

Abstract
G protein-coupled receptors (GPCRs) are conformationally dynamic proteins transmitting ligand-encoded signals in multiple ways. This transmission is highly complex and achieved through induction of distinct GPCR conformations, which preferentially drive specific receptor-mediated signaling events. This conformational capacity can be further enlarged via allosteric effects between dimers, warranting further study of these effects. Using GPCR conformation-sensitive biosensors, we investigated allosterically induced conformational changes in the recently reported F prostanoid (FP)/angiotensin II type 1 receptor (AT1R) heterodimer. Ligand occupancy of the AT1R induced distinct conformational changes in FP compared with those driven by PGF2α in bioluminescence resonance energy transfer (BRET)-based FP biosensors engineered with Renilla luciferase (RLuc) as an energy donor in the C-tail and fluorescein arsenical hairpin binder (FlAsH)-labeled acceptors at different positions in the intracellular loops. We also found that this allosteric communication is mediated through Gαq and may also involve proximal (phospholipase C) but not distal (protein kinase C) signaling partners. Interestingly, β-arrestin-biased AT1R agonists could also transmit a Gαq-dependent signal to FP without activation of downstream Gαq signaling. This transmission of information was specific to the AT1R/FP complex, as activation of Gαq by the oxytocin receptor did not recapitulate the same phenomenon. Finally, information flow was asymmetric in the sense that FP activation had negligible effects on AT1R-based conformational biosensors. The identification of partner-induced GPCR conformations may help identify novel allosteric effects when investigating multiprotein receptor signaling complexes.
Keywords: bioluminescence resonance energy transfer (BRET), cell signaling, conformational change, dimerization, G protein-coupled receptor (GPCR)
Introduction
G protein-coupled receptors (GPCRs)3 are conformationally dynamic proteins that transmit information following interaction with different ligands that can promote multiple, yet specific downstream outputs depending on cellular context. It is widely accepted that this complexity is achieved through induction of distinct conformations in the receptor, whereby distinct conformational states preferentially drive specific receptor-mediated signaling events. The available conformational capacity, and therefore possible receptor functions, can be further expanded when considering allosteric effects. Receptor signaling partners, such as heterodimer partners, may provide novel conformational space generating new signaling modalities or protein life-cycle behaviors (1, 2).
With the recent explosion in the number of GPCR structures, our understanding of the mechanics underlying GPCR function has evolved considerably. Structures of GPCRs bound to various ligands, nanobodies, and co-crystals with G proteins demonstrate the conformation complexity of individual receptors (3–7). Although X-ray crystallography provides high-resolution snapshots of the receptor, it is limited with respect to reporting on dynamic events and providing information on highly mobile protein domains or larger protein complexes. This is especially true when considering allosterically-mediated conformational changes in the context of receptor heterodimers. Although there have been some reports of receptor oligomerization observed in crystal structures, this has not been a consistent observation and it is difficult to determine whether such arrangements represent artifacts of crystal packing, or reflect physiologically relevant interactions that occur in live cells (8–12). Some might argue that the paucity of GPCR oligomers in crystal structures refutes their existence, but many other biochemical and biophysical techniques reveal structural or functional profiles consistent with receptor oligomerization (13–18). At any rate, GPCR oligomerization must be interrogated from new perspectives that overcome some of these limitations.
Resonance energy transfer (RET) has been a key technology in characterizing GPCR oligomerization. High spatial and temporal resolution and applicability in live cell systems allows for highly robust assay development. RET experiments have been used extensively to support receptor dimerization (19–24) but stringent controls are required to interpret the data (25–30). Newer fluorescence-based RET (FRET) assays have been developed that can capture intra-molecular rearrangements in GPCRs in response to agonist. These sensors make use of a small fluorescent molecule, fluorescein biarsenical hairpin binder (FlAsH), as the acceptor and report on ligand binding-associated conformational rearrangements in multiple GPCRs (31–35). Such sensors have also been used to examine conformational dynamics of GPCRs in oligomeric states, specifically a class C homodimer of mGluR1 and a class A heterodimer of α2A-adrenergic and μ-opioid receptors (36, 37). Although valuable insight has been gained through the study of class C GPCRs, they are well accepted as obligate dimers. However, the latter article (37) sheds some light on conformational cross-talk in a putative class A receptor heterodimer. These authors demonstrated that morphine, targeting the μ-opioid receptor, affected the conformation of the α2A-adrenergic receptor in the presence of its ligand norepinephrine. This effect was shown to be G protein-independent and was still detected in isolated membranes, suggesting a simple mechanism of dimerization mediated through direct GPCR/GPCR contact.
We previously reported a functional interaction between FP and AT1R in vascular smooth muscle cells (38). Furthermore, a physical interaction between the receptors was reported in HEK 293 cells heterologously expressing both receptors as well as with endogenous receptors in vascular smooth muscle cells. It was also shown that signaling pathways modulated by the putative heterodimer could be either symmetrically or asymmetrically regulated (i.e. respond or not to stimulation of either partner in a similar or dissimilar way) depending on the signaling output being measured. This differential regulation of outputs lead to a functionally relevant bias attributed to AT1R/FP heterodimerization and may constitute a new druggable molecular target. To further understand the interplay between these two receptors, we generated a panel of FlAsH/BRET-based conformation-sensitive biosensors (34, 35, 39–41) to explore the effect each of these receptors had on the conformational landscape of its dimer partner. We observed an asymmetrical transmission of conformational information from AT1R to FP whereby angiotensin II (Ang II) stimulation lead to a rearrangement in FP when tagged with biosensors engineered into the third intracellular loop with respect to its C-terminal RLuc moiety. This Ang II-induced conformational change was distinct from that driven by PGF2α. The effect of Ang II on the conformation of FP was dependent on the presence of active Gαq or Gα11 and appeared to be mostly independent of downstream signaling and conformational information flow between the two receptors seemed to be unidirectional or asymmetric. Finally, this transmission from AT1R to FP appeared to be specific as another Gαq-coupled receptor was unable to induce similar conformational rearrangements in FP.
Results
AT1R ligand binding induces a conformational change in FP
To investigate the interplay between the protomers in the FP/AT1R heterodimer, we began by co-expressing each of our previously published FP-ICL3-RLucII conformational biosensors (FlAsH “walked” through the third intracellular loop (ICL3) at 5 different positions (39)) with wild-type AT1R (AT1R-WT) in HEK 293 cells. In this configuration, the assay strictly reports on conformational rearrangements between different vantage points in ICL3 and the C terminus of FP induced in response to ligand stimulation, i.e. only changes in FP conformation are reported. We noted a similar pattern across the different conformational biosensors in response to PGF2α as previously observed when expressed alone (Fig. 1A). When comparing the position of the FlAsH tags, the ICL3 P4 biosensor was again the FlAsH tag position that showed the largest ΔBRET in response to PGF2α in the panel (39). This response was dose-dependent (supplemental Fig. S1). Interestingly, when the cells were stimulated with Ang II, we not only observed a change in the BRET across all the FP conformational biosensors in the panel but opposite in direction, suggesting that activation of the heterodimer partner caused a distinct conformational change in FP as compared with its cognate agonist (Fig. 1B). As with direct stimulation of the conformational biosensor with PGF2α, the FP ICL3 P4 sensor also reported the largest ΔBRET in response to Ang II and the response was dose-dependent (supplemental Fig. 1B). We therefore selected this biosensor as a focus for further study.
Figure 1.

Ang II binding to AT1R induces a conformational rearrangement in FP. A and B, HEK 293 cells were transfected with FP conformational biosensors with a FlAsH tag inserted at the indicated position (Px) along with AT1R-WT. Cells were stimulated with 1 μm PGF2α (A) or Ang II (B) and the change in BRET (ΔBRET) due to ligand stimulation is reported. C, and D, HEK 293 cells were transfected with FP-ICL3 P4-RLucII and AT1R-WT (C) or FP-WT (D). Cells were pre-treated with assay buffer, 10 μm AS604872, or 10 μm losartan for 30 min prior to BRET recording. Cells were stimulated with 1 μm PGF2α or Ang II and the change in BRET (ΔBRET) due to ligand stimulation is reported. Bars represent the mean of 3 independent biological replicates and error bars represent S.E. A and B, Tukey's test was performed where: *** = p < 0.001; ** = p < 0.01; and * = p < 0.05, n = 3. C and D, Dunnett's test was performed to compare treatments to buffer. No comparisons were statistically significant. Increasing n was not possible as availability of AS604872 was limited. E and F, kinetic traces of conformation biosensors in response to ligand stimulation. HEK 293 cells were transfected with FP-ICL3 P4-RLucII alone (E) or co-transfected with AT1R-WT (F). The specified ligand was injected onto the cells at the 50th repeat measure as denoted by an arrow. Traces represent the mean of 3 independent experiments.
To assess the specificity of these effects, we next pretreated cells with antagonists for either FP or AT1R (Fig. 1C). HEK 293 cells transfected with the FP-ICL3 P4-RLucII conformation sensor and AT1R-WT were pretreated with 10 μm AS604872, a FP antagonist, or 10 μm losartan, an AT1R antagonist, followed by stimulation with 1 μm PGF2α or 1 μm Ang II. AS604872 was able to block the response to PGF2α but interestingly had no effect on the response driven by Ang II, suggesting the Ang II-induced conformational rearrangements were uncoupled from the orthosteric binding pocket of FP. Pretreatment with losartan had no effect on the PGF2α-induced conformational rearrangement in FP but was able to block the effect of Ang II. To ensure the dependence of AT1R to drive conformational change in FP across the heterodimer, HEK 293 cells were transfected with only the FP-ICL3 P4-RLucII conformation-sensor and in this case, were insensitive to either Ang II or losartan (Fig. 1D).
Although we represent the data as the ligand-induced change in BRET by subtracting the average reading before stimulation from the average reading post-ligand addition for simplicity of analysis, all our data sets were also temporally resolved. We noted a rapid response of the FP-ICL3 P4-RLucII sensor in response to 1 μm PGF2α when expressed alone (Fig. 1E). This response reached a plateau and was sustained for the length of the recording, suggesting that such conformational changes are stable as long as agonist is present. When co-expressed with AT1R-WT, the response to 1 μm PGF2α was similar to FP alone, whereas the response to 1 μm Ang II had a slower time constant before reaching a plateau (Fig. 1F). Additionally, co-expression of AT1R lead to an increase in the ligand-naive BRET, suggesting expression of AT1R, independent of ligand binding, modulates the conformation of FP (compare Fig. 1, E versus F). However, how this conformational information was transmitted was not clear from these experiments.
AT1R-induced conformational change in FP is dependent on Gαq expression and activity
We next explored how the information was transmitted from AT1R to FP. Our initial focus was one step removed from the receptor, in the G proteins presumably shared by the heterodimeric receptor. Both FP and AT1R have been demonstrated to couple to Gαq, Gα12, and Gαi (42–46). To investigate involvement of different Gα subunits, we used small molecule inhibitors, microbial toxins, and cell lines with CRISPR-mediated knock-out of G proteins and then re-assessed the ability of PGF2α or Ang II to induce a conformational change in FP.
First, using the Gαq inhibitor FR900359 (47) on HEK 293 cells co-expressing the FP-ICL3 P4-RLucII conformational biosensor and AT1R-WT, we noted a slight effect on the PGF2α-induced response where the magnitude of the response was slightly larger when Gαq activity was inhibited (Fig. 2A). In contrast, the Ang II response was completely abrogated upon Gαq inhibition. We next used a HEK 293 cell line made devoid of functional Gαq, Gα11, Gα12, and Gα13 using CRISPR/Cas9 (ΔGαq/11/12/13 line, Fig. 2B) (35). This cell line was especially valuable, as we could perform rescue experiments reintroducing individual Gα subunits. Similar to our observations made using FR900359, there was minimal effect of the loss of these G proteins on the FP-ICL3 P4-RLucII response to PGF2α. As with the small molecule inhibition of Gq/11, however, the ΔGαq/11/12/13 line also showed the dependence of Ang II-mediated conformational alterations in FP on re-expression of Gαq or Gα11, highlighting the necessity of both expression and activity of Gαq/11 for Ang II to affect FP conformation. However, co-expression of either Gα12 or Gα13 did not re-establish cross-talk between FP and the AT1R in the ΔGαq/11/12/13 line. We also examined the possible involvement of Gαi in the transmission of conformational information between the AT1R and FP using pertussis toxin (PTX, Fig. 2C). Pre-treatment of the ΔGαq/11/12/13 line expressing FP-ICL3 P4-RLucII, AT1R-WT, and various co-expressed G proteins with PTX had no effect on the Ang II-induced response. Interestingly, the response to PGF2α was lost with PTX treatment only when Gαq was co-expressed suggesting a more complicated cross-talk we did not explore further here.
Figure 2.

Functional Gαq/11 is required for Ang II-induced conformational cross-talk between AT1R and FP. A, HEK 293 cells were transfected with the FP-ICL3 P4-RLucII biosensor and AT1R-WT. Cells were pre-treated with 100 nm of the Gαq inhibitor FR900359 for 1 h followed by buffer, 10 μm AS604872, or 10 μm losartan for 30 min prior to BRET recording. Cells were stimulated with 1 μm PGF2α or Ang II and the change in BRET (ΔBRET) due to ligand stimulation is reported. B, ΔGαq/11/12/13 HEK 293 cells were transfected with FP-ICL3 P4-RLucII, AT1R-WT, and pcDNA3.1 or the indicated Gα subunit. Cells were stimulated with 1 μm PGF2α or Ang II and the change in BRET (ΔBRET) due to ligand stimulation is reported. C, ΔGαq/11/12/13 HEK 293 cells were transfected with FP-ICL3 P4-RLucII, AT1R-WT, and pcDNA3.1 or the indicated Gα subunit. Cells were pre-treated with buffer or 100 ng/ml of pertussis toxin for 16 h before FlAsH labeling. Cells were stimulated with 1 μm PGF2α or Ang II and the change in BRET (ΔBRET) due to ligand stimulation is reported. Bars represent the mean of 3 independent biological replicates and error bars represent S.E. A and B, Dunnett's test was performed to compare conditions with buffer (A) or pcDNA3.1 (B), where ** = p < 0.01. C, a two-way analysis of variance was used to analyze the 2 graphs. For both graphs, the G protein factor and interaction were nonsignificant, whereas the PTX factor was significant (PGF2α = p < 0.01, Ang II = p < 0.001). Bonferroni corrected tests were used to make post hoc comparisons where: * = p < 0.05; ** = p < 0.01. D and E, kinetic traces of conformational biosensors responding to ligand in the presence and absence of G proteins. ΔGαq/11/12/13 cells were transfected with FP-ICL3 P4-RLucII and AT1R-WT (D) or FP-ICL3 P4-RLucII, AT1R-WT, and Gαq (E). The specified ligand was injected onto the cells at the 50th repeat measure as denoted by an arrow. Inset to D: offset basal traces for PGF2α and Ang II to better demonstrate differences in biosensor responses. Traces represent the mean of 3 independent experiments.
When examining the kinetic data, we noted that PGF2α induced a sustained response regardless of the expression of Gαq, whereas Ang II only induced a sustained response when Gαq was present (Fig. 2, D and E). When compared with native HEK 293 cells, an increase in the time for the PGF2α response to reach a plateau was observed (compare Fig. 1F versus 2D). The effect was more clearly seen in the inset to Fig. 2D, where we have offset the baselines to match. This was also noted when Gαq was re-introduced for both PGF2α and Ang II and therefore may be a property of the cell line as opposed to simple G protein expression per se (compare Fig. 1F versus 2E). We also observed an increase in the ligand-naive BRET when Gαq was re-introduced compared with native HEK 293 cells (compare Fig. 1F versus 2E). This may again be due differences in gene expression profiles between the cells or the differences in G protein biosynthesis from endogenous and exogenous genetic templates. Taken together, our data showed that transmission of information from AT1R leading to a conformational change in FP was dependent on both the expression and activity of Gαq/11 and independent of the other Gα proteins tested.
Ang II-induced conformational change in FP required an intact cell membrane
Knowing that specific G proteins were required to mediate transmission of conformational information between the two protomers, we next focused on the involvement of other signaling proteins downstream of the receptor. To dissociate the receptor from more distal cytosolic components associated with molecular cross-talk, we first prepared membranes from the ΔGαq/11/12/13 line expressing the FP-ICL3 P4-RLucII conformation sensor with and without AT1R-WT and Gαq. We initially examined the effect of the membrane preparation on basal BRET as reported from the FP conformational biosensor in the absence of ligand (Fig. 3A). We observed an increase in BRET in membranes that were reduced upon co-expression of Gαq. Western blot analysis on the membrane preparation samples confirmed co-expression of Gαq (Fig. 3B). We then compared intact cell and membrane preparation samples following stimulation with PGF2α (Fig. 3C) or Ang II (Fig. 3D). In response to PGF2α, we observed larger ΔBRET values in the membrane preparation samples compared with intact cells, but the effects were similar in direction. However, following stimulation with Ang II we noted a distinct ΔBRET (of opposite direction) when comparing intact cells with membranes regardless of whether or not AT1R or Gαq was co-expressed.
Figure 3.

Membrane preparation results in a loss of Ang II-induced AT1R to FP conformational cross-talk although the receptors still form a complex. A, ΔGαq/11/12/13 HEK 293 cells were transfected with FP-ICL3 P4-RLucII, the indicated wild-type receptor, and pcDNA3.1, or the indicated Gα subunit. Cells were split into intact cell or membrane preparation groups. Each group was treated as described under “Experimental procedures.” Basal BRET was recorded in the absence of receptor ligands. B, Western blot analysis demonstrating that Gαq is still present in the sample after membrane preparation. C and D, the same preparations used in A and B were stimulated with 1 μm PGF2α (C) or Ang II (D) and the change in BRET (ΔBRET) due to ligand stimulation is reported. E, Western blots demonstrating that AT1R (FLAG) can co-immunoprecipitate FP-ICL3 P4-RLucII in lysates from prepared membrane or intact ΔGαq/11/12/13 HEK 293 cell samples, independent of the expression of Gαq. A, C, and D, bars represent the mean of 3 independent biological replicates and error bars represent S.E. Tukey's test was used to compare all groups in graphs where: * = p < 0.5; ** = p < 0.01. B and E are representative blots of n = 2. E, expression of FP-ICL3 P4-RLucII in all conditions has been demonstrated based on quantification of luminescence from RLucII (data not shown).
As membrane preparation disrupted the ability of Ang II to induce ΔBRET and because Gαq/11 was required to mediate transmission of information from AT1R to FP in intact cells, we assessed whether this was correlated with disruption of a physical interaction between the receptors per se. We demonstrated that neither membrane preparation itself, nor expression or absence of Gαq, affected immunoprecipitation of FP with AT1R (Fig. 3E). These observations suggest that cell integrity (and probably proper stiochiometric association of multiple signaling partners) is necessary to manifest allosteric interactions in the dimer even though a physical interaction between the two protomers was still maintained.
Proximal but not distal signaling partners affect Ang II-induced conformational rearrangement in FP
Having identified a requirement for both Gαq expression and activity as well as an intact cell membrane to allosterically connect FP and AT1R, we wanted to more explicitly examine whether conformational cross-talk from AT1R to FP was mediated through canonical molecular cross-talk. Because the membrane preparation procedure altered communication of conformational information from AT1R to FP, we investigated involvement of downstream signaling partners. We began by examining the distal effector protein kinase C (PKC) in the Gαq signaling pathway shared by both receptors. Using either an inhibitor (Gö6983, Fig. 4A) or an activator (phorbol 12-myristate 13-acetate, Fig. 4B), we detected no effect on the ability of either PGF2α or Ang II to induce conformational changes in FP-ICL3 P4-RLucII when co-expressed with AT1R-WT in HEK 293 cells. We next examined a more proximal signaling protein in the Gαq signaling cascade, phospholipase C (PLC). When HEK 293 cells expressing the FP-ICL3 P4-RLucII conformation sensor and AT1R-WT were pre-treated with the small molecule PLC inhibitor U73122, we noted no effect on the PGF2α-mediated ΔBRET and a small but significant reduction in the response to Ang II suggesting that the receptor/Gαq/11/PLC complex may be shared by the heterodimer (Fig. 4C).
Figure 4.
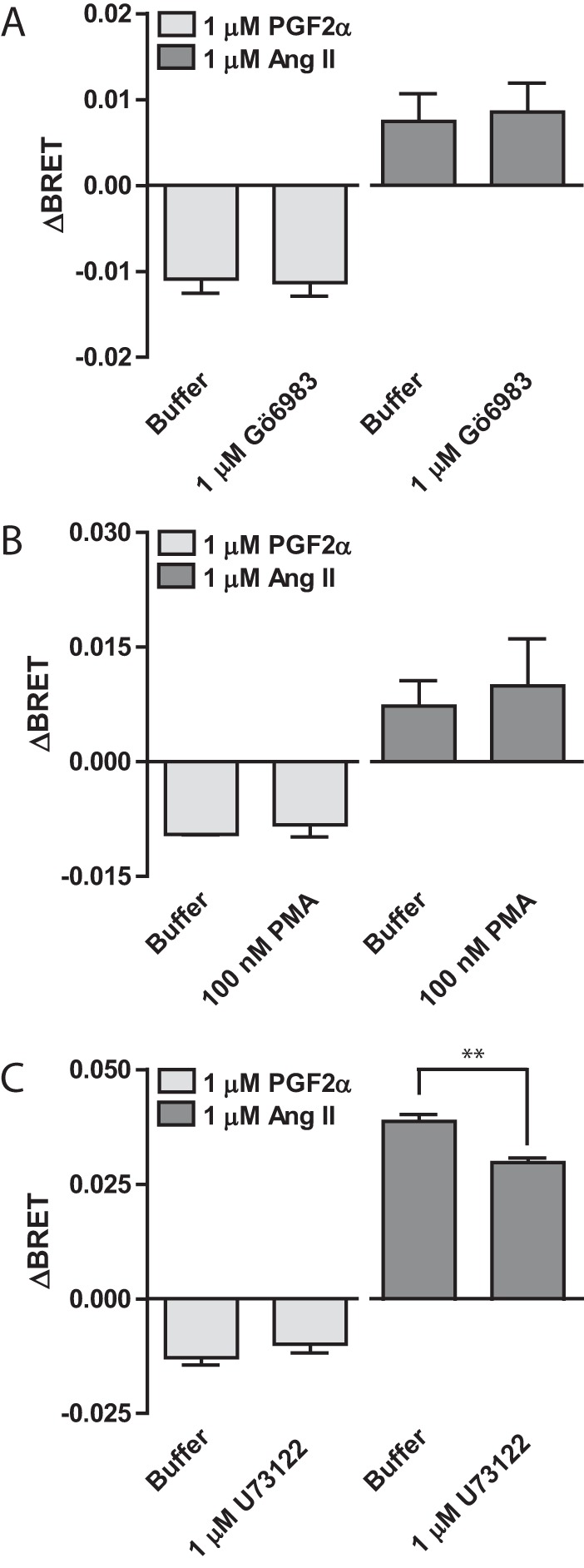
Proximal but not distal Gαq signaling cross-talk is involved in transmitting Ang II-induced conformational information to FP. A and B, HEK 293 cells were transfected with FP-ICL3 P4-RLucII and AT1R-WT. Cells were pre-treated with 1 μm of the PKC inhibitor Gö6983 for 1 h (A) or co-stimulated with 100 nm phorbol 12-myristate 13-acetate (B), a direct PKC activator. Cells were stimulated with 1 μm PGF2α or Ang II and the change in BRET (ΔBRET) due to ligand stimulation is reported. D, HEK 293 cells were transfected with FP-ICL3 P4-RLucII and AT1R-WT. Cells were pre-treated with 1 μm U73122 for 1 h. Cells were stimulated with 1 μm PGF2α or Ang II and the change in BRET (ΔBRET) due to ligand stimulation is reported. Bars represent the mean of 3 independent biological replicates and error bars represent S.E. A t test was performed to compare buffer versus treatment where ** = p < 0.01.
β-Arrestin-biased ligands of AT1R are also capable of inducing a conformational change in FP
There is considerable interest in developing β-arrestin or G protein-biased ligands as therapeutics. However, given that receptors are constitutively associated with G proteins, there is still a lot we do not understand about how G proteins might be involved in how biased ligands signal. Thus, we next used AT1R-biased ligands that selectively activate β-arrestin over Gαq (48). Using all 5 of the FP-ICL3-RLucII conformational biosensors, we screened this group of ligands to see if they could induce conformational changes in FP (Fig. 5A). Our initial screen detected the ability of both SBpA and SI, β-arrestin-biased AT1R agonists, to induce a small positive ΔBRET in the FP conformational biosensors at saturating concentrations. As with Ang II, the FP-ICL3 P4-RLucII conformational biosensor showed the largest magnitude ΔBRET across the panel of ligands and was therefore the focus of the subsequent experiment. To ensure that the heterodimerization between FP and AT1R did not alter the defined functional profile of these ligands as biased, we assessed their ability to activate Gαq using a BRET-based Gαq activation biosensor (Fig. 5A, inset). Neither SBpA nor SI were able to elicit a response in this biosensor, whereas a robust response was detected in response to Ang II. We tested SBpA and SI in the ΔGαq/11/12/13 line to assess whether there was any requirement for Gαq in mediating conformational rearrangement in FP. Interestingly, both SBpA- and SI-induced conformational changes were ablated in the ΔGαq/11/12/13 line, but could again be rescued when Gαq was reintroduced through transient co-expression (Fig. 5, B and C, comparing Gαq versus pcDNA3.1 vector control, respectively). These observations suggest that even biased ligands require Gαq to transmit information to FP even though they do not necessarily result in downstream Gαq signaling.
Figure 5.

β-Arrestin-biased AT1R agonists induce a Gαq-dependent conformational change in FP. A, HEK 293 cells were transfected with the different FP ICL3 conformational biosensors and AT1R-WT. Cells were stimulated with the indicated ligand and concentration and the change in BRET (ΔBRET) due to ligand stimulation is reported. Inset: ΔGαq/11/12/13 HEK 293 cells were transfected with FP-WT, AT1R-WT, and a BRET-based Gαq activation sensor and stimulated with the indicated ligand and concentration and the change in BRET (ΔBRET) due to ligand stimulation is reported, n = 2. B and C, ΔGαq/11/12/13 HEK 293 cells were transfected with FP-ICL3 P4-RLucII, AT1R-WT, and Gαq (B) or pcDNA3.1 (C). Cells were stimulated with the indicated ligand and concentration and the change in BRET (ΔBRET) due to ligand stimulation is reported. Bars represent the mean of 3 independent biological replicates and error bars represent S.E. Dunnett's test was performed comparing WT (A), or buffer (B and C), where ** = p < 0.01.
The Gαq-coupled OTR does not induce a similar conformational change in FP
Although one distal effector pathway was not involved in the conformational cross-talk between FP and AT1R, we did not systematically interrogate other pathways. However, to affirm receptor dimerization as a mechanism underlying the response of FP conformational biosensors to Ang II, we explored whether another Gαq-coupled GPCR could lead to a similar response in FP (49, 50) where similar patterns of molecular cross-talk might also be expected. We compared HEK 293 cells transfected with the FP-ICL3 P4-RLucII conformational biosensor and either AT1R-WT or OTR-WT (Fig. 6, A and B). We noted FP conformational responses to Ang II and not OT, although the cells were transfected with AT1R-WT or OTR-WT. To confirm that the absence of response to OT was not due to relative expression levels of the receptors, we titrated the expression of either AT1R-WT (Fig. 6C) or OTR-WT (Fig. 6D) and noted a saturating response to AT1R-WT/Ang II conditions but no response across any of the OTR-WT/OT conditions. Finally, we confirmed that both AT1R-WT (Fig. 6E) and OT-WT (Fig. 6F) constructs were able to activate a BRET-based Gαq biosensor. This demonstrated that Gαq activation per se was not driving the conformational change in FP-ICL3 P4-RLucII, suggesting a shared G protein in the context of the AT1R/FP heterodimer.
Figure 6.

Activation of Gαq by the oxytocin receptor does not induce a similar conformational change in FP. A and B, HEK 293 cells were transfected with FP-ICL3 P4-RLucII and AT1R-WT (A) or OTR-WT (B). Cells were stimulated with the indicated ligand/concentration and the change in BRET (ΔBRET) due to ligand stimulation is reported. C and D, HEK 293 cells were transfected with a constant amount of the cDNA for FP-ICL3 P4-RLucII and increasing amounts of AT1R-WT (C) or OTR-WT (D) cDNA. Cells were stimulated with 1 μm Ang II (C) or oxytocin (OT) (D) and the change in BRET (ΔBRET) due to ligand stimulation is reported. Ligand-induced changes in BRET plotted against the relative AT1R (C) or OTR (D) surface expression as assessed by in-cell western blots normalized to the highest expression level. E and F, HEK 293 cells were transfected with a BRET-based Gαq activation sensor, FP-WT, and AT1R-WT (E) or OTR (F). Cells were stimulated with the indicated ligand/concentration and the change in BRET (ΔBRET) due to ligand stimulation is reported. A, B, E, and F, bars represent the mean of 3 independent biological replicates and error bars represent S.E. Dunnett's test was performed comparing to buffer where, ** = p < 0.01. C and D, a two-way analysis of variance was performed, n = 3. For C, both factors of relative expression and stimulation as well as the interaction were significant. For D, both factors of relative expression and stimulation were significant but the interaction was not. Bonferroni corrected t-tests were performed to compare the buffer versus Ang II or OT at each relative expression level, where ** = p < 0.01 and *** = p < 0.001.
Asymmetrical transmission of conformational information between protomers of the FP/AT1R heterodimer
Finally, we considered whether ligand binding to the FP protomer could induce a similar conformational change in its AT1R partner. Using a corresponding panel of conformational biosensors build into AT1R (35), we screened whether ligand binding to WT-FP induced changes in the relative orientation between the ICL3 and C terminus of AT1R. Examining multiple vantage points in the ICL3 of AT1R demonstrated robust, position-dependent responses to orthosteric ligand (Ang II) in our AT1R conformational biosensors (Fig. 7A). Intriguingly, stimulation with PGF2α was not able to induce a response in any of the AT1R conformational biosensors suggesting that arrangement of the putative heterodimer was asymmetric (Fig. 7B). Examination of the kinetic data demonstrated a sustained response of the conformational biosensor to Ang II, similar to what we had observed with the FP sensor (Fig. 7, C and D). The effect of co-expressing FP-WT was not as large on ligand-naive BRET compared with AT1R-WT with the FP biosensor (compare Fig. 7, C versus D, and 1, E versus F). As before, we noted no change in BRET across the entire recording period when in response to PGF2α (Fig. 7D). When comparing traces from the ΔGαq/11/12/13 with and without re-introducing Gαq, we noted an increase in the sustained response to Ang II when Gαq was present (Fig. 7, E versus F). This is similar to observations we made when examining the sensor expressed alone without the co-expression of FP-WT (35).
Figure 7.

Conformational communication between protomers of the FP/AT1R heterodimer is asymmetric. HEK 293 cells were transfected with AT1R conformation sensors with a FlAsH tag inserted at the indicated position along with FP-WT. Cells were stimulated with 1 μm PGF2α (A) or Ang II (B) and the change in BRET (ΔBRET) due to ligand stimulation is reported. Bars represent the mean of 3 independent biological replicates and error bars represent S.E. Tukey's test was performed where, ** = p < 0.01 and ** = p < 0.001. C and D, kinetic traces of AT1R conformational biosensors responding to ligand. HEK 293 cells were transfected with AT1R-ICL3 P3-RLucII alone (C) or co-transfected with FP-WT (D). The specified ligand was injected onto the cells at the 50th repeat measure as denoted by an arrow. Traces represent the mean of 3 independent experiments. E and F, kinetic traces of AT1R conformational biosensors responding to ligand in the presence and absence of G proteins. ΔGαq/11/12/13 cells were transfected with AT1R-ICL3 P3-RLucII and FP-WT (E) or AT1R-ICL3 P3-RLucI, FP-WT, and Gαq (F). The specified ligand was injected onto the cells at the 50th repeat measure as denoted by an arrow. Traces represent the mean of 3 independent experiments.
Discussion
Here, we demonstrate asymmetric transmission of conformational information between protomers of the putative FP/AT1R heterodimer (summarized in Fig. 8). The AT1R-induced conformational rearrangement in FP was dependent on both expression and activation of Gαq and possible involvement of the proximal Gαq-effector PLC. This is consistent with reports showing that PLCβ is stably associated with Gαq (51). Furthermore, we demonstrate the AT1R-driven conformational change in FP was predominantly independent of a key distal downstream receptor signaling pathway. We propose that the transmission of information occurs at the level of the membrane and is most likely propagated via a shared G protein as part of a signaling complex. As we noted that even in the absence of Gαq, the AT1R/FP heterodimer remains intact, suggesting that Gαq subunits are not critical to the assembly of the receptor heterodimer, although other G protein heterotrimers might also serve this role in their absence. Gβγ subunits are also important in the formation of GPCR dimers and their associated signaling complexes, as suggested by our previous work (52, 53). Our data here suggests that Gαq acts as a conduit, allosterically connecting the two receptors once assembled into a signaling complex. Surprisingly, β-arrestin-biased AT1R ligands (48) also demonstrated a dependence on Gαq although they elicited no Gαq activation per se. This would further support the notion that Gαq plays a key structural role enabling conformational cross-talk between receptors, regardless of the nature of the bound ligand. Therefore, we demonstrate a novel mechanism in which allosteric interactions can transmit information between protomers of a GPCR heterodimer. Our observations are in contrast with a previous report demonstrating independence of the G proteins for conformational cross-talk between the receptors (37) suggesting that heterodimer-specific arrangements are possible. We have preliminary data using BRET that FP and OTR do not dimerize (data not shown).
Figure 8.
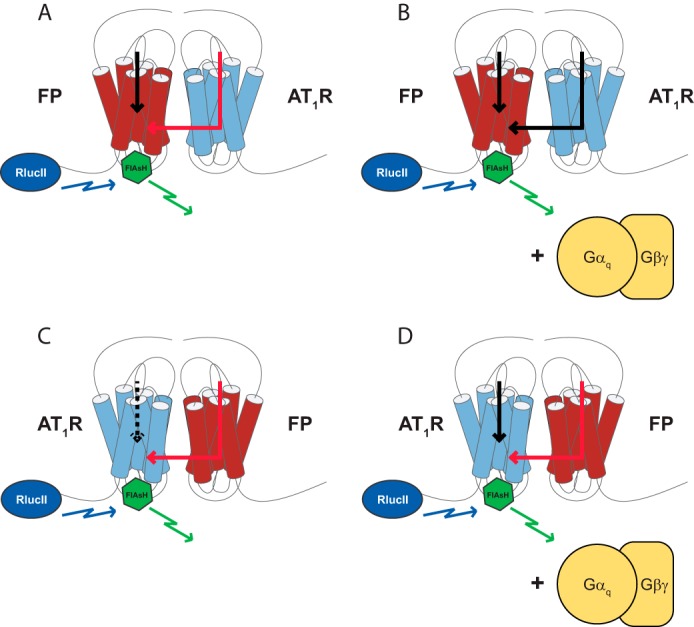
Conformational information is transmitted asymmetrically between protomers of the FP/AT1R heterodimer and is dependent on Gα. In the absence (A) of Gαq/11/12/13, PGF2α binding to the sensor receptor can elicit a conformational (black arrow), but this response was blunted for the partner AT1R (red arrow). However, when Gαq/11 is present (B), full responses to in the FP biosensor are observed in response to either receptor. When the AT1R is tagged with the conformational biosensor, responses are detected in response to Ang II but not PGF2α (C and D, respectively), indicating that such conformational responses might be asymmetric depending on the relative positions of the sensor tags and the organization of the dimer/G protein complex.
We previously showed that within the FP/AT1R complex, each receptor was capable of modulating the functional output of the other through asymmetric allosteric interactions (38). Asymmetric structural arrangements have been noted in luteinizing hormone oligomers (54), rhodopsin (55), mGluR2/3 heterodimers (56), and leukotriene B4 receptor dimers (18). These studies support the notion that individual protomers in a receptor dimer may interact with a shared G protein through distinct interfaces (see also Ref. 2), suggesting that structural asymmetries may translate into functional or conformational asymmetries. Our results here further strengthen the case for functional FP/AT1R heterodimeric complexes and provide insight into the mechanism by which the two receptors communicate. Although the precise functional consequences of AT1R-induced change in FP conformation are yet to be determined, it also appears to be asymmetric in nature. We observed asymmetry in allosteric communication between receptors, with AT1R modulating FP but not the converse. Furthermore, the AT1R to FP conformational cross-talk in the heterodimer may be biased toward Gαq/11, as no effect was observed when we altered Gα12/13 or Gαi function or levels. This could represent a coupling preference of the heterodimer or it may be possible that our biosensors are sensitive to conformations driven by particular G proteins coupled to the heterodimer. Capitalizing on such signal bias and asymmetric conformational cross-talk may provide novel venues for targeting heterodimers, ignored in most current drug discovery programs (57–59). As we have demonstrated previously, ligand binding to AT1R can modulate the functional output of FP (38). Because both AT1R and FP couple to Gαq, it is difficult to explore the functional effect of the AT1R-induced conformational effects on FP, as they share a number of common signaling outputs. It is also important to acknowledge that there is also the possibility that the induced conformation may be silent with respect to signaling (60). A larger understanding of unique and shared receptor signaling outputs may help settle this question.
FP and AT1R are important targets at the core of many biological functions. AT1R is a primary target in the treatment of hypertension with AT1R antagonists of the sartan family being widely prescribed (61). A role for FP has also been demonstrated in regulating blood pressure where its blockade has been suggested to reduce blood pressure (62). FP is involved in parturition with enhanced PGF2α signaling initiating labor by causing smooth muscle contraction of the myometrium (63, 64). The AT1R is also expressed in the myometrium with increased levels coinciding with pregnancy (65–67). Examining the receptor complex may yield novel drug targets in these tissues. An understanding as to how these two receptors communicate at a structural level could facilitate rational drug design as our data indicates that at least as regards receptor conformation, Ang II is a biased ligand for FP, whereas the converse is not true for PGF2α and the AT1R. Regardless of the mechanisms, our conformational biosensors could be used to identify new conformational and allosteric connections between known and orphan GPCRs, without the requirement for knowledge about how downstream signaling or receptor trafficking is altered. This may help identify new targets for drug discovery as ligands for one receptor may act as allosteric modulators of heterodimer partners, provide a new vantage point from which to understand receptor dynamics, and foster the development of receptor screens that are portable from cell type to cell type regardless of a priori knowledge about downstream signaling.
Experimental procedures
Reagents
Unless noted otherwise, all chemicals were reagent-grade and purchased from Sigma. FR900395 was purchased from the Institute of Pharmaceutical Biology (University of Bonn, Germany). PGF2α was from Cayman Chemical, whereas AS604872 was a kind gift from Dr. Stéphane Laporte. SBpA, SI, SII, and DVG were synthesized by Lifetein. Western Lightning Plus-ECl was purchased from PerkinElmer Life Sciences.
Constructs
pIRES-SP-FLAG-AT1R-WT, pIRES-SP-FLAG-AT1R-ICL3 WT, P2, P3, P4, P5-RLucII were described in Ref. 35. pIRESpuro3-HA-FP-WT, pcDNA3.1-SP-HA-FP ICL3 WT, P1, P2, P3, P4, P5-RLucII were previously described (39). pcDNA3.1-Gαq-EE and pcDNA3.1-Gα12-EE were obtained from the University of Missouri-Rolla cDNA Resource Center. pcDNA3.1-Gα11 and pcDNA3.1-Gα13 were obtained from Dr. Michel Bouvier. The generation of pcDNA3.1-myc-OTR-WT was as previously described (68). The polycistronic Gαq activation sensor was used as previously described (69).
Cell culture
HEK 293 SL or T cells and the ΔGαq/11/12/13 line (35) were cultured in Dulbecco's modified Eagle's medium (DMEM, Wisent) supplemented with 10% (v/v) fetal bovine serum (FBS, Wisent). All cells were cultured at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Cells were grown to ∼80% confluence in T75 flasks (Corning) at which point they were plated for transfection. Gαq/11/12/13 gene-deleted HEK 293 cells were generated as previously described (35).
Membrane preparation
FlAsH-labeled cells were resuspended in 1 ml of homogenizing buffer (10 mm Tris-HCl, pH 7.4, 5 mm EDTA). Cells were homogenized with a glass Teflon homogenizer with 20 up-down strokes on ice. The homogenate was transferred to 1.5-ml microtubes and the membrane was pelleted by centrifugation at 13,000 rpm at 4 °C for 30 min in a Microlite RF from Thermo IEC. The pelleted membrane was resuspended in TME buffer (50 mm Tris-HCl, pH 7.4, 4.8 mm MgCl2, 2 mm EDTA, 100 mm NaCl, 1 μm GDP, 30 nm GTP, 250× protease inhibitor mixture (Sigma)). Total protein was quantified using a Bradford reagent (Bio-Rad) and 10 μg of protein was loaded/well in a white 96-well plate (Corning).
Conformational biosensor assays
Data collection with the FlAsH/BRET-based conformation biosensors was previously described (39). Briefly, labeled cells in white 96-well plates were treated with 2 μm coelenterazine H (Nanolight) for 5 min. Plates were loaded into a Victor X Light Luminescence plate reader (PerkinElmer Life Sciences) and luminescence was captured through F460 and F535 filters for BRET 1. Filtered signals were collected every 1 s with an integration time of 0.2 s for 50 reads. The drug was then injected into the well using the attached injector module, and signals through each filter were collected for another 100 reads. To calculate BRET, the signal through the F535 filter was divided by that through the F460 filter. The basal BRET refers to the average BRET across the first 50 reads prior to injection and the change in BRET due to drug binding (ΔBRET) is calculated by subtracting the basal BRET from the average BRET across the last 50 reads post-injection. Kinetic traces are the raw BRET calculated from the F535/F460 at the sampling frequency.
Gαq-activation assay
As mentioned above, we used an established polycistronic Gαq activation sensor that is based on physical separation of Gα and Gγ subunits following receptor activation (69). Briefly, cells in white 96-well plates (Corning), transfected with the biosensor, and GPCR of interest were washed once and then left with 80 μl of Kreb's buffer in a white 96-well plate. Cells were left to rest for 2 h at room temperature before starting the assay. Data were collected on a Victor X Light luminescence plate reader (PerkinElmer Life Sciences) using 410/80 and 515/30 filters for BRET 2 in well mode. Prior to data collection, 5 μm coelenterazine 400a (Goldbio) was added to wells and incubated at room temperature for 5 min. The signal was collected through each filter with an integration time of 0.2 s every 0.6 s for 30 reads. The injector module then injected the drug of interest and the data were collected for another 60 reads. BRET was calculated by dividing the signal through the 515/30 filter by that captured through the 410/80 filter. The ΔBRET was then calculated by subtracting the average BRET pre-injection from the last 30 reads post-injection.
Co-immunoprecipitation
Transfected cells were lysed in solubilization buffer (25 mm HEPES, pH 7.4, 5 mm EDTA, 50 mm NaCl, 10% glycerol, 1% Triton X-100, 1/250 protease inhibitor mixture (Sigma)) for 1 h at 4 °C. Lysates were cleared by centrifugation at 13,500 rpm in a Microlite RF (Thermo IEC) microcentrifuge for 10 min. The supernatant was collected, and protein was quantified using a Bradford assay (Bio-Rad). FLAG M2 beads (Sigma) were loaded with 500 μg of protein and incubated at 4 °C by rocking overnight. The next day, beads were washed 3 times with solubilization buffer pelleting the beads at 1,600 rpm for 2 min at 4 °C between each wash. Protein was eluted from the beads in elution buffer (50 mm Tris, pH 7.4, 150 mm NaCl, and FLAG peptide (3 μl/100 μl of TBS, Sigma) for 20 min at 4 °C. Beads were spun down at 1,600 rpm for 2 min and the supernatant was collected. 20 μl of sample buffer (62.5 mm Tris pH 6.8, 16.3% glycerol, 2% SDS, 5% β-mercaptoethanol, 2 mg of bromphenol blue) was added and samples were heated to 65 °C for 15 min.
SDS-PAGE and Western blotting
Samples were loaded on a 10% polyacrylamide gel and run for 1 h at 120 V. Resolved protein was transferred to PVDF membrane under 100 V for 1 h. Membranes were blocked for 1 h in 5% milk in TBST. Anti-RLuc (Millipore) or anti-FLAG (Sigma) primary antibodies were prepared in TBST + 5% milk at a 1:2,000 dilution. The membrane was incubated with primary antibodies at 4 °C with gentle rocking overnight. The next day, the membrane was washed 3 times with TBST. Secondary antibodies conjugated to horseradish peroxidase (Sigma) were diluted at 1:20,000 in TBST + 5% milk and incubated with the membrane for 1 h at room temperature. The membrane was washed 3 times with TBST and then treated with enhanced chemiluminescence reagent (PerkinElmer Life Sciences).
In-cell Western blotting
Cells plated in black 96-well plates (Corning), transfected with the proteins of interest, were washed once with Kreb's buffer. Cells were then fixed with 2% paraformaldehyde that had been warmed to 37 °C for 10 min at room temperature. Cells were then washed 3 times with Kreb's + 1% BSA. Primary antibodies for HA and myc (Covance) and FLAG (Sigma) were diluted in Kreb's + 1% BSA at 1:200. Cells were incubated with primary antibodies for 1 h at room temperature. Cells were washed 3 times with Kreb's buffers and then incubated with anti-mouse (HA or myc) or anti-rabbit (FLAG) secondary antibodies conjugated to Alexa 488. Secondary antibodies were diluted at 1:500 in Kreb's + 1% BSA and incubated with the cells for 1 h at room temperature. Hoechst DNA stain was also added during the incubation with secondary antibodies at a concentration of 1 μg/ml. Fluorescent signals were measured using a Synergy 2 multimode plate reader (BioTek) using 360/40 excitation and 460/40 emission filters for Hoechst and 485/20 excitation and 528/20 emission filters for Alexa 488. The Alexa 488 signal was normalized to the Hoechst signal to quantify the average surface expression of the GPCR of interest/cell.
Author contributions
T. E. H., R. S., and D. D. designed the study and R. S., D. D., D. P., A. Z., K. B., and Y. S. performed experiments. R. S. and D. D. analyzed the data. R. S., D. D., Y. S., A. I., J. A., and D. P. generated figures. R. S., D. D, A. I., J. A., and T. E. H. wrote and edited the paper.
Supplementary Material
Acknowledgment
This work was initiated out of long discussions with Dr. Vic Rebois over many years and we acknowledge his wisdom with great gratitude.
This work was supported in part by a grant from the Consortium Québécois sur la Découverte du Médicament and the Canadian Institutes of Health Research Grant MOP-130309 (to T. E. H.), grants from PRESTO, Japan Science and Technology Agency (to A. I.), and AMED-CREST, AMED (to J. A.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Fig. S1.
- GPCR
- G protein-coupled receptor
- RET
- resonance energy transfer
- FlAsH
- fluorescein biarsenical hairpin
- FP
- F prostanoid
- AT1R
- angiotensin II type 1 receptor
- OTR
- oxytocin receptor
- BRET
- bioluminescence resonance energy transfer
- Ang II
- angiotensin II
- PTX
- pertussis toxin
- PLC
- phospholipase C
- PGF2α
- prostaglandin F2α
- RLuc
- Renilla luciferase
- OT
- oxytocin
- ICL3
- third intracellular loop.
References
- 1. Lane J. R., Donthamsetti P., Shonberg J., Draper-Joyce C. J., Dentry S., Michino M., Shi L., López L., Scammells P. J., Capuano B., Sexton P. M., Javitch J. A., and Christopoulos A. (2014) A new mechanism of allostery in a G protein-coupled receptor dimer. Nat. Chem. Biol. 10, 745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Han Y., Moreira I. S., Urizar E., Weinstein H., and Javitch J. A. (2009) Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat. Chem. Biol. 5, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2-adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tesmer J. J. (2016) Hitchhiking on the heptahelical highway: structure and function of 7TM receptor complexes. Nat. Rev. Mol. Cell Biol. 17, 439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang W., Manglik A., Venkatakrishnan A. J., Laeremans T., Feinberg E. N., Sanborn A. L., Kato H. E., Livingston K. E., Thorsen T. S., Kling R. C., Granier S., Gmeiner P., Husbands S. M., Traynor J. R., Weis W. I., et al. (2015) Structural insights into μ-opioid receptor activation. Nature 524, 315–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouvier M. (2013) Unraveling the structural basis of GPCR activation and inactivation. Nat. Struct. Mol. Biol. 20, 539–541 [DOI] [PubMed] [Google Scholar]
- 7. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., and Babu M. M. (2013) Molecular signatures of G protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 8. Huang J., Chen S., Zhang J. J., and Huang X. Y. (2013) Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., Sunahara R. K., Pardo L., Weis W. I., Kobilka B. K., and Granier S. (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., and Stevens R. C. (2010) Structures of the CXCR4 chemokine receptor in complex with small molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park J. H., Scheerer P., Hofmann K. P., Choe H.-W., and Ernst O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 12. Murakami M., and Kouyama T. (2008) Crystal structure of squid rhodopsin. Nature 453, 363–367 [DOI] [PubMed] [Google Scholar]
- 13. Rocheville M., Lange D. C., Kumar U., Patel S. C., Patel R. C., and Patel Y. C. (2000) Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science 288, 154–157 [DOI] [PubMed] [Google Scholar]
- 14. Bouvier M., and Hébert T. E. (2014) Crosstalk proposal: weighing the evidence for class A GPCR dimers, the evidence favours dimers. J. Physiol. 592, 2439–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lambert N. A., and Javitch J. A. (2014) Crosstalk opposing view: weighing the evidence for class A GPCR dimers, the jury is still out. J. Physiol. 592, 2443–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bouvier M., and Hébert T. E. (2014) Rebuttal from Michel Bouvier and Terence E. Hebert. J. Physiol. 592, 2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lambert N. A., and Javitch J. A. (2014) Rebuttal from Nevin A. Lambert and Jonathan A. Javitch. J. Physiol. 592, 2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Damian M., Martin A., Mesnier D., Pin J. P., and Banères J. L. (2006) Asymmetric conformational changes in a GPCR dimer controlled by G-proteins. EMBO J. 25, 5693–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pfleger K. D., and Eidne K. A. (2006) Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 3, 165–174 [DOI] [PubMed] [Google Scholar]
- 20. Calebiro D., Rieken F., Wagner J., Sungkaworn T., Zabel U., Borzi A., Cocucci E., Zürn A., and Lohse M. J. (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. U.S.A. 110, 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dorsch S., Klotz K. N., Engelhardt S., Lohse M. J., and Bünemann M. (2009) Analysis of receptor oligomerization by FRAP microscopy. Nat. Methods 6, 225–230 [DOI] [PubMed] [Google Scholar]
- 22. Angers S., Salahpour A., Joly E., Hilairet S., Chelsky D., Dennis M., and Bouvier M. (2000) Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. U.S.A. 97, 3684–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mercier J. F., Salahpour A., Angers S., Breit A., and Bouvier M. (2002) Quantitative assessment of β1- and β2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 44925–44931 [DOI] [PubMed] [Google Scholar]
- 24. Hebert T. E., Moffett S., Morello J. P., Loisel T. P., Bichet D. G., Barret C., and Bouvier M. (1996) A peptide derived from a β2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 271, 16384–16392 [DOI] [PubMed] [Google Scholar]
- 25. Felce J. H., Knox R. G., and Davis S. J. (2014) Type-3 BRET, an improved competition-based bioluminescence resonance energy transfer assay. Biophys. J. 106, L41–L43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gavalas A., Lan T. H., Liu Q., Corrêa I. R. Jr., Javitch J. A., and Lambert N. A. (2013) Segregation of family A G protein-coupled receptor protomers in the plasma membrane. Mol. Pharmacol. 84, 346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lan T. H., Kuravi S., and Lambert N. A. (2011) Internalization dissociates β2-adrenergic receptors. PloS One 6, e17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawano K., Yano Y., Omae K., Matsuzaki S., and Matsuzaki K. (2013) Stoichiometric analysis of oligomerization of membrane proteins on living cells using coiled-coil labeling and spectral imaging. Anal. Chem. 85, 3454–3461 [DOI] [PubMed] [Google Scholar]
- 29. James J. R., Oliveira M. I., Carmo A. M., Iaboni A., and Davis S. J. (2006) A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat. Methods 3, 1001–1006 [DOI] [PubMed] [Google Scholar]
- 30. Lan T. H., Liu Q., Li C., Wu G., Steyaert J., and Lambert N. A. (2015) BRET evidence that β2-adrenergic receptors do not oligomerize in cells. Sci. Rep. 5, 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zürn A., Zabel U., Vilardaga J. P., Schindelin H., Lohse M. J., and Hoffmann C. (2009) Fluorescence resonance energy transfer analysis of α2A-adrenergic receptor activation reveals distinct agonist-specific conformational changes. Mol. Pharmacol. 75, 534–541 [DOI] [PubMed] [Google Scholar]
- 32. Maier-Peuschel M., Frölich N., Dees C., Hommers L. G., Hoffmann C., Nikolaev V. O., and Lohse M. J. (2010) A fluorescence resonance energy transfer-based M2 muscarinic receptor sensor reveals rapid kinetics of allosteric modulation. J. Biol. Chem. 285, 8793–8800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziegler N., Bätz J., Zabel U., Lohse M. J., and Hoffmann C. (2011) FRET-based sensors for the human M1-, M3-, and M5-acetylcholine receptors. Bioorg. Med. Chem. 19, 1048–1054 [DOI] [PubMed] [Google Scholar]
- 34. Bourque K., Pétrin D., Sleno R., Devost D., Zhang A., and Hébert T. E. (2017) Distinct conformational dynamics of three G protein-coupled receptors measured using FlAsH-BRET biosensors. Front. Endocrinol. (Lausanne) 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Devost D., Sleno R., Pétrin D., Zhang A., Shinjo Y., Okde R., Aoki J., Inoue A., and Hébert T. E. (2017) Conformational profiling of the AT1 angiotensin II receptor reflects biased agonism, G protein coupling and cellular context. J. Biol. Chem. 292, 5443–5456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hlavackova V., Zabel U., Frankova D., Bätz J., Hoffmann C., Prezeau L., Pin J.-P., Blahos J., and Lohse M. J. (2012) Sequential inter- and intrasubunit rearrangements during activation of dimeric metabotropic glutamate receptor 1. Sci. Signal. 5, ra59–ra59 [DOI] [PubMed] [Google Scholar]
- 37. Vilardaga J. P., Nikolaev V. O., Lorenz K., Ferrandon S., Zhuang Z., and Lohse M. J. (2008) Conformational cross-talk between α2A-adrenergic and μ-opioid receptors controls cell signaling. Nat. Chem. Biol. 4, 126–131 [DOI] [PubMed] [Google Scholar]
- 38. Goupil E., Fillion D., Clément S., Luo X., Devost D., Sleno R., Pétrin D., Saragovi H. U., Thorin É., Laporte S. A., and Hébert T. E. (2015) Angiotensin II type I and prostaglandin F2α receptors cooperatively modulate signaling in vascular smooth muscle cells. J. Biol. Chem. 290, 3137–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sleno R., Pétrin D., Devost D., Goupil E., Zhang A., and Hébert T. E. (2016) Designing BRET-based conformational biosensors for G protein-coupled receptors. Methods 92, 11–18 [DOI] [PubMed] [Google Scholar]
- 40. Robertson D. N., Sleno R., Nagi K., Pétrin D., Hébert T. E., and Pineyro G. (2016) Design and construction of conformational biosensors to monitor ion channel activation: A prototype FlAsH/BRET-approach to Kir3 channels. Methods 92, 19–35 [DOI] [PubMed] [Google Scholar]
- 41. Rebois R. V., Maki K., Meeks J. A., Fishman P. H., Hébert T. E., and Northup J. K. (2012) D2-like dopamine and β-adrenergic receptors form a signaling complex that integrates Gs- and Gi-mediated regulation of adenylyl cyclase. Cell. Signal. 24, 2051–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goupil E., Tassy D., Bourguet C., Quiniou C., Wisehart V., Pétrin D., Le Gouill C., Devost D., Zingg H. H., Bouvier M., Saragovi H. U., Chemtob S., Lubell W. D., Claing A., Hébert T. E., and Laporte S. A. (2010) A novel biased allosteric compound inhibitor of parturition selectively impedes the prostaglandin F2α-mediated Rho/ROCK signaling pathway. J. Biol. Chem. 285, 25624–25636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hébert R. L., Carmosino M., Saito O., Yang G., Jackson C. A., Qi Z., Breyer R. M., Natarajan C., Hata A. N., Zhang Y., Guan Y., and Breyer M. D. (2005) Characterization of a rabbit kidney prostaglandin F2α receptor exhibiting Gi-restricted signaling that inhibits water absorption in the collecting duct. J. Biol. Chem. 280, 35028–35037 [DOI] [PubMed] [Google Scholar]
- 44. Gohla A., Schultz G., and Offermanns S. (2000) Role for G12/G13 in agonist-induced vascular smooth muscle cell contraction. Circ. Res. 87, 221–227 [DOI] [PubMed] [Google Scholar]
- 45. Rattan S., Puri R. N., and Fan Y. P. (2003) Involvement of Rho and Rho-associated kinase in sphincteric smooth muscle contraction by angiotensin II. Exp. Biol. Med. (Maywood) 228, 972–981 [DOI] [PubMed] [Google Scholar]
- 46. Lu H. K., Fern R. J., Luthin D., Linden J., Liu L. P., Cohen C. J., and Barrett P. Q. (1996) Angiotensin II stimulates T-type Ca2+ channel currents via activation of a G protein, Gi. Am. J. Physiol. 271, C1340–1349 [DOI] [PubMed] [Google Scholar]
- 47. Schrage R., Schmitz A. L., Gaffal E., Annala S., Kehraus S., Wenzel D., Bullesbach K. M., Bald T., Inoue A., Shinjo Y., Galandrin S., Shridhar N., Hesse M., Grundmann M., Merten N., et al. (2015) The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 6, 10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zimmerman B., Beautrait A., Aguila B., Charles R., Escher E., Claing A., Bouvier M., and Laporte S. A. (2012) Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signal. 5, ra33. [DOI] [PubMed] [Google Scholar]
- 49. Liedman R., Hansson S. R., Igidbashian S., and Akerlund M. (2009) Myometrial oxytocin receptor mRNA concentrations at preterm and term delivery: the influence of external oxytocin. Gynecol. Endocrinol. 25, 188–193 [DOI] [PubMed] [Google Scholar]
- 50. Kawamata M., Yoshida M., Sugimoto Y., Kimura T., Tonomura Y., Takayanagi Y., Yanagisawa T., and Nishimori K. (2008) Infusion of oxytocin induces successful delivery in prostanoid FP-receptor-deficient mice. Mol. Cell. Endocrinol. 283, 32–37 [DOI] [PubMed] [Google Scholar]
- 51. Dowal L., Provitera P., and Scarlata S. (2006) Stable association between Gαq and phospholipase C β1 in living cells. J. Biol. Chem. 281, 23999–24014 [DOI] [PubMed] [Google Scholar]
- 52. Dupré D. J., Robitaille M., Ethier N., Villeneuve L. R., Mamarbachi A. M., and Hébert T. E. (2006) Seven transmembrane receptor core signaling complexes are assembled prior to plasma membrane trafficking. J. Biol. Chem. 281, 34561–34573 [DOI] [PubMed] [Google Scholar]
- 53. Dupré D. J., Robitaille M., Rebois R. V., and Hébert T. E. (2009) The role of Gβγ subunits in the organization, assembly, and function of GPCR signaling complexes. Annu. Rev. Pharmacol. Toxicol. 49, 31–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jonas K. C., Fanelli F., Huhtaniemi I. T., and Hanyaloglu A. C. (2015) Single molecule analysis of functionally asymmetric G protein-coupled receptor (GPCR) oligomers reveals diverse spatial and structural assemblies. J. Biol. Chem. 290, 3875–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mishra A. K., Gragg M., Stoneman M. R., Biener G., Oliver J. A., Miszta P., Filipek S., Raicu V., and Park P. S. (2016) Quaternary structures of opsin in live cells revealed by FRET spectrometry. Biochem. J. 473, 3819–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Levitz J., Habrian C., Bharill S., Fu Z., Vafabakhsh R., and Isacoff E. Y. (2016) Mechanism of assembly and cooperativity of homomeric and heteromeric metabotropic glutamate receptors. Neuron 92, 143–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goupil E., Laporte S. A., and Hébert T. E. (2013) GPCR heterodimers: asymmetries in ligand binding and signalling output offer new targets for drug discovery. Br. J. Pharmacol. 168, 1101–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Khoury E., Clément S., and Laporte S. A. (2014) Allosteric and biased G protein-coupled receptor signaling regulation: potentials for new therapeutics. Front. Endocrinol. 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goupil E., Laporte S. A., and Hébert T. E. (2012) Functional selectivity in GPCR signaling: understanding the full spectrum of receptor conformations. Mini Rev. Med. Chem. 12, 817–830 [DOI] [PubMed] [Google Scholar]
- 60. Kenakin T., and Miller L. J. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Borghi C., and Rossi F. (2015) Role of the renin-angiotensin-aldosterone system and its pharmacological inhibitors in cardiovascular diseases: complex and critical issues. High Blood Pres. Cardiovasc. Prev. 22, 429–444 [DOI] [PubMed] [Google Scholar]
- 62. Yu Y., Lucitt M. B., Stubbe J., Cheng Y., Friis U. G., Hansen P. B., Jensen B. L., Smyth E. M., and FitzGerald G. A. (2009) Prostaglandin F2α elevates blood pressure and promotes atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 106, 7985–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jenkin G. (1992) Oxytocin and prostaglandin interactions in pregnancy and at parturition. J. Reprod. Fertil. Suppl. 45, 97–111 [PubMed] [Google Scholar]
- 64. Mejia R., Waite C., and Ascoli M. (2015) Activation of Gq/11 in the mouse corpus luteum is required for parturition. Mol. Endocrinol. 29, 238–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yamaleyeva L. M., Neves L. A., Coveleskie K., Diz D. I., Gallagher P. E., and Brosnihan K. B. (2013) AT(1), AT(2), and AT(1–7) receptor expression in the uteroplacental unit of normotensive and hypertensive rats during early and late pregnancy. Placenta 34, 497–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cox B. E., Ipson M. A., Shaul P. W., Kamm K. E., and Rosenfeld C. R. (1993) Myometrial angiotensin II receptor subtypes change during ovine pregnancy. J. Clin. Invest. 92, 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bird I. M., Zheng J., Cale J. M., and Magness R. R. (1997) Pregnancy induces an increase in angiotensin II type-1 receptor expression in uterine but not systemic artery endothelium. Endocrinology 138, 490–498 [DOI] [PubMed] [Google Scholar]
- 68. Devost D., and Zingg H. (2003) Identification of dimeric and oligomeric complexes of the human oxytocin receptor by co-immunoprecipitation and bioluminescence resonance energy transfer. J. Mol. Endocrinol. 31, 461–471 [DOI] [PubMed] [Google Scholar]
- 69. Namkung Y., Radresa O., Armando S., Devost D., Beautrait A., Le Gouill C., and Laporte S. A. (2016) Quantifying biased signaling in GPCRs using BRET-based biosensors. Methods 92, 5–10 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.