

Abstract
Pancreatic Cancers depend on driver molecules, oncogene proteins such as RAS. NR5A2 protein is a transcription factor and either activates or inhibits transcription through actions at hundreds of enhancers. It has unusual properties with effects appearing in multiple signaling networks. NR5A2 is a pluripotency reprogramming factor in the class nuclear receptor. Its controlling hormone is PIP3. Experiments suggest NR5A2 activation drives PDAC and inhibitors blunt cancer cell proliferation.
Keywords: Stem cell, LRH-1, β-catenin, structure-based inhibitor design
Introduction
Small molecules, biological macromolecules and cells have been proposed and tested for treatments of PDAC. The best small molecules are used in chemotherapies and are beneficial in the clinic. Few advances in molecular target validation or small molecule inhibitor discovery have been made for PDAC. We evaluate NR5A2 as a protein target for treating PDAC. NR5A2 is also called LRH-1, for liver receptor homolog, after the tissue where it was first discovered. Small molecules can alter NR5A2 activity.
Part 1 Discovery of Roles of NR5A2 in many signaling networks
NR5A2 is a scaffolding protein that attracts macromolecules to appropriate sites on DNA to regulate transcription of specific target genes. It is an unusual transcription factor of structure-class nuclear receptor. Post development, this protein is in liver, intestine, and pancreas, where it controls cholesterol and bile acid homeostasis. Secretion of pancreatic enzymes is under control of NR5A2.
Steroids in placenta, ovary and breast adipose tissues are maintained by NR5A2. In striking extension it has been found that NR5A2 is vital in early development stabilizing undifferentiated embryonic stem cells. This is done by controlling expression of the master transcription factors, OCT4 and NANOG [1].
Nuclear receptors are transcription factors that have cell-type and tissue-specific target genes. For example, in addition to its roles in steroid homeostasis and bile acid biosynthesis, NR5A2 functions with both Hedgehog and Wnt/β-catenin networks. Cell cycle genes are targets. When NR5A2 is paired with β-catenin, cell cycle genes CCND1 and CCNE1 genes and MYC genes are affected [2]. These factors control differentiation, growth, and proliferation pathways including tumor genesis. With such critical roles there are compelling arguments to find small molecules that affect, or more appropriately, differentially affect target genes under NR5A2 expression control.
Like most nuclear receptors, NR5A2 protein has three domains, in addition to its DNA binding domain that allows it to target scaffolding activity to relevant enhancers. These include the activation domain presumed to order on building transcription complexes. The hinge domain follows in sequence and through post translational modifications of phosphorylation and sumoylation, affect activity and localization in the cell. The third domain is the hormone-binding domain, the heart of NR5A2. The heart attracts numerous partner regulators. In many nuclear receptors, hormone binding aids nuclear localization and transcription. NR5A2 is unusual as it is constitutively active meaning it can activate transcription without hormone binding presumably through direct binding interactions with additional protein factors.
Structural and biochemical studies of transcription only recently proved that the hormone is phosphatidylinositol triphosphate, PIP3. The PIP3 pathway is well described in the cytoplasm. The related inactive lipid PIP2, a precursor to PIP3 is abundant in the nuclear membrane and presumably provides the precursor for PIP3 biosynthesis.
There are many transcriptional regulator proteins of NR5A2; positive coregulators steroid receptor co-activators Steroid Receptor Coactivators, CREB-binding protein, and peroxisome proliferator-activated receptor γ co-activator-1α (PGC-1 α). Multiple repressors block transcription. These include silencing mediator of retinoid and thyroid hormone receptors, SMRT; small heterodimer partner, SHP; prospero-related homeobox 1; PROX1 and dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1, DAX1. Of these coregulators, three dimensional crystal structures exist showing how the activators bind using a single motif, a 12 amino acid residue helix from the coactivator, to a site on the surface of NR5A2, termed AF2 for activation function 2 [3]. The potent oncoprotein β-catenin physically associates with NR5A2 on the hormone-binding domain. NR5A2 functions synergistically with β-catenin and the structure of NR5A2 with β-catenin suggests β-catenin coactivates by attracting coregulators to either or both oncoproteins [4]. If the AF2 site is described as being on the hip of NR5A2, β-catenin binds at the shoulder.
A single structure of NR5A2 with DAX shows that DAX1 represses by physically blocking AF2 and the PIP3 site.
Small molecules are expected to exist which would affect the associations of activator proteins or repressor proteins. The site most available to cause these effects on binding small molecules is the PIP3 site. For example synthetic antagonists might physically block PIP3 binding or bind in place of PIP3 and distort the structure of NR5A2 disfavoring binding at the AF2 coactivator site.
The first synthetic antagonists of NR5A2 are recently described and evaluation of effects on function encourages further work on the chemistry and functional affects of small molecules on transcription relating to PDAC. The results described here suggest that specific antagonists of NR5A2 could be developed for studies of biological mechanisms and for potential therapeutic treatments.
Part 2 Hints of The Role of NR5A2 In Pancreatic Cancer
NR5A2 was first discovered in liver cells where it was called LRH-1. Its roles in liver are actively studied. NR5A2 is readily shown to appear in cultures of many of the well-characterized pancreatic cancer cells. The amount of RNA found by quantitative PCR depends on cell type and qualitatively on whether the cell type is primary or metastatic origin. Non transformed pancreatic ductal epithelium shows about the same level as primary tumor cells. The levels of the NR5A2 RNA are 25times higher in AsPC-1, COLO 357. In the mouse passaged cells called L3.3 (from COLO 357) no NR52A RNA is found. Protein, measured by Westerns, is well expressed in cells derived from metastatic tumors. Interestingly expression is uncorrelated between NR5A2 and PDX-1, the well-known transcriptional regulator driving pancreas development. Expression of the PROX1 corepressor of NR5A2 is anti correlated [1].
There is a genetic link to NR5A2 having a role in PDAC. Genome wide association studies link mutations in the control regions of the NR5A2 gene (5 of 8 hits in 4000 cases) to development of pancreatic cancer [5]. Pathology of human tumor confirms the presence of NR5A2 in tissue specimens from normal and cancerous pancreas. Histochemistry staining with two antibodies revealed clear differences in the levels of NR5A2 in normal tissue and cancerous tissue. NR5A2 protein is at low levels in normal tissue. NR5A2 is found at high levels in the nuclei and cytoplasm of cancerous cells [6]. It is not known what directs NR5A2 to the cytoplasm aside from sumoylation of the hinge domain. NR5A2 is not in the stroma surrounding the lesions but infiltrating immune cells contain NR5A2. PANIN lesions associated with pancreatitis show clear presence of NR5A2 [6].
Presence of a protein in either high or low concentrations does not necessarily link it directly to neoplasia. Functions of NR5A2 can be tested in cells grown in culture by lessening RNA expression using two or more NR5A2-specific siRNA oligomers to nearly remove the normal RNA. Protein levels should also be lessened with this treatment. Quantitative PCR analysis unambiguously showed complete inhibition of transcription of the NR5A2 gene. NR5A2 protein in these cells was strongly reduced as expected [6].
With these effects it is not surprising that changes in the levels of RNA and protein for NR5A2 lessens cell viability and ability to proliferate. Specifically lowering NR5A2 strongly blocks cancer cell proliferation in multiple cell lines with an expected exception being L3.3 cells which lack NR5A2 and are NR5A2 independent.
Which target genes depending on NR5A2 might be expected to slow proliferation? With several hundred genes affected by NR5A2 it is difficult to identify the most important ones. Known targets are cyclins D1, E1 (Cyc D1, Cyc E1), and C-Myc genes which are essential for cell growth and proliferation [7]. Quantitative PCR measures of their RNA levels showed C-Myc up-regulated in all pancreatic cancer cells but is lower by use of the specific inhibitory siRNAs. Likewise transcription of cyclins D1 and E1 are much lower. The well-known stem cell maintenance factor Nanog is expressed in these conditions. This is a major difference between normal and neoplastic cells suggesting that the amplification of Nanog transcripts in a subset of cancer cells and the presence of Nanog in these cells fits with their being in a dedifferentiated state [6].
With these observations, effects on cell cycle are expected. Cell cycle arrest follows the block on NR5A2 function. The population in G0 and G1 in specifically treated cells was increased, whereas the populations of cells progressing through S phase and those entering G2 phase are decreased [6]. Based on these observations, it is expected that specific blocking of NR5A2 in pancreatic cancer cells induces a cell cycle arrest causing G0 and G1 phases to accumulate by slowing the G1 to S transition. This arrest occurs without cell killing [6].
Part 3 Small Molecules- Searching for NR5A2 specific inhibitors
The excitement about NR5A2 being potentially involved in PDAC is that unlike most transcription factors it is drugable. Finding drugs for nuclear receptors is challenging for both biophysical and biological reasons, but drugs exist for eight of the 48 human nuclear receptors. Computational chemistry has become a powerful tool to refine and narrow the search for drug like molecules that affect protein function. Software used in scanning libraries of accessible chemicals is beautifully constrained by structural data on the protein target and its binding site. Yet NR5A2 is a formidable challenge since the hormone site is unusually large and the protein is known to be active without hormone. By screening large libraries for activators a few active molecules have been discovered using a sensitive assay [8].
A key hypothesis to finding inhibitors was that finding a sufficiently tight binding molecule could harm the three-dimensional structure. In other words a tight binder might force a structural deformation to disallow attachment of coregulators known to bind the AF2 site. This hypothesis has been tested to find antiestrogen molecules that bind where estrogen receptor binds estradiol but when so positioned, helix 12, the last one in the protein floats off into solution. The significance of this disorder is that helix 12 is a footstool for holding the motif helix from a coactivator. Scans of possible poses of several million chemicals to a structure model of NR5A2 but missing helix 12 led to a proposed list of a few hundred candidates which was culled to about thirty hopefuls. These were tested in two kinds of binding studies, sticking to the protein and sticking to the protein while denying binding of peptide from a potent activator of NR5A2. The latter is a strong test of binding predicted to link to changes in function. The biophysics of this phenomenon is interesting in that binding the small molecule is tight, yet the protein is shown to be measurably made less stable to denature by heating [9].
This small molecule is available commercially and is called Cpd 3 and identified chemically as (1-(3′-{1-[2-(4-morpholinyl)ethyl]-1H-pyrazol-3-yl}-3-biphenylyl)ethanone. It binds specifically to NR5A2 at the hormone-binding site and through non physiological protein deformation and displaces coactivator peptide. This happens with an apparent dissociation constant of about 1 micromolar.
Transcription by NR5A2 in cells is dependent on many partner proteins. Cpd 3 not only binds to the NR5A2 hormone site but also deactivates the receptor upon binding. Transcription measured in HEK 293 cells expressing NR5A2, inducible by added tetracycline, showed changes in the mRNA levels for a cell cycle gene under control of NR5A2 [ 9]. The endogenous G0S2 gene switches to G1 from G0. Cpd 3 strongly inhibits the transcriptional activity. Treatments with this compound lowered the G0S2 mRNA levels with an IC50 value of 5 ± 1 μM. In noninduced HEK293 cells there are no changes in the levels of G0S2 transcripts with Cpd 3 [9]. These transcription data are expected from the binding experiment using surface plasmon resonance to show that binding of compound 3 to NR5A2 at the hormone site diminishes the receptor’s interactions with co-regulator peptide, in this case peptide DAX1–3 from the N terminal activation domain of DAX1. Both the physical binding and the transcription changes are dose-dependent [9]. These observations confirm the mechanism of receptor deactivation. The closest receptor to NR5A2 is NR5A1 and is unaffected by Cpd3.
These data and the known biological implications of NR5A2 including the siRNA experiments suggest that Cpd 3 will have effects on cancer cells. Specific antagonists should affect cell proliferation in vitro.
NR5A2 receptor-mediated anti-proliferative effects were confirmed in four different pancreatic PDAC cell lines, including AsPC-1, known to express high levels of NR5A2. There is dose-dependent inhibition of cell proliferation at concentrations about 20 micromolar. It is observed to be dependent on the NR5A2 axis since no anti-proliferative effects were observed in pancreatic cancer cells L3.3. These cells are derivatives of Colo 357 cells but do not express the LRH-1 receptor. As expected inhibition of transcription of NR5A2 target genes SHP and cyclin E1 is found in Colo357 but not in L3.3 cells following addition of Cpd3.
Cytotoxicity is critical in evaluating small molecule effects on cell proliferation. No cytotoxicity is found below doses of 40 to 50 micromolar.
Part 4 A Future for Regulation of Signaling Networks by Small Molecules
Cpd3 and other antagonists under evaluation can be used as molecular probes for elucidating the roles of the receptor in normal physiological and pathophysiological processes. The technologies already developed could be used to identify drugs for molecular targeted therapies of PDAC. There are clear dangers in manipulating functions of molecules that figure in signaling by multiple networks. This is particularly true for small molecules that regulate stem cell fate, which hangs in delicate balance. The estrogen receptor and its small molecule effectors reveal the drama. Tamoxifen is an effective therapy for breast cancer. It inhibits estrogen-dependent growth by binding estrogen receptors and altering their associations with coregulators. Tamoxifen, like compound Cpd3, check proliferation of tumor cells. Estrogen receptor like NR5A2 binds to many coregulators, so not all transcription is stopped. In the case of estrogen receptor, travel to the nucleus from the cytoplasm is an activation process. Tamoxifen induces this travel supporting a partly functional structure for the receptor which as expected has estrogenic activating effects in non breast tissues, such as bone and endometrium. Not all antiestrogens have the same effect on the structure of the receptor and its associations with coregulators. Several anti-estrogen compounds have been discovered by careful chemical and biological experiments and some have a reduced agonist profile on breast and related tissues. Selective inhibitors may have good response coupled with lower toxicity compared with tamoxifen. Clinical data for advanced breast cancer suggest safer versions of tamoxifen.
Part 5 Exploratory Chemistry Matched to Expansive Biology
NR5A2 has a rich biology that has challenges for developing chemical probes or drugs with specific functionality. Tissue and cell contexts determine which molecules are recruited to enhancer sites for establishing appropriate transcription machines. Stem cells will develop different NR5A2-directed assemblies from those functioning in normal liver or pancreas cells. Some of these processes will act in a repression context and some will direct activation. The newly discovered roles of PIP3 and repressor and activator polypeptide domains binding to the hormone-binding domain of NR5A2 [11] suggest the route forward is to discover specific and tight binding small molecules with directed biological responses focused to the targeted cell type. Activated NR5A2 is needed for developing pancreas from appropriate stem cells [] and repressed NR5A2 shows promise for slowing cell proliferation in PDAC. As found for estrogen receptor some drugs show a good spectrum of responses for treating breast cancers while sparing the dangerous effects on normal cells.
Imagine for example, that NR5A2 is a dangerous player in the PDAC cell nucleus, but beneficial in normal physiology. If PIP3, or β-catenin were the responsible activator in PDAC we would seek a small blocking PIIP3 or β-catenin binding but permit the the binding of housekeeping coregulators. This degree of response is typically unpredictable when discovering binding of a small molecule to a protein.
A recent paper presents new technologies for discovery of compounds with biological relevance. This technology reveals appropriate lead compounds at an unusually fast rate. Rates of discovery are often slow because physical and virtual screening of compound libraries of millions of small molecules is slow and expensive. The faster route is to use smaller libraries, a few thousand lower molecular weight members, with tight binding. These molecules, called fragment have the additional property of being derivatized to make bigger analogs.
A key feature is to exploit disulfide-trapping- forming a covalent –S-S- bond between the protein and the trial chemical. This technology is a new approach new for nuclear receptors which have large, but often water inaccessible and non polar binding sites for hormones.
This approach when applied to NR5A2 readily identified a lead compound that binds covalently to only the native cysteine residue, Cys346. This amino acid residue is within PIP3 binding site.
Computational modeling suggested a structure of the small molecule complex with NR5A2 and cellular assays were used to find an analog with no sulfur atom. This analog is termed PME8 and is a good activator, with A dissociation constant of 5 micromolar [10].
This technical approach could be used with protein complexes of NR5A2 such as with DAX1 peptide or β-catenin and suggest that with further development this chemical probes that are specific antagonists.
Discussion
Refer to the Figure for the following discussion. NR5A2 is normally resident in the nucleus, and its hormone-binding domain can bind to PIP3, PGC1a, SRC coactivators, and β-catenin. The potential for impacting PDAC by binding small molecules is in conflict with normal physiology which depends on NR5A2 functioning normally. Small molecules have been found that block transcription driven by the AF2 site and regulator proteins that bind there. This success suggests that compounds can be found that inhibit in other ways. One could block PIP3 binding which associates with affinity of about 80 nanomolar. A third class of NR5A2 antagonist might bind at the same PIP3 binding site but disorder the protein for binding the coactivator β-catenin. There is also potential for direct competition of the association.
Figure.
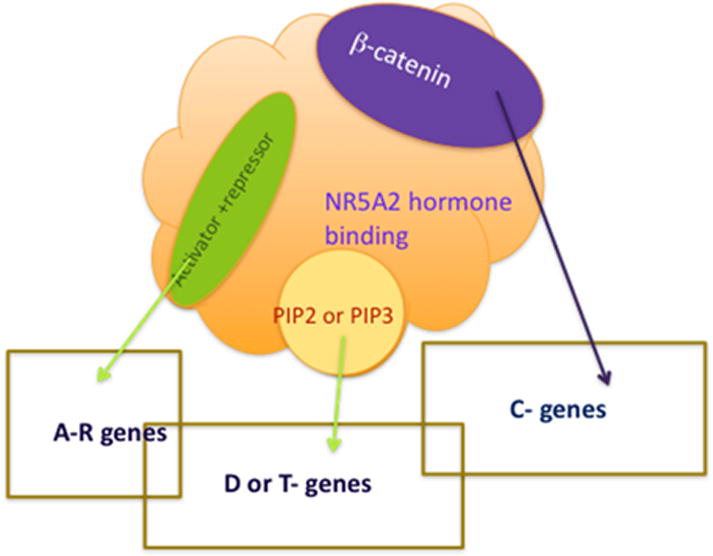
The rectangular boxes represent subsets of genes under regulatory control of NR5A2 which may relate directly to the molecules binding at the surface. The orange shape represents the hormone-binding domain. Cpd 3 binds internal to the orange surface inside the PIP3 site. PIP3 is the hormone. PIP2 also binds but less tightly and does not activate transcription. On its surface crystallography has defined binding surfaces for coregulators, the activator + repressor site in green, called AF 2 where motif helices of coactivators bind to enhance transcription. Repressors and activators compete here. DAX1 is a nuclear receptor that can activate or inhibit NR5A2. It forms a dimer with a disordered N- terminal activation domain that also contains its dimeriztion site. One monomer of the dimer of DAX1 C-terminal domain binds here and inhibits activity with a bigger footprint than an activating helix motif. The second monomer of the dimer binds at the PIP3 site blocking access to the hormone. It is thought that with PIP3 bound it becomes a key to bind additional protein activators to the exposed phosphatidyl inositol presented at the surface of NR5A2. The transcription affects are represented by the rectangles.
The β-catenin site is independent of the other surface binding sites. This site likely attracts additional factors to alter transcriptional activation.
NR5A2 is active in normal and transformed tissue. NR5A2 is not required for the development of acinar cells in the pancreas but it is required for their maintenance. Deletion of NR5A2 leads to destabilized mature acinar cells, and they cannot survive pancreatitis. Loss of NR5A2 worsens the development of oncogenesis driven by Kras [12].
Likely if antagonists to NR5A2 are to be safe in humans the effects on binding should be pronounced and biased in cancer cells or cancer stem cells. Animal studies will help evaluate the effects of NR5A2 inhibitors in tumor versus normal tissues. These studies will begin when tighter binding and specific molecules replace Cpd 3. Only about a factor of ten in affinity is needed for these studies to proceed for physiology depending on the activation site AF2. Importantly the armament of small molecules affecting functions, associations of coregulators of NR5A2 will be valuable chemical probes that define the normal and pathophysiology of this remarkable protein.
Synopsis.
Controlling aberrant transcription of gene expression that drives tumor growth may potentially treat pancreatic cancer. The transcription regulator NR5A2 may be a target for treating PDAC and is drugable.
Acknowledgments
Research Support provided from NIH and DOD grants. National Institute of Health Grants R01 DK078075 and R21 CA140751 & DOD Pancreatic Cancer Program
Footnotes
There are no disclosures of conflict by the author.
References
- 1.Molecular basis for the regulation of the nuclear receptor LRH-1. Stein S, Schoonjans K. Curr Opin Cell Biol. 2015 Apr;33:26–34. doi: 10.1016/j.ceb.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Canonical Wnt/β-catenin regulation of liver receptor homolog-1 mediates pluripotency gene expression. Wagner RT, Xu X, Yi F, Merrill BJ, Cooney AJ. Stem Cells. 2010 Oct;28(10):1794–804. doi: 10.1002/stem.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1. Sablin EP, Krylova IN, Fletterick RJ, Ingraham HA. Mol Cell. 2003 Jun;11(6):1575–85. doi: 10.1016/s1097-2765(03)00236-3. [DOI] [PubMed] [Google Scholar]
- 4.Structural basis of coactivation of liver receptor homolog-1 by β-catenin. Yumoto F, Nguyen P, Sablin EP, Baxter JD, Webb P, Fletterick RJ. Proc Natl Acad Sci U S A. 2012 Jan 3;109(1):143–8. doi: 10.1073/pnas.1117036108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Petersen GM, Amundadottir L, Fuchs CS,O, Thomas G, et al. Nat Genet. 2010 Mar;42(3):224–8. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nuclear receptor liver receptor homologue 1 (LRH-1) regulates pancreatic cancer cell growth and proliferation. Benod C, Vinogradova MV, Jouravel N, Kim GE, Fletterick RJ, Sablin EP. Proc Natl Acad Sci U S A. 2011 Oct 11;108(41):16927–31. doi: 10.1073/pnas.1112047108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, Schoonjans K. Mol Cell. 2004 Aug 27;15(4):499–509. doi: 10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Identification of small molecule agonists of the orphan nuclear receptors liver receptor homolog-1 and steroidogenic factor-1. Whitby RJ, Dixon S, Maloney PR, Delerive P, Goodwin BJ, Parks DJ, Willson TM. J Med Chem. 2006 Nov 16;49(23):6652–5. doi: 10.1021/jm060990k. [DOI] [PubMed] [Google Scholar]
- 9.Structure-based discovery of antagonists of nuclear receptor LRH-1. Benod C, Carlsson J, Uthayaruban R, Hwang P, Irwin JJ, Doak AK, Shoichet BK, Sablin EP, Fletterick RJ. J Biol Chem. 2013 Jul 5;288(27):19830–44. doi: 10.1074/jbc.M112.411686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Disulfide-Trapping Identifies a New, Effective Chemical Probe for Activating the Nuclear Receptor Human LRH-1 (NR5A2) de Jesus Cortez F, Suzawa M, Irvy S, Bruning JM, Sablin E, Jacobson MP, Fletterick RJ, Ingraham HA, England PM. PLoS One. 2016 Jul 28;11(7):e0159316. doi: 10.1371/journal.pone.0159316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Structure of Liver Receptor Homolog-1 (NR5A2) with PIP3 hormone bound in the ligand binding pocket. Sablin EP, Blind RD, Uthayaruban R, Chiu HJ, Deacon AM, Das D, Ingraham HA, Fletterick RJ. J Struct Biol. 2015 Dec;192(3):342–8. doi: 10.1016/j.jsb.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. von Figura G, Morris JP, 4th, Wright CV, Hebrok M. Gut. 2014;63(4):656–64. doi: 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]