

Abstract
Kidney disease has been linked to dysregulated signaling via PKC in kidney cells such as podocytes. PKCα is a conventional isoform of PKC and a well-known binding partner of β-catenin, which promotes its degradation. β-Catenin is the main effector of the canonical Wnt pathway and is critical in cell adhesion. However, whether other PKC isoforms interact with β-catenin has not been studied systematically. Here we demonstrate that PKCϵ-deficient mice, which develop proteinuria and glomerulosclerosis, display lower β-catenin expression compared with PKC wild-type mice, consistent with an altered phenotype of podocytes in culture. Remarkably, β-catenin showed a reversed subcellular localization pattern: Although β-catenin exhibited a perinuclear pattern in undifferentiated wild-type cells, it predominantly localized to the nucleus in PKCϵ knockout cells. Phorbol 12-myristate 13-acetate stimulation of both cell types revealed that PKCϵ positively regulates β-catenin expression and stabilization in a glycogen synthase kinase 3β-independent manner. Further, β-catenin overexpression in PKCϵ-deficient podocytes could restore the wild-type phenotype, similar to rescue with a PKCϵ construct. This effect was mediated by up-regulation of P-cadherin and the β-catenin downstream target fascin1. Zebrafish studies indicated three PKCϵ-specific phosphorylation sites in β-catenin that are required for full β-catenin function. Co-immunoprecipitation and pulldown assays confirmed PKCϵ and β-catenin as binding partners and revealed that ablation of the three PKCϵ phosphorylation sites weakens their interaction. In summary, we identified a novel pathway for regulation of β-catenin levels and define PKCϵ as an important β-catenin interaction partner and signaling opponent of other PKC isoforms in podocytes.
Keywords: β-catenin, cadherin, glycogen synthase kinase 3 (GSK-3), kidney, PKC, Wnt pathway, cytoskeleton, podocytes
Introduction
The PKC family of serine and threonine kinases is subdivided into the three subfamilies of conventional, atypical, and novel PKCs (1), depending on the involvement of diacylglycerol (DAG)2 and calcium during activation. Conventional, atypical, and novel PKCs display different roles in signal transduction via phosphorylation of their target proteins. Regarding the kidney, several studies have shown that PKCs play a pivotal role in the development of diabetic nephropathy. For PKCα and PKCβ, two members of the conventional PKC subfamily, complete depletion seems to lead to a better outcome in streptozotocin-induced diabetic mice in terms of proteinuria (2) or renal hypertrophy and glomerular injury (3). This implies that these PKC isoforms induce or exacerbate kidney injury. In contrast, we have shown previously that knockdown of PKCϵ in mice leads to a renal phenotype with glomerulosclerosis, indicating that PKCϵ may protect from diabetic nephropathy (4, 5).
PKCϵ belongs to the subfamily of novel PKCs and is activated via DAG, different from conventional isoforms, which are activated by calcium and DAG. PKCϵ was shown previously to play an essential role in cell proliferation, migration, invasion, and survival, and lack of PKCϵ is linked to reduced cardioprotective effects (6) and neurotransmission (7). Moreover, PKCϵ has been well characterized as an important binding protein of actin and, thus, has a regulatory function for the cytoskeleton (8, 9). β-Catenin is the key component in the highly conserved canonical Wnt pathway, regulating cell-cell adhesion as well as serving as a transcription co-factor (10). Widely expressed, β-catenin is also found in podocytes and plays a pivotal role in cell adhesion and differentiation. Depending on phosphorylation, β-catenin exists in an active (non-phosphorylated) and an inactivated (phosphorylated) state. Early studies demonstrated that overexpression of β-catenin alone can cause or aggravate damage in podocytes (11), whereas, in apparent contradiction, a later study showed that β-catenin knock-out mice under diabetic conditions display more albuminuria than control mice in long-term studies (>15 weeks) (12). From their conflicting data, Kato et al. (12) went on to show that a balanced expression of active and inactive β-catenin might be what is important for cell function .
The presence of β-catenin is strongly regulated via glycogen synthase kinase 3β (GSK3β), which forms a degradation complex with adenomatous polyposis coli (APC), Axin, and CK-1 upon Wnt absence. The resulting N-terminal phosphorylation of β-catenin leads to ubiquitination and proteasome-mediated reduction of the cellular β-catenin level. In contrast, activation of Wnt, a group of secreted glycolipoproteins, leads to stabilization and accumulation of β-catenin in the cytoplasm and translocation into the nucleus, which then results in Wnt target gene expression (13).
Other so-called non-canonical Wnt signaling pathways, especially the Wnt/Ca2+ pathway, are involved in PKC signaling (14); many studies have demonstrated the alternative degradation of β-catenin by PKC isoforms such as PKCα (15, 16). PKCα has been shown to mediate GSK3β-independent β-catenin down-regulation by phosphorylation of the N-terminal serine residues Ser-33, Ser-37, and Ser-45 of β-catenin. This finding is consistent with the known common phosphorylation sites of GSK3β (17).
The aim of this study was to analyze the relationship between β-catenin and PKCϵ in podocytes. We used a murine PKCϵ knockout podocyte cell line to perform in vitro immunofluorescence staining and characterize β-catenin expression under phorbol 12-myristate 13-acetate (PMA) stimulation and adenoviral constructs to investigate the effects of overexpression of β-catenin in PKCϵ knockout podocytes. Furthermore, we created β-catenin mutants and validated these in vitro and in vivo to investigate which domain of β-catenin may contribute to the interaction/function with PKCϵ.
Results
PKCϵ knockout in mice leads to proteinuria and reduced expression of β-catenin
To evaluate podocytic expression of β-catenin in mice, we performed immunofluorescence co-staining of the podocyte marker synaptopodin and β-catenin on glomeruli of 12-week-old wild-type and PKCϵ−/− mice. β-Catenin expression was detected in the nucleus as well as in the cytoplasm in wild-type mice, whereas, in PKCϵ−/− glomeruli, we observed predominantly nuclear localization. Further, β-catenin expression was less intense in PKCϵ−/− than in wild-type mice (Fig. 1A). Semiquantitative analysis of β-catenin in fluorescent cells in the glomeruli of mice (≥10 glomeruli were included for each genotype) confirmed that β-catenin expression in WT mice was significantly higher than in PKCϵ−/− mice at 12 weeks of age (Fig. 1B; ****, p < 0.0001). Of note, Coomassie staining of urine revealed that glomerular barrier function was already impaired, displaying mild albuminuria onset at 12 weeks of age in PKCϵ-deficient mice (Fig. 1C).
Figure 1.

β-Catenin expression in glomeruli of wild-type and PKCϵ knockout mice. A, immunofluorescence staining of murine glomeruli at 12 weeks of age in PKCϵ+/+ and PKCϵ−/− mice. Co-staining with β-catenin (green), the podocyte marker synaptopodin (red), and DAPI (blue) shows expression and localization within glomeruli. B, semiquantitative analysis of the number of cells with β-catenin expression in PKCϵ+/+ and PKCϵ−/− mice (****, p < 0.0001). C, SDS-PAGE/Coomassie gel staining of urine from wild-type and PKCϵ knockout mice at 9 and 12 weeks (w). BSA at 1, 5, and 10 μg/ml served both as a control and standard. Data are mean ± S.D. of at least three different independent experiments.
PKCϵ influences the subcellular distribution of β-catenin and its activity
Because we detected a difference in podocytic β-catenin expression between glomeruli of wild-type and PKCϵ−/− mice, we wanted to investigate expression in vitro in wild-type and PKCϵ−/− podocytes. Interestingly, PKCϵ−/− podocytes displayed a very unique phenotype in tissue culture, as they were significantly smaller than wild-type podocytes, detached very easily, and never reached a confluent growth pattern. Western blot analysis using antibodies against active and total β-catenin showed similar results as those in vivo, displaying increased expression from day 0 to day 10 (Fig. 2A), with stronger up-regulation in wild-type cells than in knockout cells. The up-regulation supported findings in other cell lines (18, 19), which indicated that the presence of β-catenin is critical for cell differentiation. An immunofluorescent staining time course (0, 4, and 8 days after differentiation) revealed reverse localization of β-catenin in wild-type and PKCϵ−/− podocytes during differentiation (Fig. 2B). In wild-type cells, the localization of β-catenin expression changed from predominant expression in the perinuclear areas, exhibited on day 0, to expression in nuclei and cell junctions during differentiation (on days 4 and 8). In contrast, β-catenin in PKCϵ−/− podocytes translocated from the nuclei (day 0) to the perinuclear areas and cell junctions (days 4 and 8). We quantified the shift and intensity of the localization of β-catenin by expressing the ratio of the mean fluorescence of the nuclei in relation to the perinuclear areas. The quantification supported the observation that β-catenin expression changes significantly from perinuclear areas to nuclei in wild-type podocytes during differentiation (Fig. 2, B and C; ****, p < 0.0001), whereas, in PKCϵ−/− podocytes, β-catenin switches from nuclei to perinuclear areas (Fig. 2, B and C; ***, p < 0.0002). These data suggest that PKCϵ orchestrates the subcellular localization of β-catenin in podocytes, which significantly shifts during a differentiation time course (days 0 and 8; ****, p < 0.0001; *, p < 0.05). Interestingly, on day 4, no difference was observed between both genotypes, implying a PKCϵ-dependent checkpoint of β-catenin during podocyte differentiation.
Figure 2.
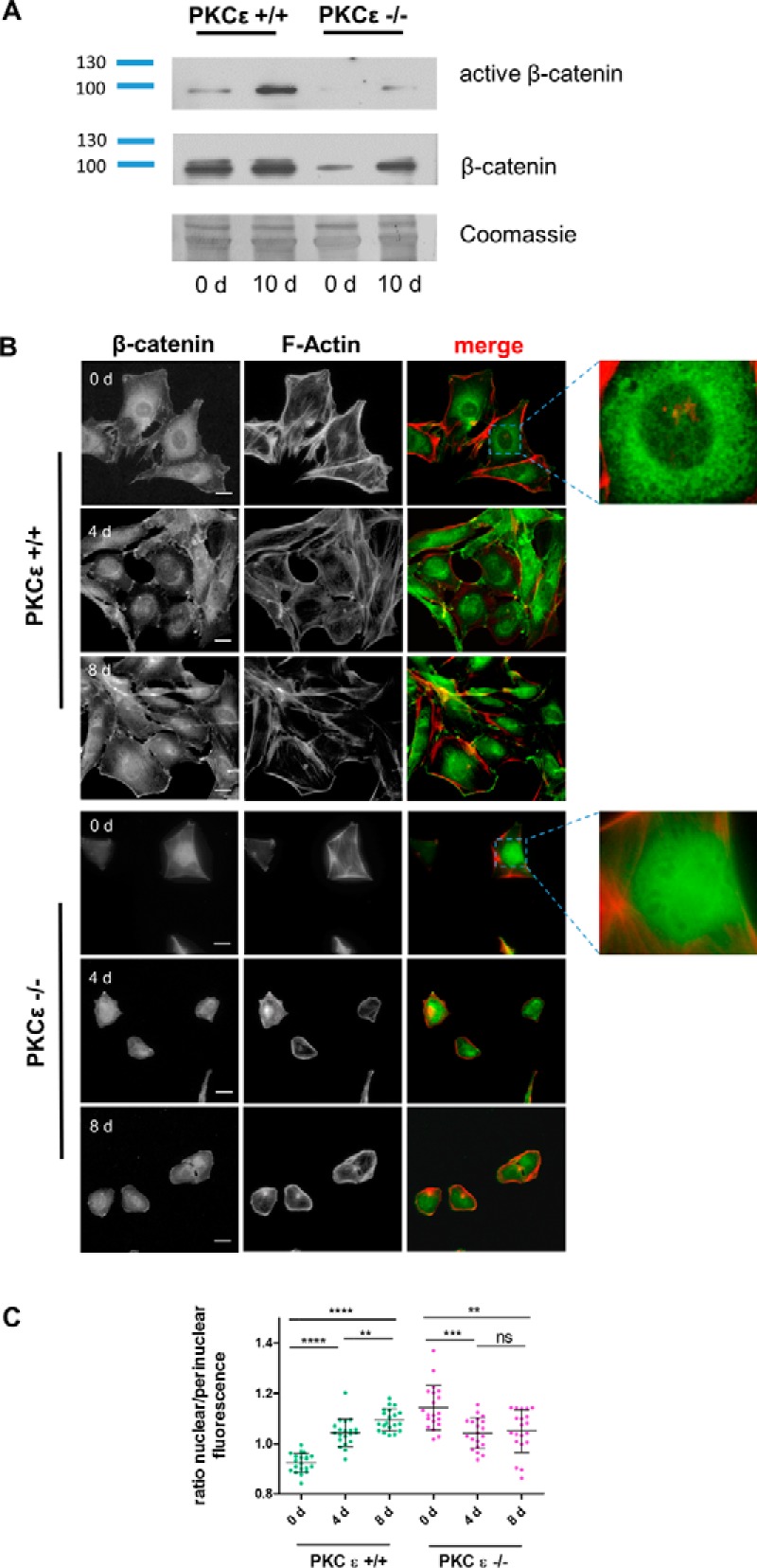
PKCϵ influences subcellular β-catenin expression and activity. A, Western blot analysis of active β-catenin expression in murine podocytes from day (d) 0 and day 10. B, immunofluorescent staining of PKCϵ+/+ and PKCϵ−/− podocytes at different time points of differentiation (days 0, 4, and 8) shows the subcellular localization of β-catenin (green). Phalloidin staining labels the actin cytoskeleton (red). Scale bars = 40 μm. C, the ratio of nuclear to perinuclear β-catenin was determined by measuring the mean fluorescence intensity with ImageJ. The graph shows the ratio of nuclear to perinuclear β-catenin fluorescence intensity, displaying the reversed shift of subcellular localization of β-catenin during the differentiation (*, p < 0.05; **, p < 0.05; ***, p < 0.0001; ns, not significant; Student's t test). Data are represented as mean ± S.D. of at least three different independent experiments.
PKCϵ stabilizes active β-catenin levels in a GSK3β-independent manner
To further explore the effects of PKCϵ on β-catenin activity, PMA (a reversible and highly potent PKC activator) was used to stimulate murine wild-type and PKCϵ knockout podocytes. Normalization of Western blot results indicated that, after differentiation for 8 days, PMA stimulation for 30 min and 1, 2, 4, 8, and 24 h results in an increase of active (non-phosphorylated) β-catenin in wild-type podocytes (Fig. 3A) up to 2 h, followed by continuous reduction over the 24-h time course. In contrast, PKCϵ−/− podocytes showed no temporary up-regulation of β-catenin; instead, we noted a gradual diminishment of active β-catenin from the start of the time course to its end, indicating accelerated degradation of active β-catenin in PKCϵ−/− podocytes. The impact of PMA stimulation on active β-catenin expression was significantly higher (*, p < 0.05; **, p < 0.01) in wild-type podocytes than in PKCϵ-deficient cells (Fig. 3B).
Figure 3.

PKCϵ stabilized β-catenin independent from GSK3β. A, differentiated PKCϵ+/+ and PKCϵ−/− podocytes were treated with PMA for 0.5, 1, 2, 4, 8, and 12 h and then harvested for Western blot analysis. Untreated cells (0 h) served as a control. B, active β-catenin expression was normalized to GAPDH. Two-way analysis of variance of active β-catenin expression of PKCϵ+/+ and PKCϵ−/− cells under PMA stimulation at 1, 2, 4, 8, and 24 h was performed. ns, not significant. *, p < 0.05; **, p < 0.05. C, PKCϵ+/+ and PKCϵ−/− podocytes were treated as in A and analyzed for GSK3β and p-GSK3 α/β expression. D, GSK3β protein level was normalized to GAPDH. Data represent mean ± S.E. of three different independent experiments in B and D.
To investigate whether the diminished β-catenin levels resulted from a rising activation of GSK3β, the main inhibitor of the canonical Wnt pathway, we analyzed GSK3 activity after PMA stimulation. GSK3β levels were detected at a constant expression level in both wild-type and PKCϵ knockout podocytes (Fig. 3, C and D). Because the phosphorylation level of GSK3 α/β at Ser-9/21 leads to decreased activity (20), we also looked at p-GSK3 after PMA stimulation. p-GSK3β increased in both groups in a similar fashion during the time course (Fig. 3C), indicating that the reduced β-catenin expression in the PKCϵ−/− podocytes is independent of GSK3β expression and activity.
β-Catenin overexpression rescues the impaired actin cytoskeleton of PKCϵ-deficient podocytes
As shown previously (21), PKCϵ-deficient murine podocytes in culture display a malfunction in the organization of the actin cytoskeleton. F-actin and focal adhesion marker paxillin staining revealed an overall smaller cell size, less stress fibers, and reduced size and number of focal adhesions in PKCϵ−/− podocytes (Fig. 4A). Adenoviral transduction with a PKCϵ wild-type construct in PKCϵ-deficient podocytes indicated complete rescue of the phenotype, as expected, and the cells displayed a rearranged cytoskeleton and higher levels of paxillin. Interestingly, adenoviral transduction with a wild-type β-catenin construct in PKCϵ knockout cells led to a similar recovery as the rescue with the PKCϵ-wildtype construct. Measurement of the average cell size performed with ImageJ demonstrated that PKCϵ knockout cells transfected with PKCϵ and β-catenin reached cell sizes similar to PKC wild-type cells and a similar distribution pattern of paxillin expression with elongated focal contacts (Fig. 4, A and B).
Figure 4.
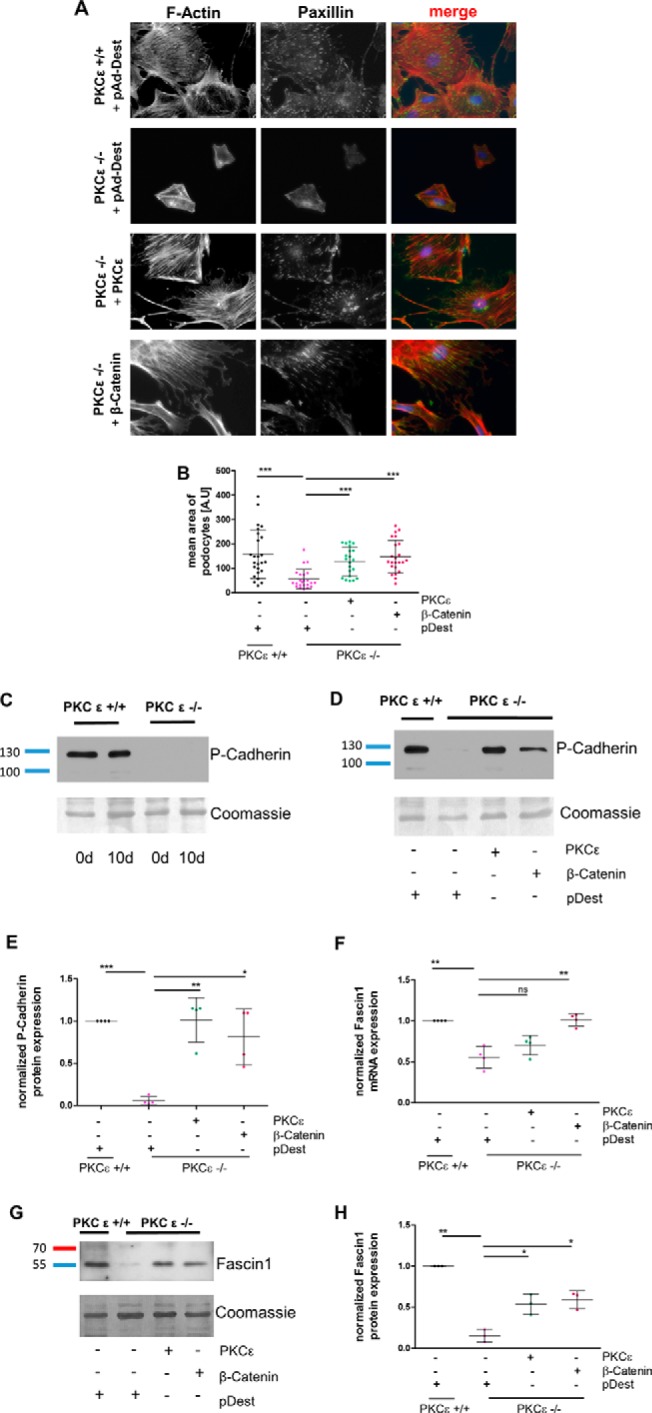
Overexpression of β-catenin rescues the phenotype of PKCϵ knockout podocytes. A, murine PKCϵ−/− podocytes were transduced with adenoviral vectors expressing either pAd-Dest, PKCϵ, or β-catenin. Immunofluorescence staining of the actin cytoskeleton with phalloidin and against paxillin was performed. B, the cell size of phalloidin-marked podocytes was assessed in ImageJ for determination of the mean area (A.U, area unit). C, Western blot analysis of P-cadherin expression in murine podocytes from day (d) 0 and day 10. D, PKCϵ−/− podocytes were transduced with adenoviral vectors expressing either pAd-Dest, PKCϵ, or β-catenin and then analyzed for P-cadherin expression with Western blot analysis. E, quantification of P-cadherin band intensity using total protein analysis with Coomassie staining of the membrane. F, quantitative RT-PCR of RNA extracted from cells of the same experiment as in A was used to assess fascin1 mRNA expression and normalized to HPRT. G, Western blot analysis of the same experiment as in A for fascin1 protein expression. H, quantification of band intensity by total protein analysis with Coomassie staining (*, p < 0.05; **, p < 0.05; ***, p < 0.0001; ns, not significant; Student's t test). Data are represented as mean ± S.D. of at least three different independent experiments.
Because β-catenin plays a major role in cell adhesion with its binding partner P-cadherin (22), and PKCϵ knockout podocytes in culture detach very easily, we examined the impact of PKCϵ deficiency on P-cadherin expression during cell differentiation. As depicted in Fig. 4C, P-cadherin expression was drastically reduced in knockout cells. As expected, we could restore normal P-cadherin expression by overexpressing a wild-type PKCϵ construct (Fig. 4, D and E). Interestingly, overexpressing a wild-type β-catenin construct in PKCϵ knockout podocytes also led to a significant increase in P-cadherin expression (*, p < 0.05), which was not detected in cells transduced with a pAd-Dest vector alone.
To explore how the actin cytoskeleton is restored by β-catenin, we considered known protein targets of β-catenin that are downstream in the Wnt pathway and that may interfere with the cytoskeleton. This led us to discover reduced mRNA expression levels of fascin1 in PKCϵ-deficient podocytes. The fascin1 level significantly increased when cells were transduced with either a PKCϵ or a β-catenin construct (Fig. 4F). Fascin1, an actin filament-bundling protein (23), is known to bind to β-catenin at the cellular edges and possesses a PKC binding site at serine 39 (24, 25). Western blot analysis of lysates from the adenovirus-transfected cells confirmed the quantitative RT-PCR results (Fig. 4, G and H). The knockout cells that were rescued by overexpression of PKCϵ or β-catenin exhibited significantly higher fascin1 protein expression than mock-(pAd-Dest)-transduced PKCϵ−/− podocytes (**, p < 0.05). These data suggest that β-catenin mediates the podocytic actin cytoskeleton via regulation of fascin1 expression.
PKCϵ-specific phosphorylation sites in β-catenin are indispensable for filtration barrier function
After the discovery that β-catenin and PKCϵ expression levels influence each other, we wanted to explore whether PKCϵ is a binding partner of β-catenin, as reported previously for PKCα (15, 16). To this end, we performed co-immunoprecipitation experiments in HEK cells transfected with GFP-tagged PKCϵ (or PKCα as a positive control) and FLAG-tagged β-catenin constructs. Indeed, after immunoprecipitation of FLAG-tagged β-catenin, we detected an interaction with PKCϵ as well as with PKCα (Fig. 5A).
Figure 5.
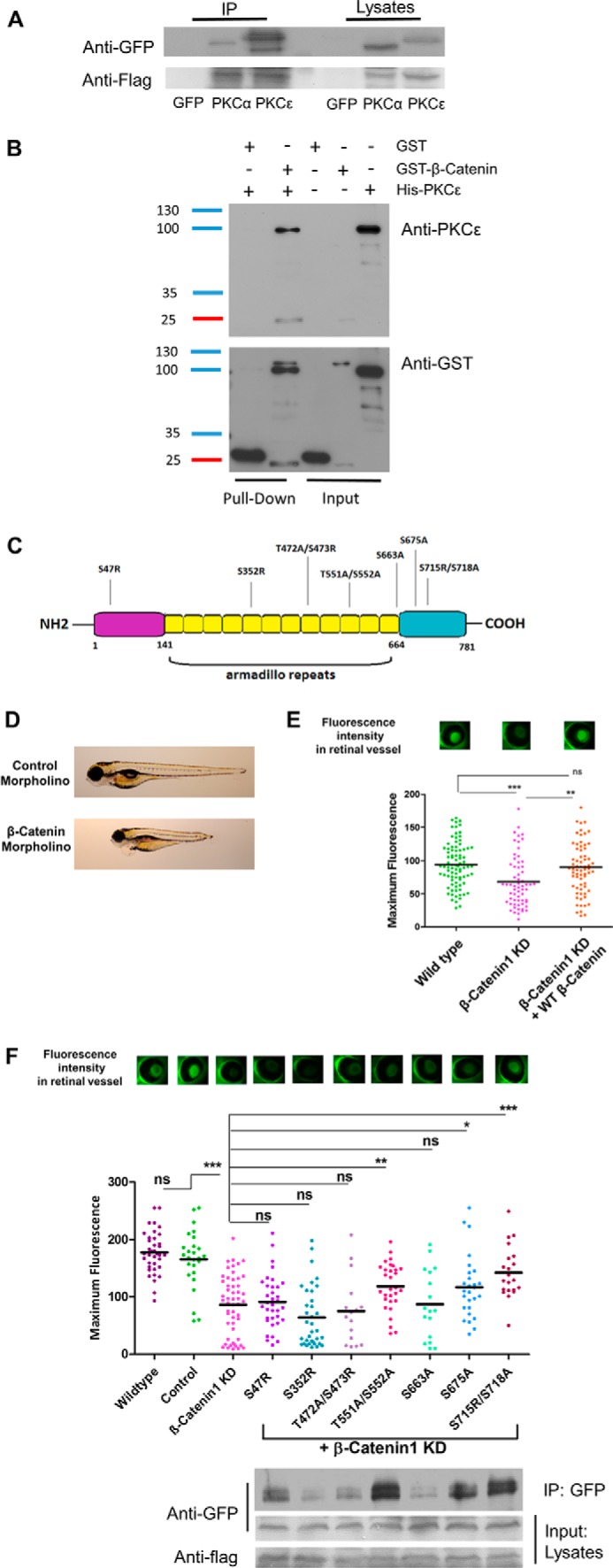
PKCϵ is a functional binding partner of β-catenin, and the interaction is indispensable for filtration barrier function. A, FLAG immunoprecipitation (IP) of HEK cells transduced with a FLAG-tagged β-catenin construct and either a GFP, GFP-tagged PKCα (positive control), or GFP-tagged PKCϵ construct. Immunoblotting against FLAG shows successful transfection. B, in vitro direct interaction between β-catenin and PKCϵ was examined in a GST pulldown experiment with pure recombinant GST-β-catenin and His-PKCϵ. His-PKCϵ was incubated with either GST or GST-β-catenin. The pulldown fractions were analyzed by Western immunoblotting against PKCϵ and GST. C, schematic illustrating β-catenin protein structure, which is divided into the N-terminal, Armadillo repeats, and C-terminal parts. The suggested phosphorylation sites of PKCϵ are shown. D, individual larvae at 120 hpf, indicating control morpholino–injected fish with no edema and β-catenin morpholino–injected fish with edema and a dorsalized phenotype. E, murine β-catenin-mutants were tested in the zebrafish model by injecting the capped mRNA into fertilized zebrafish eggs at one- to four-cell–stage embryos. The transgenic zebrafish produce a vitamin D-binding protein fused with GFP that, under normal conditions, accumulates in the retina and is quantified 120 hpf by measuring the fluorescence level. Reduced fluorescence indicates a disturbed glomerular filtration barrier. The graph shows GFP fluorescence intensity of the eye assay of wild-type, β-catenin1 morpholino and co-injection of β-catenin1 morpholino with a human wild-type RNA construct. F, group mean fluorescence intensities of the eye assay. A comparison of the combined injection of β-catenin morpholino plus β-catenin-mutant results was conducted against β-catenin morpholino injection alone (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant). Binding of each β-catenin mutant to GFP-tagged PKCϵ was tested in vitro with FLAG immunoprecipitation in transfected HEK cells. Immunoblotting was performed against GFP and FLAG.
To further characterize whether the interaction between PKCϵ and β-catenin derives from a direct or indirect linkage, we performed a pulldown assay with pure recombinant GST-β-catenin and His-PKCϵ. These experiments confirmed a direct interaction between the two proteins in vitro (Fig. 5B).
To elucidate which domain of β-catenin is important and functionally altered by its interaction with PKCϵ, we searched for specific phosphorylation sites with phosphomotif-predicting programs such as PhosphoNET, HPRD release 9, dbPTM 3.0, SysPTM 2.0, and UniProt. We chose seven phosphomotifs with matches in at least three databases (Fig. 5C). We performed site-directed mutagenesis to ablate these phosphorylation sites in β-catenin (the amino acid serine was either mutated to alanine or arginine, and threonine was switched to alanine) and performed zebrafish experiments to explore the biological relevance of these mutations of potential PKCϵ binding sites. To accomplish this, we first established the knockdown model of zebrafish β-catenin1 expression using a β-catenin1 morpholino. Zebrafish express two isoforms of β-catenin that seem not to be functionally redundant during development (26). We decided to use specific morpholinos for the β-catenin1 isoform for our knockdown experiments because there is a significantly higher percentage of sequence similarity of this isoform to the mammalian and Xenopus protein sequences (27). As depicted in Fig. 5D, reduction of β-catenin in zebrafish leads to a phenotype with edema of the yolk sac, pericardial effusion, and a shorter tail compared with control morpholino-injected fish. The dorsalized phenotype thus displayed the typical features of β-catenin1 knockdown also seen in other studies (26, 27). Using our previously described eye assay, we measured the level of proteinuria in zebrafish larvae (28). In brief, the eye assay is an indirect method for determining proteinuria. The experimental zebrafish are transgenic for a liver promoter-driven, GFP-labeled vitamin D-binding protein with a molecular mass of 78 kD that accumulates in the circulation under normal conditions and can be easily quantified in the retinal blood vessel plexus of the fish. Decreased fluorescence levels, as displayed by the β-catenin knockdown fish, indicated a significant loss of high-molecular-weight proteins from the circulation of the fish (Fig. 5E). To verify the specificity of the β-catenin1 knockdown, we also performed a cross-species rescue experiment with a wild-type mRNA construct, leading to full recovery of the proteinuria phenotype (Fig. 5E). Next we performed cross-species rescue experiments by co-injecting the β-catenin zebrafish morpholino and cRNA of different murine β-catenin mutant constructs. The mutants S47R, T551A/S552A, S675A, and S715R/S718A showed partial or full rescue of the proteinuria phenotype with increased levels of circulating fluorescence in the fish. The rescue was considered partial when the mean fluorescence level was significantly higher than that of the β-catenin knockdown zebrafish (*, p < 0.05) and full when the statistical significance was ***, p < 0.001. In contrast, the mutants S352R, T472A/S473R, and S663A were not able to rescue the β-catenin phenotype, indicating a functional role of these phosphorylation sites. Interestingly, co-immunoprecipitation experiments using anti-FLAG beads and HEK cells transfected with GFP-tagged PKCϵ and with these FLAG-tagged β-catenin mutants also confirmed a lower binding affinity in the β-catenin mutants that did not exhibit rescue activity in the zebrafish experiments (Fig. 5F). These observations indicated that the ablated phosphorylation sites are relevant for the interaction between PKCϵ and β-catenin in vivo and in vitro. As a control, to ensure that the constructs would not influence glomerular filtration function, we also performed overexpression of all constructs in developing fish larvae and detected no difference in fluorescence levels compared with scrambled morpholino-injected control fish (data not shown). These data indicate that the interaction between PKCϵ and β-catenin is indispensable for proper filtration barrier function and depends on several binding sites.
Discussion
We and others have shown previously that the dynamics of the actin cytoskeleton are crucial for podocyte function and often depend on a single player in the network of cytoskeletal signaling components (29). Here we demonstrate that PKCϵ is a key player in this context via its interaction and regulation of β-catenin (Fig. 6). PKCϵ dictates the subcellular localization of β-catenin; immunofluorescence staining of PKCϵ-deficient cells showed that β-catenin translocation from the nucleus to perinuclear areas and the membrane during differentiation depends on PKCϵ. PKCϵ shifted β-catenin into the cytosol and to the cellular membrane. Under normal conditions in undifferentiated cells in culture, β-catenin first accumulates in the cytoplasm and is localized to the membrane, whereas, during the differentiation process, excessive β-catenin enters the nucleus to stimulate Wnt target signaling (30).
Figure 6.

Schematic overview of β-catenin regulation by PKCϵ in wild-type and PKCϵ−/− podocytes. In wild-type podocytes, PKCϵ regulates cytoplasmic β-catenin level in balance with other PKC isoforms: PKCα phosphorylates β-catenin and, thus, leads to its degradation. PKCϵ inhibits degradation via its interaction sites (Ser-352, Thr-472/Ser-473, and Ser-663) in a GSK3β-independent manner and leads to a predominantly cytosolic localization of β-catenin, which then might bind to the actin-bundling protein fascin1 in filopodia of the podocytes or localize to the membrane, forming the cell adhesion complex with P-cadherin and α-catenin, resulting in a stabilized actin cytoskeleton and cell adhesion. In PKCϵ-deficient podocytes, this balance is disturbed, leading to degradation of β-catenin, abolished P-cadherin expression, and lower fascin1 expression, which then results in impaired cell adhesion and actin bundling and, thus, in a weakened actin cytoskeleton.
Use of PMA stimulation as a PKC activator in tissue culture revealed a more detailed picture. PKCϵ knockout decreased active β-catenin, whereas increasing β-catenin levels were found in control cells. PMA triggers not only PKCϵ activity (31) but also other C1 domain–containing isoforms such as PKCα and PKCβ. PKCα has been described previously as a negative regulator of β-catenin, as its specific inhibition by the small molecule A23187 results in β-catenin up-regulation (15), and our data indicate that PKCϵ might counterbalance the influence of PKCα on β-catenin. We showed that β-catenin and PKCϵ interact in a GSK3β-independent manner. These results are in line with those of previous studies in other cell types, indicating that PKCα also regulates β-catenin expression separately from GSK3β, when β-catenin is reduced after withdrawing glucose from the medium (32). Our findings also support our previous observation suggesting an antagonistic role of PKCα and PKCβ compared with PKCϵ in the context of diabetic nephropathy (3). Under physiological conditions, β-catenin levels are highly regulated via their phosphorylation sites. The major pathway of β-catenin degradation is via GSK3β, which relies on phosphorylation of β-catenin by CK1 (33). Besides this pathway, alternative pathways of β-catenin degradation are not well characterized. In the Ca2+/DAG-dependent pathway, PKCα knockdown leads to an accumulation of β-catenin (16), indicating that this conventional PKC plays a direct role in β-catenin degradation. In the planar cell polarity pathway, inhibition of PKCδ, another novel PKC, has been shown to induce stabilization of β-catenin (16). Our data define PKCϵ as an antagonistic regulator of β-catenin stability in both signaling pathways.
PKCϵ deficient podocytes in vitro show a distinct phenotype, displaying an abnormally small cell size, disturbed actin cytoskeleton dynamics, a higher tendency to undergo spontaneous apoptosis, and lower expression of podocyte differentiation markers (21). Adenoviral transfection of PKCϵ−/− podocytes with a PKCϵ wild-type construct led to complete recovery of the cells. Interestingly, viral transduction of knockout cells with a human β-catenin construct could also overrule the effects of the PKCϵ deficiency. The podocytes appeared normal in size and exhibited a normalized actin cytoskeleton arrangement (Fig. 4A).
P-cadherin, one major factor of the cell-cell adhesion complex of podocytes, was down-regulated in PKCϵ podocytes throughout the differentiation process (Fig. 4C). This finding certainly could explain the poor cell adhesion of PKCϵ deficient podocytes because β-catenin and P-cadherin together form a complex, and both show reduced expression. Transfection with PKCϵ rescued the phenotype as expected. β-Catenin overexpression in PKCϵ-deficient podocytes also induced up-regulation of P-cadherin expression not detected in control-transfected cells. This observation surprised us because it is established that aberrant E-cadherin expression promotes accumulation of an unbound cytoplasmic β-catenin pool. This excessive β-catenin can then further act as a transcription co-factor (34). Our findings suggest that there is a mutual relationship between the two proteins, leading to up-regulation of P-cadherin expression by ectopic expression of β-catenin. Further, our results indicate that PKCϵ is a protein that leads to P-cadherin enhancement rather than down-regulation. These results further supplement the described relationship between novel PKCϵ and P-cadherin, in addition to findings by other groups (35), describing novel PKCs as important regulators of endocytosis and recycling of E-cadherin in other cell types.
Fascin1, a downstream target of the canonical Wnt/β-catenin signaling pathway, is an actin filament-bundling protein. It is well-known for binding to β-catenin at the cellular edges and possesses a PKC binding site at Ser-39 that has been described for conventional PKCs such as PKCα (24). Larrson (8) reported that fascin1 phosphorylation by PKCα leads to release of fascin1 from actin bundles, presumably enabling cell spreading. However, so far, an interaction between PKCϵ and fascin1 has not been presented. Our data suggest an interdependence among PKCϵ, β-catenin, and fascin1 that supports a healthy actin cytoskeleton (Fig. 6). Whether fascin1 up-regulation solely relies on increased β-catenin expression mediated per PKCϵ or could also be induced by PKCϵ alone needs further investigation, as the rescue of PKCϵ−/− podocytes with the PKCϵ construct did not restore fascin1 mRNA expression levels present in wild-type cells, whereas Western blot analysis suggests a normalized fascin1 protein presence.
The question remains whether rescue of the PKCϵ knockout phenotype by β-catenin overexpression is derived from enhanced Wnt-signaling activity or from accumulation at the cell adhesion complex. We will address this in the future.
We could identify three different PKCϵ binding/phosphorylation sites in β-catenin, located in its central domain, within the armadillo repeats. This location differs from those for GSK3β and PKCα, which have been demonstrated previously to regulate β-catenin via their phosphorylation sites in the N-terminal domain (Ser-33, Ser-37, Thr-41, and Ser-45) (36). Further studies in vivo in the zebrafish model demonstrate that knockdown of β-catenin via morpholino and coinjection of β-catenin RNA lacking Ser-352, Thr-472/Ser-473, or Ser-663 cannot rescue the proteinuria phenotype, suggesting that these binding sites are indispensable for phosphorylation of β-catenin by PKCϵ, leading to its stabilization.
Conclusion
In summary, our data indicate that PKCϵ deficiency leads to low β-catenin expression and defective actin cytoskeleton organization and contributes to disrupted podocyte function, leading to an impaired glomerular filtration barrier. Our results further support the hypothesis that balanced β-catenin levels are important for normal kidney function (12). The novel binding sites of PKCϵ identified here and the role of PKCϵ in β-catenin regulation, opposing those of GSK3β and other PKCs, might represent a new way of counterbalancing physiological β-catenin levels. Therefore, our study could be the basis for further pharmacological intervention studies targeting these binding sites, which will be explored in future studies.
Experimental procedures
Urine analysis
Murine spontaneous spot urine samples were produced by abdomen massage and collected on parafilm. The samples were analyzed by SDS-PAGE and followed by Coomassie Blue staining.
Antibodies and reagents
Primary antibodies used for Western blot, immunoprecipitation, and immunofluorescent staining were as follows: mouse anti-active-β-catenin (Merck Millipore, Temecula, CA); rabbit anti-β-catenin (Abcam, Cambridge, UK); rabbit anti-phospho-GSK3α/β, rabbit anti-GSK3β, rabbit anti-GFP, and rabbit anti-FLAG (Cell Signaling Technology, Cambridge, UK); rabbit anti-Fascin1 (Abcam, Cambridge, UK), rabbit anti-Paxillin (Merck Millipore); goat anti-P-cadherin (Novus Biologicals, Littleton, CO); and rabbit anti-GST (Cell Signaling Technology). Secondary antibodies for Western blotting included the following: goat anti-mouse IgG horseradish peroxidase, goat anti-rabbit IgG horseradish peroxidase, and donkey anti-goat IgG horseradish peroxidase (all from Santa Cruz Biotechnology, Santa Cruz, CA). The secondary antibody for immunofluorescent staining was mouse Alexa Fluor 488 from Invitrogen. Another reagent used was Alexa Fluor 546 phalloidin (Invitrogen). Immunoprecipitation and the pulldown assay were performed with FLAG and magnetic GST beads (Sigma-Aldrich, St. Louis, MO).
Western blot analysis
For protein extractions, podocytes were lysed in radioimmune precipitation assay buffer (50 mm Tris (pH 7.5), 150 mm NaCl, 0.5% sodium desoxycholate, 1% Nonidet P-40, and 0.1% SDS). The lysates were stored at −80 °C overnight and centrifuged at 11,000 rpm for 15 min at 4 °C. Afterward, the supernatant was collected and transferred into a new tube. Protein concentrations were then determined with the BCA protein assay kit (Thermo Scientific, Rockford, IL) according to the manual. Equal amount of proteins were separated by 10% SDS-PAGE and electrotransferred to a PVDF membrane (Immobilon-P, Millipore). After blocking in 2% BSA (SERVA Electrophoresis GmbH, Heidelberg, Germany), the membrane was sequentially probed with the first antibody (1:1000) and HRP-conjugated secondary antibody (1:10,000). Visualization of the HRP signals was achieved with the enhanced chemiluminescence kit (Pierce). As a loading control, either GAPDH or total protein analysis via Coomassie staining of the membrane was used. For Coomassie staining, the membrane was incubated in 0.1% Coomassie 250G (w/v) in 50% (v/v) methanol for 1 min, destained for 15 min in a solution of water/ethanol/acetic acid in proportions of 4:5:1, and then air-dried overnight.
Cell culture and drug treatment
Cell culture of conditionally immortalized mouse podocytes was performed by following the description of Mundel et al. (37). Under permissive conditions, podocyte proliferation was induced in the presence of 10 units/ml γ-interferon (Cell Sciences, Canton, MA) in RPMI 1640 medium (Biochrom AG, Berlin, Germany) including 10% FCS and 1% penicillin/streptomycin (both from Gibco/Invitrogen) at 33 °C. The podocytes were allowed to differentiate at the nonpermissive temperature of 37 °C in the same medium but without γ-interferon. All flasks used for podocytes were coated with collagen I (BD Biosciences), which was mixed in 20 mm sodium acetate (pH 4.7). After being cultured at 37 °C for 9 days, the podocytes were pretreated in starvation medium containing 1% FCS and 1% penicillin/streptomycin for 16 h. On the 10th day of differentiation, the podocytes were treated with PMA (Sigma-Aldrich) and harvested at different time points. For immunocytochemistry, the podocytes were plated on collagen I-coated coverslips in a 24-well plate. To analyze differentiation, podocytes were cultured under nonpermissive conditions and harvested at different time points (30 min, 1 h, 2 h, 4 h, 8 h, and 24 h). HEK 293T cells were cultured in DMEM (Invitrogen) containing 10% FCS and 1% antimycotic solution (Gibco/Invitrogen) at 37 °C and plated in 10-cm dishes for transfection.
Transfection
HEK 293T cells were seeded in 10-cm dishes 2 days prior to transfection. According to the user manual, 1–2 μg of plasmid and 6 μl of FuGENE HD transfection reagent (Promega Corp., Fitchburg, WI) were mixed gently in 200 μl of serum-free medium and incubated for 20 min at room temperature. Afterward, the mixture was added dropwise into every dish and incubated for 48 h.
Immunoprecipitation
About 48 h after transfection, HEK293T cells were washed with ice-cold PBS and lysed in 1 ml of ice-cold radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 7.5), 200 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, and 0.25% desoxycholic acid sodium salt) with protease inhibitor and phosphatase inhibitor (Roche Diagnostics GmbH, Mannheim, Germany) on ice. The lysate was collected and rotated for 1 h at 4 °C and then centrifuged at 11,000 rpm for 15 min at 4 °C. The supernatant was transferred into new tubes, and 50 μl of FLAG beads (Sigma, 50% slurry in Triton buffer) was added to each tube. The tubes were then rotated at 4 °C for 3 h. The beads were collected and centrifuged at 3000 rpm for 3 min, washed with 1 ml of radioimmune precipitation assay buffer, and rotated for 5 min at 4 °C three times. After detachment from the beads by adding loading buffer and boiling at 95 °C, the proteins were separated by SDS-PAGE and analyzed by Western blotting as described above.
Pulldown assay
30 μl of magnetic GST beads (Sigma) was first washed in washing buffer (50 mm TBS, 138 mm NaCl, and 2.7 mm KCl (pH 8.0)) before adding either 1 μg of pure recombinant GST or GST-β-catenin (Sigma) protein and rotating the mixture for 30 min at room temperature. After washing the mixture twice for 1 min each time with 300 μl of washing buffer, pure recombinant PKCϵ protein was added and rotated at 4 °C overnight. After washing three times, 120 μl of elution buffer (TBS with 15 mm reduced glutathione (pH 8.0)) was added to the mixture and rotated for 30 min. The eluate was then used for separation by SDS-PAGE and analyzed by Western blot analysis.
Immunofluorescence staining
The immortalized mouse podocytes were plated on coverslips for differentiation and fixed with 4% paraformaldehyde at different time points. After permeabilization with 0.1% Triton X-100, podocytes were blocked in 10% donkey serum (Jackson ImmunoResearch Laboratories, Suffolk, England) for 30 min and incubated with the primary antibody at 4 °C overnight, followed by incubation with Alexa Fluor 488 donkey anti-rabbit IgG and Alexa-Fluor 546 phalloidin for 1 h (Invitrogen). Afterward, glass coverslips were mounted in Aquapolymount medium (Polysciences Inc., Warrington, PA) with DAPI.
For immunohistochemistry, murine kidney sections were blocked in 10% donkey serum and incubated with the primary antibody at 4 °C overnight. Then the sections were rinsed with TBS three times, incubated with the Alexa Fluor 488 donkey anti-rabbit IgG for 1 h, and mounted with DAPI in Aquapolymount medium. A Zeiss Axioplan-2 imaging microscope and digital image processing software (Axio Vision 4.6, Zeiss, Jena, Germany) were used for analysis of the images.
Real-time PCR
Following the manual of the RNeasy Mini Kit (Qiagen, Hilden, Germany), total RNA was extracted from the cultured mouse podocytes. Reverse transcription was performed using 1 μg of total RNA, Moloney murine leukemia virus reverse transcriptase, oligo(dT)15, and random primers (Promega). The amplification reaction of cDNA was achieved by using Fast Start Taq polymerase (Roche Diagnostics), SYBR Green (Molecular Probes, Eugene, OR), and gene-specific primers in the following thermal cycle: 95 °C for 5 min, followed by 45 cycles for 10 s at 95 °C, 10 s at 60 °C, and 10 s at 72 °C. Each reaction was performed in triplicate and normalized to the constitutive gene mouse hypoxanthine phosphoribosyl transferase 1 (mHPRT-1). Melting curve analysis was used to verify the specificity of the PCR product. The primers used for amplification were as follows: HPRT-1, 5′-CAGTCCCAGCGTCGTGATTA-3′ and 5′-AGCAAGTCTTTCAGTCCTGTC-3′; fascin1, 5′-AACGTGTCCACGCGCC-3′ and 5′-GCAGCTGGCGTTCTTGGT-3′.
Site-directed mutagenesis
After searching in five phosphorylation databases (PhosphoNET, HPRD release 9, dbPTM 3.0, SysPTM 2.0, and UniProt), predicted phosphosites were chosen and compared in five species (human, mouse, zebrafish, Drosophila, and Caenorhabditis elegans) to determine the most conserved ones. Finally, seven promising phosphomotifs were selected as candidates, and corresponding site-specific mutations of mouse-derived DNA were introduced into the plasmid with overlapping PCR. The primers used were as follows: S47R forward, 5′-CAGCTCCTTCTCTGAGA(T)GGTAAAGGCAATCCTG-3′; S47R reverse, 5′-CAGGATTGCCTTTACCT(A)CTCAGAGAAGGAGCTG-3′; S352R forward, 5′-GCTATCTGTCTGCTCTAGA(T)AATAAGCCGGCTATTGTAG-3′; S352R reverse, 5′-CTACAATAGCCGGCTTATTT(A)CTAGAGCAGACAGATAG-C-3′; T472A and S473R forward, 5′-CTCTTCGTCATCTGG(A)CCAG G(C)CGAC-ACCAAGAAGC-3′; T472A and S473R reverse, 5′-GCTTCTTGGTGTCGC(G)CTGGC(T)CAGATGACGAAGAG-3′; T551A and S552A forward, 5′-CCCAGCGCCGTG(A)CGG(T)CCATGGGTGGG-3′; T551A and S552A reverse, 5′-CCCACCCATGGC(A)CGC(T)ACGGCGCTGGG-3′; S663A forward, 5′-GCTGTTTTGTTCCGAATGG(T)CTGAGGACAAGCCACAAG-3′; 663A reverse, 5′-CTTGTGGCTTGTCCTCAGC(A)CATTCGGAACAAAACAGC-3′; S675A forward, 5′-GATTACAAGAAACGGCTTG(T)CAGTTGAGCTGACCAGC-3′; S675A reverse, 5′-GCTGGTCAGCTCAACTGC(A)AAGCCGTTTCTTGTAATC-3′; S715R and S718A forward, 5′-GCCAGGATGATCCTAGG(C)TATCGTG(T)CTTTTCACTCTGGTGG-3′; and S715R and S718A reverse, 5′-CCACCAGAGTGAAAAGC(A)ACG ATAC(G)CTAGGATCATCCTGGC-3′.
The original bases are shown in parentheses. The site-directed mutagenesis was performed with the QuikChange site-directed mutagenesis kit following the instructions recommended by the manufacturer. All constructs were sequenced to verify the nucleotide sequences.
Adenoviral production and infection
Gateway technology (Invitrogen) was used for the generation of adenoviral vectors. With BP Clonase II, donor vectors were established by cloning wild-type or site-directed mutant β-catenin into the pDONR221 vectors. Afterward, adenoviral constructs were generated by recombining pDONR221 with pAd/CMV/V5-DEST using LR Clonase II. All reactions were accomplished by following the manual of the manufacturer. All constructs were confirmed by DNA sequencing. Adenoviral expression and amplification were performed in HEK293 cells. Adenoviral transduction of PKCϵ−/− podocytes with either pDest, PKCϵ, or β-catenin constructs was conducted on day 5 of differentiation. About 24 h later, the medium was changed, and 72 h after transduction, the cells were harvested or fixed for further analysis.
Cell size
ImageJ was used to quantify the total area of phalloidin-stained podocytes. At least 30 single cells (verified by DAPI staining) of every subgroup were photographed in black-and-white format.
Zebrafish experiments
We followed the method described by Hentschel et al. (28). Zebrafish (L-FABP:DBP-EGFP) were mated and housed at 28.5 °C in embryo rearing medium (E3). After having been embedded in 1.2% agarose, one-cell– to four-cell–stage fertilized embryos were injected using a Nanoject II injection device (Drummond Scientific, Broomall, PA). For overexpression experiments, mRNA of wild-type or site-directed mutant β-catenin was 1:1 diluted with injection buffer (20 mm HEPES, 200 mm KCl, and 0.01% phenol red) and injected at a final concentration of 30 ng/μl in a total volume of 4.6 nl. For rescue experiments, wild-type or site-directed mutant β-catenin mRNA was mixed with β-catenin morpholino, which was diluted in injection buffer, and injected at final concentrations of 30 ng/μl mRNA and 100 μm morpholino in a total volume of 4.6 nl.
Scrambled morpholino was injected as a control. Morpholino sequences were designed and ordered from GeneTools (Philomath, OR) as follows: standard control sequence, 5′-CCTCTTACCTCAGTTACAATTTATA-3′; β-catenin sequence, CTGTGTCAAAAGCTGTATATTCCTG. The β-catenin morpholino sequence was blasted to ensure no off-target splice junction or start codon annealing occurred for sequence matches greater than 14 nt. 120 h post-fertilization (hpf), zebrafish larvae were anesthetized with a 1:28 dilution of 4 mg/ml tricaine (MESAB, 1% Na2HPO4 (pH 7.0)) and photographed at ×10 magnification. The fluorescence of GFP-labeled vitamin D-binding protein in the pupil of the zebrafish eye was measured and analyzed with ImageJ. The animal protocol was approved by the Animal Care and Use Committee of the Mount Desert Island Biological Laboratory (Protocol 14-06).
Statistical analysis
Data are shown as means ± S.D. and were compared with unpaired Student's t tests. PMA stimulation experiments were compared by using two-way analysis of variance. Prism 6 was used for data analysis. Differences were considered significant at p < 0.05.
Ethics statement
Animal work, which was performed following the guidelines of the American Physiologic Society, was approved by Institutional Animal Care and Use Committee of Hannover Medical School and the animal welfare authorities of Lower Saxony. All efforts were made to minimize the number of animals used and their suffering.
Author contributions
M. D., X. Y., and B. T. conducted all experiments and analyzed the results. P. S. was essential for the zebrafish experiments. M. D. and M. S. wrote the paper. P. S. and S. E. edited the manuscript and together with H. H. provided helpful discussions. M. S., B. T., and M. D. conceived the idea for the project and wrote the paper with M. D. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by German research council Grant Schi587/3-6 and German Federal Ministry for Education and Research (BMBF) Grant 01GM1518A (to M. S.). Zebrafish experiments were supported by Institutional Development Awards (IDeA) from NIGMS of the National Institutes of Health under Grants P20GM0103423 and P20GM104318. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DAG
- diacylglycerol
- PMA
- phorbol 12-myristate 13-acetate
- hpf
- hours post-fertilization
- HPRT
- hypoxanthine phosphoribosyl transferase
- MESAB
- ethyl 3-aminobenzoate methanesulfonate methanesulfonate acid salt.
References
- 1. Mochly-Rosen D., Das K., and Grimes K. V. (2012) Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 11, 937–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Menne J., Meier M., Park J. K., Boehne M., Kirsch T., Lindschau C., Ociepka R., Leitges M., Rinta-Valkama J., Holthofer H., and Haller H. (2006) Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C α signaling in vivo. Kidney Int. 70, 1456–1462 [DOI] [PubMed] [Google Scholar]
- 3. Meier M., Park J. K., Overheu D., Kirsch T., Lindschau C., Gueler F., Leitges M., Menne J., and Haller H. (2007) Deletion of protein kinase C-β isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes 56, 346–354 [DOI] [PubMed] [Google Scholar]
- 4. Meier M., Menne J., Park J. K., Holtz M., Gueler F., Kirsch T., Schiffer M., Mengel M., Lindschau C., Leitges M., and Haller H. (2007) Deletion of protein kinase C-ϵ signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J. Am. Soc. Nephrol. 18, 1190–1198 [DOI] [PubMed] [Google Scholar]
- 5. Teng B., Duong M., Tossidou I., Yu X., and Schiffer M. (2014) Role of protein kinase C in podocytes and development of glomerular damage in diabetic nephropathy. Front. Endocrinol. (Lausanne) 5, 179, 10.3389/fendo.2014.00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inagaki K., Churchill E., and Mochly-Rosen D. (2006) ϵ Protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc. Res. 70, 222–230 [DOI] [PubMed] [Google Scholar]
- 7. Obis T., Besalduch N., Hurtado E., Nadal L., Santafe M. M., Garcia N., Tomàs M., Priego M., Lanuza M. A., and Tomàs J. (2015) The novel protein kinase C ϵ isoform at the adult neuromuscular synapse: location, regulation by synaptic activity-dependent muscle contraction through TrkB signaling and coupling to ACh release. Mol. Brain 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Larsson C. (2006) Protein kinase C and the regulation of the actin cytoskeleton. Cell. Signal. 18, 276–284 [DOI] [PubMed] [Google Scholar]
- 9. Prekeris R., Mayhew M. W., Cooper J. B., and Terrian D. M. (1996) Identification and localization of an actin-binding motif that is unique to the ϵ isoform of protein kinase C and participates in the regulation of synaptic function. J. Cell Biol. 132, 77–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Voronkov A., and Krauss S. (2013) Wnt/β-catenin signaling and small molecule inhibitors. Curr. Pharm. Des. 19, 634–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai C., Stolz D. B., Kiss L. P., Monga S. P., Holzman L. B., and Liu Y. (2009) Wnt/β-catenin signaling promotes podocyte dysfunction and albuminuria. J. Am. Soc. Nephrol. 20, 1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kato H., Gruenwald A., Suh J. H., Miner J. H., Barisoni-Thomas L., Taketo M. M., Faul C., Millar S. E., Holzman L. B., and Susztak K. (2011) Wnt/β-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J. Biol. Chem. 286, 26003–26015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Willert K., and Nusse R. (1998) β-Catenin: a key mediator of Wnt signaling. Curr. Opin. Genet. Dev. 8, 95–102 [DOI] [PubMed] [Google Scholar]
- 14. Sheldahl L. C., Park M., Malbon C. C., and Moon R. T. (1999) Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr. Biol. 9, 695–698 [DOI] [PubMed] [Google Scholar]
- 15. Gwak J., Cho M., Gong S. J., Won J., Kim D. E., Kim E. Y., Lee S. S., Kim M., Kim T. K., Shin J. G., and Oh S. (2006) Protein-kinase-C-mediated β-catenin phosphorylation negatively regulates the Wnt/β-catenin pathway. J. Cell Sci. 119, 4702–4709 [DOI] [PubMed] [Google Scholar]
- 16. Hernández-Maqueda J. G., Luna-Ulloa L. B., Santoyo-Ramos P., Castañeda-Patlán M. C., and Robles-Flores M. (2013) Protein kinase C δ negatively modulates canonical Wnt pathway and cell proliferation in colon tumor cell lines. PLoS ONE 8, e58540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gwak J., Jung S. J., Kang D. I., Kim E. Y., Kim D. E., Chung Y. H., Shin J. G., and Oh S. (2009) Stimulation of protein kinase C-α suppresses colon cancer cell proliferation by down-regulation of β-catenin. J. Cell. Mol. Med. 13, 2171–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davidson K. C., Adams A. M., Goodson J. M., McDonald C. E., Potter J. C., Berndt J. D., Biechele T. L., Taylor R. J., and Moon R. T. (2012) Wnt/β-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc. Natl. Acad. Sci. U.S.A. 109, 4485–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otero J. J., Fu W., Kan L., Cuadra A. E., and Kessler J. A. (2004) β-Catenin signaling is required for neural differentiation of embryonic stem cells. Development 131, 3545–3557 [DOI] [PubMed] [Google Scholar]
- 20. Fang X., Yu S. X., Lu Y., Bast R. C. Jr, Woodgett J. R., and Mills G. B. (2000) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 97, 11960–11965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schiffer M., Teng B., Gu C., Shchedrina V. A., Kasaikina M., Pham V. A., Hanke N., Rong S., Gueler F., Schroder P., Tossidou I., Park J. K., Staggs L., Haller H., Erschow S., et al. (2015) Pharmacological targeting of actin-dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat. Med. 21, 601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reiser J., Kriz W., Kretzler M., and Mundel P. (2000) The glomerular slit diaphragm is a modified adherens junction. J. Am. Soc. Nephrol. 11, 1–8 [DOI] [PubMed] [Google Scholar]
- 23. Jayo A., and Parsons M. (2010) Fascin: a key regulator of cytoskeletal dynamics. Int. J. Biochem. Cell Biol. 42, 1614–1617 [DOI] [PubMed] [Google Scholar]
- 24. Anilkumar N., Parsons M., Monk R., Ng T., and Adams J. C. (2003) Interaction of fascin and protein kinase Cα: a novel intersection in cell adhesion and motility. EMBO J. 22, 5390–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li A., Dawson J. C., Forero-Vargas M., Spence H. J., Yu X., König I., Anderson K., and Machesky L. M. (2010) The actin-bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr. Biol. 20, 339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bellipanni G., Varga M., Maegawa S., Imai Y., Kelly C., Myers A. P., Chu F., Talbot W. S., and Weinberg E. S. (2006) Essential and opposing roles of zebrafish β-catenins in the formation of dorsal axial structures and neurectoderm. Development 133, 1299–1309 [DOI] [PubMed] [Google Scholar]
- 27. Hao J., Ao A., Zhou L., Murphy C. K., Frist A. Y., Keel J. J., Thorne C. A., Kim K., Lee E., and Hong C. C. (2013) Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 4, 898–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hentschel D. M., Mengel M., Boehme L., Liebsch F., Albertin C., Bonventre J. V., Haller H., and Schiffer M. (2007) Rapid screening of glomerular slit diaphragm integrity in larval zebrafish. Am. J. Physiol. Renal Physiol. 293, F1746–F1750 [DOI] [PubMed] [Google Scholar]
- 29. Perico L., Conti S., Benigni A., and Remuzzi G. (2016) Podocyte-actin dynamics in health and disease. Nat. Rev. Nephrol. 12, 692–710 [DOI] [PubMed] [Google Scholar]
- 30. Behrens J. (2005) The role of the Wnt signalling pathway in colorectal tumorigenesis. Biochem. Soc. Trans. 33, 672–675 [DOI] [PubMed] [Google Scholar]
- 31. Newton P. M., and Messing R. O. (2010) The substrates and binding partners of protein kinase Cϵ. Biochem. J. 427, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Choi S. W., Song J. K., Yim Y. S., Yun H. G., and Chun K. H. (2015) Glucose deprivation triggers protein kinase C-dependent β-catenin proteasomal degradation. J. Biol. Chem. 290, 9863–9873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luna-Ulloa L. B., Hernández-Maqueda J. G., Castañeda-Patlán M. C., and Robles-Flores M. (2011) Protein kinase C in Wnt signaling: implications in cancer initiation and progression. IUBMB Life 63, 915–921 [DOI] [PubMed] [Google Scholar]
- 34. Orsulic S., Huber O., Aberle H., Arnold S., and Kemler R. (1999) E-cadherin binding prevents β-catenin nuclear localization and β-catenin/LEF-1-mediated transactivation. J. Cell Sci. 112, 1237–1245 [DOI] [PubMed] [Google Scholar]
- 35. Le T. L., Joseph S. R., Yap A. S., and Stow J. L. (2002) Protein kinase C regulates endocytosis and recycling of E-cadherin. Am. J. Physiol. Cell Physiol. 283, C489–C499 [DOI] [PubMed] [Google Scholar]
- 36. Liu C., Li Y., Semenov M., Han C., Baeg G. H., Tan Y., Zhang Z., Lin X., and He X. (2002) Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 [DOI] [PubMed] [Google Scholar]
- 37. Mundel P., Reiser J., and Kriz W. (1997) Induction of differentiation in cultured rat and human podocytes. J. Am. Soc. Nephrol. 8, 697–705 [DOI] [PubMed] [Google Scholar]