

Abstract
FOXM1 is a transcription factor of the Forkhead family that is required for cell proliferation of normal cells. However, FOXM1 is repeatedly overexpressed in a variety of human cancers and it has been implicated in all major hallmarks of cancer delineated by Hanahan and Weinberg. It has been postulated that the oncogenic potential of FOXM1 is determined by its capacity to trans-activate target genes that are implicated in different phases of cancer development. However, FOXM1 may also play an oncogenic role by interacting with other proteins such as beta-catenin or SMAD3 to induce oncogeneic WNT and TGF-β signaling pathways, respectively. In this review, I will discuss the protein-protein interactions of FOXM1 that are critical for cancer development and may represent novel targets for anticancer drugs.
Keywords: cancer, FOXM1, NPM, MELK, Pin-1, STAT3, NF-kB, β-catenin, SMAD3, HSP70
Introduction
Mammalian transcription factor Forkhead Box M1 (FOXM1) is one of the members of the Forkhead family proteins that share a 100 amino acid long winged-helix DNA binding domain (1). There are four FOXM1 isoforms: FOXM1A, B, C and D generated by the alternative splicing. FOXM1A is a transcriptional repressor that locates in cytoplasm. FOXM1C is transcriptional activator overexpressed in some tumors and FOXM1D predominantly locates in cytoplasm and may promote metastasis. In this review we are focused on FOXM1B that we refer as FOXM1. We believe that that focus on FOXM1B is fully justified since FOXM1B (FOXM1), but not other isoforms is dominantly overexpressed in human cancers and may represent attractive target for anticancer drugs. Overexpression of FOXM1 has been detected in a broad range of cancer types, suggesting that FOXM1 is essential for tumorigenesis (1-3). While in normal cells FOXM1 is mainly responsible for cell proliferation (3), in cancer cells it contributes to all hallmarks of cancer described by Hanahan and Weinberg (3, 4). Suppression of FOXM1 in tumor cells by RNA interference led to reduced cell proliferation, anchorage-independent growth (5) migration, invasion and angiogenesis (reviewed in (3), suggesting that FOXM1 regulates these properties of cancer cells. In addition, FOXM1 evades the action of tumor suppressor p53. We and others showed that FOXM1 expression increased after mutating or deleting p53, (6, 7), suggesting that p53 inactivation in human tumors leads to the overexpression of FOXM1. FOXM1 also increases the resistance of cancer cells to apoptosis and genomic instability (3).
Recently, it has been shown that the FOXM1 regulatory network is a major predictor of adverse outcomes in 18,000 cancer cases across 39 human malignancies (8) confirming the important role of FOXM1 in cancer. However, it is still not fully understood how exactly FOXM1 exerts its oncogenic activity in human cells. One mechanism is linked to the transcriptional activation of FOXM1 targets that leads to several pro-tumorigenic effects including enhanced cell proliferation (3). Alternatively, FOXM1 may act as an oncogene by interacting with other proteins thus supporting distinct oncogenic pathways. In this review, I will describe the known interactions of FOXM1 in cancer and I will discuss how this information may help to develop novel specific FOXM1 inhibitors.
1. Interaction between NPM and FOXM1 is required for FOXM1 expression and may explain the outcome of AML treatment
Using mass spectrometric analysis we found earlier that FOXM1 interacts with multifunctional protein nucleophosmin (NPM) that shuttles between the nucleus and cytoplasm and is overexpressed in a variety of human tumors (3, 5). Immunofluorescence microscopy confirmed the interaction between FOXM1 and NPM in the nucleus of human cancer and immortal cells. Furthermore, knockdown of NPM in immortal and cancer cells led to significant down-regulation of FOXM1, suggesting that NPM might stabilize FOXM1 protein.
In a ground-breaking research paper published in 2005 (9) it has been shown that in 30% of AML patients NPM1 gene is mutated in exon 12 leading to re-localization of NPM protein to the cytoplasm and to significantly better prognosis for AML patients after chemotherapy. However, the explanation to this phenomenon was lacking. We found that in the AML cell line OCI-AML3 with NPM1 exon 12 mutation (10) FOXM1 protein co-localized with NPM in the cytoplasm (5). We hypothesized that the improved outcomes in this subset of NPM1mut AML are associated with the cytoplasmic localization and consequent inactivation of oncogenic transcription factor FOXM1. To prove this notion, we demonstrated predominantly cytoplasmic localization of FOXM1 and NPM only in AML primary patient samples with NPM1 mutations (11). In addition, we investigated the effects of nuclear FOXM1 on chemoresistance to cytosine arabinoside (cytarabine), the backbone of AML chemotherapy and we found that stable knockdown of FOXM1 in AML cell lines with nuclear FOXM1 resulted in increased sensitivity to this chemotherapeutic agent (11).
Residues 187-295 AA of NPM have been identified as essential for the interaction with FOXM1 (5). We investigated whether the truncated NPM corresponding to 187-295 AA inhibits the interaction of full length NPM and FOXM1 (365-748 AA). In NMR experiments we observed a reduction in the chemical shift changes upon addition of this truncated NPM indicating that the FOXM1/NPM interaction was significantly inhibited. These data suggested that an NPM peptide that interacts with FOXM1 competes with NPM for FOXM1 binding and inhibits FOXM1/NPM interaction, and FOXM1 expression in human cancer cells. Overall, in cancer cells NPM interacts with FOXM1 and their interaction is required for sustaining the level and localization of FOXM1 (5). In AML cells mutant NPM drives FOXM1 to the cytoplasm leading to its inactivation (11). Generally speaking, interaction of nuclear FOXM1 with NPM may be targeted by peptides or small molecules in human cancer.
2. MELK binds and activates FOXM1 in glioma cells
Maternal embryonic leucine-zipper kinase (MELK) is a serine/threonine kinase that is overexpressed in several human cancers, including glioma (12). MELK directly binds to FOXM1 and regulates its phosphorylation and activation in glioma stem-like cells (13). Interestingly, that activation of FOXM1 was also dependent on the binding and phosphorylation by PLK1 kinase, because the FOXM1 mutant that could only bind PLK1 but could not get phosphorylated by it was not activated by MELK (13). Using mouse neural progenitor cells it has been found that overexpression of FOXM1 accelerated neurosphere formation, while FOXM1 expression increased as cells progressed to pre-tumorigenic progenitors and glioma stem-like cells (13). The authors suggested that FOXM1 is involved in the development of glioblastoma multiforme (GBM) and MELK-FOXM1 interaction is a potential therapeutic target in glioma (13). Unfortunately, the drug Siomycin A the authors of this work used to illustrate the targeting of FOXM1/MELK did not inhibit FOXM1/MELK interaction, but inhibits FOXM1 expression as a proteasome inhibitor (14, 15). Therefore, it is too early to predict whether the strategy to target FOXM1/MELK interaction with objection to inhibit cancer growth will be successful.
3. Pin1–FOXM1 interaction induces metastatic melanoma
Pin1 is a phospho-specific peptidyl-prolyl isomerase that facilitates substrate isomerization through interaction with phosphorylated Ser/Thr-Pro motifs. In malignant melanoma FOXM1 activity correlated with Pin1 expression (16). Moreover, Pin1 acted as a main regulator of FOXM1 activity in the melanoma cells through physical binding with FOXM1 and the driver of melanoma, oncogenic BRAFV600E, stimulated this interaction. Depletion of FOXM1 by RNAi in melanoma cells or inhibition of binding by cell-permeable Pin1-FOXM1-blocking peptides repressed FOXM1 activity and inhibited melanoma cell proliferation ex vivo (16). These data suggest that the physical interaction of FOXM1-Pin1 may increase FOXM1 activity in melanoma cells and Pin1-FOXM1 inhibitors may work as anticancer drugs in melanoma.
4. STAT3 interacts with FOXM1 in glioblastoma after radiation treatment
Radiation of glioblastoma cells led to the phosphorylation of STAT3 and the induction of FOXM1. Increase of FOXM1 expression was linked to the direct interaction of FOXM1 with STAT3 (17). Co-localization of FOXM1 and STAT3 in the nucleus of radiation-treated cells was also confirmed by immunofluorescence (17) and suppression of STAT3 repressed FOXM1 expression. In addition, FOXM1 inhibition sensitized glioblastoma cells to radiation. The authors suggested that the FOXM1/STAT3 interaction may contribute to the activation of FOXM1 expression and resistance to radiation in glioma stem cells (17).
5. Interaction of p65 with FOXM1/β-catenin is critical in CML
It has been shown that NF-kB (p65) protein physically interacts with FOXM1/β-catenin and transcriptionally induces FOXM1 expression in chronic myelogenous leukemia (CML) cells (18). At the same time β-catenin activation promotes nuclear translocation of p65. Silencing of FOXM1 led to the suppression of p65 and β-catenin, while silencing of β-catenin led to the suppression of FOXM1 and p65 (18). These data indicate mutual positive regulation between p65 and FOXM1/ β-catenin in CML. Disruption of the p65-FOXM1/ β-catenin positive feedback loop in CML cells impaired the self-renewal capacity and survival of CML stem cells (18). These data suggest that p65-FOXM1 interaction has oncogenic activity in CML cells.
6. The long noncoding RNA (lncRNA) PVT1 binds FOXM1 protein and post-translationally stabilizes FOXM1 protein
LncRNA-PVT1 is abnormally overexpressed in different cancer types and may promote tumor growth and metastasis in gastric cancer (19). Using RNA-protein pull-down assay it has been shown that PVT1 directly binds to FOXM1 protein and inhibits its degradation in gastric cells cells (19). In addition, FOXM1 binds to PVT1 promoter and activates its transcription. In vitro metastasis assays showed that FOXM1 knockdown partially diminished the effects of PVT1 overexpression on gastric cancer cell metastasis. Similarly, it has been demonstrated that FOXM1 knockdown could to some extent abolish the increased tumor size initiated by the overexpression of PVT1 in mouse xenograft models (19). These data do not give clear answer whether PVT1 or FOXM1 is the effector or the stabilizing cofactor of tumor growth and metastasis in gastric cancer. Additional experiments are required to resolve this question.
7. FOXM1 determines β-catenin nuclear localization in glioma cells
Wnt signaling is the primary signaling system in stem/progenitor and cancer cells that leads to the stabilization of β-catenin. The transcriptional complex of β-catenin with TCF/LEF in the nucleus activates the expression of Wnt target genes important for cancer development (20). Activation of β-catenin has been implicated in the development of GBM, the most malignant form of glioma (21). It turned out that FOXM1 directly binds to β-catenin in the nucleus of glioma cells and enhances β-catenin transcriptional activity (22). Deletion of FOXM1 or use of FOXM1 mutants that abolish interaction with β-catenin prevented β-catenin nuclear localization and inhibited Wnt signaling in glioma cells (22). Conversely, knockdown of β-catenin in glioma cells diminished their tumorigenicity indicating that the oncogenic activity of FOXM1 in glioma depends on β-catenin. The authors of this work suggested that FOXM1 regulates two facets of β-catenin nuclear function: nuclear accumulation and assembly of a transcription activation complex. In contrast to the majority of other cancers where FOXM1 directly induces oncogenesis as a transcription factor, in glioma FOXM1 contributes to tumorigenesis via direct interaction with β-catenin, acting as a co-factor that stabilizes β-catenin. These data suggest that direct FOXM1–β-catenin interaction that enhances β-catenin expression and Wnt signaling is required for tumorigenesis in glioma (22).
8. FOXM1 interacts with SMAD3 and regulates the nuclear retention of the SMAD3/SMAD4 complex in the TGF-β signaling pathway
It has been shown that FOXM1 directly interacts with SMAD3 in breast cancer cells and induces TGF-β/SMAD3-mediated transcription (23). In contrast, the ubiquitin-protein ligase transcriptional intermediary factor 1γ (TIF1γ) binds to SMAD4, induces its mono-ubiquitination, and disruption of SMAD3/SMAD4 complex (23). Xue et al. demonstrated that FOXM1 interferes with the SMAD3/TIF1γ interaction and consequently with SMAD4/TIF1γ binding thus inhibiting the degradation of SMAD4 and stabilizing the SMAD3/SMAD4 complex (23). Moreover, they found that FOXM1/SMAD3 interaction was required for TGF-β–induced breast cancer invasion (24), as a result of SMAD3/SMAD4-dependent transcriptional activation of factor Slug (Snail2), a C2H2-type zinc finger transcription factor that has been widely expressed in aggressive human cancers (25). Because the inhibitory effects of shFOXM1 on the metastatic ability of breast cancer cells was rescued by the shRNA-resistant FOXM1 R286A/H287A mutant, which is incapable of DNA binding, the effect of FOXM1 on metastasis in breast cancer was not dependent on its transcriptional activity. Instead, FOXM1 acted as a SMAD3 interacting partner that protected SMAD4 from degradation thus inducing TGF-β signaling and the development of metastasis in breast cancer.
9. HSP70 inhibits FOXM1 activity and expression
Previously, we have shown that proteasome inhibitors suppress FOXM1 transcriptional activity and as a result of the FOXM1 positive auto-regulatory loop (26) they also inhibit FOXM1 mRNA and protein expression (14, 15). Later we demonstrated that HSP90 inhibitor PF-4942847 and heat-shock also suppress FOXM1 in several human cancer cell lines (27). It turned out that all these treatments have something in common: they all induce HSP70. Moreover, when we used HSP70 inhibitor 9AA or HSP70-siRNA in combination with proteasome/HSP90 inhibitors we found that these treatments reversed suppression of FOXM1 (27), suggesting that HSP70 is a specific FOXM1 inhibitor. To investigate if FOXM1 inhibition linked to HSP70/FOXM1 interaction we performed reciprocal co-immunoprecipitations and found that FOXM1 interacts with HSP70 (27). In addition, we found that HSP70 is able to interfere with the transcriptional activity of FOXM1 (unpublished data). Furthermore, quantitative ChIP assays revealed that HSP70 reduces up to 2-fold the ability of exogenous FOXM1 to bind to its regulatory elements, while the levels of exogenous FOXM1 was not affected (27). These data suggest that the primary effect of HSP70 on FOXM1 is the inhibition of its transactivation via binding and that the suppression of FOXM1 expression is a secondary effect.
10. Synergy between FOXM1 and CENPF to stimulate prostate cancer
CENPF (centromere protein F) encodes a protein that associates with the centromere-kinetochore complex(28) and COUP-TFII an orphan nuclear receptor (29). The localization of CENPF points toward its role in chromosome segregation during mitosis. It turned out that simultaneous FOXM1 and CENPF overexpression in prostate cancer is linked to the loss of microRNAs, miR-101 and miR-27a. FOXM1 and CENPF are direct targets of COUP-TFII, which is targeted by miR-101 and miR-27a. Overexpression of FOXM1 and CENPF in prostate cancer is indirectly regulated by the loss of miR-101 and miR-27a as a primary event leading to activation of COUP-TFII that induces FOXM1 and CENPF (30). Computational analysis of genome-wide regulatory networks for human and mouse prostate cancer has identified FOXM1 and CENPF as synergistic master regulators of prostate cancer (28). When FOXM1 and/or CENPF was silenced individually or together in human prostate cell lines or in prostate xenograft tumors the anticancer response was synergistic (28). Interestingly, co-expression of FOXM1 and CENPF in prostate cancer patient samples is a predictor of poor prognosis. While FOXM1 does not directly interact with CENPF, they synergistically induce prostate cancer via up-regulation PI3K and MAPK signaling pathways (28).
Summary
Oncogenic transcription factor FOXM1 is overexpressed in the majority of human cancers and contributes to all hallmarks of cancer (3). Typically, FOXM1 induces cancer development by the transcriptional activation of its targets. Several proteins or RNA interact with FOXM1 and stabilize FOXM1 expression, and increase FOXM1 activity in cancer cells (Fig 1A). However, now it became clear that FOXM1 may also act as a cofactor for the activation or the enhancement of other oncogenic pathways. For example, FOXM1 stabilizes β-catenin to induce Wnt-pathway (22) and SMAD3 to induce TGF-β pathway in cancer cells (23) (Fig 1B). In addition, direct interaction between HSP70 and FOXM1 leads to suppression of FOXM1 (Fig 1C). The information from this paper will help to devise novel FOXM1 inhibitors that either will hinder FOXM1 protein-protein/RNA interactions or will mimic FOXM1 repressive proteins and will be valuable anticancer drugs.
Figure 1.
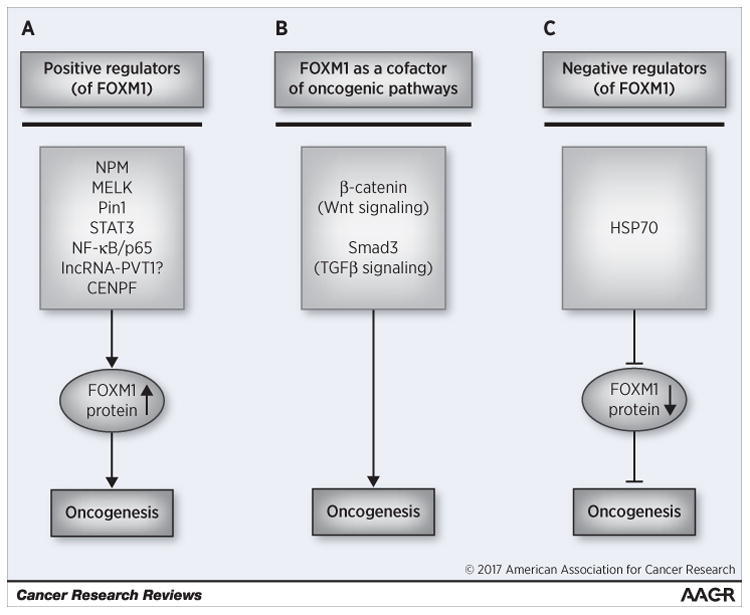
Different examples of FOXM1 interactions in cancer. (A) Several proteins stabilize FOXM1 and increase its oncogenic activity. (B) FOXM1 as an assembly factor contributes to Wnt and TGFβ signaling. (C) Proteotoxic stress suppresses FOXM1 via up-regulation of HSP70 and its interaction with FOXM1.
Acknowledgments
I thank Marianna Halasi (UIC) for her valuable comments and for the preparation of the figure.
Financial support: NIH grants 1R01CA129414, 5R01CA138409, 1R21CA134615 and 5R21CA194608.
Footnotes
Conflict of interest: The author declared no conflict of interest.
References
- 1.Laoukili J, Stahl M, Medema RH. FoxM1: At the crossroads of ageing and cancer. Biochim Biophys Acta. 2007;1775:92–102. doi: 10.1016/j.bbcan.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004;6:744–50. doi: 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halasi M, Gartel AL. FOX(M1) News--It Is Cancer. Mol Cancer Ther. 2013;12:245–54. doi: 10.1158/1535-7163.MCT-12-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Bhat UG, Jagadeeswaran R, Halasi M, Gartel AL. Nucleophosmin interacts with FOXM1 and modulates the level and localization of FOXM1 in human cancer cells. J Biol Chem. 2011;286:41425–33. doi: 10.1074/jbc.M111.270843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandit B, Halasi M, Gartel AL. p53 negatively regulates expression of FoxM1. Cell Cycle. 2009;8:3425–7. doi: 10.4161/cc.8.20.9628. [DOI] [PubMed] [Google Scholar]
- 7.Barsotti AM, Prives C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene. 2009;28:4295–305. doi: 10.1038/onc.2009.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–66. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 10.Quentmeier H, Martelli MP, Dirks WG, Bolli N, Liso A, Macleod RA, et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005;19:1760–7. doi: 10.1038/sj.leu.2403899. [DOI] [PubMed] [Google Scholar]
- 11.Khan I, Halasi M, Zia MF, Gann P, Gaitonde S, Mahmud N, Gartel AL. Nuclear FOXM1 drives chemoresistance in AML. Leukemia. 2017;31:251–255. doi: 10.1038/leu.2016.270. [DOI] [PubMed] [Google Scholar]
- 12.Ganguly R, Hong CS, Smith LG, Kornblum HI, Nakano I. Maternal embryonic leucine zipper kinase: key kinase for stem cell phenotype in glioma and other cancers. Mol Cancer Ther. 2014;13:1393–8. doi: 10.1158/1535-7163.MCT-13-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells. 2013;31:1051–63. doi: 10.1002/stem.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a Chemical Inhibitor of the Oncogenic Transcription Factor Forkhead Box M1. Cancer Res. 2006;66:9731–35. doi: 10.1158/0008-5472.CAN-06-1576. [DOI] [PubMed] [Google Scholar]
- 15.Bhat UG, Halasi M, Gartel AL. FoxM1 is a general target for proteasome inhibitors. PLoS One. 2009;4:e6593. doi: 10.1371/journal.pone.0006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruiswijk F, Hasenfuss SC, Sivapatham R, Baar MP, Putavet D, Naipal KA, et al. Targeted inhibition of metastatic melanoma through interference with Pin1-FOXM1 signaling. Oncogene. 2016;35:2166–77. doi: 10.1038/onc.2015.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maachani UB, Shankavaram U, Kramp T, Tofilon PJ, Camphausen K, Tandle AT. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget. 2016;7(47):77365–7737. doi: 10.18632/oncotarget.12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin B, Wang C, Li J, Du X, Ding K, Pan J. Anthelmintic niclosamide disrupts the interplay of 65 and FOXM1/beta-catenin and eradicates leukemia stem cells in chronic myelogenous leukemia. Clin Cancer Res. 2017;23(3):789–803. doi: 10.1158/1078-0432.CCR-16-0226. [DOI] [PubMed] [Google Scholar]
- 19.Du X, Xu MD, Wang Y, Weng WW, Wei P, Qi P, et al. A positive feedback loop of lncRNA-PVT1 and FOXM1 facilitates gastric cancer growth and invasion. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-0742. [DOI] [PubMed] [Google Scholar]
- 20.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 21.Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, et al. miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein. Cell Stem Cell. 2010;7:694–707. doi: 10.1016/j.stem.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H. FoxM1 Promotes beta-Catenin Nuclear Localization and Controls Wnt Target-Gene Expression and Glioma Tumorigenesis. Cancer Cell. 2011;20:427–42. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M, et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-beta-dependent cancer metastasis. J Clin Invest. 2014;124:564–79. doi: 10.1172/JCI71104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundqvist A, Ten Dijke P, van Dam Key signaling nodes in mammary gland development and cancer: Smad signal integration in epithelial cell plasticity. Breast Cancer Res. 2012;14:204. doi: 10.1186/bcr3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan Y, Li J, Zhang Y, Wang N, Liang H, Liu Y, Zhang CY, Zen K, Gu H. Slug-upregulated miR-221 promotes breast cancer progression through suppressing E-cadherin expression. Sci Rep. 2016;6:25798. doi: 10.1038/srep25798. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halasi M, Gartel AL. A novel mode of FoxM1 regulation: positive auto-regulatory loop. Cell Cycle. 2009;8:1966–7. doi: 10.4161/cc.8.12.8708. [DOI] [PubMed] [Google Scholar]
- 27.Halasi M, Varaljai R, Benevolenskaya E, Gartel AL. A Novel Function of Molecular Chaperone HSP70: SUPPRESSION OF ONCOGENIC FOXM1 AFTER PROTEOTOXIC STRESS. J Biol Chem. 2016;291:142–8. doi: 10.1074/jbc.M115.678227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014;25:638–51. doi: 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu M, Qin J, Tsai SY, Tsai MJ. The role of the orphan nuclear receptor COUP-TFII in tumorigenesis. Acta Pharmacol Sin. 2015;36:32–6. doi: 10.1038/aps.2014.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin SC, Kao CY, Lee HJ, Creighton CJ, Ittmann MM, Tsai SJ, et al. Dysregulation of miRNAs-COUP-TFII-FOXM1-CENPF axis contributes to the metastasis of prostate cancer. Nat Commun. 2016;7:11418. doi: 10.1038/ncomms11418. [DOI] [PMC free article] [PubMed] [Google Scholar]