, via Cajal bodies WRAP53β targets telomerase to telomeres, thereby regulating their elongation.4 Third, WRAP53β helps orchestrate the repair of DNA double-strand breaks by recruiting the ubiquitin ligase RNF8 (Ring finger protein 8) to DNA breaks important for both homologous recombination (HR) and non-homologous end joining (NHEJ).5,6
The significance of WRAP53β for tissue homeostasis is demonstrated clearly by the finding that inherited mutations in this protein lead to dyskeratosis congenita, a syndrome characterized by failure of the bone marrow and a predisposition to develop cancer.7 Moreover, certain single nucleotide polymorphisms (SNPs) in the gene or downregulation of this protein predisposes individuals to various sporadic forms of cancer, including breast, ovarian and head-neck cancer, and are also correlated with shorter survival of such patients and resistance of head-neck tumors to radiotherapy.6,8-10 In addition, overexpression of WRAP53β has been detected in some types of tumor, including head-neck,11,12 lung13 and rectal14 cancer. Even though recent observations demonstrate that overexpression of WRAP53β leads to more efficient repair of DNA double-strand breaks,15 the clinical relevance of such overexpression in connection with cancer remains vague. Furthermore, the exact manner in which the different functions and regulators of WRAP53β are coordinated is not yet clear.
As with so many other processes, post-translational modifications of proteins, including phosphorylation play a crucial signaling role in the orchestration of cellular responses to DNA damage. The protein kinases related to phosphoinositide 3-kinase (PI3K), including ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related) and DNA-PK (DNA-dependent protein kinase) initiate the damage cascade by phosphorylating nearby molecules of histone H2AX (at serine 139) to form γH2AX, a well-established marker of DNA damage and repair.
Although these kinases all recognize Serine-Glutamine (SQ) and Threonine-Glutamine (TQ) motifs,16,17 with a preference for phosphorylating serine over threonine, their co-factors and the types of damage by which they are activated differ. For example, ATM senses double-strand breaks induced by ionizing radiation (IR), whereas ATR responds primarily to single-strand breaks, replication stress and bulky lesions induced by ultraviolet (UV) light.18 While often functioning in a manner similar to ATM, DNA-PK is also distinct in acting mainly together with the Ku proteins of the NHEJ repair pathway.19,20 In addition to H2AX, a proteomic screen following induction of DNA damage by IR revealed 700 other potential substrates for ATM/ATR.21
We reported previously that following exposure of cells to IR, WRAP53β is recruited to DNA double-strand breaks by a process that requires MDC1, γH2AX and ATM.5 At these sites, WRAP53β acts as a scaffold for interactions between RNF8 and MDC1, thereby mediating ubiquitylation of damaged chromatin and promoting recruitment of downstream repair factors (RNF168, 53BP1, BRCA1 and RAD51).5 In the current investigation, we demonstrate that upon DNA damage WRAP53β is phosphorylated on serine 64 by ATM and this phosphorylation promotes its localization to DNA double-strand breaks, its interaction with γH2AX and, in addition, its role in the repair of these lesions.
Results
WRAP53β is phosphorylated in response to DNA damage
A proteomic screen previously identified serine residue 64 (S64) on WRAP53β, as a putative site for phosphorylation by ATM/ATR (Fig. 1A).21 Comparative analysis revealed maintenance of this site and the subsequent glutamine (the SQ motif) throughout evolution, an indicator of its biological importance (Fig. 1B). To examine whether in cells WRAP53β is phosphorylated at S64, we generated a phosphorylation-specific antibody targeting this site (pWRAP53βS64). The specificity of this antibody was first confirmed by expressing a WRAP53β construct in which serine 64 was mutated to alanine and indeed binding was only obtained to the wild-type (WT) and not the phospho-incompetent variant (S64A) of WRAP53β (Fig. 1C). No major change in protein stability was observed between WT and the S64A mutant of WRAP53β (Fig. 1D). Strikingly, this antibody revealed S64-phosphorylation of Flag-WRAP53β in response to DNA damage triggered by a variety of agents, including IR, UV, hydroxyurea, camptothecin and mitomycin C (Fig. 1E).
Figure 1.
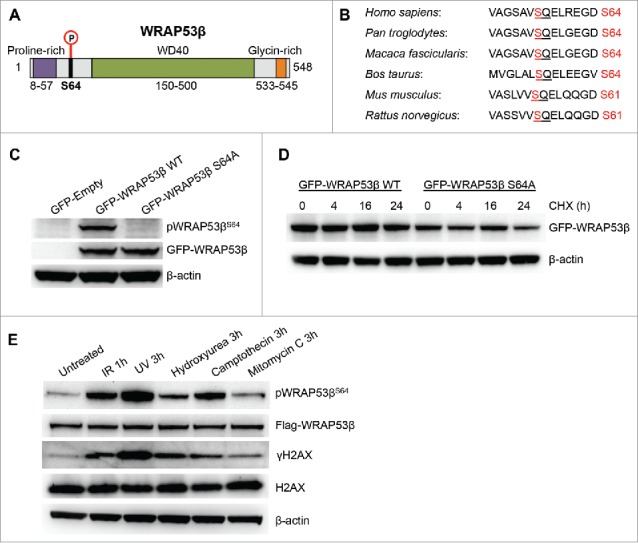
WRAP53β is phosphorylated in response to DNA damage. (A) Schematic illustration of the WRAP53β protein, the organization of its domains and its S64 phosphorylation site. (B) Conservation analysis of the S64 phosphorylation site in different species. The SQ motif is underlined and the S phosphorylation site marked in red. (C) U2OS cells were transfected with an empty plasmid or a plasmids encoding wild-type (WT) or S64A WRAP53β tagged with GFP for 16 h; irradiated (6 Gy) and harvested 1 h later for western blotting of pWRAP53βS64, WRAP53β and β-actin. (D) Representative western blots of protein levels of WT or S64A GFP-WRAP53β in irradiated U2OS cells. Cells were transfected with indicated plasmids for 16 h; irradiated (6 Gy) then immediately treated with cycloheximide (CHX, 50 μg/ml) for the periods indicated, after which they were harvested for western blotting with GFP and β-actin antibodies. (E) U2OS cells stably expressing Flag-WRAP53β were either left untreated or exposed to 6 Gy IR followed by 1 h recovery; 30 J/m2 UV followed by 3 h recovery; 2 mM hydroxyurea followed by 3 h recovery; 1 μM camptothecin followed by 3 h recovery or 6 μg/ml mitomycin C followed by 3 h recovery, after which they were harvested for western blotting with the antibodies indicated.
ATM mediates phosphorylation of WRAP53β in response to DNA damage induced by IR and UV
Since the role of WRAP53β in the repair of DNA damage induced by IR is well known, we utilized this damaging agent for further characterization. Human U2OS cancer cells that stably overexpress Flag-tagged WRAP53β were initially used to examine phosphorylation of WRAP53β.2,15 Following irradiation, WRAP53β was rapidly phosphorylated, a process that continued for 24 h (Fig. 2A) with a time-course resembling the kinetics of WRAP53β accumulation at DNA lesions.5
Figure 2.
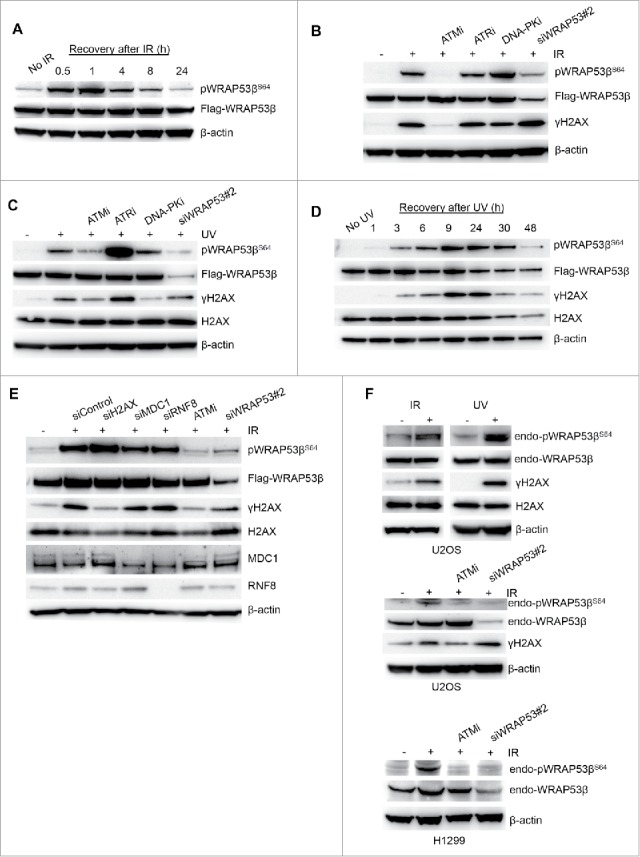
ATM phosphorylates WRAP53β upon IR and UV exposure. (A) U2OS cells stably overexpressing Flag-WRAP53β were irradiated (6 Gy) and harvested at the time-points indicated for western blotting with the antibodies indicated. (B) U2OS cells stably expressing Flag-WRAP53β were treated with the inhibitors indicated for 16 h or siWRAP53 for 48 h, exposed to 6 Gy IR, and harvested 1 h later for western blotting with the antibodies indicated. (C) U2OS cells stably expressing Flag-WRAP53β were treated with the inhibitors indicated for 24 h or siWRAP53 for 48 h, exposed to 30 J/m2 UV, and harvested 3 h later for western blotting with the antibodies indicated. (D) U2OS cells stably overexpressing Flag-WRAP53β were exposed to 30 J/m2 UV and harvested at the time-points indicated for western blotting with the indicated antibodies. (E) U2OS cells stably expressing Flag-WRAP53β were treated with the siRNAs indicated for 48 h or an inhibitor of ATM (ATMi) for 24 h, exposed to 6 Gy IR, and harvested 1 h later for western blotting with the antibodies indicated. (F) Parental U2OS cells were exposed to 6 Gy IR (1 hour recovery) or 30 J/m2 UV (9 hours recovery) and harvested for western blotting with the antibodies indicated. H1299 cells were treated with ATMi for 16 h or siWRAP53 for 48 h, irradiated with 6 Gy and harvested 30 min later for western blotting of pWRAP53βS64, WRAP53β and β-actin.
The recruitment of WRAP53β to DNA double-strand breaks is totally dependent on ATM, partially dependent on ATR, but independent of DNA-PK.5 Strikingly, inhibition of ATM completely abolished the phosphorylation of WRAP53β at S64 following IR treatment (Fig. 2B), whereas inhibition of ATR or DNA-PK had no effect (Fig. 2B). Since ATR responds mainly to other types of DNA damage, we also examined phosphorylation WRAP53β after UV treatment, which confirmed that ATM, and not ATR or DNA-PK mediate this phosphorylation (Fig. 2C). Furthermore, knockdown of WRAP53β attenuated such phosphorylation (Fig. 2B and 2C), again confirming the specificity of our antibody. WRAP53β phosphorylation was more rapid following exposure to IR than UV, in agreement with the relative kinetics of induction of DNA damage by these agents,22 and as reflected in the slower formation of γH2AX after UV (Fig. 2D). Depleting cells of γH2AX, MDC1 or RNF8, which are known to interact with WRAP53β,5,23 did not influence phosphorylation of WRAP53β, indicating that these proteins are not involved in the formation of pWRAP53βS64 (Fig. 2E).
Phosphorylation of endogenous WRAP53β was also observed after UV and IR in parental U2OS and H1299 cells, the latter of which express high levels of WRAP53β (Fig. 2F). However, detection of phosphorylated WRAP53β was weak in comparison to total WRAP53β, indicating that only a small proportion of this protein is normally phosphorylated at S64. Together, these observations reveal that in response to DNA damage, ATM rapidly phosphorylates WRAP53β at S64.
Phosphorylated WRAP53β accumulates at DNA double-strand breaks
To determine whether phosphorylated WRAP53β is recruited to sites of DNA damage, we employed three different approaches. The first of these was based on U2OS cells in which site-specific DNA double-strand breaks can be induced by FokI endonuclease. These cells are stably transfected with a reporter containing several hundred repeats of the Lac operator (LacO) and expression of an mCherry-tagged Lac repressor (LacI) fused to FokI (mCherry-LacI-FokI) results in binding of LacI to the LacO, in which the nonspecific FokI endonuclease creates double-strand breaks (Fig. 3A),24,25 as confirmed here by the formation of γH2AX at these sites (Fig. 3B). Interestingly, upon formation of DNA breaks in the LacO array, endogenous pWRAP53βS64 was recruited to this array (Fig. 3C), a pattern observed with different antibodies against WRAP53β (Fig. 3C). Next, following laser micro-irradiation, endogenous phosphorylated WRAP53β was observed to rapidly re-localize to the sites of DNA damage (laser stripes) (Fig. 3D). Thus, endogenous pWRAP53βS64 is recruited to DNA damaged sites induced by FokI or laser micro-irradiation.
Figure 3.
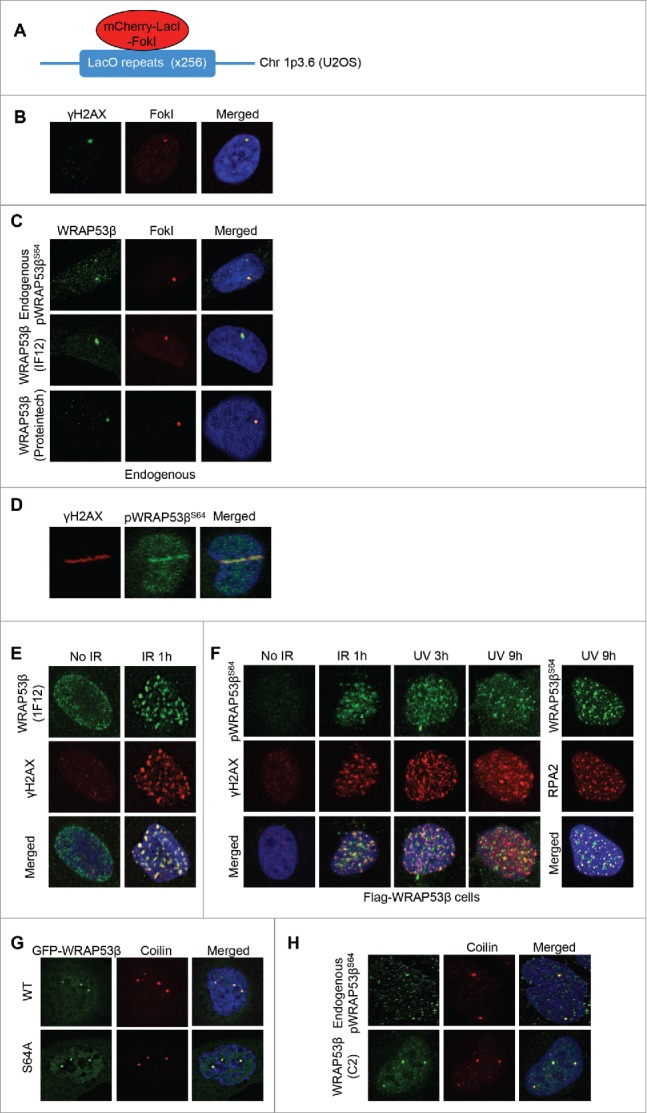
Phosphorylated WRAP53β accumulates at sites of DNA damage. (A) Schematic illustration of the LacO-LacI-FokI system in U2OS cells. (B) Following a 5 h induction of the mCherry-LacI-FokI fusion protein, the U2OS-FokI cells were fixed and immunostained for γH2AX to confirm the generation of double-strand breaks. (C) Following a 5 h induction of double-strand breaks by the mCherry-LacI-FokI fusion protein, the U2OS-FokI cells were fixed and immunostained with the antibodies indicated. (D) Parental U2OS cells were micro-irradiated, fixed 15 min later and immunostained for γH2AX and pWRAP53βS64. (E) Parental U2OS cells were either left untreated or irradiated with 6 Gy (1 hour recovery), pre-extracted using CSK buffer, fixed and immunostained for γH2AX and endogenous WRAP53β using the 1F12 antibody. (F) U2OS cells stably expressing Flag-WRAP53β were either left untreated or exposed to 6 Gy IR or 30 J/m2 UV, allowed to recover for periods of time indicated, then immunostained for pWRAP53βS64, γH2AX or RPA2. (G) U2OS cells were transiently transfected with the plasmids indicated for 24 h, fixed and stained for the Cajal body marker protein coilin. (H) U2OS cells were irradiated with 6 Gy (1 hour recovery), pre-extracted using CSK buffer, fixed and immunostained for WRAP53β using indicated antibodies and for coilin. In all immunofluorescent stainings, nuclei were stained with DAPI.
Finally, we examined whether pWRAP53βS64 accumulated in repair foci, which previously has been observed for endogenous WRAP53β and as also confirmed here (Fig. 3E).5 While we were unable to detect foci formation of endogenous pWRAP53βS64 upon DNA damage, phosphorylated WRAP53βS64 formed foci in irradiated U2OS cells that stably overexpress Flag-tagged WRAP53β (Fig. 3F) and these foci clearly overlapped with γH2AX foci (Fig. 3F). The fact that IR-induced foci of pWRAP53βS64 were only detected in cells stably overexpressing WRAP53β could be due to the weak detection of phosphorylated endogenous WRAP53β. Foci formation of WRAP53βS64 overlapping both γH2AX and the repair factor RPA2 (replication protein A2) was also observed in cells overexpressing Flag-WRAP53β after UV exposure (Fig. 3F). Together, these findings reveal that phosphorylated WRAP53βS64 localizes to sites of DNA damage induced by IR, UV or FokI, indicating that it performs a function there.
WRAP53β is highly enriched in Cajal bodies and to examine whether this localization depends on phosphorylation of WRAP53β, cells were transfected transiently with WT or the phospho-incompetent mutant of WRAP53β tagged with green fluorescent protein (GFP). This revealed that the phospho-incompetent mutant of WRAP53β accumulated in these bodies to the same extent (or often to a greater extent) as the WT protein (Fig. 3G). This demonstrates that phosphorylation of WRAP53β is dispensable for its localization to Cajal bodies, in agreement with the fact that this protein accumulate in these organelle also in non-irradiated or unstressed cells where the level of pWRAP53βS64 should be low.
We also explored whether phosphorylated WRAP53βS64 localizes to Cajal bodies. Although co-staining between pWRAP53βS64 and the Cajal body marker protein coilin revealed localization of pWRAP53βS64 in Cajal bodies (Fig. 3H), this localization was observed less frequently using the pWRAP53βS64 antibody (pWRAP53βS64 detected in around 50% of Cajal bodies) compared to antibodies targeting total WRAP53β (total WRAP53β detected in 100% of Cajal bodies).
Phosphorylation of WRAP53β promotes its interaction with γH2AX and stimulates its functional role in repair of double-strand breaks
Since recruitment of WRAP53β to sites of DNA damage requires interaction with its partners γH2AX and MDC1,5,23 we next tested whether phosphorylation of WRAP53β influences these interactions. Strikingly, substitution of serine 64 in WRAP53β with alanine prevented its interaction with γH2AX in response to DNA damage, but not with MDC1 (Fig. 4A). The same extent of interaction was also observed between the phospho-mutant of WRAP53β and RNF8 (Fig. 4A). These results are consistent with our previous findings that binding of WRAP53β to MDC1 and RNF8 is independent of DNA damage and ATM.5,23 Thus, phosphorylation of WRAP53β at S64 by ATM promotes its interaction with γH2AX, but not with MDC1 or RNF8.
Figure 4.
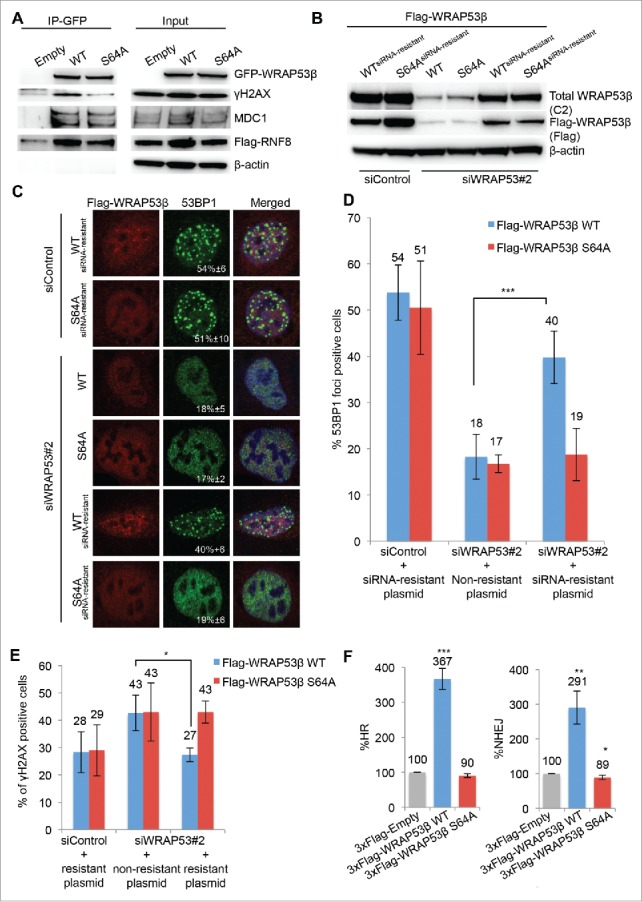
Phosphorylation of WRAP53β promotes its interaction with γH2AX, but not its association with MDC1 or RNF8 and promotes its role in double-strand break repair. (A) U2OS cells were transiently transfected with the plasmids indicated for 16 h, irradiated with 6 Gy and subjected to immunoprecipitation with a GFP antibody 15 minutes later followed by immunoblotting with the antibodies indicated. (B-D) U2OS cells were transfected with siControl or siWRAP53#2 oligonucleotides for 24 h followed by transfection with the 1xFlag-plasmids indicated for another 24 h, exposed to 6 Gy IR and harvested 1 h later for (B) western blotting with the antibodies indicated or (C) immunostaining for 53BP1 and Flag and quantification. The numbers in white depict the percentages of 100–200 Flag-transfected cells whose nuclei contained >10 IR-induced 53BP1 foci. Means ± SD are shown, n = 3. (D) The graph shows the quantification described in (C). *p < 0.05, **p < 0.01, ***p < 0.001, as determined by a non-paired 2-tailed Student's t-test. Similar results were obtained using 3xFlag-plasmids, i.e. restoration of 53BP1 foci by re-introduction of 3xFlag-WRAP53β WT but not S64A into cells depleted of WRAP53β (data not shown). (E) U2OS cells were transfected with siControl or siWRAP53#2 oligonucleotides for 8 h followed by transfection with the 1xFlag-plasmids indicated for another 16 h, exposed to 6 Gy IR and fixed 24 h later for immunostaining for γH2AX and Flag and quantification. The graph shows the percentages of 100–200 Flag-transfected cells whose nuclei contained >10 IR-induced γH2AX foci. Means ± SD, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by a non-paired 2-tailed Student's t-test. Similar results were obtained using 3xFlag-plasmids (data not shown). (F) FACS analysis of the efficiency of HR and NHEJ following transient transfection of U2OS cells with the 3xFlag-Empty, 3xFlag-WRAP53β WT or 3xFlag-WRAP53β S64A vectors for 48 hours. In all cases the values presented are means ± SD, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by Student's t-test.
It is known that WRAP53β facilitates recruitment of repair factors (RNF8, RNF168, 53BP1, BRCA1 and RAD51) to double-strand breaks5,9 and we wanted to explore the influence of WRAP53β phosphorylation in this context as well. Loss of this protein impairs accumulation of these factors at DNA lesions and we examined whether WRAP53β S64A could correct this defect. When constructs encoding siRNA-resistant forms of WT or S64A WRAP53β were introduced into cells depleted of endogenous WRAP53β (Fig. 4B), notably, only the WT could restore 53BP1 foci induced by IR (Fig. 4C and 4D), clearly indicating that phosphorylation of WRAP53β stimulates its recruitment of 53BP1 to double-strand breaks.
Loss of WRAP53β also results in increased amounts of residual γH2AX foci 24 h after IR, due to impaired DNA repair (Fig. 4E).5 While re-introduction of WT WRAP53β restored clearance of IR-induced γH2AX foci, re-introduction of the S64A mutant did not (Fig. 4E), again indicating that phosphorylation of WRAP53β stimulates its function in repair of double-strand breaks.
WRAP53β has recently been shown to be a rate-limiting factor in the repair of double-strand breaks and overexpression of this protein results in more efficient HR and NHEJ repair and fewer DNA breaks.15 Therefore, we asked whether phosphorylation of WRAP53β influences HR and NHEJ employing reporter assays that measure restoration of a functional GFP protein by these processes. The HR assay involves U2OS cells stably carrying a construct with the direct repeat (DR)-GFP sequence, in which a single DNA double-strand break can be introduced by expression of exogenous I-SceI endonuclease. Repair of this break by HR creates a functional GFP gene and its expression level provides an accurate measure of the efficiency of HR.26 The NHEJ assay is similar, but involves a GFP reporter construct (EJ5-GFP) with two I-SceI sites flanking a puro gene. Following cleavage of these sites by I-SceI, the puro gene is removed and repair of these double-strand breaks by NHEJ places a promoter adjacent to the GFP gene and allows expression of GFP.27 Strikingly, overexpression of WT or S64A of WRAP53β in these cells revealed that only the WT had an effect on the efficiency of HR and NHEJ, enhancing these approximately 4 and 3-fold, respectively (Fig. 4F). Thus, ATM-dependent phosphorylation of WRAP53β promotes the role of this protein in the repair of double-strand breaks, possibly by facilitating its recruitment to these lesions and interaction there with γH2AX (Fig. 5).
Figure 5.
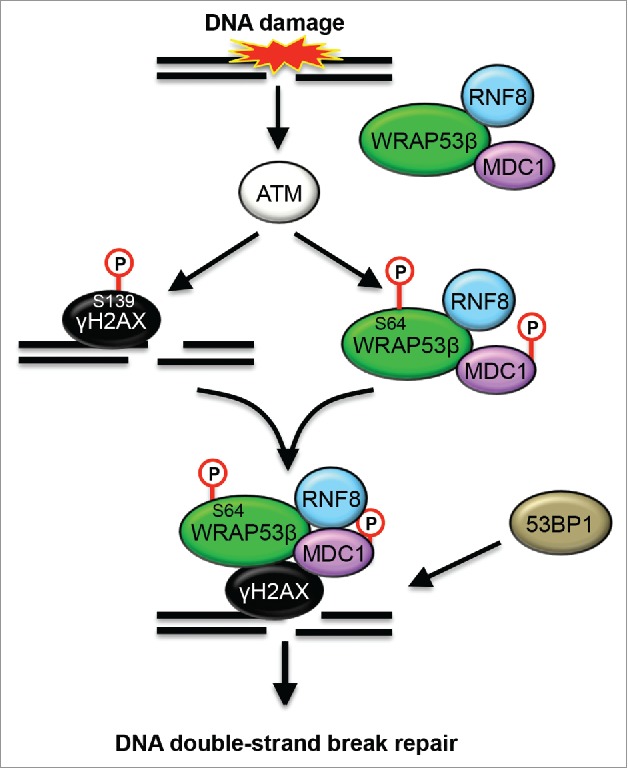
Schematic model of how phosphorylation of WRAP53β promotes role in the repair of DNA double-strand breaks. In response to DNA damage, ATM phosphorylates WRAP53β, H2AX and MDC1. This phosphorylation of WRAP53β (at S64) promotes its interaction with γH2AX, but not with MDC1 or RNF8. Since unphosphorylated WRAP53β still interacts with both MDC1 and RNF8, it appears like WRAP53β forms a complex with these proteins prior to DNA damage and that phoshporylation of WRAP53β triggers recruitment of this protein complex to DNA lesions. Phosphorylation of WRAP53β also stimulates the recruitment of 53BP1 to site of DNA damage and the following repair of DNA double-strand breaks by HR and NHEJ.
Discussion
In addition to controlling recruitment of factors to and maintenance of Cajal bodies, WRAP53β safeguards genome integrity by regulating both telomere elongation and repair of DNA double-strand breaks. Regulation of WRAP53β itself in connection with these different processes has been unknown, but here we demonstrate that phosphorylation of S64 by ATM controls its role in the repair of DNA double-strand breaks. In response to various DNA damaging agents, WRAP53β is phosphorylated with a time-course that, in the case of IR treatment, parallels the accumulation of this protein at DNA double-strand breaks. Inhibition of the upstream protein kinases ATM, ATR and DNA-PK revealed that only inhibition of ATM completely abolished this phosphorylation in response to IR and UV-induced DNA damage.
Our findings clearly demonstrate that phosphorylated WRAP53β is recruited to sites of DNA damage, induced by the FokI endonuclease, IR or UV, indicating that it plays a functional role at these sites. Indeed, phosphorylated, but not unmodified WRAP53β interacts with γH2AX, a known interaction partner of WRAP53β upon DNA damage. Notably, the known involvement of WRAP53β in promoting the interaction between the ubiqutin ligase RNF8 and its upstream partner MDC1 at double-strand breaks does not appear to be influenced by phosphorylation of WRAP53β, since unmodified WRAP53β still interacts with both MDC1 and RNF8. Our finding that the the mutant form of WRAP53β that could not be phoshporylated was fully capable of interacting with both RNF8 and MDC1 is in agreement with our previous observations that this binding is independent of both DNA damage and ATM.23 Furthermore, this indicates that WRAP53β forms a complex with MDC1 and RNF8 prior to DNA damage and subsequent phoshorylation of WRAP53β triggers recruitment of this protein complex to DNA lesions. Moreover, we previously showed that this recruitment of WRAP53β also requires MDC1,5 as well as RNF8 (data not shown), and these proteins may thus be necessary for stable association of WRAP53β with sites of DNA damage.
To unravel the function of pWRAP53βS64 at DNA lesions, we monitored accumulation of the repair protein 53BP1 (formation of 53BP1 repair foci) at these breaks, a process known to require WRAP53β.5 53BP1 is involved in the selection of repair pathway employed upon double-strand break formation and promotes repair via NHEJ.28 Strikingly, re-introduction of WT, but not S64A of WRAP53β into irradiated cells depleted of this protein could restore formation of these foci, clearly demonstrating that phosphorylation of WRAP53β by ATM is required for its role in the repair of double-strand breaks. Moreover, only the WT, but not S64A of WRAP53β could resolve residual γH2AX foci induced by irradiation of cells lacking endogenous WRAP53β. In addition, overexpression of WRAP53β was recently shown to results in more efficient HR and NHEJ repair. Our current findings demonstrate that only the WT, and not the phospho-incompetent mutant of WRAP53β had any effect on the efficiency of HR and NHEJ repair, again indicating that phosphorylation of WRAP53β promotes the role of this protein in the repair of double-strand breaks.
Our finding that only a small proportion of WRAP53β is phosphorylated in response to DNA damage, provides support for our hypothesis that the protein modified in this manner by ATM plays a specific role in DNA repair rather than the other functions of WRAP53β. For instance, phosphorylation could trigger re-distribution of WRAP53β from Cajal bodies to DNA breaks.
Indeed, accumulation of pWRAP53βS64 in Cajal bodies was detected less frequently compared to the localization of total WRAP53β in Cajal bodies, although this could also be due to low amounts of pWRAP53βS64 at this site below the threshold of detection. Moreover, S64A WRAP53β accumulated to a greater extent in Cajal bodies than the WT protein, indicating that an inability to become phosphorylated traps the protein at this location.
In summary, we demonstrate here that in response to DNA damage the Cajal body protein WRAP53β is phosphorylated by ATM and that this phosphorylation is important for the recruitment of WRAP53β to DNA lesions, its interaction with γH2AX, subsequent localization of the downstream repair factor 53BP1 to DNA breaks, as well as for HR and NHEJ repair. The present results represent a first step toward understanding how this multifunctional protein is regulated and, in particular the signals that govern its involvement in repair of DNA damage.
Materials and methods
Cells and culture conditions
Mock and Flag-WRAP53β U2OS cells were maintained in McCoy's 5A medium (HyClone, Thermo Scientific), selected with 10μg/ml Blasticidine S (InvivoGen), U2OS cells were maintained in McCoy's 5A medium (HyClone, Thermo Scientific), U2OS-FokI cells were maintained in DMEM with GlutaMAX (Gibco, Life Technologies) and H1299 cells (human non-small cell lung carcinoma cell line) were maintained in Dulbecco's modified medium (HyClone, Thermo Scientific) supplemented with 10% fetal bovine serum (HyClone) and 2,5 µg/mL Plasmocin (InvivoGen) at 37°C in 5% CO2 humidified incubators. The stable Flag-WRAP53β cells overexpress the open-reading frame of the protein tagged with 1xFlag.
Ionizing radiation
γ-irradiation was performed with a 137Cs source (Scanditronix, Uppsala, Sweden) at the Karolinska Institutet, Stockholm, at a photon dose rate of 0.5 Gy·minutes−1. Dosimetry was done with an ionization chamber as well as with ferro sulfate.
UV
UV irradiation was carried out with 30 J/m2 in a UV cross-linker (model UVC-500; Hoefer).
FokI system
The U2OS-FokI cells contain a stably integrated LacO array and stably express the mCherry-LacI-FokI fusion protein fused to a destabilization domain (DD) and a modified estradiol receptor (ER) (ER-mCherry-LacI-FokI-DD). This enable inducible nuclear expression of ER-mCherry-LacR-FokI-DD after administration of the small molecule Shield-1 ligand (stabilizes the DD-domain) and 4-hydroxytamoxifen (4-OHT; induce nuclear translocation of ER-mCherry-LacR-FokI-DD). To induce site-specific double-strand breaks by FokI, these cells were incubated with 1 µM Shield-1 (cat. no. 632189, Clontech) and 1 µM 4-OHT (cat. no. H7904, Sigma-Aldrich) for 5h.
Laser micro-irradiation
Localized DNA damage was generated by exposure of cells to a UV-A laser. U2OS cells were pre-sensitized with 10 mM 5-Bromo-2′-deoxyuridine (BrdU) for 24 hours at 37°C. Prior to microscopy the medium was replaced for a phenol red-free medium. Micro-irradiation was performed with a confocal microscope equipped with a 365-nm UV-A laser.
Treatment with drugs or inhibitors
2 mM Hydroxyurea (cat. no. H8627, Sigma-Aldrich), 1 μM camptothecin (cat. no. C9911, Sigma-Aldrich) or 6 μg/ml mitomycin C (cat. no. 10107409001, Roche) were added to cells for 3 h. ATM (KU55933) and DNA-PK (NU7441) inhibitors were obtained from TOCRIS bioscience. The ATR inhibitor (VE-821) was obtained from Axon MedChem (cat# Axon 1893). Where appropriate, 10 μM ATMi, 2 μM DNA-PKi and 2.5 μM ATRi were added to the culture medium 24 h prior to IR or UV treatment.
Cycloheximide chase
Cycloheximide was added directly to the culture medium after irradiation to give a final concentration of 50 µg/ml.
Antibodies
Primary antibodies: rabbit α-WRAP53-C2 (cat. no PA-2020-100, Innovagen AB, Sweden), mouse monoclonal α-WDR79 (clone 1F12, cat. no. H00055135-M04; Abnova), rabbit α-pWRAP53βS64 (cat. no PA-2230-100, Innovagen AB), mouse monoclonal α-WDR79 (clone 1F12, cat. no. H00055135-M04; Abnova), rabbit α-WRAP53 (cat. no 14761-1-AP, Proteintech), mouse α-γH2AX (cat. no 05-636, Millipore), rabbit α-γH2AX (cat. no 2577, Cell Signaling), rabbit α-H2AX (cat. no. ab11175, Abcam), mouse α-MDC1 (cat. no ab50003, abcam), mouse α-RNF8 (cat. no sc-271462, Santa Cruz Biotechnology), mouse RPA32/RPA2 [9H8] (cat. no ab2175, abcam), rabbit α-53BP1 (cat. no NB100-904, Novus Biologicals), mouse α-coilin (cat. no. sc-56298, Santa Cruz Biotechnology), mouse α-β-actin (cat. no A5441, Sigma), rabbit α-GFP (cat. no ab290, abcam), mouse α-Flag (cat. no F1804, Sigma-Aldrich), normal rabbit IgG (cat. no sc-2027, Santa Cruz Biotechnology) and normal mouse IgG (cat. no sc-2025, Santa Cruz Biotechnology).
Secondary antibodies: goat α-rabbit HRP (cat. no 7074, Cell Signal), horse α-mouse HRP (cat. no 7076, Cell Signal), goat α-rabbit Alexa Fluor 488 (cat. no A11008, Life technologies), goat α-mouse Alexa Fluor 488 (cat. no A11029, Life technologies) and donkey α-mouse Alexa Fluor 594 (cat. no A21203, Life technologies).
Western blotting
Cell extracts for western blot analysis: cells were harvested, washed and lysed in ice cold lysis buffer (100 mM Tris-HCL pH 8, 150mM NaCl, 1% NP-40, 1% protease inhibitor cocktail) for 30 minutes on ice followed by sonication. Lysates were centrifuged at 14000 rpm for 15 minutes at 4°C and protein concentrations were determined using Bradford assay (Biorad). Western blotting was performed according to standard procedures.
Immunoprecipitation
Cells were lysed in NP40 buffer (150 mM NaCl, 50 mM Tris-HCL pH 8,0, 1% NP40, 1% protease inhibitor cocktail) for 15 minutes on ice, followed by 3×10 seconds sonication. Protein lysates were spun down at 6000 rpm for 5 minutes and protein concentrations were quantified by Bradford assay (Biorad). Proteins were immunoprecipitated with 1 µg antibody per 1 mg protein and 10 µl Dynabeads Protein G (Life technologies) overnight at 4°C. The beads were washed 4 × 15 minutes in 1 ml NP40 buffer and prepared for western blotting.
Immunofluorescence microscopy
Cells grown on sterilized cover slips were fixed with 4% paraformaldehyde for 15 minutes at room temperature. They were then permeabilized with 0.1% Triton X-100 for 5 minutes at room temperature, followed by 30 minutes of blocking in blocking buffer (2% BSA, 5% glycerol, 0.2% Tween20, 0.1% NaN3). The coverslips were subsequently incubated for 1 hour in primary antibody followed by 40 minutes in secondary antibody, both diluted in blocking buffer, and finally mounted with Vectashield mounting medium containing DAPI (4′,6-diamidino-2-phenylindole, Vector laboratories). Images were acquired with a LSM700 confocal microscope (Zeiss), mounted on Axio observer.Z1 (Zeiss) equipped with Plan-Apochromat 63x/1.4 oil immersion lens, and processed with using Zen 2012 Black (Zeiss).
Pre-extraction: The cells were first washed with PBS and then incubated for 3 minutes at room temperature with cytoskeleton buffer (CSK) (10 mM Pipes, pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2 and 0.7% Triton X-100) and thereafter for another 3 minutes with the same CSK buffer supplemented with 0.3 mg/ml RNase A (CSK+R). Following these treatments, the cells were washed once again with PBS and then fixed in 4% paraformaldehyde.
siRNA
siRNA oligonucleotides used: siWRAP53#2 (cat. no SI00388948, Qiagen), siH2AX (cat. no SI00032844, Qiagen), siMDC1 (cat. no L-003506-00-0005, Dharmacon), siRNF8 (cat. no L-006900-00-0005, Dharmacon). 10 nM of siRNA was transfected into cells using HiPerfect (Qiagen) transfection reagent in accordance with the supplier's recommendations.
Plasmids
Plasmid transfections were performed using Lipofectamine 2000 Reagent (Invitrogen) according to the manufacturer's recommendations. The GFP-WRAP53β S64A, Flag-WRAP53β S64A, Flag-WRAP53β S64AsiRNA resistant and Flag-WRAP53β WTsiRNA resistant mutants were generated by Stratagene's QuikChange XL II site-directed mutagenesis kit (cat. no 200521). All primers used for PCR amplifications are listed in Table 1. Flag vector corresponds to the 1xFlag vector pCMV-Tag2 vector (Invitrogen). Generation of the GFP- and Flag-WRAP53β WT (same as EGFP/Flag-WRAP53β FL) was described previously.5
Table 1.
Primers used in this study.
| Mutagenesis primers to generate the S64A mutation | |
|---|---|
| PCR primer name | Sequence 5′– 3′ |
| WRAP53βS64A F | CTGGCTCAGCTGTGGCCCAGGAGCTACG |
| WRAP53βS64A R | CGTAGCTCCTGGGCCACAGCTGAGCCAG |
| Mutagenesis primers to generate the siWRAP53#2 resistant mutations | |
| PCR primer name | Sequence 5′– 3′ |
| WRAP53βsiRNA resistant F | GCAAACGGGAGTCTCTCTGAAGAAGAAGC |
| WRAP53βsiRNA resistant R | GCTTCTTCTTCAGAGAGACTCCCGTTTGC |
HR and NHEJ assays
300 000 cells were seeded into 6-well plates. 24 hours later cells were transfected with an I-SceI vector together with a vector expressing Flag-Empty, Flag-WRAP53β WT or Flag-WRAP53β S64A using Lipofectamine 2000 (Invitrogen). 3xFlag vector corresponds to p3xFlag-CMV-9 (Sigma-Aldrich). The next day media was changed and 24 hours after this cells were harvested by trypsination, washed with PBS and the GFP signal arising from the recombination event was measured by flow cytometry on a FACS Calibur, with fluorescence detected in the FL1-H channel (logarithmic scale). The frequency of repair in cells transfected with the various plasmids was calculated relative to cells transfected with the empty plasmid. Each data point represents the mean ± standard deviation from 3 independent experiments.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank Dr Roger Greenberg (University of Pennsylvania) for the U2OS-FokI cell line. This work was supported by grants from the Swedish Cancer Society (Cancerfonden), the Swedish Research Foundation (VR), the Strategic Research Program in Cancer (StratCan), Worldwide Cancer Research (former AICR), the Swedish Childhood Cancer Society (Barncancerfonden), the Cancer Society of Stockholm (Cancerföreningen) and the Karolinska Institutet.
References
- 1.Mahmoudi S, Henriksson S, Corcoran M, Mendez-Vidal C, Wiman KG, Farnebo M. Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol Cell 2009; 33:462-71; PMID:19250907; https://doi.org/ 10.1016/j.molcel.2009.01.028 [DOI] [PubMed] [Google Scholar]
- 2.Mahmoudi S, Henriksson S, Weibrecht I, Smith S, Soderberg O, Stromblad S, Wiman KG, Farnebo M. WRAP53 is essential for Cajal body formation and for targeting the survival of motor neuron complex to Cajal bodies. PLoS Biol 2010; 8:e1000521; PMID:21072240; https://doi.org/ 10.1371/journal.pbio.1000521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tycowski KT, Shu MD, Kukoyi A, Steitz JA. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol Cell 2009; 34:47-57; PMID:19285445; https://doi.org/ 10.1016/j.molcel.2009.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, Artandi SE. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 2009; 323:644-8; PMID:19179534; https://doi.org/ 10.1126/science.1165357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henriksson S, Rassoolzadeh H, Hedstrom E, Coucoravas C, Julner A, Goldstein M, Imreh G, Zhivotovsky B, Kastan MB, Helleday T, et al.. The scaffold protein WRAP53beta orchestrates the ubiquitin response critical for DNA double-strand break repair. Genes Dev 2014; 28:2726-38; PMID:25512560; https://doi.org/ 10.1101/gad.246546.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henriksson S, Farnebo M. On the road with WRAP53beta: guardian of Cajal bodies and genome integrity. Frontiers Genetics 2015; 6:91; PMID:25852739; https://doi.org/ 10.3389/fgene.2015.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, Artandi SE. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 2011; 25:11-6; PMID:21205863; https://doi.org/ 10.1101/gad.2006411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garvin S, Tiefenbock K, Farnebo L, Thunell LK, Farnebo M, Roberg K. Nuclear expression of WRAP53beta is associated with a positive response to radiotherapy and improved overall survival in patients with head and neck squamous cell carcinoma. Oral Oncol 2015; 51:24-30; PMID:25456005; https://doi.org/ 10.1016/j.oraloncology.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 9.Hedstrom E, Pederiva C, Farnebo J, Nodin B, Jirstrom K, Brennan DJ, Farnebo M. Downregulation of the cancer susceptibility protein WRAP53beta in epithelial ovarian cancer leads to defective DNA repair and poor clinical outcome. Cell Death Dis 2015; 6:e1892; PMID:26426684; https://doi.org/ 10.1038/cddis.2015.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silwal-Pandit L, Russnes H, Borgen E, Skarpeteig V, Moen Vollan HK, Schlichting E, Karesen R, Naume B, Borresen-Dale AL, Farnebo M, et al.. The Sub-cellular localization of WRAP53 has prognostic impact in breast cancer. PLoS One 2015; 10:e0139965; PMID:26460974; https://doi.org/ 10.1371/journal.pone.0139965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun CK, Luo XB, Gou YP, Hu L, Wang K, Li C, Xiang ZT, Zhang P, Kong XL, Zhang CL, et al.. TCAB1: a potential target for diagnosis and therapy of head and neck carcinomas. Mol Cancer 2014; 13:180; PMID:25070141; https://doi.org/ 10.1186/1476-4598-13-180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao X, Huang D, Sui X, Liu G, Song X, Xie J, Huang D. Overexpression of WRAP53 is associated with development and progression of esophageal squamous cell carcinoma. PLoS One 2014; 9:e91670; PMID:24626331; https://doi.org/ 10.1371/journal.pone.0091670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y, Yang C, Chen J, Song X, Li Z, Duan M, Li J, Hu X, Wu K, Yan G, et al.. Overexpression of WDR79 in non-small cell lung cancer is linked to tumour progression. J Cell Mol Med 2016; 20:698-709; PMID:26849396; https://doi.org/ 10.1111/jcmm.12759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Wang DW, Adell G, Sun XF. WRAP53 is an independent prognostic factor in rectal cancer- a study of Swedish clinical trial of preoperative radiotherapy in rectal cancer patients. BMC Cancer 2012; 12:294; PMID:22805008; https://doi.org/ 10.1186/1471-2407-12-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rassoolzadeh H, Bohm S, Hedstrom E, Gad H, Helleday T, Henriksson S, Farnebo M. Overexpression of the scaffold WD40 protein WRAP53beta enhances the repair of and cell survival from DNA double-strand breaks. Cell Death Dis 2016; 7:e2267; PMID:27310875; https://doi.org/ 10.1038/cddis.2016.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 1999; 274:37538-43; PMID:10608806; https://doi.org/ 10.1074/jbc.274.53.37538 [DOI] [PubMed] [Google Scholar]
- 17.O'Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, Abraham RH, Lai JH, Hill D, Shiloh Y, Cantley LC, et al.. Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem 2000; 275:22719-27; PMID:10801797; https://doi.org/ 10.1074/jbc.M001002200 [DOI] [PubMed] [Google Scholar]
- 18.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462-7; PMID:11571274; https://doi.org/ 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- 19.Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 2004; 64:2390-6; PMID:15059890; https://doi.org/ 10.1158/0008-5472.CAN-03-3207 [DOI] [PubMed] [Google Scholar]
- 20.Jette N, Lees-Miller SP. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Progress Biophys Mol Biol 2015; 117:194-205; PMID:25550082; https://doi.org/ 10.1016/j.pbiomolbio.2014.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al.. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160-6; PMID:17525332; https://doi.org/ 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- 22.Staszewski O, Nikolova T, Kaina B. Kinetics of gamma-H2AX focus formation upon treatment of cells with UV light and alkylating agents. Environmental Mol Mutagenesis 2008; 49:734-40; PMID:18800352; https://doi.org/ 10.1002/em.20430 [DOI] [PubMed] [Google Scholar]
- 23.Rassoolzadeh H, Coucoravas C, Farnebo M. The proximity ligation assay reveals that at DNA double-strand breaks WRAP53beta associates with gammaH2AX and controls interactions between RNF8 and MDC1. Nucleus 2015; 6:417-24; PMID:26734725; https://doi.org/ 10.1080/19491034.2015.1106675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010; 141:970-81; PMID:20550933; https://doi.org/ 10.1016/j.cell.2010.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 2013; 20:317-25; PMID:23377543; https://doi.org/ 10.1038/nsmb.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13:2633-8; PMID:10541549; https://doi.org/ 10.1101/gad.13.20.2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunn A, Stark JM. I-SceI-Based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol 2012; 920:379-91; PMID:22941618; https://doi.org/ 10.1007/978-1-61779-998-3_27 [DOI] [PubMed] [Google Scholar]
- 28.Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 2014; 34:1380-8; PMID:24469398; https://doi.org/ 10.1128/MCB.01639-13 [DOI] [PMC free article] [PubMed] [Google Scholar]