

Summary
Interaction between the endoplasmic-reticulum protein STIM1 and the plasma-membrane channel Orai1 generates calcium signals that are central for diverse cellular functions. How STIM1 binds and activates Orai1 remains poorly understood. Using electrophysiological, optical and biochemical techniques, we examined the effects of mutations in the STIM1-Orai1 activating region (SOAR) of STIM1. We find that SOAR mutants that are deficient in binding to resting Orai1 channels are able to bind to and boost activation of partially activated Orai1 channels. We further show that the STIM1 binding regions on Orai1 undergo structural rearrangement during channel activation. The results suggest that activation of Orai1 by SOAR occurs in multiple steps. A first step in which SOAR binding to Orai1 partially activates the channel and induces a rearrangement in the SOAR binding site of Orai1. That rearrangement of Orai1 then permits sequential steps of SOAR binding, via distinct molecular interactions, to fully activate the channel.
Graphical Abstract
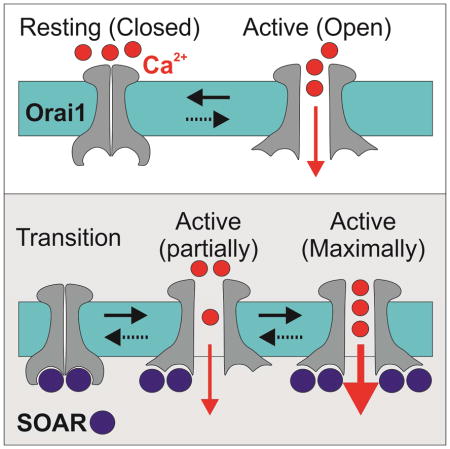
Store operated Ca2+ entry (SOCE) is a specialized cellular mechanism that relies on a dynamic assembly of endoplasmic reticulum-plasma membrane (ER-PM) bridges to translate the depletion of Ca2+ from the ER to the opening of Ca2+ channels in the plasma membrane (Hogan et al., 2010; Putney, 1986). The prototypical mediator of this pathway is the Ca2+ Release Activated Ca2+ (CRAC) channel (Hoth and Penner, 1992; Lewis and Cahalan, 1989; Zhang and McCloskey, 1995). The best characterized components of CRAC channels are STIM1, a single span membrane protein that mostly localizes to the ER membrane(Soboloff et al., 2006a; Spassova et al., 2006), and Orai1 (also known as CRACM1(Vig et al., 2006)), a pore forming subunit that multimerizes to form channels in the plasma membrane(Feske et al., 2006; Liou et al., 2005; Mercer et al., 2006; Peinelt et al., 2006; Prakriya et al., 2006; Roos et al., 2005; Soboloff et al., 2006b; Vig et al., 2006; Zhang et al., 2005, 2006). STIM1 and Orai1 are ubiquitously expressed and contribute to diverse cellular functions such as secretion, gene expression and cell differentiation and proliferation(Hogan et al., 2010). The activation of CRAC channels is a subject of intense investigation and several steps in this process are becoming well understood. Depletion of Ca2+ from the ER causes Ca2+ release from the luminal facing STIM1 EF-hand Ca2+ binding domain and elicits dimerization of adjacent SAM domains(Stathopulos et al., 2006). These changes prompt reorganization of the STIM1 trans-membrane region that, in turn, drives an extensive conformational change in the cytosolic facing carboxyl-terminal region(Covington et al., 2010; Luik et al., 2008; Muik et al., 2011; Stathopulos et al., 2006; Yu et al., 2011, 2013; Zhou et al., 2013). The conformational change of STIM1 exposes a short region that has been broken down into three largely overlapping segments, which are necessary and sufficient for interaction with Orai1: the CAD (amino acids 342–448), CCb9 (amino acids 339–444) and SOAR (amino acids 344–442) (Kawasaki et al., 2009; Park et al., 2009; Yuan et al., 2009). We refer to the activation region hereafter as SOAR. Activated STIM1 molecules form oligomers and translocate to cortical regions of the ER that are in close proximity to the plasma membrane(Lioudyno et al., 2008; Luik et al., 2006, 2008; Wu et al., 2006; Xu et al., 2006). In these ER-PM microdomains, STIM1 binds to Orai1 through cooperative interactions with the N and C termini regions of Orai1, to gate the channel open(Lis et al., 2010; McNally et al., 2013; Palty and Isacoff, 2016; Palty et al., 2015; Park et al., 2009; Zheng et al., 2013; Zhou et al., 2010). The process of coupling STIM1 binding to induction of channel opening is, however, not well understood.
X-ray crystallography of the Drosophila melanogaster dOrai revealed that the channel consists of six subunits(Hou et al., 2012). In the resting channel, in the absence of STIM, the C terminal regions from neighboring dOrai subunits dimerize through hydrophobic coiled-coil interactions to generate an internal trimeric symmetry within each dOrai hexamer(Hou et al., 2012). Structural and biochemical studies have shown that SOAR is a functional dimer(Park et al., 2009; Yang et al., 2012; Yuan et al., 2009). Recently, an NMR study showed that short fragments of SOAR assemble into a dimer that binds to two Orai1 C terminal segments thereby suggesting that STIM1 binds to Orai1 at 1:1 STIM1 to Orai1 stoichiometry (Stathopulos et al., 2013). Investigations of the stoichiometric requirements for Orai1 channel activation by STIM1 showed, however, that a 1:1 STIM1 to Orai1 ratio yields only partial, ~25%, channel activation compared to the full activation obtained at a 2:1 STIM1 to Orai1 ratio (Hoover and Lewis, 2011; Li et al., 2011). In line with a 2:1 STIM1 to Orai1 stoichiometry, a recent study suggested that STIM1 dimers couple to Orai1 monomers to activate the channel(Zhou et al., 2015). Nevertheless, it is not known whether the mechanism of STIM1 binding is the same for all STIM1 molecules that bind the Orai1 channel at saturation and the molecular nature of partial and full activation is not understood. In this work we address these issues by studying the effect of mutations in the α1, α2 and α4 helical regions of SOAR on binding to Orai1 and induction of the partial and maximal activation states. We show that SOAR mutants with deficient binding to and activation of resting Orai1 channel are fully capable of boosting to maximum the activity of a channel that is partially activated by covalent fusion to SOAR, by pharmacological stimulation or by a mutation of Orai1. We further show that channel activation is accompanied by a rearrangement of the STIM1 binding sites in Orai1. Our findings suggest that the resting Orai1 channel is partially activated following binding to STIM1 and that this process induces a rearrangement in the STIM1 binding site of Orai1 that permits binding, via a distinct set of molecular interactions, to additional STIM1 molecules that induce full channel activation.
Results
Mutant SOAR ligands interact with Orai1 channels in a state dependent manner
The α1, α2 and α4 regions of SOAR were recently shown to play important roles in the binding of STIM1 to Orai1 and Orai1 activation (Maus et al., 2015; Wang et al., 2014). We asked whether these interactions mediate both partial activation, which occurs at 1:1 STIM1 to Orai1 ratio, and full activation, which occurs at 2:1 STIM1 to Orai1 ratio(Hoover and Lewis, 2011; Li et al., 2011). Within the SOAR helical regions mutation of four lysine residues in α1 (382KIKKK386mutated to 382AIAAA386 herein and referred to henceforward as 4KA), F394H in α2 or R429C in α4 were each shown to abolish the ability of either full length STIM1 or SOAR-like constructs of STIM1 to bind and activate Orai1 (Calloway et al., 2010; Jha et al., 2013; Korzeniowski et al., 2010; Stathopulos et al., 2013; Wang et al., 2014; Yang et al., 2012). We created SOAR-like constructs (residues 342–465, referred to as S1C henceforward, Figure 1A) harboring these mutations. Single molecule analysis showed that all mutants retain the ability to form dimers (Figure S1i). Western blot analysis showed that WT, the 4KA or the F394H S1C mutants express at similar levels (Supplemental text and Figure S1i). Expression of S1C R429C, however, required increased transcription in order to reach these expression levels (achieved by a four-fold increase in plasmid concentration of S1C R429C, Figure S1i). Consistent with the earlier studies (Korzeniowski et al., 2010; Maus et al., 2015; Park et al., 2009; Yuan et al., 2009; Zhou et al., 2015), whole cell patch-clamp electrophysiology showed that cells co-expressing Orai1 with S1C have large currents, with a characteristic CRAC current-voltage relationship, whereas there was little or no such current in cells expressing Orai1 with one of the mutant S1Cs (Figure 1B and see Supplemental text and Figure S1ii). The disruption of Orai1 channel activation by these mutations of S1C was also seen in an assay of nuclear NFAT localization, an established downstream effector of store-operated Ca2+ entry (Figure S1iiB, red bars).
Figure 1. SOAR mutants 4KA or F394H interact with Orai1 channels in a state dependent manner.

(A) Cartoon representation of Orai1 and S1C mutants used in this work. (B) Summary of current densities recorded from cells co-expressing the indicated mCherry-S1C mutants together with Orai1-EGFP. (C) Summary of current densities recorded from cells co-expressing the indicated mCherry-S1C construct together with Orai1-S-EGFP. Note that mutations are introduced to S1C domains but not to the S domain. (D–F) Western blot analysis of cell lysate or immunoprecipitated material (IP) prepared from cells expressing Orai1-EGFP (D), Orai1-S-EGFP (E) or the indicated Orai1-SEGFP constructs (F) together with the indicated FLAG-mCherry-S1C constructs. Agarose beads conjugated to anti-mCherry nanododies were used to immunoprecipitate mCherry tagged proteins and antibodies against Orai1, FLAG and GFP were used for protein detection. Full blots of images shown in (D–F) and statistical analysis of data shown in (B–C) are displayed in Figure S1iii and in Figure S1ii, respectively.
Having confirmed that the published mutations in the α1, α2 and α4 of STIM1 each disrupt the activation of Orai1, we asked if they would boost activity in a partially activated channel. To induce partial activation, we relied on an earlier approach (Li et al., 2011) for generating a defined sub-maximal SOAR to Orai1 ratio of 1:1 by fusing a single SOAR-like domain (residues 340–485, referred to henceforward as S) to Orai1 (Figure 1C). Consistent with earlier studies (Li et al., 2011; McNally et al., 2012; Palty et al., 2015), and indicative of partial channel activation, cells expressing Orai1-S alone exhibited an average current density of about ~25–30% of that measured when wt S1C was co-expressed together with either Orai1 or with Orai1-S (Figure 1C and Figure S1ii). Strikingly, cells expressing Orai1-S together with a soluble version of either the 4KA or F394H mutant of S1C had a wildtype current level (Figure 1C and Figure S1ii). This suggests that, like wildtype SOAR, these SOAR mutants are able to interact with the partially activated channel and boost its level of activation to the maximum. In contrast, S1C R429C did not boost the current of Orai1-S (Figure 1C and Figure S1ii), suggesting that this mutation disrupts not only interaction with the resting channel but also with the partially activated Orai1-S channel. Furthermore, while similar currents where recorded from cells co-expressing S1C and Orai1-S wt or with Orai1-S in which the S domain carried the F394H mutation, little to no current was recorded when cells co-expressed Orai1-S and S1C constructs in which both SOAR domains carried the F394H mutation, suggesting that partial activation of the Orai1-S channel is critical for interaction with the S1C F394H mutant.
We next directly tested binding of the mutant SOARs to two versions of the channel: Orai1 on its own and the partially activated Orai1-S. To do this, we co-expressed an EGFP-tagged Orai1 (Orai1-EGFP) or Orai1-S (Orai1-S-EGFP) with wildtype or mutant versions of FLAG- and mCherry-tagged S1C (FLAG-mCherry-S1C) and performed co-immunoprecipitation (coIP) analysis. In line with our electrophysiology results, wt S1C bound robustly to Orai1, whereas the 4KA mutant bound weekly and the F394H or R429C mutants did not bind. However, wt S1C and the 4KA and F394H mutant versions of S1C all bound to Orai1-S (Figure 1D & E and Figure S1iii). Consistent with our electrophysiological analysis, the S1C with the R429C mutation did not bind to either Orai1 or Orai1-S (Figure 1D & E and Figure S1iii). We reasoned that because of their enforced proximity, adjacent S domains in each hexameric Orai1-S channel would form three dimers of SOAR. Nonetheless, we also considered the possibility that the S domains of Orai1-S could alternatively dimerize with soluble S1C mutant subunits thereby creating a SOAR heterodimers that may be fully capable of activing Orai1 channels as recently shown(Zhou et al., 2015). To address this possibility, we generated two Orai1-S constructs, in the first we deleted the Orai1 C terminal region (residues 273–301, Orai1 ΔC) and in the second we introduced the point mutation L273S to the C terminal region. Since these Orai1 mutations abolish interaction with soluble S1C we asked whether fusion of the S domain to these Orai1 mutant channels will be sufficient to fully rescue physical interaction with soluble S1C. Arguing against dimerization between soluble S1C and S domains found on Orai1-S, S1C bound robustly to wt Orai1-S but only weakly to Orai1-S L273S and not at all to Orai1-S ΔC. The indication that soluble S1C do not readily heterodimerize with channel fused S domains is further supported by the finding that S1C R429 failed to interact with Orai1-S (Figure 1E) despite its ability to form heterodimers with WT SOAR(Maus et al., 2015) or with S1C subunits (see Figure S1i).
Taken together, these results suggest that binding of SOAR to Orai1 at 1:1 SOAR to Orai1 ratio partially activates the channel and enables binding of additional SOAR molecules, which is required to fully activate the channel.
The CRAC channel modulator 2-APB partially activates Orai1 and enables liganding by mutant SOARs
In order to further understand the interaction of SOAR mutants with the partially active Orai1 channel, we undertook a second approach to modify the active state of Orai1 by using the CRAC channel modulator 2-APB(Peinelt et al., 2008). Previous work showed that the cytoplasmic C-terminal domain of STIM1 or the F394H mutant of SOAR exhibit little or no interaction with Orai1, respectively, but are recruited to the channel by 2-APB, which also transiently activated the channel (Wang et al., 2009; Zhou et al., 2015). We reasoned that 2-APB may enable the binding of these deficient STIM1 ligands to Orai1 by transiently activating Orai1 channels. To address this hypothesis we asked whether 2-APB would rescue interaction between Orai1 and the 4KA or R429C SOAR mutants. We expressed mCherry-Orai1 alone or together with a GFP-tagged version of wt S1C or a version of S1C carrying either the 4KA, F394H or R429C S1C mutation and studied changes in the cellular distribution of S1C and the level of intracellular Ca2+ following application of 2-APB. At rest (in absence of 2-APB), the EGFP-S1C mutants were mostly distributed in the cytosol but, strikingly, following addition of 2-APB (50μM) this fluorescence redistributed to the plasma membrane (Figure 2A–C and Figure S2). In line with deficient binding of the S1C mutants to resting Orai1 channels, Ca2+ addback experiments revealed a strong rise in intracellular Ca2+ levels in cells co-expressing Orai1 and wt S1C but not in cells expressing Orai1 alone or together with either of the S1C mutants. Addition of 2-APB after Ca2+ addition inhibited Ca2+ fluxes in cells co-expressing Orai1 and wt S1C. However, addition of 2-APB induced a strong and transient Ca2+ rise in cells co-expressing Orai1 with mutant mCherry-S1C, but only a weak and transient increase in cells expressing Orai1 alone (Figure 2D and Figure S2). Thus, these results are consistent with earlier studies(Peinelt et al., 2008; Wang et al., 2014, 2009; Zhou et al., 2015) and indicate that Orai1 is weakly and transiently activated by 2-APB and that this activation is massively potentiated by binding of the mutant S1C constructs. Importantly, the result demonstrate that the SOAR R429C mutant that carries a structural impairment (Maus et al., 2015) and fails to activate Orai1 channels or partially active Orai1-S channels is not a dead ligand but rather a ligand that operates in a specific state dependent manner. Thus, these results suggest that transient activation of Orai1 channels by 2-APB induces a transition that enables the mutant SOAR ligands to bind.
Figure 2. The CRAC channel modulator 2-APB facilitates interaction between mutant S1C and Orai1 channels.

(A–C) Representative fluorescent images of mCherry-Orai1 and the indicated EGFP-S1C construct before and after application of 2-APB (50μM). Note that the cellular distribution of EGFP-S1C mutants is changed following addition of 2-APB. (D) Averaged intracellular Ca2+ responses in cells expressing Orai1 alone (n=25) or together with the indicated S1C construct (wt n=17, 4KA n=41, F394H n=96, R429C n=46) following addition of Ca2+ (2mM) and 2-APB (50μM) to the extracellular solution as marked by arrows. Individual traces are shown in Figure S2.
The P245L mutation in Orai1 induces partial activated channels and interaction with S1C mutants
Thus far, we have seen that two methods of partially activating Orai1 (by enforcement of a 1:1 SOAR to Orai1 ratio or by exposure to 2-APB) induce a change in the channel that enables it to be liganded and further activated by mutant SOARs that cannot bind or activate Orai1 in its resting state. To further test this, we asked if a mutant version of Orai1, which is constitutively partially activated by mutation to a conserved proline residue at the bend near the middle of TM4 (P245L) (Palty et al., 2015), would show the same behavior. Indicative of interaction between mutant S1C’s and P245L Orai1, we found that GFP-tagged versions of S1C carrying either the F394H or 4KA mutation were localized both to plasma membrane and cytosol in cells co-expressing mCherry-Orai1 P245L (Figure S3). Furthermore, co-immunoprecipitation analyses revealed similar interaction between Orai1 P245L and either wt S1C or the 4KA mutant S1C, as well as substantial, albeit reduced, interaction with the F394H and R429C mutant versions of S1C (Figure 3A and Figure S3). In line with co-IP analysis, currents recorded from cells expressing Orai1 P245L together with wt S1C or either mutant were significantly larger than currents recorded in cells that expressed Orai1 P245L alone (Figure 3B). Thus, unlike wt Orai1, the partially active P245L mutant of Orai1 binds to and is further activated by the mutant versions of S1C.
Figure 3. Orai1 P245L interacts with S1C mutants.

(A) Western blot analysis of cell lysate or IP material prepared from cells expressing the indicated EGFP-S1C constructs together with mCherry-Orai1 P245L. Agarose beads conjugated to anti-GFP antibodies were used to immunoprecipitate EGFP tagged S1C proteins and antibodies against FLAG and mCherry were used for protein detection. (B) Time course of averaged currents (left) and typical plots of the current-voltage relationship (right) of currents recorded from cells co-expressing mCherry-orai1 P245L alone or with EGFP-S1C construct. Full blots of images shown in (A) and statistical analysis of data shown in (B) are displayed in Figure S3
The N and C terminal regions of Orai1 contribute to the interface that binds mutant SOAR when the channel is partially activated
Our results so far show that partial activation of Orai1, by any one of three manipulations, induces a change in the channel that enables liganding and further activation by mutant SOAR ligands that neither bind to nor activate the resting channel. This suggests that the mutant SOAR binds to an interface in Orai1 that is exposed in the partially activated state of the channel. SOAR has been shown to interact with two sites in Orai1, a weak interaction with the N terminal region and a strong interaction with the C terminal region (Derler et al., 2013; McNally et al., 2013; Palty and Isacoff, 2016; Park et al., 2009; Yuan et al., 2009; Zhou et al., 2010). To study the contributions of the N and C terminal regions of Orai1 to SOAR interaction in the partially activated state, we separately examined key mutations in the Orai1 N terminal region (K85E) and C terminal region (L273S) that diminish or prevent, respectively, binding of soluble SOAR fragments to the resting channel(Lis et al., 2010; Muik et al., 2008; Navarro-Borelly et al., 2008). We asked whether partial activation by 2-APB would restore binding of SOAR to the mutant Orai1 channels. We expressed EGFP-S1C together with mCherry-Orai1 in which Orai1 was either wt or mutated in the N or C terminal region and examined the ability of 2-APB to change the cellular distribution of EGFP-S1C. Under basal conditions, before addition of 2-APB, EGFP-S1C fluorescence was found almost exclusively on the plasma membrane in cells expressing wt Orai1 and the addition of 2-APB did not change this distribution (Figure 4A). This is consistent with the high affinity and maximal degree of binding of SOAR to the wt channel (McNally et al., 2013; Palty and Isacoff, 2016; Park et al., 2009; Yuan et al., 2009). In cells co-expressing mCherry-Orai1 K85E, EGFP-S1C was found in both the plasma membrane and cytosol and this distribution was not affected by 2-APB (Figure 4B). This is consistent with the reduced affinity of S1C binding to the N-terminal Orai1 mutant (Lis et al., 2010; McNally et al., 2013; Palty and Isacoff, 2016) and shows that the remaining interaction of wt S1C, presumably with the C terminal region of Orai1, is not further boosted by 2-APB. In striking contrast, in cells co-expressing Orai1 L273S, at basal conditions EGFP-S1C fluorescence was localized entirely to the cytosol but addition of 2-APB redistributed a substantial fraction of the fluorescence to the plasma membrane (Figure 4C). The redistribution of S1C to the PM in cells co-expressing S1C and Orai1 L273S also triggered strong Ca2+ influx compared to cells expressing Orai1 L273S alone. This shows that although soluble S1C cannot bind to the C-terminal mutated Orai1 when the channel is in the resting state, it does bind and boosts channel activation when the channel is partially activated. The observation that the K85E mutation in Orai1 eliminates the enhancing effect of 2-ABP on SOAR-Orai1 interaction suggests that the N-terminal region may constitute an important element in the interface in Orai1 that is exposed in the active state of the channel. The observation that 2-APB restores only a moderate degree of interaction between SOAR with Orai1 L273S compared to the level seen with wt Orai1 suggests that the C terminal region of Orai1 may also contribute to the exposed active-state site.
Figure 4. The Orai1 N and C terminal regions are required for recruitment of mutant S1C to Orai1-S channels.

(A–C, Right panels) Representative fluorescent images of the indicated EGFP-S1C (wt) construct co-expressed with the indicated mCherry-Orai1 construct before and after application of 2-APB (50μM). (D-G, Left panels) The averaged ratio of the EGFP–S1C fluorescence harboring the indicated mutation ((D)- wt, (E)-4KA, (F)-F394H and (G)-R429C) and co-expressed with the indicated Orai1 construct at the plasma membrane normalized to that in the cytosol from multiple corresponding images (n=8–13 cells).
Since mutation in the α1, α2 or α4 regions of SOAR all prevent binding to Orai1 and 2-APB restores a moderate degree of SOAR-Orai1 binding, we used the S1C mutants as sensitive tools to further elucidate whether both the N and C terminal regions contribute to the SOAR-binding active state interface of Orai1. We reasoned that if a mutant SOAR fragment interacts solely with either the N or the C terminal region of the partially activated Orai1 channel then mutating the other terminal region will not affect this interaction. When co-expressed with Orai1 L273S, the cellular distribution of all S1C mutants was unchanged following the addition of 2-APB, indicating that L273 in Orai1 is critical for interaction with the S1C mutants (Figure 4E–G and Figure S4) and suggesting that L273 contributes the interface in Orai1 that is exposed in the active state of the channel. When co-expressed with Orai1 K85E and following the addition of 2-APB the cellular distribution of all S1C mutants did not recapitulate the pattern of distribution seen when either mutant was co-expressed with wt Orai1 (Figure 4E–G and Figure S4). The 4KA S1C mutant did not redistribute to plasma membrane regions following 2-APB application (Figure 4E and Figure S4). A small fraction of R429C and a moderate fraction S1C F394H reallocated to plasma membrane regions after addition of 2-APB (Figure 4F–G and Figure S4). Compared to cells expressing Orai1 K85E alone, the moderate redistribution of S1C F394H to the plasma membrane in cells co-expressing Orai1 K85E, however, did not elicit a stronger Ca2+ influx (Figure 4S). These results are consistent with a dual role of the N terminal region of Orai1 in binding to STIM1(McNally et al., 2013; Palty and Isacoff, 2016; Palty et al., 2015) and in channel gating(Lis et al., 2010) and further suggest that this Orai1 region contributes to interaction with each of the S1C mutants when the channel transits to a partially activated state. Thus, taken together, results from Figure 4 show that sites from both the N and C terminal regions of Orai1 affect binding of SOAR to partially activated Orai1 channels and therefore suggest that both terminal regions of Orai1 are involved in forming the partially activated state interface that engages SOAR.
Rearrangement of the STIM binding sites in Orai1 is required for binding to SOAR mutants
Having shown that residues from both N and C terminal regions of Orai1 contribute to the channel’s partially activated-state interface that engages SOAR, we next asked if the exposure of this interface was mediated by a molecular rearrangement of the Orai1 terminal domains. In resting Orai1 channels adjacent C-termini from neighboring subunits dimerize through anti-parallel coiled-coil interaction to form a putative interaction site for SOAR (Hou et al., 2012; Stathopulos et al., 2013; Tirado-Lee et al., 2015). Since the Orai1 N terminal site and the monomeric or dimeric Orai1 C terminal sites are architecturally distinct, we investigated the interaction between wt or mutant S1C and isolated N or C terminal regions of Orai1. To test for interaction between S1C and the Orai1 N terminal region we co-expressed S1C constructs tagged with EGFP together with fragments of the Orai1 N terminal region (residues 66–91) tagged with FLAG and mCherry and performed co-immunoprecipitation analysis. Results from this analysis revealed similar levels of interaction between wt or mutant S1C and the N terminal region of Orai1 (Figure 5A and Figure S5). To study interaction between S1C and the C terminal region of Orai1, free of constraints imposed by the hexameric assembly of the channel (assessed in Figure 1D), we expressed wt or mutant FLAG-mCh-S1C together with a short fragment of the Orai1 C terminal region (residues 264–301) tagged with EGFP and repeated the co-immunoprecipitation analysis. We found clear interaction between wt S1C and the Orai1 C terminal region (Figure 5A). Strikingly, all three mutants S1C had reduced interaction with the Orai1 C terminal region compared to wt S1C, but were able to maintain some binding (Figure 5B and Figure S5). Since 2-APB has been previously shown to exert functional and structural effects on STIM1 (DeHaven et al., 2008; Muik et al., 2011; Peinelt et al., 2008), we further asked whether the compound would modify interaction between S1C and the Orai1 C terminal fragments. Treatment with 2-APB did not change the interaction between S1C mutants and Orai1 C terminal proteins (Figure S5C), suggesting that the ability of 2-APB to rescue interaction between S1C mutants and full length Orai1 is due to modulation of Orai1. The finding that SOAR mutants F394H and R429C retain binding to isolated N and C terminal fragments of Orai1 (Figure 5A–B), interact with the partially activated full length Orai1 (Figure 1–3), but do not interact with the resting full-length Orai1 (Figure 1), suggests that during channel activation the cytosolic facing regions of Orai1 may rearrange to permit interaction with the mutant SOAR’s.
Figure 5. Interaction of SOAR with the cytosolic facing domains of Orai1.

(A) Western blot analysis of cell lysate or immunoprecipitated material (IP) prepared from cells expressing the indicated FLAG-mCherry-S1C constructs together with EGFP tagged Orai1 C terminal fragment (residues 264–301). (B) Western blot analysis of cell lysate or IP material prepared from cells expressing the indicated FLAG and mCherry tagged Orai1 N terminal fragment (residues 66–91) together with the indicated EGFP-S1C constructs.
To test the model that partial activation involves a conformational rearrangement of the N or C terminal regions of Orai1 we expressed SNAP-tagged versions of Orai1in HEK293 cells. The SNAP-tag was added to the N or C terminal regions of Orai1 (SNAP-Orai1 or Orai1-SNAP). We used cell-permeable benzylguanine (BG) versions of green (Oregon Green) and red (6-carboxytetramethylrhodamine, TMR) fluorophores to selectively and covalently label SNAP (Figure 6A). In cells expressing SNAP-Orai1 and stained with a mixture of BG-Oregon green and BG-TMR, green and red fluorescence was localized to intracellular structures and seen only faintly at the plasma membrane (Figure S6A), indicating that SNAP-Orai1 is impaired and precluding further evaluation. However, cells expressing Orai1-SNAP exhibited selective labeling of the plasma membrane (Figure 6B), permitting further study. Analyses of the cellular distribution of S1C and NFAT in cells co-expressing Orai1-SNAP indicated that the SNAP tag does not interfere with S1C binding or Orai1 activation (Figure S6B). Based on the close proximity of the C terminal regions of neighboring Orai1 subunits, which the crystal structure (Hou et al., 2012) suggests reside within the Forster resonance energy transfer (FRET) distance of this donor-acceptor pair, we used FRET as a readout of conformational changes in the Orai1 C-terminal following channel activation by 2-APB or STIM1. We found that 2-APB, but not vehicle alone (DMSO), induces a transient increase in donor (green) emission and decrease in acceptor (red) emission, indicating a transient decrease in FRET (Figure 6C and Figure S6) and consistent with a conformational change in the C terminal region of Orai1. Orai1 channel activation by STIM1 triggered a distinct FRET response in the channels C termini. Store depletion with thapsigargin (Tg), an inhibitor of endoplasmic reticulum Ca2+ pump, caused a gradual FRET increase in cells that co-express Orai1-SNAP and STIM1 but not in cells expressing Orai1-SNAP alone (Figure 6D), indicating that the rise in FRET depended on STIM1 and on Ca2+ stores. Since binding of STIM1 to Orai1, which elicits maximal channel activation, and treatment with 2-APB, which elicits transient and partial channel activation, induced different FRET changes, we further analyzed changes in the configuration of the Orai1 C terminal region. We used the recovery of donor fluorescence after acceptor photobleaching to asses FRET under additional conditions that induce partial and maximal Orai1 channel activation. We found a strong increase in FRET efficiency (Figure 6E) in cells co-expressing S1C together with Orai1-SNAP and a moderate increase in cells expressing the Orai1-SNAP P245L mutant alone. In contrast, treatment with 2-APB reduced the average FRET efficiency compared to non-treated cells or to cells treated with vehicle (DMSO), however, this reduction did not reach statistical significance (Figure 6E and Figure S6). Taken together, results from Figure 6 indicate that the transition from resting to partially and fully active states of Orai1 channel involves changes in the configuration of the channels C terminal region.
Figure 6. Conformational dynamics of the cytosolic facing C termini of Orai1.

(A) Cartoon illustrations of Orai1-SNAP construct and labeling with benzylguanine (BG) derivatives. (B) Representative fluorescent images of cells expressing Orai1-SNAP and labeled with BG-Oregon-Green (OG) and BG-TMR. (C) Time course of donor (OG) and acceptor (TMR) fluorescence intensity and their ratio in cells expressing Orai1-SNAP and co-labeled with BG-OG and BG-TMR following addition of 2-APB (50μM, grey background) to the extracellular solution. Inset (Lower panel) shows the distribution of FRET change (2-APB n=21 cells, DMSO n=10, p<0.0001), estimated by calculating the difference in the acceptor/donor ration before and after addition of 2-APB or DMSO (as indicated by ΔR). (D) Time course of averaged donor and acceptor ratios as in (C) in cells expressing Orai1-SNAP alone or together with STIM1 following addition of Thapsigargin (Tg, 1μM) to the extracellular solution (Orai1 alone n=10, Orai1 + STIM1 n=8). (E) Summary of FRET efficiency values measured in cells expressing Orai1-SNAP P245L, Orai1-SNAP alone (control) or together with S1C, labeled with the SNAP dyes and subjected to treatment with DMSO or 2-APB, as indicated. Individual measurements and statistical analysis of data shown in (E) are displayed in Figure S6.
Discussion
The amplitude, duration and frequency of oscillation of internal Ca2+ concentration exert specific effects on cell behavior. Orai1 channels contribute in an important way to the spatio-temporal Ca2+ pattern in several types of immune cells, including T-cells and B-cells(Hogan et al., 2010). Previous work on the relationship between STIM1 and Orai1 stoichiometry and the degree of Orai1 channel activation revealed that maximal channel activation occurs at a 2:1 STIM1:Orai1 subunit ratio (Hoover and Lewis, 2011; Li et al., 2011).
However, recent structural evidence by Ikura and colleagues (Stathopulos et al., 2013) showed that the interaction between isolated peptides from SOAR and the C terminal region of Orai1, the major determinants for STIM1-Orai1 protein-protein interactions, saturate at a 1:1 ratio, a stoichiometry which does not maximally activate the channel (Hoover and Lewis, 2011; Li et al., 2011).
Our results shed light on this conundrum by revealing distinct forms of STIM1-Orai1 interaction that arise when Orai1 channels transit from the resting state to a partially activated state. This sequential-step interaction was revealed through our study of SOAR mutants that are deficient in binding to resting Orai1 channels. We found that these mutant SOARs bind to the partially activated state of Orai1. This behavior was observed whether the Orai1 channel was partially activated by fusion of each of its subunits to one wt SOAR or by exposure to the gating modifier 2-APB or by mutation of a key hinge proline near the internal end of the Orai1 TM4. In each of these cases a mutant SOAR, which had no binding to or effect on the resting state Orai1, bound to the partially activated state Orai1 and boosted its activity toward the fully activated level.
How does the activation state of Orai1 affect SOAR binding? Although detailed structural information regarding the interaction between STIM1 and Orai1 is still lacking previous studies have shown that both the N and C terminal regions of Orai1 cooperate in binding to SOAR, with the C terminal region contributing to high affinity binding(McNally et al., 2013; Palty and Isacoff, 2016; Park et al., 2009; Zhou et al., 2010). Recent structural studies have reported different conformations of the C terminal region of Orai1. Two crystal structures of dOrai, obtained in the absence of STIM proteins, propose significant flexibility in the region that begins with the fourth transmembrane segment and continues C terminally. In the first published structure of dOrai, the pore appears closed, TM4 is twisted and the C terminal extension of TM4 from neighboring subunits self-associate (Hou et al., 2012). Notably, the configuration of the Orai1 C terminal regions in this structure(Hou et al., 2012) is analogous to their configuration in the NMR structure of the STIM1-Orai1 complex(Stathopulos et al., 2013), suggesting that the conformation of the resting Orai1 C terminal region is primed for STIM1 binding at 1:1 STIM1 to Orai1 stoichiometry(Stathopulos et al., 2013). In a second more recent but yet unpublished structure, in which the pore also appears to be closed, the TM4s are straight and the C terminal regions extend into the cytosol (Hou and Long, 2015). The apparent closed state of the pore in this recent dOrai structure (Hou and Long, 2015) may reflect a low open probability of an activated state.
The finding that SOAR mutants with deficient binding to and activation of resting Orai1 are able to bind to and boost activation of partially activated Orai1 channels suggest that binding of SOAR to resting or activated Orai1 channels involves different interaction interfaces. This is further supported by our finding that the SOAR mutants that do not interact with full-length Orai1 in the resting state do interact with isolated Orai1 C termini fragments.
Several lines of evidence from this work and from earlier studies support a model in which Orai1 C termini undergo rearrangements during channel gating. By monitoring FRET between Orai1-CFP and Orai1-YFP, Navarro-Borelly et al(Navarro-Borelly et al., 2008) had shown that the Orai1 C termini undergo conformational changes during STIM1 binding and channel activation. Our FRET analysis is consistent with that study and further shows that the conformation of the channel C terminal region differs between the resting channel and the channel when it is bound to SOAR. Notably, however, although both studies employed Orai1 constructs tagged C terminally with fluorescent molecules, we observe an increase in FRET while Navarro-Borelly et al (Navarro-Borelly et al., 2008) observed a decrease in FRET following Orai1 interaction with STIM1. These differences may be explained by differences in chromophore position between the fluorescent proteins employed in the earlier study(Navarro-Borelly et al., 2008) and our SNAP-tag substrate dyes. The model is further supported by the finding that a mutation of proline P245 to leucine in the TM4 region of Orai1 , which is predicted to straighten TM4 and hence contribute to disassociation of adjacent C termini, modulates the configuration of the Orai1 C termini and drives the channel into a partially activated state(Palty et al., 2015). We note that while both the Orai1 P245L mutant and Orai1 that is activated by S1C have a higher FRET between C termini compared than what is seen in the resting Orai1 channel, FRET efficiency is lower in Orai1 P245L than in Orai1 that is activated by S1C. This difference could reflect two distinct conformations, one corresponding to a partially active channel and the other to the maximally active channel. Alternatively, it may arise if the P245L mutant infrequently occupies an active conformation that is similar to the conformation stabilized when S1C engages the channel.
Finally, we find that treatment with 2-APB, in the absence of STIM1, induces a conformational change in the C terminal region of Orai1 and partially activates the channel. However, although both the P245L mutation and treatment with 2-APB induce partial activation, the FRET responses differ, indicating distinct channel conformations. We currently do not know the structural basis for this difference. It may arise from the complex effects of 2-APB, which include activation and block, or reflect the existence of more than one partially open state. The small (~2.4%) difference in FRET between resting and 2-APB treated channels suggests that the conformation stabilized by 2-APB reflects only a slight change in the Orai1 C termini from the resting channel conformation. Hence, this conformation may be similar to the one recently captured in an NMR structure at a 1:1 SOAR to Orai1 ratio in which the Orai1 C-termini retain an antiparallel configuration but do not self-associate (Stathopulos et al., 2013). We and others (Navarro-Borelly et al., 2008) failed to detect a transient FRET level associated with this transition state, possibly because it is masked by the larger rearrangement and FRET change associated with full activation. Indeed, binding of STIM1 to Orai1 and subsequent treatment with 2-ABP induce opposing changes to Orai1 C termini FRET (Figure S6 and (Navarro-Borelly et al., 2008)). These 2-APB dependent changes are correlated with an increase in STIM1-Orai1 FRET but they do not induce an apparent change in the number of interacting molecules(Navarro-Borelly et al., 2008), suggesting that 2-APB stabilizes a particular STIM1-Orai1 interaction state. Lastly, additional support for multiple distinct open states of Orai1 comes from the finding that the S1C R429C mutant does not bind to or affect channel activation of Orai1-S channels but binds to and contributes to activation of Orai1 P245L or following treatment of Orai1 with 2-APB.
In summary, we propose a sequential-step model to describe how interaction of the Orai1 channel with SOAR activates the channel. We suggest that the antiparallel configuration formed by neighboring Orai1 C termini in the resting conformation of the channel (Hou et al., 2012) establishes an initial binding site for the SOAR dimer. In the first step of activation, a dimer of SOAR interacts with this dimer of Orai1 C-termini to yield the 1:1 stoichiometry state. We propose that this dimeric interaction leads to one or more partially active transition states. One such state may have been captured in the NMR structure(Stathopulos et al., 2013), in which the Orai1 C-termini break their interactions but retain an antiparallel configuration. A subsequent state then proceeds to a conformation in which the Orai1 TM4 segments straighten out, the C termini extend towards the cytosol and the pore opens a small fraction of the time. Consistent with this, a recent study by Prakriya and colleagues (Tirado-Lee et al., 2015) found that cross-linking neighboring Orai1 C termini impairs channel activation. Finally, we propose that the dissociated Orai1 C termini of the partially active channel each present a new class of binding site with a distinct interaction surface for SOAR that enables binding of each SOAR dimer per single Orai1 subunit and yields the fully liganded 2:1 stoichiometry and maximal channel activation.
Experimental Procedures
Cell culture and transfection
HEK293 cells were cultured in DMEM as previously described (Palty et al., 2012). Plasmid transfection of cells was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer protocol. For electrophysiological experiments, cells were plated onto 18 mm cover glass coated with L-polylysine 6–8 hours after plasmid transfection and 12–15 hours before the start time of experiments. For NFAT translocation, FRET and intracellular Ca2+ imaging experiments, cells were plated onto 18 mm cover glass coated with L-polylysine and culture media was replaced to wash the transfection reagent away 6–8 hours after plasmid transfection. To avoid constitutive Ca2+ level elevation in cells expressing Orai1-SS or co-expressing Orai1 or Orai1-S together with S1C, cells were cultured in high glucose, Ca2+ free DMEM supplemented with 50μM La3+.
Fluorescent Measurements of Intracellular Ca2+ Ions
On the day of the experiments, the cover glass was mounted on an imaging chamber and washed with Ringer solution. In order to load cells with Flou-4, cells were incubated for 1 hour in Ringers solution containing 5μM of the Ca2+ indicator and for additional 30 minutes without the dye. Cytosolic Ca2+ levels were recorded from flou-4 loaded cells, excited at wavelengths of 488nm and emission collected from 493 to 524 nm. Images were acquired at 0.5 Hz. For all single cell imaging experiments, traces of individual or averaged responses, recorded from 17 to 96 cells in each experiment, were plotted with KaleidaGraph. Individual experiments were repeated twice and all data are shown as average ± SEM.
Confocal microscopy
Mid-planes sections of transfected cells were captured using an LSM 780 confocal microscope (Zeiss) controlled by Zen imaging software (Zeiss). EGFP or BG-Oregon Green were excited at 488 nm and emission collected from 493 to 540 nm, and mCherry or BG-TMR were excited at 561 nm and emission collected from 583 to 685 nm. The parameters used for image acquisition were kept constant across individual sets of experiments and analysis was restricted to cells with similar EGFP and mChery fluorescence to ensure similar ratios of S1C/Orai1 expression. Line-scans of regions spanning the PM and cytosol were analyzed from individual images using Image-J. As indicated in Supplemental Fig. 2, the relative amount of S1C in plasma membrane and cytoplasmic regions was calculated by dividing the mean intensities of EGFP in regions of interest drawn around the plasma membrane by those around the cytosol. This ratio was used to compare the relative amount of S1C that is bound to Orai1 channels on the plasma membrane.
Electrophysiological recordings
Membrane currents were recorded under voltage-clamp conditions using the whole-cell patch-clamp configuration on an Axopatch 200B amplifier (Axon Instruments). Patch pipettes were fabricated from borosilicate glass capillaries (5–10 MΩ). Signals were analog filtered using a 2 kHz low-pass Bessel filter. Data acquisition and analysis were performed using pCLAMP 10 software (Axon Instruments). Voltage protocols consisted of a 100-ms ramp from −100 to +100 mV delivered alone or subsequent to a 100-ms voltage step to −100mV every 1 or 4 seconds from a holding potential of 0 mV. Current densities were calculated by normalizing currents measured at −90mV to cell capacitance. The internal solution contained 150 mM Cs aspartate, 8 mM MgCl2, 10 mM HEPES (pH 7.2 with CsOH). To chelate intracellular Ca2+ either 8 mM BAPTA or 1.2mM EGTA was included in the intracellular solution. External Ringer’s solution contained 145 mM NaCl, 2.8 mM KCl, 10 mM HEPES and 10 mM Glucose (pH 7.4 with NaOH) and either 10 mM CaCl or 10 mM MgCl2 was added to the external solution for high-Ca2+ or Ca2+-free solutions, respectively. All data were leak-corrected using the current elicited in high Ca2+ Ringer’s solution supplemented with 10–100μM La3+ or in Ca2+ free Ringer’s solution as appropriate.
Statistical analysis
Statistical significance of differences between data groups was calculated using one-way ANOVA with bonferroni correction (Kaleidagraph, Synergy Software) and unpaired two-tailed Student's t test when comparing two dataset groups (Microsoft Excel 2010).
Supplementary Material
Acknowledgments
We would like to thank Cherise Stanley and Holly Aaron (UC Berkeley Molecular Imaging Center) for their technical assistance. This work was supported by the Technion-Israel Institute of Technology Women's Division Career Advancement Chair program (to RP) and by a grant from the National Institutes of Health (R01 NS35549 to EYI).
Footnotes
Additional experimental procedures are included in Supplemental Experimental Procedures.
Author contributions
R.P and E.Y.I. designed research and wrote the paper and R.P. performed research and analyzed data. Z.F performed western blot analyses.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Calloway N, Holowka D, Baird B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry (Mosc) 2010;49:1067–1071. doi: 10.1021/bi901936q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell. 2010;21:1897–1907. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW. Complex Actions of 2-Aminoethyldiphenyl Borate on Store-operated Calcium Entry. J Biol Chem. 2008;283:19265–19273. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Plenk P, Fahrner M, Muik M, Jardin I, Schindl R, Gruber HJ, Groschner K, Romanin C. The Extended Transmembrane Orai1 N-terminal (ETON) Region Combines Binding Interface and Gate for Orai1 Activation by STIM1. J Biol Chem. 2013;288:29025–29034. doi: 10.1074/jbc.M113.501510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover PJ, Lewis RS. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci. 2011;108:13299–13304. doi: 10.1073/pnas.1101664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hou X, Long SB. Functional Reconstitution and Structural Flexibility of the CRAC Channel Orai. Biophys J. 2015;108:178a. [Google Scholar]
- Hou X, Pedi L, Diver MM, Long SB. Crystal structure of the calcium release-activated calcium channel Orai. Science. 2012;338:1308–1313. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Maleth J, Moreno CM, Yuan JP, Kim MS, Muallem S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J Cell Biol. 2013;202:71–79. doi: 10.1083/jcb.201301148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniowski MK, Manjarres IM, Varnai P, Balla T. Activation of STIM1-Orai1 Involves an Intramolecular Switching Mechanism. Sci Signal. 2010;3:ra82-ra82. doi: 10.1126/scisignal.2001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu L, Deng Y, Ji W, Du W, Xu P, Chen L, Xu T. Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res. 2011;21:305–315. doi: 10.1038/cr.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol CB. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioudyno MI, Kozak JA, Penna A, Safrina O, Zhang SL, Sen D, Roos J, Stauderman KA, Cahalan MD. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci U S A. 2008;105:2011–2016. doi: 10.1073/pnas.0706122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Zierler S, Peinelt C, Fleig A, Penner R. A single lysine in the N-terminal region of store-operated channels is critical for STIM1-mediated gating. J Gen Physiol. 2010;136:673–686. doi: 10.1085/jgp.201010484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus M, Jairaman A, Stathopulos PB, Muik M, Fahrner M, Weidinger C, Benson M, Fuchs S, Ehl S, Romanin C. Missense mutation in immunodeficient patients shows the multifunctional roles of coiled-coil domain 3 (CC3) in STIM1 activation. Proc Natl Acad Sci U S A. 2015;112:6206–6211. doi: 10.1073/pnas.1418852112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Yamashita M, Prakriya M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482:241–245. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Jairaman A, Yamashita M, Prakriya M. The C- and N- terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J Physiol. 2013;591:2833–2850. doi: 10.1113/jphysiol.2012.250456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R. Dynamic Coupling of the Putative Coiled-coil Domain of ORAI1 with STIM1 Mediates ORAI1 Channel Activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- Muik M, Fahrner M, Schindl R, Stathopulos P, Frischauf I, Derler I, Plenk P, Lackner B, Groschner K, Ikura M. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 2011;30:1678–1689. doi: 10.1038/emboj.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1–Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Isacoff EY. Cooperative Binding of Stromal Interaction Molecule 1 (STIM1) to the N and C Termini of Calcium Release-activated Calcium Modulator 1 (Orai1) J Biol Chem. 2016;291:334–341. doi: 10.1074/jbc.M115.685289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Raveh A, Kaminsky I, Meller R, Reuveny E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell. 2012;149:425–438. doi: 10.1016/j.cell.2012.01.055. [DOI] [PubMed] [Google Scholar]
- Palty R, Stanley C, Isacoff EY. Critical role for Orai1 C-terminal domain and TM4 in CRAC channel gating. Cell Res. 2015;25:963–980. doi: 10.1038/cr.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJS, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet J-P. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol. 2008;586:3061–3073. doi: 10.1113/jphysiol.2008.151365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrimgeour N, Litjens T, Ma L, Barritt GJ, Rychkov GY. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J Physiol-Lond. 2009;587:2903–2918. doi: 10.1113/jphysiol.2009.170662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol CB. 2006a;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006b;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- Spassova MA, Soboloff J, He L-P, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci U S A. 2006;103:4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Li G-Y, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Schindl R, Fahrner M, Zheng L, Gasmi-Seabrook GM, Muik M, Romanin C, Ikura M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat Commun. 2013;4 doi: 10.1038/ncomms3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado-Lee L, Yamashita M, Prakriya M. Conformational Changes in the Orai1 C-Terminus Evoked by STIM1 Binding. PloS One. 2015;10:e0128622. doi: 10.1371/journal.pone.0128622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, Gill DL. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat Commun. 2014;5 doi: 10.1038/ncomms4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Deng X, Zhou Y, Hendron E, Mancarella S, Ritchie MF, Tang XD, Baba Y, Kurosaki T, Mori Y. STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci. 2009;106:7391–7396. doi: 10.1073/pnas.0900293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- Yang X, Jin H, Cai X, Li S, Shen Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci. 2012;109:5657–5662. doi: 10.1073/pnas.1118947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Sun L, Courjaret R, Machaca K. Role of the STIM1 C-terminal domain in STIM1 clustering. J Biol Chem. 2011;286:8375–8384. doi: 10.1074/jbc.M110.188789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Sun L, Hubrack S, Selvaraj S, Machaca K. Intramolecular shielding maintains the ER Ca2+ sensor STIM1 in an inactive conformation. J Cell Sci. 2013;126:2401–2410. doi: 10.1242/jcs.117200. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi Y-J, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, McCloskey MA. Immunoglobulin E receptor-activated calcium conductance in rat mast cells. J Physiol. 1995;483(Pt 1):59–66. doi: 10.1113/jphysiol.1995.sp020567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH-F, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Zhou M-H, Hu C, Kuo E, Peng X, Hu J, Kuo L, Zhang SL. Differential Roles of the C and N Termini of Orai1 Protein in Interacting with Stromal Interaction Molecule 1 (STIM1) for Ca2+ Release-activated Ca2+ (CRAC) Channel Activation. J Biol Chem. 2013;288:11263–11272. doi: 10.1074/jbc.M113.450254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Meraner P, Kwon HT, Machnes D, Oh-hora M, Zimmer J, Huang Y, Stura A, Rao A, Hogan PG. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 2010;17:112–116. doi: 10.1038/nsmb.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, Stathopulos PB, Ikura M, Rao A, Hogan PG. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat Struct Mol Biol. 2013;20:973–981. doi: 10.1038/nsmb.2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang X, Wang X, Loktionova NA, Cai X, Nwokonko RM, Vrana E, Wang Y, Rothberg BS, Gill DL. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat Commun. 2015;6:8395. doi: 10.1038/ncomms9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.