

SUMMARY
Merlin encoded by the Nf2 gene is a bona fide tumor suppressor that has been implicated in regulation of both the Hippo-Yap and the Rac1-Pak1 pathways. Using genetically engineered murine liver models, we show that co-deletion of Rac1 with Nf2 blocks tumor initiation but paradoxically exacerbates hepatomegaly induced by Nf2 loss, which can be suppressed either by treatment with pro-oxidants or by co-deletion of Yap. Our results suggest that while Yap acts as the central driver of proliferation during Nf2 tumorigenesis, Rac1 primarily functions as an inflammation switch by inducing ROS that on one hand induces NFkB signaling and expression of inflammatory cytokines, and on the other activates p53 checkpoint and senescence programs dampening the cyclinD1-pRb-E2F1 pathway. Interestingly, senescence markers are associated with benign NF2 tumors but not with malignant NF2 mutant mesotheliomas, suggesting that senescence may underlie the benign nature of most NF2 tumors.
Keywords: NF2, Rac1, Yap, Senescence, ROS, inflammation, DNA damage response
INTRODUCTION
Merlin (Moesin, ezrin, and radixin like protein), a member of the Band 4.1 ERM family of proteins, is encoded by the Neurofibromin 2 (NF2) gene whose mutations underlie Neurofibromatosis type 2 (NF2), an autosomal dominant genetic disorder characterized by the development of benign vestibular schwannomas and meningiomas (Rouleau et al., 1993; Trofatter et al., 1993). Somatic mutations of NF2 occur frequently in sporadic schwannomas, meningiomas, ependymomas and mesotheliomas, and at lower rates in thyroid cancer, melanoma, kidney cancer and other common cancers (Petrilli and Fernandez-Valle, 2015). Merlin is primarily localized to the cell:cell junctions where it mediates contact dependent inhibition of proliferation (Petrilli and Fernandez-Valle, 2015), although it has can also translocate into the nucleus (Li et al., 2014; Li et al., 2010). Inactivation of Merlin has been linked to deregulation of numerous signaling molecules, including most prominently Yes-associated protein (Yap) and Ras-related C3 botulinum toxin substrate 1 (Rac1).
Yap is a transcriptional co-activator that partners with the Tead family of transcription factors to regulate the expression of genes involved in cell proliferation and survival (Hansen et al., 2015). Merlin forms scaffolding complexes with WW45 and Kibra, which recruit the Mst1/2 and Lats1/2 kinases to the cell junctions upon cell:cell contact, initiating an Mst1/2-Lats1/2-Yap phosphorylation cascade that results in cytoplasmic retention and proteasomal degradation of Yap (Hamaratoglu et al., 2006; Yin et al., 2013; Zhang et al., 2010). In addition, Merlin has been shown to prevent ubiquitination/degradation of Lats1/2 by the CRL4-DCAF1 E3 ubiquitin complex in the nucleus (Li et al., 2014).
Rac1 is a Rho family small GTPase that regulates a plethora of signaling pathways including JNK, MAPK, NFkB, mTOR, and reactive oxygen species (ROS) production (Bosco et al., 2009). Merlin is a bona fide substrate of p21-activated kinases - direct downstream effectors of Rac1 (Kissil et al., 2002; Rong et al., 2004; Xiao et al., 2002). It has been shown that myelination arrest caused by Rac1 inactivation in Schwann cells can be rescued by co-deletion of Nf2, supporting Merlin functioning downstream of Rac1 (Guo et al., 2012). Existing evidence also supports Merlin functioning upstream of the Rac1-Pak axis. Primary schwannoma cells isolated from NF2 patients exhibit elevated Rac1-GTP levels compared to normal Schwann cells (Kaempchen et al., 2003). Merlin has been found to block the recruitment of Rac1-GTP to the plasma membrane upon cell-cell contacts (Das et al., 2015; Okada et al., 2005). Dominant-active Rac1 or Pak can reverse Merlin’s inhibition of MAPK signaling (Morrison et al., 2007). We have shown that Merlin inhibits Rac1 and the MAPK pathway by releasing Rich1 (a Rac1/Cdc42 GTPase activating protein) from its inhibitor Angiomotin at the tight junctions (Yi et al., 2011).
In vivo modeling of NF2 has been challenging. Homozygous Nf2 knockout in mice is embryonically lethal, whereas heterozygous deletion produces tumors that do not recapitulate human disease (McClatchey et al., 1998; McClatchey et al., 1997). Targeted deletion of Nf2 in Schwann or arachnoidal cells in mice leads to tumors with low penetrance and long latency (Giovannini et al., 2000; Kalamarides et al., 2002), limiting their usefulness. In recent years, a highly penetrant liver system utilizing Cre-recombinase driven by the Albumin promoter (Alb-cre) to specifically knockout Nf2 in parenchymal liver epithelial cells has been adopted to model NF2 tumorigenesis (Benhamouche et al., 2010; Postic et al., 1999; Zhang et al., 2010). Nf2 deletion in the murine liver leads to the expansion of Sox9+CK19+ liver progenitor cells (LPCs) and development of cholangiocarcinoma (CC) as well as hepatocellular carcinoma (HCC) in a Yap-dependent manner (Liu-Chittenden et al., 2012; Yimlamai et al., 2014; Zhang et al., 2010). The importance of Yap as a downstream effector of Merlin has been subsequently confirmed in NF2-relevant cell types (Guerrant et al., 2016; Lavado et al., 2013; Serinagaoglu et al., 2015), underscoring the value of the liver model in elucidating the molecular mechanisms of Nf2 tumorigenesis.
Even though Nf2 mutations are rare in human hepatic malignancies, deregulation in Hippo-Yap signaling is commonly associated with both human HCC and CC. An estimated 5–10% of human HCCs harbor genomic amplification of the YAP locus (Zender et al., 2006), whereas a study identified focal deletion in the coding regions of the SAV1 gene (encodes the WW45 protein) in 12% of human CCs in a mutually exclusive manner to KRAS and BRAF mutations (Sia et al., 2013). Given that the Alb-Cre-mediated liver knockout of Nf2 largely phenocopies that of other components of the Hippo pathway (Lee et al., 2010; Lu et al., 2010; Nishio et al., 2016; Song et al., 2010; Zhou et al., 2009), this model system also provides a useful platform to interrogate the role of the Hippo-Yap pathway in liver cancer.
Using this model, we previously showed that Angiomotin, which functions both upstream of Rac1 and Yap, is also required for tumorigenesis induced by Nf2 loss (Yi et al., 2013). In the current study, we again utilize this well-established system to resolve the role of Rac1 in Nf2 tumorigenesis and dissect signaling hierarchies between Merlin, Rac1 and Yap.
RESULTS
Inactivation of Rac1 aggravates hepatomegaly while impeding tumorigenesis in Nf2-deleted mouse livers
Liver-specific knockout of the Hippo pathway genes leads to the expansion of LPCs, hepatomegaly, and development of frank (macroscopically visible) tumors in mice that resemble human CC (Lee et al., 2010; Lu et al., 2010; Nishio et al., 2016; Song et al., 2010; Zhang et al., 2010). Consistent with a previous report (Sia et al., 2013), analysis of The Cancer Genome Atlas (TCGA) CC dataset revealed the presence of mutations or deletions in the Hippo pathway genes including NF2, SAV1, and FAT1-4 in significant fractions of the tumors (Figure 1A), underscoring the relevance of this pathway to human CC.
Figure 1. Co-deletion of Rac1 suppresses tumorigenesis but exacerbates hepatomegaly induced by Nf2 ablation.
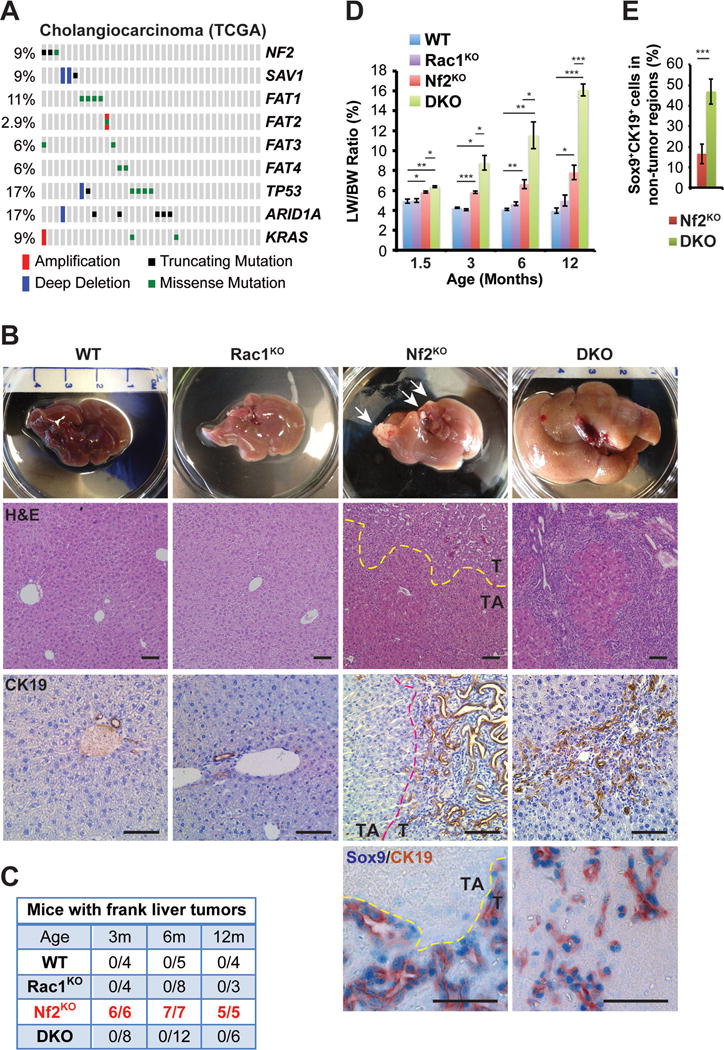
(A) Mutation and copy number status of indicated genes in TCGA cholangiocarcinoma collection (N=35).
(B) Representative images of gross livers, H&E staining, and CK19 immunohistochemistry (IHC) of livers from 6-month-old WT, Rac1KO, Nf2KO and DKO mice, and Sox9 (blue)/CK19 (brown) double IHC of livers from 3-month-old Nf2KO and DKO mice. Arrows indicate frank tumor lesions. Dotted lines mark the borders between tumor (T) and tumor adjacent (TA) areas in the Nf2KO livers. Scale bars = 100 μm.
(C) The numbers of WT, Rac1KO, Nf2KO, and DKO mice with frank liver tumors at 3, 6 and 12 months of age. The tumor numbers in Nf2KO mice are highlighted in red.
(D) Liver weight to body weight ratios (LW/BW) of 1.5-, 3-, 6- and 12-month-old WT, Rac1KO, Nf2KO and DKO mice (1.5 months: WT n=8, Rac1KO n=4, Nf2KO n=4, DKO n=6; 3 months: WT n=4, Rac1KO n=4, Nf2KO=6, DKO n=8; 6 months: WT n=5, Rac1KO n=8, Nf2KO=7, DKO n=12; 12 months: WT n=4, Rac1KO n=3, Nf2KO=5, DKO n=6). Data are represented as mean +/− SEM. *P<0.05, **P<0.005, ***P<0.0005.
(E) Average percentage of Sox9+CK19+ cells in 3-month-old Nf2KO_TA regions (n=4) and DKO (n=6) livers. Data are represented as mean +/− SD. ***P<0.0005.
See also Figure S1.
Besides deregulation of Hippo signaling, MERLIN/NF2 loss also causes hyperactivation of RAC1 (Das et al., 2015; Kaempchen et al., 2003; Okada et al., 2005). To elucidate the role of RAC1 in Nf2 tumorigenesis, we generated liver-specific Nf2:Rac1 double knockout (Alb-cre;Nf2flox/flox;Rac1flox/flox, DKO) mice and compared them anatomically and histopathologically with wild type (WT), Nf2KO (Alb-cre;Nf2flox/flox) and Rac1KO (Alb-cre;Rac1flox/flox) littermates. Consistent with the previous reports (Benhamouche et al., 2010; Zhang et al., 2010), we found that Nf2KO livers were significantly enlarged compared to WT at all time points, and developed frank bile duct hamartomas (BDH) composed mostly of Sox9+CK19+ LPCs with 100% penetrance as early as 1.5 months (Figures 1B–1E, and Figure S1A). Despite the early tumor onset, Nf2KO tumors were slow growing and the size of the Nf2KO tumors relative to the size of the livers did not significantly change over time (Figures S1B–C). Compared to Nf2KO livers, DKO livers exhibited diffuse expansion (hyperplasia) of CK19+Sox9+ LPC populations and significant further increase in liver size (Figures 1B, 1D and 1E). By 6 months to 1 year, LPC hyperplasia originating from the portal veins became so prevalent in DKO livers that they began to overpopulate, squeezing hepatocytes into small islands (Figure 1B). Paradoxically, in spite of the extensive LPC hyperplasia, DKO livers remained free of frank tumors for as long as 1 year (Figures 1B–1C).
The hyperplastic phenotype of DKO mice was not caused by compensatory upregulation of other Rac1-related proteins Rac2, Rac3 or Cdc42, as their mRNA levels were reduced or unchanged in DKO livers when compared to Nf2KO livers (Figure S1D and Data S1). It also did not result from additive effects of Rac1 deletion, as Rac1KO mice developed normally, lived a normal lifespan, and displayed normal liver histology, size, transcriptional profiles, and response to 3,5-diethoxycarbonyl-l,4-dihydrocollidine (DDC) induced liver injury (Figures 1B–1D, Figures S1E–S1F, and Data S1) (Bopp et al., 2013). These results indicate that instead of playing a general anti-proliferative role in LPCs, Rac1 functions in the context of Nf2 inactivation in restricting LPC proliferation and liver size.
Knockout of Rac1 does not affect the Hippo and Notch pathway inactivation and hepatocyte de-differentiation induced by Nf2 loss
In agreement with the original report (Zhang et al., 2010), we found that heterozygous Yap deletion (Alb-cre:Nf2flox/flox:Yapflox/+, Nf2KO:Yaphet) was just as effective as homozygous Yap knockout (Alb-cre:Nf2flox/flox:Yapflox/flox, Nf2KO:YapKO) in suppressing liver enlargement and tumorigenesis (Figure S1G and data not shown).
To investigate whether Rac1 affects the Hippo-Yap pathway, we carried out western blot, immunohistochemistry (IHC), microarray and quantitative real time PCR (qRT-PCR) analyses with WT, Rac1KO, Nf2KO (both tumor and tumor adjacent regions, Nf2KO_T and Nf2KO_TA) and DKO livers. Compared to WT and Rac1KO livers, both Nf2KO_T and DKO livers exhibited decreased levels of pLats1 (inhibitory kinase of Yap) and pYap (Figure 2), increased nuclear Yap expression in CK19+ cells (Figures S1H–S1I), and correspondingly increased expression of Hippo-Yap pathway regulated genes, including Amot and Cyr61 (Figure 2, Figures S2A–S2B, and Data S1). While the raw expression of Hippo pathway genes is lower in the DKO livers compared to Nf2KO_T regions, this likely reflects the lower percentage of CK19+Sox9+ LPC population in the DKO livers compared to Nf2KO_T regions (Figure S2B and Data S1). Indeed, when adjusted to CK19 levels, the majority of Hippo pathway genes showed no significant change or even increased expression in DKO liver cells compared to Nf2KO_T regions (Figure S2C).
Figure 2. Deletion of Nf2, Rac1 or Yap alone or in combination has differential effects on cell cycle, DNA damage checkpoint, NFkB, Akt, Erk and Stat3 signaling.
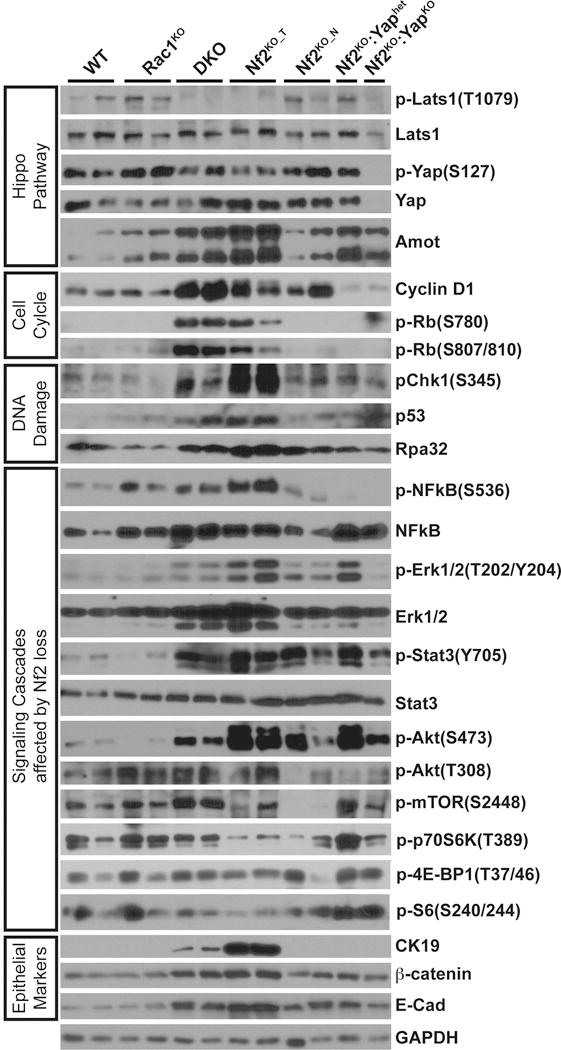
Western blot analysis of WT, Rac1KO, DKO, Nf2KO_T, Nf2KO_TA, Nf2KO;Yaphet, and Nf2KO;YapKO liver tissues with antibodies against components of the Hippo-Yap pathway, cell cycle, DNA damage response, signaling cascades affected by Nf2, Rac1 and/or Yap loss, and epithelial cell markers as indicated. GAPDH was used as loading control. Shown is representative of three replicated experiments. All samples were run on the same gel for each of the blots.
See also Figures S2–S3.
Inactivation of Nf2 or activation of Yap induces hepatocyte-ductal metaplasia by activating Sox9 and Notch signaling (Yimlamai et al., 2014). Compared to WT and Rac1KO livers, both Nf2KO and DKO livers displayed reduced expression of hepatocyte-associated genes while increasing expression of genes associated with the Notch pathway as well as ductal/progenitor cells (Figures S2A–S2B, and Data S1). Correspondingly, DKO livers showed similar distribution of Sox9+CK19+, Sox9+CK19−, and Sox9−CK19+ cell populations as Nf2KO livers (Figure S1J). Thus, co-deletion of Rac1 does not interfere with the inactivation of Hippo and Notch signaling and hepatocyte-ductal metaplasia caused by Nf2 inactivation.
Ablation of Rac1 in the Nf2 deficient liver further enhances cell proliferation by hyperactivating the CyclinD-Rb-E2F1 pathway
In line with their overall liver growth rates (Figure 1D), DKO livers contain persistently higher percentage of PCNA+ proliferating cells compared to Nf2KO livers (both Nf2KO_T and Nf2KO_TA regions) up until 6 months of age (Figure S2D). Correspondingly, the percentage of proliferating CK19+ cells are also significantly higher in DKO livers compared to Nf2KO livers (Figures 3A–3B).
Figure 3. Deletion of Rac1 synergizes with Nf2 loss to induce the expression of cell cycle genes in the mouse liver.
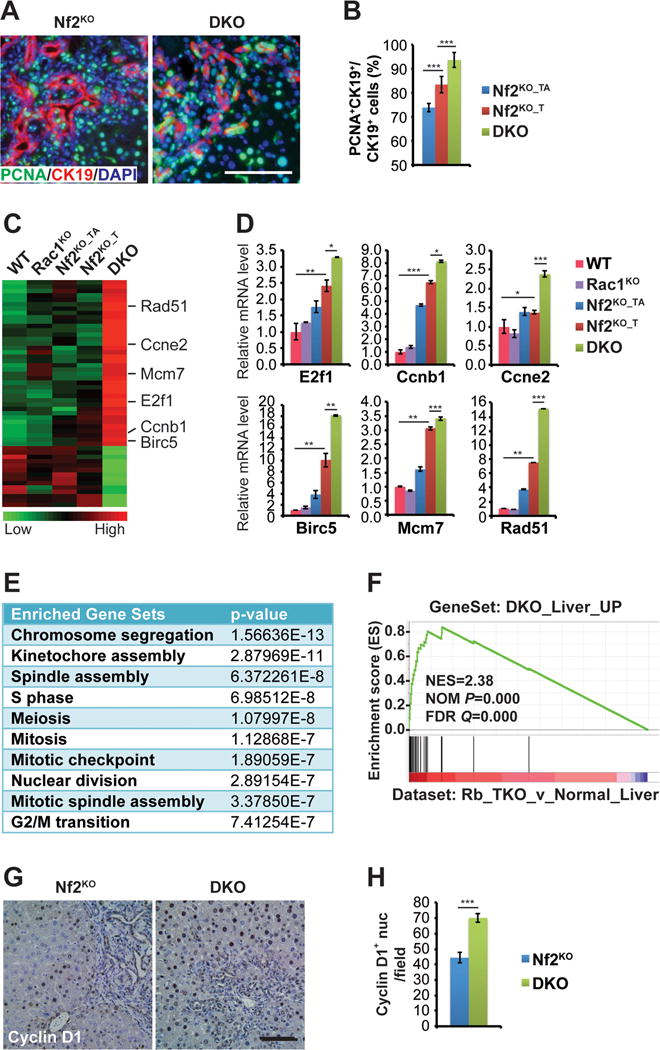
(A) Representative images of immunofluorescence (IF) staining of PCNA (green) with CK19 (red) on livers from 3-month-old Nf2KO and DKO mice. Nuclei are counterstained with DAPI (blue). Scale bars = 100 μm.
(B) Quantification of the average percentage of CK19+ cells that are also PCNA+ in 3-month-old Nf2KO and DKO (n=8) liver tissues. Nf2KO_T (n=7) and Nf2KO_TA (n=7) regions were quantified separately. Data are represented as mean +/− SD. ***P<0.0005.
(C) Heatmap of genes differentially expressed in DKO livers compared to the rest of the cohorts from microarray analysis of WT, Rac1KO, the tumor adjacent (Nf2KO_TA) and tumor (Nf2KO_T) regions of Nf2KO, and DKO liver tissues in 3-month-old mice (n=3 for all groups).
(D) Quantitative real-time PCR analysis of the indicated cell cycle genes from WT, Rac1KO, Nf2KO_TA, Nf2KO_T, and DKO liver tissues from 3-month-old mice (n=3 for all groups). Data are represented as mean +/− SEM. ***P<0.0005; **P<0.005, *P<0.05.
(E) The top 10 enriched pathways from GO enrichment analysis of genes from (C).
(F) GSEA by comparing upregulated genes from (C) (DKO_Liver_Up) with a published Rb family triple knockout dataset (GSE19004) (Viatour et al., 2011).
(G) Representative images of CyclinD1 IHC staining of livers from 3-month-old Nf2KO and DKO mice. Scale bars = 100 μm.
(H) Quantification of the number of CyclinD1-positive nuclei per microscopic field. (Nf2KO n=10, DKO n=9). Data are represented as mean +/− SD. ***P<0.0005.
See also Figure S2, and Data S1.
To probe for the underlying molecular mechanism, we conducted pathway profiling of genes differentially expressed between DKO and Nf2KO livers from our microarray dataset (Data S1, GSE70742). Strikingly, the top 10 gene sets enriched for the DKO-upregulated genes (DKO_Liver_Up) were involved in cell cycle regulation, including genes such as E2f1, Ccnb1, Ccne2, Birc5, Mcm7 and Rad51 that were also validated by qRT-PCR (Figures 3C–2E, and Figure S2E). Moreover, gene set enrichment analysis (GSEA) demonstrated that DKO_Liver_Up genes were strongly induced in the Rb family triple knockout (Rb_TKO) livers (Figure 3F) (Viatour et al., 2011), suggesting that induction of cell-cycle-associated genes in DKO livers was likely caused by Rb inactivation. Indeed, we detected marked further increase in the protein levels of pRb and Cyclin D1 in DKO livers compared to Nf2KO livers (Figures 2, 3G, and 3H). In contrast, either heterozygous or homozygous Yap deletion blocked Rb phosphorylation and Cyclin D1 accumulation (Figure 2), consistent with their similar ability to suppress liver enlargement caused by Nf2 loss (Figure S1G). These data suggest that Rac1 suppresses cell cycle progression in Nf2-deleted liver cells by dampening the activation of CyclinD1-pRb-E2F1 pathway, opposite of Yap.
The status of Erk, Akt and Stat3 signaling does not correlate with liver size or tumorigenesis in Nf2 deficient livers
Inactivation of NF2 has been shown to activate ERK, AKT and STAT3 signaling (Ammoun et al., 2008; Benhamouche et al., 2010; Hilton et al., 2009; James et al., 2009; Scoles et al., 2002; Yi et al., 2011). To examine whether these pathways were the sources of the increased proliferative signals in the DKO liver, we analyzed the phosphorylation status of Stat3, Erk, and Akt via western blot or Immunofluorescence (IF).
As expected, we observed increased activating phosphorylation of Erk (T202/Y204) and Stat3 (Y705) in the tumor regions of Nf2KO livers (Figure 2). IF analysis showed that within Nf2KO_T regions activation of Erk primarily occurs in the CK19+ tumor cells, whereas Stat3 activation occurs in subsets of tumor cells as well as stromal cells (Figures S3A–S3B). Unexpectedly, the activation of these two pathways was suppressed in DKO livers in spite of their hyperproliferative state (Figure 2, and Figures S3A–S3B). These results suggest that while Rac1 is required for activation of the Erk and Stat3 pathways upon Nf2 loss, these pathways are not necessary for sustaining proliferation and liver enlargement in DKO livers. In further support of a lesser role for these pathways in driving proliferation and tumorigenesis caused by Nf2 loss, Erk and Stat3 signaling remained high in Nf2KO:Yaphet livers that are tumor free (Figure 2). This was in sharp contrast to pRB and CyclinD1, which were effectively blocked by either heterozygous or homozygous Yap ablation in Nf2 deficient livers (Figure 2).
Full activation of AKT requires both PDK1-mediated phosphorylation at T308 and TORC2-mediated phosphorylation at S473 (Dibble and Cantley, 2015). Interestingly, the tumor regions of Nf2KO livers exhibited robust induction of phosphorylation of Akt at S473, but not at T308 (Figure 2), suggesting that inactivation of Nf2 is insufficient in fully activating Akt in the mouse liver. Indeed, Nf2 deletion also did not induce phosphorylation of mTOR or its downstream targets p70S6K, 4E-BP1 and S6 (Figure 2), again suggesting that the Akt-mTOR pathway does not play a major role in in Nf2 deficient livers.
Inactivation of Nf2 induced p53 expression and DNA damage checkpoint in a Rac1-dependent manner
In spite of hyperactivation of the CyclinD1-pRb-E2f1 pathway and cell cycle genes, progression to frank tumors was suppressed in DKO livers. We hypothesized that genes differentially expressed between Nf2KO and WT livers but with a reversed profile in DKO livers (DKO_Liver_Reversed, Figure S4A) could be important for Nf2 tumorigenesis. Pathway profiling showed that DNA damage response (DDR) and cell death were among the top 10 pathways enriched with the DKO_Liver_Reversed gene set (Figure S4B). Interestingly, GSEA showed this set of genes also significantly associated with genes upregulated in human vestibular schwannomas (VS) (Torres-Martin et al., 2013) despite the tissue and cell type differences (Figure S4C), implying their potential relevance to NF2.
DNA damage induces CHK1/2 phosphorylation, which in turns activates p53 (Lavin and Gueven, 2006). Indeed, the protein levels of pChk1, p53 as well as Rpa32 were induced in Nf2KO tumors to a much higher extent than in DKO livers (Figure 2). Correspondingly, we also observed a robust increase in transcript levels of other DNA damage-induced genes including Cdkn1a (p21), Cdkn2a (p16), Cdkn2b (p15), Gadd45a, and Birc3 in Nf2KO tumors compared to DKO livers (Figure 4A, Figure S4D). These results suggest that Rac1-mediated induction of DDR and p53 serves as a negative feedback mechanism limiting cell cycle progression, LPC expansion and tumor growth in Nf2KO livers. By contrast, failure to fully activate DDR and the p53 checkpoint due to Rac1 loss results in uncontrolled cell cycle progression and pervasive LPC hyperplasia in DKO livers. However, the extensive LPC hyperplasia never progressed into frank tumors in DKO livers, suggesting that besides activating p53 checkpoint that restricts tumor growth, Rac1 likely induces additional pro-tumorigenic signaling that are required for tumor development in Nf2 deficient livers.
Figure 4. Co-deletion of Rac1 suppresses DNA damage and senescence markers induced by Nf2 loss.
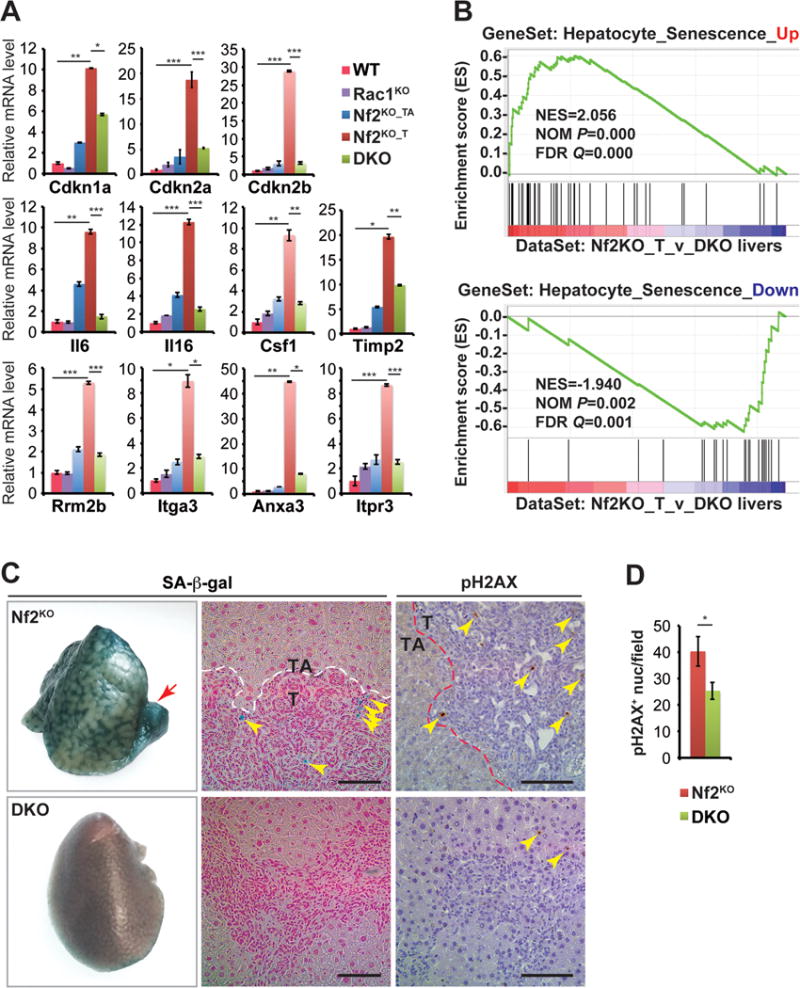
(A) Quantitative real-time PCR analysis of senescence-associated genes in WT, Rac1KO, Nf2KO_TA, Nf2KO_T, and DKO liver tissues from 3-month-old mice (n=3 for all groups). Data are represented as mean +/− SEM. ***P<0.0005; **P<0.005; *P<0.05.
(B) GSEA by comparing a high-confidence senescence-associated gene signature (Aravinthan et al., 2014; Yildiz et al., 2013) with genes ranked by expression differences between Nf2KO_T and DKO liver tissues.
(C) Representative images of senescence associated β-galactosidase (SA-β-gal) staining (left panel), sections from SA-β-gal stained livers counterstained with Nuclear Fast Red Solution (middle panel), and pH2AX IHC staining of livers from 3-month-old Nf2KO and DKO mice (right panel). Red arrow indicates a frank tumor lesion in the Nf2KO liver. Yellow arrowheads indicate positive deposits of SA-β-gal and pH2AX staining. Scale bars = 100 μm.
(D) Average number of pH2AX+ nuclei per 10× microscopic field in 3-month-old Nf2KO (n=5) and DKO (n=7) liver tissues. Nf2KO_T and Nf2KO_TA regions were quantified separately. Data are represented as mean +/− SD. *P<0.05.
See also Figures S4–S5, and Tables S1.
Nf2 inactivation induces Rac1-mediated activation of the NFkB pathway and senescent phenotypes
In addition to cell cycle arrest, DDR and p53 can induce senescence or apoptosis. However, we detected no cleaved Caspase-3 (commonly used apoptosis marker) signals in either Nf2KO or DKO livers, while DDC-treated WT and DDC-treated Rac1KO livers showed robust Caspase-3 staining (Figure S4E).
Prompted by the observation of specific upregulation of common senescence drivers p53, p21, p16 and p15 in Nf2KO tumors, we examined our liver cohorts for evidence of the so-called senescence-associated secretory phenotype (SASP) (Coppe et al., 2010). Significantly higher levels of SASP-associated proinflammatory cytokines including Il6, Il16, Csf2 and Timp2 as well as immune infiltrates were present in Nf2KO_T regions compared to WT, Rac1KO, DKO livers or Nf2KO_TA regions (Figure 4A, and Figures S5A–S5B), matching the expression patterns of p53, p21, p16 and p15 (Figures 2 and 4A).
As senescence involves global transcription reprogramming, we next compiled a high confidence hepatocyte senescence gene signature (HSGS) by intersecting genes differentially expressed in hepatocytes that had undergone H2O2 or replication induced senescence from two independent studies (66 up and 42 down) (Aravinthan et al., 2014; Yildiz et al., 2013). GSEA showed that the upregulated genes within our HSGS (Hepatocyte_Senescence_Up) were strongly enriched with genes expressed higher in Nf2KO tumors than in DKO livers, whereas the downregulated genes within the HSGS (Hepatocyte_Senescence_Down) significantly coincided with genes with decreased expression in Nf2KO tumors compared to DKO livers (Figure 4B). Finally, compared to DKO livers, Nf2KO livers expressed significantly higher levels of senescence-associated β-galactosidase (SA-β-gal) and pH2AX, particularly within the tumor nodules (Figures 4C and 4D).
NFkB signaling has been reported to be the major driver of SASP (Salminen et al., 2012). Western blot, IHC, subcellular fractionation, GSEA, and qRT-PCR analyses showed that NFkB signaling and the expression of NFkB target genes were significantly elevated in Nf2KO_T regions compared to WT livers or Nf2KO_TA regions, and inhibited by deletion of either Rac1 or Yap (Figures 2 and 5A–5E). Notably, NFkB IHC showed specific increase in NFkB nuclear staining along ductal structures within Nf2KO_T regions indicative of activation of NFkB within tumor cells (Figure 5A–B), although it cannot be ruled out that infiltrating stromal cells might also contribute to increased NFkB signal in Nf2KO livers.
Figure 5. Inactivation of Nf2 induces NFkB signaling in a Rac1-dependent manner.
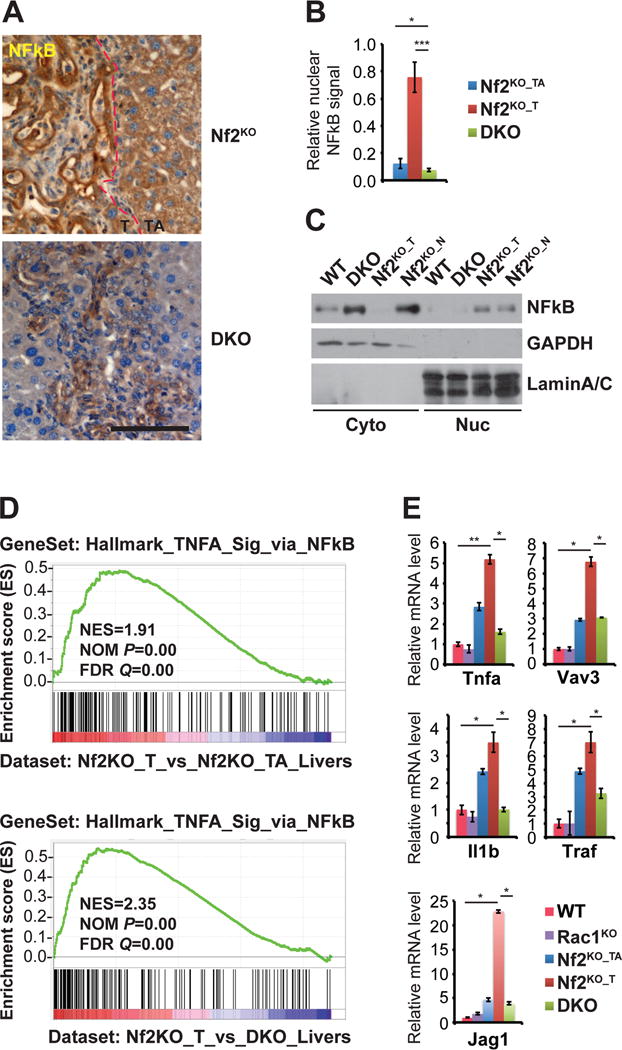
(A)Representative images of NFkB IHC of livers from 3-month-old Nf2KO and DKO mice. Scale bars = 100 μm.
(B) Quantification of the relative NFkB staining intensity within the nucleus from (A). Nf2KO tissue quantified separately as Nf2KO_T and Nf2KO_TA. Data are represented as mean +/− SD. P<0.0005, *P<0.05.
(C) Western blot analysis of NFkB expression in cytoplasmic and nuclear fractions of isolated liver cells from 3-month-old WT, DKO, and Nf2KO mice. The tumor and non-tumor tissues from the Nf2KO liver were processed separately as Nf2KO_T and Nf2KO_N. GAPDH and Lamin A/C were used as markers for cytoplasmic and nuclear fractions.
(D) GSEA by comparing the Hallmark NF-kB gene signature geneset to genes specifically upregulated in Nf2KO_T livers vs DKO livers.
(E) Quantitative real-time PCR analysis of NF-kB signature genes in 3-month-old WT, Rac1KO, Nf2KO_TA, Nf2KO_T, and DKO livers tissues (n=3 for all groups). Data are represented as mean +/− SEM. *P< 0.05; **P< 0.005.
Taken together, our data demonstrate that inactivation of Nf2 induces NFkB signaling and senescent phenotypes in a Rac1-dependent manner.
DNA damage/senescence signature is present in NF2 associated human schwannomas and meningiomas
Similar to the Nf2KO mouse livers, human schwannomas and meningiomas carrying NF2 mutations are generally benign and slow growing. We postulated that a p53-mediated DNA damage/senescence program might also constrict the growth of human NF2 tumors. To test this hypothesis, we conducted GSEA with our HSGS on three published human schwannomas and meningiomas microarray datasets (Clark et al., 2013; Tabernero et al., 2009; Torres-Martin et al., 2013). Despite the difference in cell types, we observed significant enrichment of the Hepatocyte_Senescence_Up gene set among genes upregulated in NF2-associated schwannomas compared to normal nerves or in NF2-associated meningiomas (NF2) compared to meningiomas that do not contain NF2 mutations (nonNF2) (Figure 6A). In contrast, we detected no significant association between our HSGS and genes differentially expressed in NF2 mutant mesotheliomas (Bott et al., 2011) (Figure 6A), a highly malignant form of cancer frequently carrying p16 mutations (Sekido, 2013). In further support of p53-mediated cellular senescence underlying the benign nature of NF2-associated tumors, we noted that human schwannoma samples were distinguishable from normal nerves or human Schwann cells (HSCs) by the expression of 20 validated senescence/p53 associated genes (Figure S7). Consistent with findings from the gene expression analysis, IHC staining of five human NF2 schwannomas and one meningioma revealed that a significant percentage of tumor cells are positive for pH2AX (Figure 6B). Likewise, wide spread expression of pH2AX was detected in schwannomas developed in the uterus of P0-cre;Nf2flox/flox mice, but not in WT uterus (Figure 6B). These results imply that NF2 inactivation may also cause DNA damage/senescence in benign NF2 tumors.
Figure 6. DNA damage/senescence signature is present in benign NF2-associated schwannomas and meningiomas, but not in NF2 mutant malignant mesotheliomas.
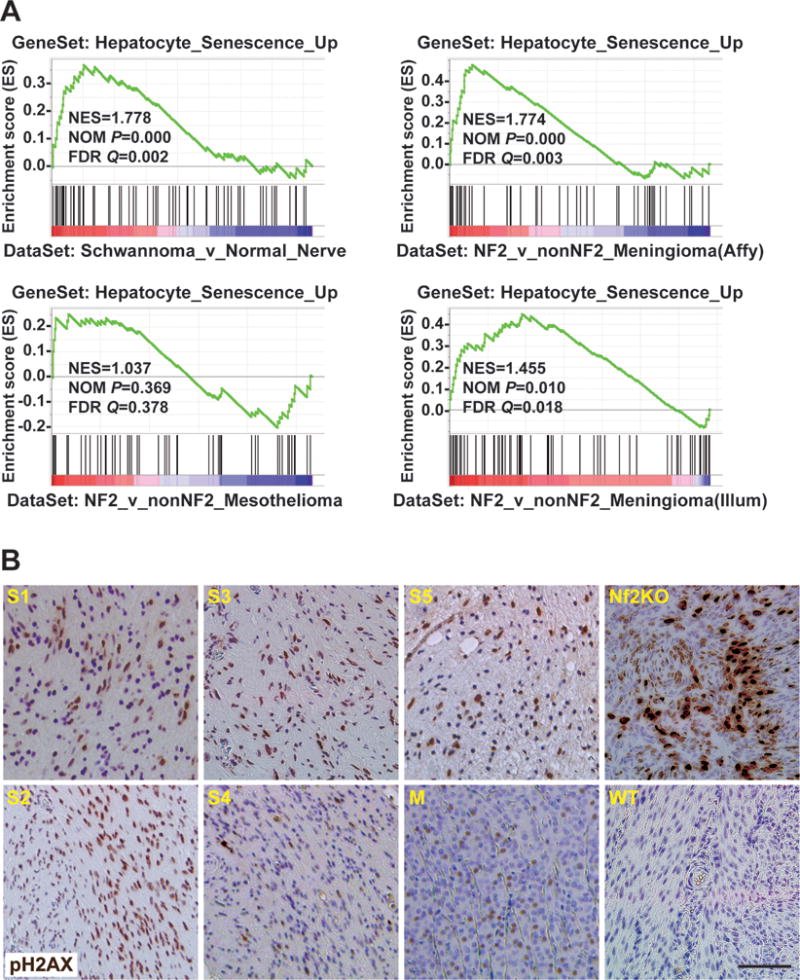
(A) GSEA by comparing a high-confidence senescence associated gene set with published datasets of human schwannomas (GSE39645) (Torres-Martin et al., 2013), meginingiomas (GSE43290 and GSE58037) (Clark et al., 2013; Tabernero et al., 2009), or mesotheliomas (GSE29354) (Bott et al., 2011) as indicated.
(B) Representative images of pH2AX IHC of five schwannomas (S1–S5) and one meningiomas (M) from NF2 patients, and of uteri from 15-month-old P0-Cre;Nf2flox/flox (Nf2KO) and WT mice.
See also Figure S6, and Table S1.
Production of ROS in the Nf2-deleted mouse liver is dependent on Rac1
Previous reports have shown that Rac1 is required for the production of ROS both in vitro and in vivo (Daugaard et al., 2013; Myant et al., 2013; Sundaresan et al., 1996). We compared the levels of ROS in Nf2KO and DKO livers by staining liver sections with a ROS-responsive dye, Dihydroethidium (DHE), which turns fluorescent red and intercalates with DNA upon oxidation by ROS (Carter et al., 1994). Nf2KO livers, particularly regions containing hyperplastic LPCs, exhibited significantly higher nuclear DHE staining when compared to DKO livers (Figures 7A and 7B), suggesting that ROS overproduction in Nf2KO livers is Rac1-induced.
Figure 7. Ablation of Rac1 suppresses ROS production induced by Nf2 loss and ROS-inducer APAP treatment activates p53 and DNA damage and reverses hyper-proliferation in Nf2:Rac1 double knockout livers.
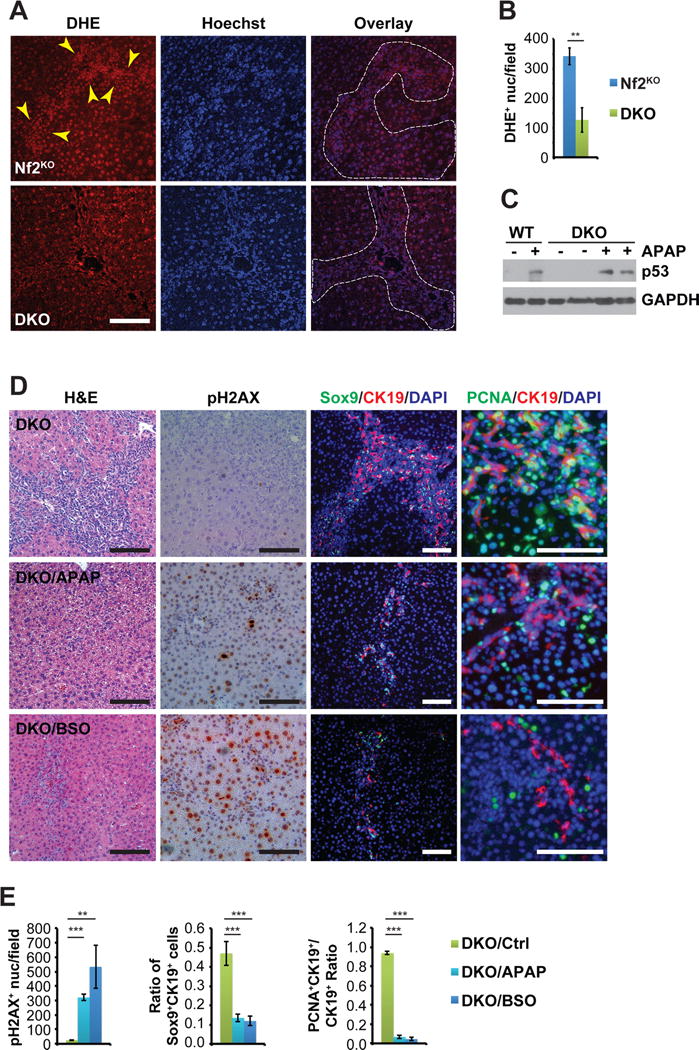
(A) Representative images of DHE and Hoechst double staining of Nf2KO_TA regions and DKO livers from 6-month-old mice. Dotted lines denote the LPC expansion areas in both Nf2KO_TA and DKO livers. Arrowheads indicate the stronger nuclear DHE staining within the LPC expansion region compared to surrounding areas in Nf2KO_TA. Scale bars = 100 μm.
(B) Quantification of the average number of DHE+ nuclei per microscopic field in Nf2KO and DKO livers (Nf2KO n=5, DKO n=6) as visualized in (A). Data are represented as mean +/− SEM. **P<0.005.
(C) Western blot analysis of p53 expression in WT and DKO liver tissues from age-matched untreated and APAP-treated mice. GAPDH was used as loading control.
(D) Representative images of H&E staining, pH2AX IHC, and Sox9 (green)/CK19 (red) and PCNA (green)/CK19 (red) double IF of livers from 3-month-old DKO mice fed with regular diet/drinking water (Ctrl), diet mixed with APAP for 1.5 months (APAP), or drinking water containing BSO for 1 month (BSO). Nuclei are counterstained with DAPI (blue). Scale bars = 100 μm.
(E) Quantification of the average number of pH2AX+ nuclei per microscopic field, the average percentage of Sox9+CK19+ cells, and the average percentage of CK19+ cells that are also PCNA+ as visualized in (D) in liver tissues from 3-month-old untreated (n=7), APAP-treated (n=9), or BSO-treated (n=6) DKO mice. Data are represented as mean +/− SD. ***P<0.0005, **P<0.005.
See also Figure S7.
Modulation of ROS levels affects LPC expansion in Nf2 and Nf2:Rac1 knockout mouse livers
Acetaminophen (N-acetyl-p-aminophenol, APAP), when used above the clinically recommended dose, has been shown to raise ROS levels and cause oxidative stress in the liver by draining the ROS scavenger glutathione (GSH) in both human and animal models (Jaeschke et al., 2011; McGill and Jaeschke, 2013). To test whether reduced ROS levels are responsible for blocking DDR and p53 activation, and for exacerbating LPC hyperplasia and liver enlargement in DKO mice, we sought to raise the ROS levels in the livers of WT and DKO mice by supplementing their diet with subtoxic doses of APAP. As expected, APAP treatment induced the accumulation of p53 and DNA damage marker pH2AX in both WT and DKO livers (Figures 7C–7E). Correspondingly, DKO livers exhibited dramatic decrease in the overall numbers and proliferation rates of CK19+Sox9+ LPCs (Figures 7D–E) and did not develop any frank tumors even after three months of APAP treatment. To corroborate this finding, we treated DKO mice with another pro-oxidant, buthionine sulfoximine (BSO) that acts by blocking GSH synthesis. Similar to APAP, BSO treatment induced massive accumulation of pH2AX and rapidly blocked the proliferation of CK19+Sox9+ LPCs, resulting in gradual decrease in the percentage of CK19+Sox9+ LPCs over time (Figures 7D–E, and Figure S6A). These data suggest that Rac1 limits the proliferative potentials of Nf2 deficient LPCs at least in part through ROS-mediated activation p53 and DDR.
To further confirm that elevated ROS levels limit the proliferation of LPCs in Nf2KO livers, we treated Nf2KO mice from birth with ROS inhibitor N-acetylcysteine (NAC) for 1.5 months. As shown in Figures S6B–C, compared to age-matched untreated Nf2KO livers, NAC-treated Nf2KO livers exhibited increased hyperplasia and corresponding increase in the percentage of PCNA+CK19+ cells within both the tumor or non-tumor regions. Interestingly, NAC treatment also induces increased buildup in SMA+ fibroblasts in Nf2KO livers (Figures S6B–C), suggesting that the effects of ROS depletion are not limited to the liver epithelial cells.
The hyperproliferative capacity of Nf2:Rac1 double knockout liver cells is dependent on Yap
Given the high Yap activity in DKO livers as evidenced by their robust expression of nuclear Yap and the strong association of DKO_Liver_Up genes with genes induced in Yap-overexpressing (Yap_OE) livers (Yimlamai et al., 2014) (Figures S1H and S2F), we generated Alb-cre;Nf2flox/flox;Rac1flox/flox;Yapflox/+ (TKO) mice to evaluate the contribution of Yap to the hyperproliferative phenotype of DKO livers. Histopathological and IHC analyses showed that with reduction in nuclear Yap, the extent of CK19+Sox9+ LPC hyperplasia and Cyclin D1 staining (both in terms of cell numbers and intensity) significantly decreased in TKO livers compared to DKO livers (Figures 8A and 8B). Correspondingly, TKO livers significantly diminished in size and expressed reduced levels of cell cycle genes compared to DKO livers (Figures 8C and 8D). In contrast, knockout of Yap did not significantly affect the expression of senescence-associated genes (Figure S5C). These findings indicate that while Rac1 is the primary driver of inflammation in Nf2 deficient hepatic cells, they depend on Yap to proliferate regardless of the Rac1 status.
Figure 8. Co-deletion of Yap represses biliary epithelial cell expansion and hepatomegaly induced by Nf2:Rac1 double knockout.
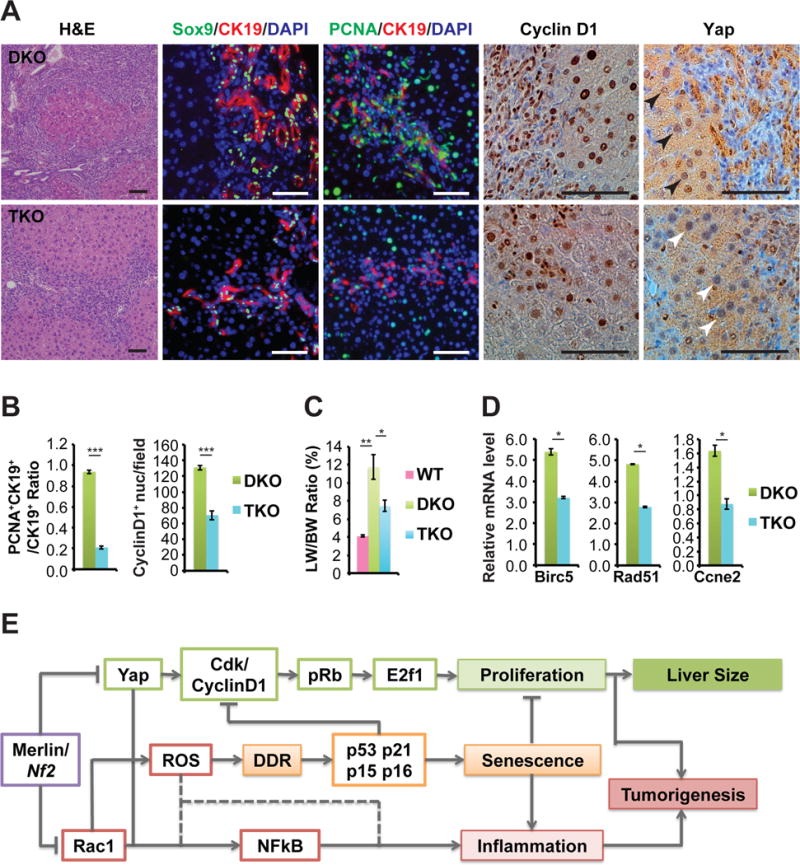
(A) Representative images of H&E, Sox9 (green) with CK19 (red), PCNA (green) with CK19 (red), CyclinD1, and Yap IF and IHC staining of DKO and TKO livers from 6-month-old mice. Scale bars = 100 μm.
(B) Ratio of CK19+ cells that are also PCNA+ nuclei from DKO and TKO mice at 6 months of age. Quantification of CyclinD1 positive nuclei in DKO and TKO livers from 6-month-old mice. (DKO n=9, TKO n=13). Data are represented as mean +/− SD. ***P<0.0005; NS: not significant.
(C) LW/BW ratio of 6-month-old WT, DKO, and TKO mice (WT n=5, DKO n=13, TKO n=2). Data are represented as mean +/− SEM.*P<0.05, **P<0.005.
(D) Quantitative real-time PCR analysis of indicated cell cycle genes in 6-month-old DKO and TKO livers (DKO n=4, TKO n=3). Data are represented as mean +/− SEM. *P<0.05.
(E) A working model of regulation of liver size and tumorigenesis by a signaling network controlled with Rac1 and Yap functioning downstream of Merlin/Nf2 as inflammation and proliferation switches, respectively.
DISCUSSION
Using genetically engineered mouse models, we have demonstrated that Rac1 is necessary for tumorigenesis, but unexpectedly also limits LPC hyperplasia, liver enlargement and tumor growth in Nf2KO livers. Mechanistically, we show that Rac1 promotes the accumulation of ROS, which triggers and p53-mediated DDR, dampening the CyclinD-Rb-E2F1 pathway while inducing senescence in Nf2 deficient tumor cells (Figure 8E). On the other hand, Rac1 drives NFkB activation and the expression of inflammatory cytokines that promote tumor growth (Figure 8E). These findings thus point to a complex role of Rac1 during Nf2 tumorigenesis, in which Rac1 can be growth suppressive or pro-tumorigenic depending on the context or disease stage. We propose that during early stage of Nf2 tumorigenesis, Rac1-ROS-p53-mediated cell cycle checkpoints and senescence serve as gatekeepers limiting the rate of tumor growth. With time, persistent DNA damage and inflammation associated with high Rac1 and ROS activities cause bypass of the p53 checkpoint in Nf2KO tumors, resulting in eventual disease progression. This model could also explain why Rac1 signaling has been found largely pro-proliferative in previous studies using immortalized cell lines, as these cells have had to overcome replicative senescence by inactivating p16 or p53 in order to indefinitely propagate in vitro.
Interestingly, we find that a senescence/p53 gene signature enriched in Nf2KO murine livers is also present in NF2-associated benign schwannomas and meningiomas, but not in malignant mesotheliomas that carry NF2 mutations. Consistent with our finding, several recent IHC studies of large cohort of human schwannomas and meningiomas that have detected elevated p53 expression in the majority of NF2 tumors (Chen et al., 2014; Pavelin et al., 2014). Moreover, p53 mutations and loss of p16 have both been linked to progression in human meningiomas (Chang et al., 2009; Kim et al., 2014). While it is tempting to speculate that Rac1-ROS-p53-driven checkpoints and senescence programs may also underlie the benign nature of NF2, it needs to be confirmed using disease relevant mouse models.
Despite the slow growing nature of early BDH developed in Nf2KO livers, progression to CC as well as HCC does eventually occur (Benhamouche et al., 2010; Zhang et al., 2010). In contrast, DKO livers remained tumor free by one year of age in spite of its hyperproliferative state. Unfortunately, we were not able to follow DKO mice beyond one year due to morbidity associated with their excess liver overgrowth, and therefore could not determine whether DKO mice would eventually develop tumors if allowed to survive longer. Notably, triple knockout of Rb family members or overexpression of E2F1 in the mouse liver has been shown to induce HCC development (Conner et al., 2000; Viatour et al., 2011), suggesting that sustained activation of the cell cycle machinery is sufficient in inducing liver tumorigenesis in Rac1WT background. It would be interesting to see whether Rac1 is required for tumorigenesis in these models.
Previous studies from us and others have identified Rac1 as a mediator of signaling between Merlin and the MAPK and AKT pathways (Morrison et al., 2007; Okada et al., 2005; Yi et al., 2011). Indeed, ablation of Rac1 reduces the activation of these two pathways in Nf2 deficient livers. However, despite the downregulation of these pathways, Nf2:Rac1 DKO livers grew at a significantly faster rate compared to Nf2KO livers. Moreover, removal of one allele of Yap effectively inhibits both proliferation and tumorigenesis associated with Nf2 deficiency without lowering the activities of these pathways. These results underscore the need to reexamine the contribution of these pathways to NF2 tumorigenesis using relevant in vivo models.
Rac1 has been demonstrated to induce both ROS and NFkB signaling in Apc mutant intestine stem cells (Myant et al., 2013). We observed similar Rac1-dependent co-activation of NFkB signaling with ROS in Nf2 knockout livers. However, contrary to their finding that ablation of Rac1 or inhibition of ROS blocks the proliferation of Apc mutant intestine stem cells (Myant et al., 2013), we show that Rac1 inactivation enhances the proliferation of Nf2 deficient LPCs by reducing ROS-mediated activation of p53 checkpoints. These discrepancies could be due to differences in mutations, tissue/cell types or other factors. Our preliminary analysis indicates that Rac1 and Yap converge to activate NFkB in Nf2 deficient livers, implying a potential role for NFkB signaling during NF2 tumorigenesis, particularly involving inflammation. Given the extensive crosstalk between ROS and the NFkB pathway (Morgan and Liu, 2011), it will be also interesting to dissect the signaling hierarchy between Rac1, ROS, NFkB signaling, p53 and DDR.
We have found that reducing Yap levels by just 50% is sufficient to block the activation of the CyclinD1-pRb-E2f1 pathway and cell cycle progression in both Nf2 single and Nf2:Rac1 double knockout livers. These results suggest that Nf2 deficient cells are addicted to Yap for continued proliferation even when p53 and p16 are inactivated. Indeed, YAP has also been shown to be required for the proliferation of NF2 deficient malignant mesothelioma cells (Mizuno et al., 2012; Yokoyama et al., 2008). In addition to driving proliferation, Yap is also required for the activation of NFkB and thus may also contribute to inflammation in NF2 deficient tumors. These findings underscore the promise of exploiting this exquisite sensitivity to YAP activity levels by NF2 deficient cells to treat NF2 and other cancers carrying NF2 mutations.
EXPERIMENTAL PROCEDURES
Mouse Strains and Histopathological Analysis
Genetically engineered mouse strains Nf2tm2Gth (Nf2flox/flox), Rac1tm1Tyb (Rac1flox/flox), Yap1tm1.1Dupa (Yapflox/flox), and Tg(Alb-cre)21Mgn (Alb-Cre) were interbred to generate all liver cohorts (Postic et al., 1999; Walmsley et al., 2003; Zhang et al., 2010). All liver animal experiments were conducted according to protocol #11-046 and #14-051 approved by the Institutional Animal Care and Use Committee (IACUC) at Georgetown University. P0-Cre;Nf2flox/flox mice were generated under protocol #2014-010 approved by IACUC at UCLA as previously described (Giovannini et al., 2000). All histopathological analysis was performed blind under the guidance of an experienced pathologist.
APAP Treatment
Beginning at 1.5 months of age, mice were administered 4-acetamidophenol (N-acetyl-p-aminophenol, APAP) (Alfa Aesar, Ward Hill, MA) via powdered food at 7.5g/kg until euthanasia at 3 months or 6 months of age.
BSO Treatment
Mice were administered L-buthionine-SR sulfoximine (BSO) (Abcam, Cambridge, MA) via drinking water at 20 mM for 1 week or 1 month until euthanasia at 3 months of age.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
Standard IHC and IF protocols were conducted for all experiments. Antigen retrieval conditions and a list of primary antibodies can be found in Supplementary Experimental Methods, Antibodies used in immunohistochemistry and immunofluorescence analysis, along with additional details regarding staining protocols. All histopathological analysis was performed blind under the supervision of a trained pathologist.
Image Quantification
Representative images of the stained slides with anonymous filenames were analyzed in ImageJ for the percentage area covered by stain or by manually counting positively stained cells. Images were initially color thresholded before thresholding was applied to identify areas for quantification. Particles of all circularity and small pixel size exclusion were applied in all measurements.
Western blot
Proteins were extracted from mouse livers using Urea buffer and western blot analysis was performed as previously described (Yi et al., 2013). Primary antibody dilutions are as listed in Supplementary Experimental Methods, Antibodies used in Western blot analysis.
qRT-PCR
Reverse transcription was performed with iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Using iTaq Universal SYBR Green Supermix (Bio-Rad), cDNA products were amplified and quantified. Reactions were conducted in triplicates. Relative gene expression was calculated as a unit value of 2−ΔCt=2−[Ct(HPRT)-Ct(Gene of Interest))], with Ct being threshold cycle value defined as the fractional cycle number at which the target fluorescent signal passes a fixed threshold above baseline. Data is represented by mean of all replicates within the genotypes. P-values displayed in qRT-PCR data represent significance between WT and Nf2KO_T or Nf2KO_T and DKO, unless otherwise indicated. Primer sequences of the genes analyzed are listed in Supplementary Experimental Methods, Primer sequences of genes analyzed by qRT-PCR.
Microarray/Genomic Analysis
Total mRNA were amplified, labeled, and hybridized to Illumina MouseRef-8 v2.0 Expression BeadChips at the UCLA Neuroscience Genomics Core, Los Angeles, California. Raw intensity data (deposited in GEO under GSE70742) was background corrected, normalized, and differential expression was calculated in R using the limma package from Bioconductor (see Data S1). A cutoff of p < 0.05 and FC < 2 were used to determine differentially expressed genes. Previously published datasets (GSE19004, GSE47739, GSE17546, GSE3964, GSE43290, GSE58037, GSE29354) were downloaded from GEO as raw intensity data and reprocessed in R using the oligo (for Affymetrix data) or limma (for Illumina data) package. Heatmaps were generated using the pheatmap package and, where applicable, Ward hierarchical clustering was used on Euclidean distance matrices. Pathway profiling was conducted using Pathway Studio. The GSEA Java-based software package from The Broad Institute was used to determine enrichment scores, significance of enrichment, and to generate enrichment plots. Mutation and copy number variations of TCGA cholangiocarcinomas were compiled using cBioPortal. The high confidence hepatocyte senescence gene signature (HSGS) listed in Table S1 was compiled by intersecting genes differentially expressed in hepatocytes that had undergone H2O2 or replication induced senescence from two independent studies (Aravinthan et al., 2014; Yildiz et al., 2013).
Statistical Analysis
Minimum three separate mice or samples were used for all the experiments unless otherwise indicated. Student’s t-test was used to determine significance unless otherwise indicated. Significance is defined as a p value of 0.05 or less. Error bars on all graphs are standard error of the mean or standard deviation as indicated.
Additional methods are in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Drs. DJ Pan and VL Tybulewicz for providing us with the mouse strains used in study, Drs. A Wellstein and R Yarden for critical commenting on our manuscript. We are thankful of Dr. B Kallakury (board certified pathologist) for his supervision of histopathological examinations, Dr. DL Berry, V Canales, E Permaul, and S Sen of Histopathology & Tissue Shared Resource (HTSR) for IHC/IF service, Dr. P Johnson of Microscopy & Imaging Shared Resource (MISR) for assistance in microscope imaging. We thank the staff of the Division of Comparative Medicine (DCM) at Georgetown University for advice and support in our animal studies.
Funding: C Yi is receiving funding from the NIH, V foundation, and Advocure NF2 foundation. Y Shi and S Bollam received summer fellowships from Georgetown Undergraduate Research Opportunities Program (GUROP). The Lombardi Cancer Center Shared Resource is supported by a Cancer Center Support Grant, CA051008.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, C.Y.; Methodology, Y.S., S.R.B., and C.Y; Software, G.T.G. and C.Y.; Validation, Y.S. and S.R.B.; Investigation, Y.S., S.R.B., S.M.W., S.Z.L., M.W., H.C., C.N., J.V., M.G.; Original Draft, Y.S., S.R.B., and C.Y.; Review & Editing, S.W., G.T.G., J.T.; Supervision and Project Administration, C.Y.; Funding Acquisition, C.Y.
References
- Ammoun S, Flaiz C, Ristic N, Schuldt J, Hanemann CO. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer research. 2008;68:5236–5245. doi: 10.1158/0008-5472.CAN-07-5849. [DOI] [PubMed] [Google Scholar]
- Aravinthan A, Shannon N, Heaney J, Hoare M, Marshall A, Alexander GJ. The senescent hepatocyte gene signature in chronic liver disease. Experimental gerontology. 2014;60:37–45. doi: 10.1016/j.exger.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, Giovannini M, McClatchey AI. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 2010;24:1718–1730. doi: 10.1101/gad.1938710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopp A, Wartlick F, Henninger C, Kaina B, Fritz G. Rac1 modulates acute and subacute genotoxin-induced hepatic stress responses, fibrosis and liver aging. Cell death & disease. 2013;4:e558. doi: 10.1038/cddis.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009;66:370–374. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, Creaney J, Lake RA, Zakowski MF, Reva B, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WO, Narayanan PK, Robinson JP. Intracellular hydrogen peroxide and superoxide anion detection in endothelial cells. Journal of leukocyte biology. 1994;55:253–258. doi: 10.1002/jlb.55.2.253. [DOI] [PubMed] [Google Scholar]
- Chang Z, Guo CL, Ahronowitz I, Stemmer-Rachamimov AO, MacCollin M, Nunes FP. A role for the p53 pathway in the pathology of meningiomas with NF2 loss. Journal of neuro-oncology. 2009;91:265–270. doi: 10.1007/s11060-008-9721-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang ZY, Wu H. P14ARF deficiency and its correlation with overexpression of P53/MDM2 in sporadic vestibular schwannomas. European archives of oto-rhino-laryngology : official journal of the European Federation of Oto-Rhino-Laryngological Societies. 2014 doi: 10.1007/s00405-014-3135-y. [DOI] [PubMed] [Google Scholar]
- Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, Avsar T, Li J, Murray PB, Henegariu O, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner EA, Lemmer ER, Omori M, Wirth PJ, Factor VM, Thorgeirsson SS. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene. 2000;19:5054–5062. doi: 10.1038/sj.onc.1203885. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T, Safferling K, Rausch S, Grabe N, Boehm H, Spatz JP. A molecular mechanotransduction pathway regulates collective migration of epithelial cells. Nat Cell Biol. 2015;17:276–287. doi: 10.1038/ncb3115. [DOI] [PubMed] [Google Scholar]
- Daugaard M, Nitsch R, Razaghi B, McDonald L, Jarrar A, Torrino S, Castillo-Lluva S, Rotblat B, Li L, Malliri A, et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nature communications. 2013;4:2180. doi: 10.1038/ncomms3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini M, Robanus-Maandag E, van der Valk M, Niwa-Kawakita M, Abramowski V, Goutebroze L, Woodruff JM, Berns A, Thomas G. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14:1617–1630. [PMC free article] [PubMed] [Google Scholar]
- Guerrant W, Kota S, Troutman S, Mandati V, Fallahi M, Stemmer-Rachamimov A, Kissil JL. YAP Mediates Tumorigenesis in Neurofibromatosis Type 2 by Promoting Cell Survival and Proliferation through a COX-2-EGFR Signaling Axis. Cancer Res. 2016;76:3507–3519. doi: 10.1158/0008-5472.CAN-15-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Moon C, Niehaus K, Zheng Y, Ratner N. Rac1 controls Schwann cell myelination through cAMP and NF2/merlin. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:17251–17261. doi: 10.1523/JNEUROSCI.2461-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27–36. doi: 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton DA, Ristic N, Hanemann CO. Activation of ERK, AKT and JNK signalling pathways in human schwannomas in situ. Histopathology. 2009;55:744–749. doi: 10.1111/j.1365-2559.2009.03440.x. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Williams CD, Ramachandran A. Current issues with acetaminophen hepatotoxicity–a clinically relevant model to test the efficacy of natural products. Life Sci. 2011;88:737–745. doi: 10.1016/j.lfs.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, Gusella JF, Ramesh V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29:4250–4261. doi: 10.1128/MCB.01581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaempchen K, Mielke K, Utermark T, Langmesser S, Hanemann CO. Upregulation of the Rac1/JNK signaling pathway in primary human schwannoma cells. Human molecular genetics. 2003;12:1211–1221. doi: 10.1093/hmg/ddg146. [DOI] [PubMed] [Google Scholar]
- Kalamarides M, Niwa-Kawakita M, Leblois H, Abramowski V, Perricaudet M, Janin A, Thomas G, Gutmann DH, Giovannini M. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002;16:1060–1065. doi: 10.1101/gad.226302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Kim KH, Lee EH, Lee YM, Lee SH, Kim HD, Kim YZ. Results of immunohistochemical staining for cell cycle regulators predict the recurrence of atypical meningiomas. J Neurosurg. 2014;121:1189–1200. doi: 10.3171/2014.7.JNS132661. [DOI] [PubMed] [Google Scholar]
- Kissil JL, Johnson KC, Eckman MS, Jacks T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. The Journal of biological chemistry. 2002;277:10394–10399. doi: 10.1074/jbc.M200083200. [DOI] [PubMed] [Google Scholar]
- Lavado A, He Y, Pare J, Neale G, Olson EN, Giovannini M, Cao X. Tumor suppressor Nf2 limits expansion of the neural progenitor pool by inhibiting Yap/Taz transcriptional coactivators. Development. 2013;140:3323–3334. doi: 10.1242/dev.096537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell death and differentiation. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:8248–8253. doi: 10.1073/pnas.0912203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, Snuderl M, Ladanyi M, Hanemann CO, Zhou P, et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell. 2014;26:48–60. doi: 10.1016/j.ccr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140:477–490. doi: 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 2010;107:1437–1442. doi: 10.1073/pnas.0911427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchanda PK, Jones GN, Lee AA, Pringle DR, Zhang M, Yu L, La Perle KM, Kirschner LS. Rac1 is required for Prkar1a-mediated Nf2 suppression in Schwann cell tumors. Oncogene. 2013;32:3491–3499. doi: 10.1038/onc.2012.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, Jacks T. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev. 1998;12:1121–1133. doi: 10.1101/gad.12.8.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI, Saotome I, Ramesh V, Gusella JF, Jacks T. The Nf2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes Dev. 1997;11:1253–1265. doi: 10.1101/gad.11.10.1253. [DOI] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30:2174–2187. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y, Akatsuka S, Toyokuni S, Yokoi K, Osada H, et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31:5117–5122. doi: 10.1038/onc.2012.5. [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison H, Sperka T, Manent J, Giovannini M, Ponta H, Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007;67:520–527. doi: 10.1158/0008-5472.CAN-06-1608. [DOI] [PubMed] [Google Scholar]
- Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D, et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell stem cell. 2013;12:761–773. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Sugimachi K, Goto H, Wang J, Morikawa T, Miyachi Y, Takano Y, Hikasa H, Itoh T, Suzuki SO, et al. Dysregulated YAP1/TAZ and TGF-beta signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc Natl Acad Sci U S A. 2016;113:E71–80. doi: 10.1073/pnas.1517188113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Lopez-Lago M, Giancotti FG. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J Cell Biol. 2005;171:361–371. doi: 10.1083/jcb.200503165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelin S, Becic K, Forempoher G, Tomic S, Capkun V, Drmic-Hofman I, Mrklic I, Lusic I, Pogorelic Z. The significance of immunohistochemical expression of merlin, Ki-67, and p53 in meningiomas. Applied immunohistochemistry & molecular morphology : AIMM / official publication of the Society for Applied Immunohistochemistry. 2014;22:46–49. doi: 10.1097/PAI.0b013e318289f490. [DOI] [PubMed] [Google Scholar]
- Petrilli AM, Fernandez-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2015 doi: 10.1038/onc.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- Rong R, Surace EI, Haipek CA, Gutmann DH, Ye K. Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene. 2004;23:8447–8454. doi: 10.1038/sj.onc.1207794. [DOI] [PubMed] [Google Scholar]
- Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP) Cell Signal. 2012;24:835–845. doi: 10.1016/j.cellsig.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Scoles DR, Nguyen VD, Qin Y, Sun CX, Morrison H, Gutmann DH, Pulst SM. Neurofibromatosis 2 (NF2) tumor suppressor schwannomin and its interacting protein HRS regulate STAT signaling. Hum Mol Genet. 2002;11:3179–3189. doi: 10.1093/hmg/11.25.3179. [DOI] [PubMed] [Google Scholar]
- Sekido Y. Molecular pathogenesis of malignant mesothelioma. Carcinogenesis. 2013;34:1413–1419. doi: 10.1093/carcin/bgt166. [DOI] [PubMed] [Google Scholar]
- Serinagaoglu Y, Pare J, Giovannini M, Cao X. Nf2-Yap signaling controls the expansion of DRG progenitors and glia during DRG development. Dev Biol. 2015;398:97–109. doi: 10.1016/j.ydbio.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, Peix J, Sole M, Tovar V, Alsinet C, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology. 2013;144:829–840. doi: 10.1053/j.gastro.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci U S A. 2010;107:1431–1436. doi: 10.1073/pnas.0911409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, Goldschmidt-Clermont PJ, Finkel T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. The Biochemical journal. 1996;318(Pt 2):379–382. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabernero MD, Maillo A, Gil-Bellosta CJ, Castrillo A, Sousa P, Merino M, Orfao A. Gene expression profiles of meningiomas are associated with tumor cytogenetics and patient outcome. Brain pathology. 2009;19:409–420. doi: 10.1111/j.1750-3639.2008.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Martin M, Lassaletta L, San-Roman-Montero J, De Campos JM, Isla A, Gavilan J, Melendez B, Pinto GR, Burbano RR, Castresana JS, et al. Microarray analysis of gene expression in vestibular schwannomas reveals SPP1/MET signaling pathway and androgen receptor deregulation. Int J Oncol. 2013;42:848–862. doi: 10.3892/ijo.2013.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, Pulaski K. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;75:826. doi: 10.1016/0092-8674(93)90501-g. [DOI] [PubMed] [Google Scholar]
- Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, Zmoos AF, Mazur PK, Schaffer BE, Ostermeier A, et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. The Journal of experimental medicine. 2011;208:1963–1976. doi: 10.1084/jem.20110198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley MJ, Ooi SK, Reynolds LF, Smith SH, Ruf S, Mathiot A, Vanes L, Williams DA, Cancro MP, Tybulewicz VL. Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science. 2003;302:459–462. doi: 10.1126/science.1089709. [DOI] [PubMed] [Google Scholar]
- Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. The Journal of biological chemistry. 2002;277:883–886. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC, Liu Q, Shimono A, Sudol M, Holmgren L. The 130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Science signaling. 2013;6:ra77. doi: 10.1126/scisignal.2004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila JL, Christian N, Persson NL, Shimono A, Speicher DW, Marmorstein R, et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell. 2011;19:527–540. doi: 10.1016/j.ccr.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz G, Arslan-Ergul A, Bagislar S, Konu O, Yuzugullu H, Gursoy-Yuzugullu O, Ozturk N, Ozen C, Ozdag H, Erdal E, et al. Genome-wide transcriptional reorganization associated with senescence-to-immortality switch during human hepatocellular carcinogenesis. PloS one. 2013;8:e64016. doi: 10.1371/journal.pone.0064016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154:1342–1355. doi: 10.1016/j.cell.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama T, Osada H, Murakami H, Tatematsu Y, Taniguchi T, Kondo Y, Yatabe Y, Hasegawa Y, Shimokata K, Horio Y, et al. YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation. Carcinogenesis. 2008;29:2139–2146. doi: 10.1093/carcin/bgn200. [DOI] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Developmental cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.