

Abstract
Gi/o-coupled G protein-coupled receptors can inhibit neurotransmitter release at synapses via multiple mechanisms. In addition to Gβγ-mediated modulation of voltage-gated calcium channels (VGCC), inhibition can also be mediated through the direct interaction of Gβγ subunits with the soluble N-ethylmaleimide attachment protein receptor (SNARE) complex of the vesicle fusion apparatus. Binding studies with soluble SNARE complexes have shown that Gβγ binds to both ternary SNARE complexes, t-SNARE heterodimers, and monomeric SNAREs, competing with synaptotagmin 1(syt1) for binding sites on t-SNARE. However, in secretory cells, Gβγ, SNAREs, and synaptotagmin interact in the lipid environment of a vesicle at the plasma membrane. To approximate this environment, we show that fluorescently labeled Gβγ interacts specifically with lipid-embedded t-SNAREs consisting of full-length syntaxin 1 and SNAP-25B at the membrane, as measured by fluorescence polarization. Fluorescently labeled syt1 undergoes competition with Gβγ for SNARE-binding sites in lipid environments. Mutant Gβγ subunits that were previously shown to be more efficacious at inhibiting Ca2+-triggered exocytotic release than wild-type Gβγ were also shown to bind SNAREs at a higher affinity than wild type in a lipid environment. These mutant Gβγ subunits were unable to inhibit VGCC currents. Specific peptides corresponding to regions on Gβ and Gγ shown to be important for the interaction disrupt the interaction in a concentration-dependent manner. In in vitro fusion assays using full-length t- and v-SNAREs embedded in liposomes, Gβγ inhibited Ca2+/synaptotagmin-dependent fusion. Together, these studies demonstrate the importance of these regions for the Gβγ-SNARE interaction and show that the target of Gβγ, downstream of VGCC, is the membrane-embedded SNARE complex.
Keywords: exocytosis, G protein, G protein-coupled receptor (GPCR), neurotransmitter release, SNARE proteins
Introduction
Release of neurotransmitter into the synapse is an intricate synchronized process involving core exocytotic machinery proteins, ion channels, calcium sensors, presynaptic inhibitory Gi/o-coupled receptors (GPCRs),3 and accessory proteins that each play a role in facilitating or inhibiting the docking, priming, and fusion of synaptic vesicles (1–3). The core exocytotic machinery consists of three members of a group of proteins known as soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) (2, 4, 5). On the vesicle, the SNARE protein (v-SNARE) is vesicle-associated membrane protein-2 (VAMP2), also known as synaptobrevin. Within its sequence is a SNARE motif that forms an α-helix that binds to the coiled-coil SNARE motifs in the dimer of two target membrane SNARE proteins (t-SNAREs), syntaxin1A and SNAP-25. SNAP-25 has within its sequence two SNARE motifs, so the full ternary SNARE complex comprises syntaxin1A, SNAP-25, and VAMP2 through association of these four α-helical SNARE motifs (5).
Many other proteins have been shown to interact with either the SNARE proteins individually, the t-SNARE dimer, or the full ternary SNARE. The components of minimal membrane fusion are thought to be the SNAREs, synaptotagmin, the SM proteins (nSec1, Munc18), Munc13, and complexin (2, 3). Calcium sensor proteins respond to the increase in calcium concentration resulting from the activation of voltage-gated calcium channels and promote the fusion of the vesicle with the target membrane. One group of calcium sensors of particular interest is the synaptotagmins. In neurons, it is currently believed that a major calcium sensor for exocytosis is synaptotagmin 1, containing an N-terminal intraluminal domain, a transmembrane domain, and a tandem set of calcium-binding C2-domains termed C2A and C2B (6–8). Whereas synaptotagmin can bind syntaxin1A and SNAP25 with low affinity in the absence of calcium, its affinity increases markedly in the presence of elevated calcium levels (9–11). Synaptotagmin 1 binds lipids in addition to SNAREs, with both phosphatidylserine and PIP2 essential for the function of the full-length protein (9, 12–14). The lipid-binding and SNARE-binding functionalities of synaptotagmin are both essential for its role in mediating fast synchronous release at the synapse.
Activation of inhibitory Gi/o-coupled GPCRs causes inhibition of exocytosis through several mechanisms. GPCRs signal through heterotrimeric G proteins, with both the guanine nucleotide-binding Gα subunit and the Gβγ subunit implicated in discrete signaling pathways (1, 15). Although the best-studied mechanism involves the inhibition of adenylyl cyclase by Gαi, the Gβγ heterodimer is also capable of inhibiting exocytosis in several ways. A large number of independent groups have shown that Gβγ can inhibit calcium currents through direct binding to CaV2 (N- P/Q- and R-type) voltage-gated calcium channels (16–19). This mechanism of inhibition is found at a large number of synapses, including certain subtypes of GPCRs thought to work entirely through it (20–22). Inhibition of exocytosis by Gi/o-coupled GPCRs downstream of calcium entry is also known to occur (23–26). It has been shown by multiple independent groups that Gβγ binds to the SNARE complex to inhibit exocytosis downstream of calcium entry (27–31). Electrophysiological and in vitro protein-binding studies have shown that the C terminus of SNAP-25 is a critical binding site for this interaction (30–32). We propose that Gβγ competes with synaptotagmin 1 for binding sites on SNAP-25, disrupting the ability of synaptotagmin 1 to promote vesicle fusion in response to elevated intracellular calcium (28–30). This hypothesis is in line with current crystallographic structures of the synaptotagmin–SNARE interaction, featuring significant overlap between the known synaptotagmin 1-binding site and key Gβγ-binding residues on the second helix of SNAP-25 that would be expected to produce a steric clash (30, 33, 34). Each inhibitory GPCR may signal via one or more of these mechanisms to achieve precise spatial and temporal control over neurotransmitter release; for example, the GABAB receptor in the CA1 axons of the hippocampus signals via modulation of calcium currents, whereas the 5-HT1B receptor signals only downstream of calcium entry (20). This mechanism downstream of calcium entry has been reported to occur at a variety of inhibitory Gi/o-coupled GPCRs in a variety of secretory cell types (35–41), demonstrating its importance for the regulation of exocytosis.
Prior protein biochemical studies elucidating the interplay between Gβγ, synaptotagmin, and SNAREs have been conducted in aqueous environments in the presence of detergent, and it has never been determined whether Gβγ can bind to SNAREs and directly inhibit vesicle fusion in lipid bilayers (28–31). In addition, the molecular requirements for the binding of SNARE proteins to Gβγ are not well understood. It has been demonstrated that Gβ1γ2 has a higher affinity than Gβ1γ1 for t-SNARE complexes and is more efficacious at inhibiting fusion in permeabilized PC12 cells (28), but the regions on Gγ2 responsible for the 20-fold tighter interaction are not known. Although a number of residues on SNARE have been shown to be important for Gβγ binding (30, 31), the individual residues on Gβγ implicated in the SNARE interaction have yet to be identified.
Here, we have further explored the complex interplay between Gβγ and synaptotagmin 1 for the regulation of SNARE-driven fusion. We expand on previous differences noted between Gβγ isoforms in this mechanism and highlight the importance of a single residue, Trp-332, on Gβ, for the interaction of Gβγ not only with SNAREs but also voltage-gated calcium channels. Furthermore, we examine the ability of Gβγ to compete with synaptotagmin for association with SNARE-containing liposomes as well as demonstrate a role for this inhibition as it relates to fusion in vitro.
Results
Previous studies of Gβγ–SNARE interactions used recombinant soluble SNARE complexes in aqueous solution to show that Gβγ binds to ternary SNARE complexes, t-SNARE heterodimers, and the monomeric SNARE proteins SNAP25, syntaxin1A, and VAMP2 (28–30). To examine binding of Gβγ to full-length t-SNARE complexes embedded in lipid bilayers, we developed an assay using total internal reflection (TIRF) fluorescence intensity and anisotropy (Fig. 1A). Purified Gβγ subunits fluorescently labeled at primary amine residues with Alexa Fluor 488 N-hydroxysuccinimide ester were applied from a pipette over a t-SNARE-containing (syntaxin1A and SNAP-25) bilayer on a coverslip illuminated with TIRF or epifluorescence. During pulses of Gβ1γ1, there was a small increase in TIRF fluorescence measured with a photomultiplier in the absence of t-SNARE complexes; if t-SNAREs were present, this increase was an order of magnitude larger (Fig. 1B). To confirm that this interaction represented binding of Gβγ to a target in the lipid bilayer, anisotropy of the fluorescence TIRF signal was measured (Fig. 1B). Laser TIRF excitation was polarized, and emission polarization was detected parallel and orthogonal to this excitation polarization. Immediately after Gβγ pressure ejection (which lasted for 1 s; Fig. 1B, gray bar) no increase in anisotropy of the fluorescence signal was observed if no t-SNARE was present, whereas in the presence of t-SNAREs there was a large increase in anisotropy (Fig. 1B, inset). The difference between the two conditions was significant.
Figure 1.

Supported lipid bilayer assay of Gβγ–t-SNARE interactions in membranes. A, fluorescently labeled Gβγ was assayed over a lipid bilayer using TIRF to isolate the fluorescence field to a band of ∼100 nm above a coverslip. Anisotropy of the fluorescence emission was also assayed to confirm a Gβγ interaction occurred. B, Gβ1γ1 pressure ejected in a 1-s pulse (gray bar) over the bilayer containing t-SNAREs (produced from liposomes containing 130 t-SNAREs/vesicle) evoked a sustained increase in TIRF fluorescence. A similar pressure application over a bilayer lacking t-SNAREs showed a much smaller signal. Inset, increase in anisotropy after the pressure application ceased as flow in the chamber removed aqueous Gβγ but no increase in anisotropy after application over a bilayer lacking t-SNAREs (response 10.5 ± 1.2%). The experiment was repeated 23 times. The mean change in anisotropy post-injection was r = 0.13 ± 0.04 for 8.8 mm Gβ1γ1 for membranes containing SNAREs. For membranes lacking SNARE, the magnitude of the change was Dr = 0.016 ± 0.004, with a significance value of p = 0.02 between the two conditions; Student's t test. C, the amplitude of fluorescence transients were measured first under epifluorescence to calibrate Gβγ concentrations applied from pressure ejection pipettes. Ci, output gave a signal proportional to the concentration of Gβγ applied, and the signal ceased as the pressure application stopped. Cii, in TIRF, pressure application of Gβγ revealed a longer-lived response. This signal was normalized to the pressure applied concentration of Gβγ by dividing by the signal in Ci. D, dose-response curves were constructed from this normalized data (e.g. Cii) plotted against the dose calculated from Ci. Gβ1γ1 is depicted in blue (EC50 = 2.08 ± 0.023 μm, n = 4 technical replicates), Gβ1γ2 is depicted in green (EC50 = 147 ± 71 nm, n = 5), and Gβ1 K78A/W332Aγ2 is depicted in red (EC50 = 86 ± 50 nm, n = 5, p = 0.045, one-way ANOVA with Tukey's HSD test for each curve).
The amplitude of the TIRF fluorescence response to Gβγ pressure application over the bilayer was used to quantify t-SNARE interactions of various Gβγ subtypes. Gβγ concentration at the lipid bilayer was calculated using epifluorescence of Gβγ at known concentrations in the solution above the lipid bilayer and comparing that value to epifluorescence of pressure-ejected Gβγ (Fig, 1Ci). Dosing to saturation was obtained by increasing concentrations of Gβγ in the pressure ejection pipette. A concentration-response curve was constructed comparing Gβ1γ1 to Gβ1γ2. We observed a 14-fold difference in affinity between Gβ1γ1 and Gβ1γ2 isoforms. These results confirmed the results obtained for binding in aqueous solution (28, 29). Gβγ subunits containing Ala mutations of two residues on the Gα-binding surface of Gβ, Lys-78 and Trp-332, inhibited exocytosis at a significantly higher potency than wild type (28). To determine if this was due to increased affinity for t-SNAREs, we tested this mutant Gβ1γ2 subunit in the TIRF assay. Gβ1 K78A/W332Aγ2 (Fig. 1D, red curve) exhibited a significantly higher 1.7-fold increase in affinity for t-SNARE complexes compared with wild-type Gβ1γ2. Consistent with saturation of binding, leaving excess unbound Gβγ, anisotropy of these fluorescence signals decreased as the pressure-ejected Gβγ concentrations were increased and the intensity signal saturated (data not shown).
To determine whether synaptotagmin could compete with Gβ1γ1 in lipid membranes, we labeled recombinant synaptotagmin 1 C2AB with Alexa Fluor 488-C5-maleimide selectively on the lone Cys residue present in the sequence (Cys-277). Alexa Fluor 488-synaptotagmin 1 was applied to coverslips coated with t-SNARE complexes consisting of purified recombinant syntaxin1A and SNAP-25 embedded in lipid membranes illuminated by TIRF. The anisotropy of the fluorescence TIRF signal produced via the binding of fluorescent synaptotagmin 1 to t-SNARE was recorded (Fig. 2A). Application of Ca2+ increased the anisotropy of the fluorescent TIRF signal in a saturable manner produced by application of fluorescent synaptotagmin 1 to the t-SNARE containing bilayer (Fig. 2B). The rise in anisotropy produced via the binding of synaptotagmin 1 to t-SNARE complexes could be reversed in a concentration-dependent manner by purified Gβ1γ1 (Fig. 2C).
Figure 2.
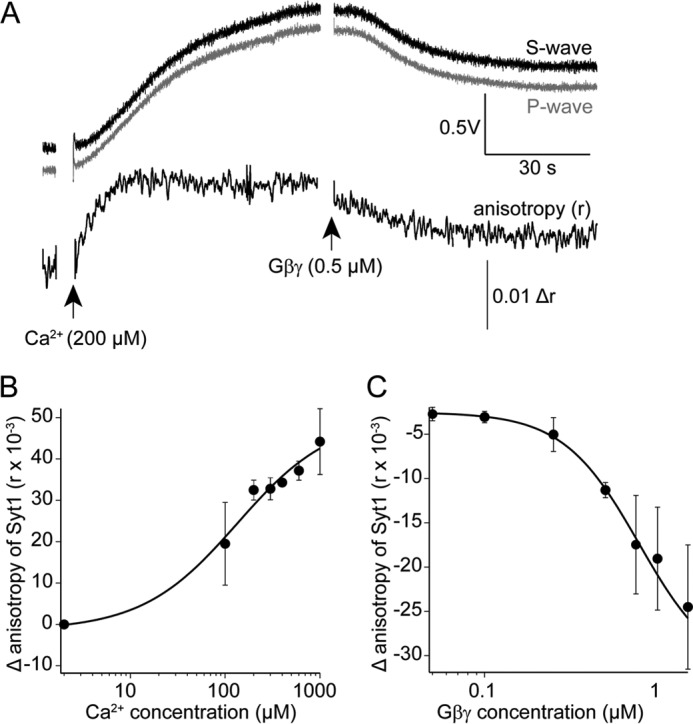
Ca2+ enhances synaptotagmin 1 C2AB-binding to t-SNARE-containing lipid bilayers, whereas Gβγ inhibits this interaction. Recombinant synaptotagmin 1 C2AB domains were labeled on the single Cys residue in the primary sequence with Alexa Fluor 488-C5-maleimide. A, a lipid bilayer consisting of 55% phosphatidylcholine, 15% phosphatidylserine, 29% PE, and 1% DiO containing t-SNARE complexes of syntaxin1A and SNAP-25 was maintained under solution containing Alexa Fluor 488-labeled syt1 (1 μm) and imaged using polarized laser TIRF microscopy. Emission was detected with two PMTs orthogonally placed with respect to the polarization of the excitation beam. The traces show an example after the addition of 200 μm Ca2+ to the solution. Absolute fluorescence of the TIRF field increased as did anisotropy of the emission signal, indicating binding of syt1 to the t-SNARE membrane. The subsequent addition of purified bovine Gβ1γ1 (0.5 μm) reduced both anisotropy and absolute fluorescence. B, Ca2+ enhances the anisotropy signal of Alexa Fluor 488-labeled synaptotagmin 1 C2AB in a concentration-dependent manner (EC50 = 130 ± 81 μm). The experiment was performed three times for a total of three technical replicates. C, concentration-dependent inhibition of the anisotropy signal produced by Alexa Fluor 488-synaptotagmin 1 C2AB binding to t-SNARE complexes consisting of syntaxin1A and SNAP25 embedded in lipid membranes as in Fig. 2A at a Ca2+ concentration of 175 ± 25 μm. The IC50 value for Gβ1γ1 was 0.7 ± 0.3 μm. The experiment was performed three times for a total of three technical replicates. Error bars represent mean ± S.E.
To investigate whether the ability of Gβ1 K78A/W332Aγ2 and its corresponding single-Ala mutants to inhibit voltage-gated calcium currents was correspondingly enhanced, we conducted electrophysiological studies in HEK cells expressing CaV2.2 (N-type) calcium channels along with wild-type Gβ1γ2, Gβ1 K78A/W332Aγ2, Gβ1 K78Aγ2, or GβW332Aγ2 (Fig. 3). Gβγ-mediated inhibition of CaV2.2 channels is voltage-dependent; the inhibition is less pronounced at more depolarized test potentials and is transiently reversed by a conditioning pre-pulse to very depolarized potentials (42). This reversal (also called pre-pulse facilitation) is thought to reflect transient dissociation of Gβγ from the channel at the depolarized membrane potential. The magnitude of pre-pulse facilitation can, therefore, be used to quantify the extent of Gβγ-mediated inhibition of ICa. We used a double-pulse protocol (Fig. 3A) in which cells voltage-clamped at −80 mV were stimulated by two identical test pulses (P1 and P2) to various potentials (−10, 0, +10, and +20 mV). The second test pulse (P2) was preceded by a conditioning prepulse to +120 mV to maximally reverse any Gβγ-mediated inhibition. In control conditions lacking Gβγ overexpression, there was a slight prepulse facilitation due to the presence of endogenous Gβγ (43). Robust prepulse facilitation and inhibition of ICa was observed with co-expression of Gβγ. The magnitude of prepulse facilitation was diminished at more depolarized test potentials (Fig. 3B), consistent with the known reduction of Gβγ-mediated inhibition at depolarized test potentials (42). Compared with cells expressing wild-type Gβγ, prepulse facilitation was only modestly affected in cells expressing the K78A mutant but was abolished in cells expressing the W332A mutant (Fig. 3B). The K78A/W332A double mutant produced slight prepulse facilitation of ICa that was not statistically different from control cells (Fig. 3B). These studies, in tandem with those conducted in Fig. 1, suggest that the W332A mutation and the K78A/W332A double mutant dramatically reduce the ability of Gβ1γ2 to interact with CaV2.2 channels and that the increased potency of the Gβ1 K78A/W332Aγ2 double mutant in the cracked PC12 cell assay is due to the enhanced interaction of Gβ1 K78A/W332Aγ2 with SNAREs.
Figure 3.

Gβ1 mutants that bind better to SNARE are less potent at inhibiting voltage-gated calcium channel currents. HEK cells were transiently transfected with CaV2.2 channels alone (control) or CaV2.2 channels with wild-type Gβ1, Gβ1 K78A (K78A), Gβ1 W332A (W332A), or Gβ1 K78A/W332A (K78A/W332A) in addition to Gγ2. A, representative whole cell currents (ICa) recorded from a cell expressing CaV2.2 channels and wild-type Gβ1γ2. The upper panel shows the voltage protocol consisting of two identical 20-ms steps to 0 mV (P1 and P2) and the conditioning prepulse (50 ms step to +120 mV) before P2. The lower panel shows ICa. Note the current after the prepulse (P2) was much larger than before the prepulse (P1) due to reversal of the tonic Gβγ-mediated inhibition. Also note the slow activation kinetics in P1, another characteristic of Gβγ-mediated inhibition that is reversed by the prepulse. The inset (bottom) shows superimposed P1 and P2 currents (normalized to the peak of P2) from a control cell and cells expressing the indicated Gβγ. B, prepulse facilitation was determined at several test potentials (potential of P1 and P2 step) in control cells (no exogenous Gβγ) (n = 7) and cells transfected with Gβγ variants. Box and whisker plots denote the range of the individual data points (box denotes 25%, median, 75%; whiskers denote standard deviation of mean). At a test potential of −10 mV there was statistically significant prepulse facilitation produced by wild-type Gβγ (n = 12, p < 0.001) and K78A (n = 5, p < 0.05) but not by W332A (n = 5) or K78A/W332A (n = 13) (one-way ANOVA with Dunnett's multiple pairwise comparison to control cells); at 0-mV test potential there was significant prepulse facilitation produced by wild type (n = 12, p < 0.001) and K78A (n = 5, p < 0.05) but not by W332A (n = 5) or K78A/W332A (n = 13) (one-way ANOVA with Dunnett's multiple pairwise comparison to control); at +10-mV test potential only wild-type Gβγ produced significant prepulse facilitation (n = 12, p < 0.01) but not K78A (n = 5), W332A (n = 5), or K78A/W332A (n = 13) (one-way ANOVA with Dunnett's multiple pairwise comparison to control); at +20-mV test potential only wild-type Gβγ produced significant prepulse facilitation (n = 12, p < 0.01) but not K78A (n = 5), W332A (n = 5), or K78A/W332A (n = 13) (one-way ANOVA with Dunnett's multiple pairwise comparison to control). ns denotes not significantly different; *, p < 0.05; **, p < 0.01; ***, p < 0.001; one-way ANOVA followed by Dunnett's multiple pairwise comparison to control).
To identify important regions on Gβ and Gγ for the binding of SNAP25, we utilized a peptide-competition approach to examine whether peptides derived from the primary sequence of Gβ1, Gγ1, or Gγ2 could disrupt the interaction between full-length Gβγ and SNAP25. We measured the ability of peptides to disrupt the interaction between full-length Gβ1γ2 with SNAP25 using the Alphascreen competition-binding assay (31). Gβ1 peptide 328–337, which contains Trp-332, is a reasonably potent inhibitor of Gβ1γ2 SNAP25 interaction (Fig. 4A); however, peptides corresponding to Gβ1 peptides 86–98 and 243–251 did not inhibit the interaction. Peptides from Gγ subunits could also compete with Gβ1γ2–SNAP-25 interactions; a peptide corresponding to the N-terminal 2–24 residues of Gγ2 was much more potent than a corresponding peptide on Gγ1 (Fig. 4, B and C). Peptides corresponding to residues 8–25 on Gγ1 inhibited the interaction with equivalent potency to residues 9–28 on Gγ2. In contrast, no inhibition was observed with peptides corresponding to residues 32–48 on Gγ1 or 29–45 on Gγ2. These studies suggest that the region 328–337, including Trp-332, on Gβ1 is important for SNAP-25 binding, and the N terminus of Gγ2 is responsible for Gβ1γ2's increased affinity for t-SNARE heterodimers. In Fig. 5, regions on Gβ1, Gγ1, and Gγ2 shown to be involved in interactions with SNAP-25 are illustrated in red in the 3-dimensional X-ray crystallographic structures of the Gβγ subunit (44–46), whereas individual residues Lys-78 and Trp-332 are depicted in blue.
Figure 4.

Gβγ-derived peptides can perturb binding of full-length Gβ1γ2 to SNAP25. Alphascreen competition-binding assays in which 20 nm biotinylated SNAP25 reacts with an EC80 concentration of Gβ1γ2 (180 nm) to produce a luminescent signal (20) were conducted in the presence of peptides corresponding to primary sequences within Gβ1 (A), Gγ1 (B), and Gγ2 (C) at varying concentrations (3.16 nm to 100 μm) dissolved in DMSO. A value of 100% was assigned to the average of all conditions tested containing only DMSO as a positive control. The primary sequence of each peptide is depicted above the graph for each condition. Experiments were repeated three times. The following IC50 values and 95% confidence intervals were observed: β1, 328–337 IC50 = 26 μm (95% confidence interval (CI): 17.0–37.8 μm); Gγ2, 2–24: IC50 = 3.81 μm (95% CI: 24.4-5.95 μm); Gγ1, 2–24 (IC50 = 83.7 μm (95% CI: 27.7 μm to an upper limit greater than the highest concentration tested); Gγ1, 8–25 IC50 = 21.1 μm (95% CI: 12.2–36.2 μm); Gγ2, 9–28 IC50 = 17.7 μm (95% CI: 11.6–26.8 μm).
Figure 5.
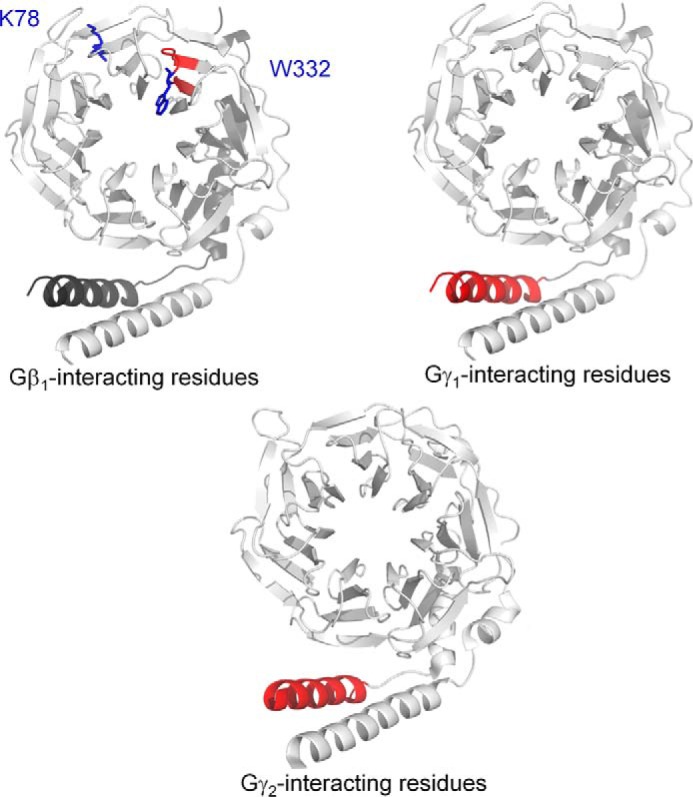
Key regions for SNARE interaction upon Gβγ. Three-dimensional X-ray crystallographic structure of either Gβ1γ1 (45) (upper panels) or Gβ1γ2 (46) (lower panel) containing key SNARE-binding regions obtained from Fig. 3 and Fig. 4 highlighted in red. The key inhibitory residues Lys-78 and Trp-332 on Gβ1 are highlighted in blue (upper left).
To determine whether Gβγ subunits can inhibit SNARE-catalyzed liposome fusion in the presence of synaptotagmin-1, we performed reconstituted fusion assays similar to those conducted in previous studies (47–50). Liposomes containing v-SNAREs were prepared using recombinant full-length VAMP2 and a FRET donor-acceptor pair consisting of NBD-phosphatidylethanolamine (PE) and rhodamine-PE. t-SNARE liposomes were prepared containing full-length recombinant rat syntaxin 1A and SNAP-25B heterodimers. As the quenched FRET-pair–containing liposomes fuse with the unlabeled liposomes, the concentration of the FRET acceptor rhodamine in the liposome is lowered, resulting in an increase in NBD donor fluorescence, as measured with an excitation wavelength of 460 nm and an emission wavelength of 538 nm (Fig. 6A). The cytoplasmic domain of synaptotagmin 1 (C2AB, 10 μm; Ref. 50) stimulated fusion in the presence of 1 mm Ca2+ (Fig. 6B, black lines). The extent and maximum rate of fusion obtained in the presence of both Ca2+ and synaptotagmin 1 was substantially greater than in the absence of either component (data not shown). Purified bovine Gβ1γ1 as well as recombinant His-tagged Gβ1γ2 purified from SF9 cells inhibited Ca2+-synaptotagmin-1 triggered fusion in a concentration-dependent manner with reductions in both the slope and maximal levels of fusion (Fig. 6B, blue and green lines). The potency for Gβ1γ2 inhibition of Ca2+ and SNARE-dependent synaptotagmin 1-driven lipid mixing was 14.5-fold higher than for Gβ1γ1 (Fig. 6C). Maximal concentrations of Gβ1γ1 reduced fusion to a baseline level, comparable with conditions lacking synaptotagmin 1 or Ca2+. In the absence of synaptotagmin 1, Gβ1γ1 did not additionally inhibit fusion (Fig. 6D), indicating that the effect of Gβγ is to inhibit synaptotagmin 1-stimulated fusion.
Figure 6.

Gβ1γ1 inhibits liposome fusion triggered by synaptotagmin 1 and Ca2+ in a concentration-dependent manner. A, diagram showing assay principle. Synaptobrevin-bearing liposomes (containing ∼50 copies/vesicle) containing the FRET pair NBD and rhodamine, covalently attached to PE, fuse with unlabeled liposomes containing t-SNARE complexes (containing ∼130 copies/vesicle) consisting of syntaxin1A and SNAP-25. The increased surface area of the fused liposome reduces the quenching of NBD fluorescence by rhodamine and NBD fluorescence increases as a result. B, NBD fluorescence traces over time for synaptotagmin 1 (C2AB domain; 10 μm)- and Ca2+ (1 mm)-dependent liposome fusion in the presence of a series of concentrations of purified bovine Gβ1γ1 (concentrations from 100 nm to 10 μm were tested). C, maximum fluorescence values obtained for each concentration of Gβ1γ1 and Gβ1γ2. An IC50 value of 1.782 μm was obtained for Gβ1γ1-mediated inhibition of synaptotagmin-1-regulated fusion (95% confidence interval (CI) 1.334–2.382 μm), whereas the potency was higher for Gβ1γ2 with an IC50 value of 156.7 nm (95% CI 36.18–678.1 nm). Conditions containing 0 nm Gβ1γ1 were plotted at the 100 nm point. Experiments were performed three or four times for a total of three or four technical replicates for each condition tested. D, the condition containing no synaptotagmin 1 or Gβ1γ1 was not different from the condition containing no synaptotagmin 1 and 10 μm Gβ1γ1 (p = 0.10, Student's t test with Welch's correction). The condition lacking synaptotagmin 1 or Gβ1γ1 was performed 11 times for 11 technical replicates, whereas the corresponding experiment with 10 μm Gβ1γ1 was performed four times for four technical replicates. E, maximum fluorescence values obtained for each concentration of Gβ1γ1 in the presence of either 10 μm synaptotagmin 1 C2AB (blue line) or 3.16 μm synaptotagmin 1 C2AB (orange line). Gβ1γ1 was significantly more potent at 3.16 μm synaptotagmin 1 C2AB, with an IC50 value of 987.5 μm (95% confidence interval 733 nm to 1.330 μm).
To investigate whether Gβγ had higher potency at alternative concentrations of synaptotagmin 1, we performed reconstituted fusion assays similar to those in Fig. 6C with a lower concentration of synaptotagmin 1 (3.16 μm) at 1 mm Ca2+. Gβ1γ1 was found to inhibit fusion 1.8-fold more potently in a concentration-dependent manner at this reduced concentration of synaptotagmin 1 (Fig. 6E).
Discussion
Multiple independent groups have reported that the Gβγ-SNARE interaction is one of several important mechanisms through which Gi/o-coupled GPCRs inhibit exocytosis (35–38) along with inhibition of Ca2+ influx by Gβγ through voltage-gated calcium channels. Mutagenesis studies have provided some of the strongest arguments in favor of this hypothesis; mutant forms of SNAP-25 that are unable to efficiently bind Gβγ are also unable to support Gi/o-coupled GPCR-mediated inhibition of exocytosis (30), and partial loss-of-function SNAP-25 mutants, with decreased Gβγ binding, show concomitant partially reduced Gi/o-coupled GPCR-mediated inhibition in the same populations of neurons (31). The heterogeneous nature of cellular systems prevents ruling out the possibility that an alternative effector for exocytosis that uses the same residues in SNAP-25 is similarly perturbed. Here, we provide strong evidence against this idea by showing that Gβγ inhibits synaptotagmin-1-regulated, SNARE-catalyzed liposome fusion in a purified system lacking any other proteinaceous components. A concentration dependence for Gβ1γ2 inhibition was observed, with an IC50 of 157 nm. Other components, such as voltage-gated calcium channels and adenylyl cyclases, are entirely absent. From these and prior studies, it can be concluded that Gβ1γ1 and Gβ1γ2 both bind SNAREs, including monomeric syntaxin1A, SNAP25, t-SNARE heterodimers, and/or VAMP2, incorporated into lipid bilayers, as determined here by fluorescence polarization or previously in aqueous solution (28, 29), and inhibits membrane fusion. The inhibitory effect of Gβγ on liposome fusion is only observed in the presence of Ca2+-synaptotagmin 1. The binding sites of Gβγ on SNAP-25 and synaptotagmin 1 on ternary SNAREs are known, encompassing overlapping but not identical regions (10, 30, 33). It has previously been shown that Gβγ competes with synaptotagmin 1 for binding sites on t-SNARE heterodimers and SNAP-25 in solution (28, 29). From these prior studies in tandem with the competition-binding assay studies conducted in Fig. 2 and our data showing that Gβγ has minimal effects on fusion in the absence of synaptotagmin 1 (Fig. 6D), we conclude that the inhibition of liposome fusion occurs directly via Gβγ binding to membrane-embedded SNAREs in a competitive manner with synaptotagmin 1. This observation corroborates and extends our previously published work (28–32). Despite this, it is clear that inhibition of voltage-gated calcium currents is an important mechanism through which Gβγ inhibits exocytosis for some, but not all, Gi/o-coupled GPCRs (20, 42).
Although a considerable amount is known regarding the SNAP-25 residues that mediate binding to Gβγ (29–32), comparatively little is known concerning which residues of Gβγ mediate binding to SNARE proteins. It has been hypothesized that the SNARE-binding residues are located on the Gα-binding surface of Gβ, as heterotrimeric Gαβγ is incapable of binding SNAREs (29). Here, we validate and expand upon those results by demonstrating the role of Trp-332 of Gβ in interaction with the SNARE complex and modulation of voltage-dependent calcium channels. This is a key Gα-binding residue required for heterotrimer assembly and receptor interaction as well as adenylyl cyclase activation (51). Strikingly, however, Ala mutation of this residue increases the potency of Gβγ inhibition of fusion in permeabilized PC12 cells (28), while abrogating modulation of voltage-dependent calcium channels. Strong linkage between this observation and the Gβγ–SNARE interaction has been generated here with the K78A/W332A double mutant of Gβ1γ2-binding lipid–embedded t-SNAREs with significantly higher affinity than wild-type Gβ1γ2. Peptide binding studies expanded upon these studies, with a peptide Gβ1 328–337 corresponding to this region on Gβ1-binding t-SNARE. Importantly, this peptide also inhibits the interaction of Gβ1γ2 with SNAP25 in a concentration-dependent manner, whereas peptides corresponding to other regions of Gβ1do not.
Two key regions of interaction were identified on Gγ. In both this article (Fig. 1) and previous work (28), Gβ1γ2 was shown to bind to t-SNARE complexes consisting of Stx1A and SNAP25 20-fold more tightly than Gβ1γ1. In Fig. 4, peptide competition data suggests that there is a SNAP-25-binding site in residues 9–28 of Gγ1 and 8–25 of Gγ2, but a larger 2–24-residue peptide is 4.66-fold more potent than the 8–25-residue peptide. This suggests that the N-terminal residues 2–7 of Gγ2 may be partially responsible for the increased ability of Gβ1γ2 to bind t-SNARE.
The reduced ability of W332A mutants to inhibit voltage-gated calcium channels further supports the role of the Gβγ–SNARE binding in inhibition of exocytosis. Our results echo prior studies showing the W332A mutant dramatically reduced the Gβγ-mediated inhibition of ICa (43, 51)(Fig. 3). The double mutant K78A/W332A produced only slight inhibition of ICa, with facilitation ratios not significantly different from control (no exogenous Gβγ) at any of the voltages tested (Fig. 3B). The K78A mutant has also been reported to reduce the inhibition of ICa (51), although we found only a modest effect (Fig. 3B). That said, the inhibition produced by K78A was more sensitive to membrane potential; it produced significant inhibition (prepulse facilitation) at hyperpolarized test potentials but not at the more depolarized test pulses (Fig. 3B). This might suggest a modest reduction in binding affinity with K78A, but we did not investigate this further in the present study.
The bulky tryptophan residue Trp-332 has previously been shown to be inhibitory to certain classes of Gβ effectors, with enhanced βARK interaction previously reported for W332A mutants (51). In the X-ray crystallographic structure of βARK in complex with Gβ1γ2 (46, 52), Trp-332 is in close proximity to the α-helical C terminus of βARK. Potentially, steric clashes may occur between Trp-332 and Lys-K663 or Asn-666 of βARK, which may be relieved in the W332A mutant. The enhanced binding affinity of W332A-containing mutant Gβγ for SNARE in in vitro binding assays supports a hypothesis that the side chain of Trp-332 and/or Lys-78 inhibits the interaction through steric clash and/or electrostatic repulsion with one of the Lys or Arg residues on the SNARE complex, such as Arg-135, Arg-136, Arg-142, Arg-161, Arg-198, or Lys-201 of SNAP25 (30). In line with this hypothesis, a peptide corresponding to residues 548–671 of βARK blocks Gβγ–SNARE–mediated inhibition of exocytosis (27). Charge-reversal of Gβγ-binding residues from Lys/Arg to Glu at the C terminus of SNAP25 is far more destructive than neutral mutation to Ala (30, 31), implying that negatively charged residues on Gβ or Gγ in addition to Lys-78/Trp-332 may contribute to the interaction.
Both direct binding assays and cell-based studies highlight the contribution of Gγ to the interaction, as Gβ1γ2 binds SNAREs and inhibits exocytosis with an order of magnitude higher affinity than Gβ1γ1 (28) despite the presence of identical Gβ in each complex. Two explanations for this phenomenon were hypothesized: specific residues on Gγ2 have higher affinity for SNARE than those on Gγ1, or the C-terminal geranylgeranyl modification of Gγ2 (53, 54) contributes to the interaction more so than the farnesyl modification of Gγ1. We have localized the SNARE-binding residues to the N terminus of Gγ2 (Fig. 5); thus, our data are supportive of the former hypothesis. Despite these studies, we have limited insights as to the specific binding mode of SNARE upon Gβγ. It is clear that X-ray crystallographic studies of the complete Gβγ–SNARE complex would yield tremendous insights as to the specific interplay of individual residues for the interaction.
In summary, we show that Gβγ subunits bind SNARE complexes in a lipid environment and inhibit fusion in a system containing only Gβγ, SNAREs, synaptotagmin 1, calcium, and lipids. We highlighted residues and regions of importance on Gβ and Gγ for SNARE binding. These studies provide further evidence for the Gβγ–SNARE hypothesis and highlight its importance in lipid bilayer-containing environments that more closely approximate the environment of the presynaptic active zone.
Experimental procedures
Plasmids
The open reading frames for the SNARE component proteins were subcloned into the glutathione S-transferase (GST) fusion vector, pGEX6p1 (GE Healthcare), for expression in bacteria. The dual-expression vector pRSF-Duet1 with subcloned N-terminal His-tagged full-length rat SNAP-25 and full-length rat syntaxin1A was previously described (55). Full-length bovine Gβ1 and His-tagged Gγ2 were incorporated in Sf9 vectors, described previously (51). The plasmid of a GST fusion with rat synaptotagmin1 (56) and the plasmid for the high-affinity Gβ1γ2 (K78A/W332A) were previously described (30). Voltage-gated calcium channel plasmids were as follows: bovine N-type CaV2.2 (GenBankTM number NM174632; rat brain β2a (GenBankTM number M80545); rat α2δ (GenBankTM number M86621).
Chemicals
Unless otherwise specified, chemicals were obtained from Sigma. Accudenz® A.G. Cell Separation Media was obtained from Accurate Chemical & Scientific Co.
Preparation and purification of recombinant proteins in Escherichia coli
Recombinant bacterially expressed syntaxin1A and His6-SNAP-25 were expressed in pRSF-Duet vector was transformed into E. coli strain BL21. His6VAMP2 was expressed in the plasmid pTW2. After the initial starter culture overnight in LB media with 50 μg/ml kanamycin (or ampicillin for pTW2), 4 liters of LB with antibiotic were inoculated with the starter culture and placed on a shaker at 37 °C until an A600 of 0.8 was obtained. Protein expression was induced with 0.4 mm isopropyl β-d-thiogalactoside for an additional 4 h at 37 °C. Bacterial cultures were pelleted and then resuspended in 10–15 ml of resuspension buffer (25 mm HEPES-KOH, pH 8.0, 400 mm KCl, 10 mm imidazole, and 5 mm β-mercaptoethanol). After adding protease inhibitors (aprotinin, leupeptin, pepstatin, and phenylmethylsulfonyl fluoride, cells were sonicated with a sonic dismembrator at 4 °C for 2 cycles of 45 s with 45 s of rest in between cycles. After sonication, 3 ml of 25% Triton X-100 was added to each tube and then incubated at 4 °C on a rotator for 3–4 h. These samples were then centrifuged to remove the insoluble material (25 min in a Beckman SW-34 rotor at 26,000 rpm). The supernatant was then purified via bulk affinity chromatography by mixing at 4 °C overnight with cobalt resin (Talon), prewashed, and equilibrated in resuspension buffer with the addition of 1% Triton X-100. The next day the beads were pelleted and washed twice with 5 volumes of OG wash buffer (25 mm HEPES-KOH, pH 8.0, 400 mm KCl, 20 mm imidazole, 5 mm β-mercaptoethanol, and 1% n-octylglucoside). After the second pelleting, the beads were then mixed at 4 °C on a rotator with 5 volumes of elution buffer (25 mm HEPES-KOH, pH 8.0, 400 mm KCl, 200 mm imidazole, 5 mm β-mercaptoethanol, 1% n-octylglucoside, and 10% glycerol) for 2 h. The samples were pelleted to remove beads from the eluted protein. The protein concentrations were approximately determined with the Pierce 660 nm Protein Assay (#22660, Thermo Scientific) and then confirmed for purity and concentration by SDS/PAGE analysis and comparison to a bovine serum albumin standard curve from the same gel. For synaptotagmin 1 purification, recombinant synaptotagmin 1 C2AB was prepared as a GST fusion according to previously published methods (47), substituting the imidazole for recombinant human rhinovirus 3C protease to liberate the synaptotagmin 1 from the GST tag.
Gβγ purification
Gβ1γ1 was purified from bovine retina as described previously (57). Recombinant Gβ1γ2 was expressed in Sf9 cells and purified according to the method of Kozasa and Gilman (58) with the following exceptions; frozen Sf9 cell pellets were lysed via sonication with a duty cycle of 10 s followed by a resting period of 20 s for 3 min at 30% intensity at 0 °C. Gβ1·His6-γ2 dimers were affinity-purified twice from detergent-solubilized crude cell membrane using Talon® cobalt resin (Clontech) followed by 3 rounds of dialysis for a minimum of 2 h in the following buffer: 20 mm Na-HEPES, pH 8.0, 100 mm NaCl, 10 mm 2-mercaptoethanol, 0.8% n-octylglucoside, 10% glycerol.
Gβγ and synaptotagmin 1 C2AB labeling
Purified Gβγ subunits were buffer-exchanged into 20 mm HEPES, pH 7.5, 100 mm NaCl, 1 mm MgCl2, 10 mm 2-mercaptoethanol, and 0.8% n-octylglucoside. Recombinant purified synaptotagmin 1 was buffer-exchanged into 25 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, and 10% glycerol. Alexa Fluor 488 NHS Ester (A20000, Invitrogen) or Alexa Fluor 488-C5-maleimide (A10254, Invitrogen) were prepared as a 10 mm solution in DMSO. Proteins were labeled at a 20:1 probe:protein ratio for 1 h for all labeling reactions. For Gβγ, the reaction was then quenched via the addition of 50 mm Tris-HCl and filtered through a 0.2 μm polyethersulfone filter before being purified on a TSKgel G2000SW gel filtration column (TOSOH Biosciences). Fractions were collected and concentrated on an Amicon Ultra 10,000 molecular weight cutoff centrifugal filter unit (Millipore) before being placed in a storage buffer consisting of 50 mm HEPES, pH 7.6, 100 mm NaCl, 5 mm 2-mercaptoethanol, and 10% glycerol. For synaptotagmin 1 C2AB, the reaction was quenched by the addition of 5 mm 2-mercaptoethanol and dialyzed to remove excess probe into 25 mm HEPES, pH 7.8, 150 mm NaCl, 1 mm DTT, and 10% glycerol. Labeling stoichiometry was ∼0.7–1 for synaptotagmin 1 C2AB and Gβγ.
Preparation of liposomes for fusion and TIRF assays
Protein-free and t-SNARE-embedded liposomes were made as described previously (47, 48). Briefly, a mixture of 55% POPC (1-palmitoyl-2-Oleoyl-sn-glycero-3-phosphocholine), 15% DOPS (1,2-dioleoyl-sn-glycero-3-phospho-l-serine (sodium salt) and 30% POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) in chloroform that would be equal to 15 mm of lipids in 100 μl was dried down. All lipids were purchased from Avanti Polar Lipids (Alabaster, AL). To this, either elution buffer (25 mm HEPES-KOH, pH 7.8, 400 mm KCl, 500 mm imidazole, 10% glycerol, 5 mm 2-mercaptoethanol, 1% n-octylglucoside) alone or with 0.4 mg of t-SNARE dimer was added to each tube of lipids to a final volume of 500 μl. For v-SNARE-containing liposomes, 1.5% 1.5% N-(7-nitro-2–1,3-benzoxadiazol-4-yl)-1,2-dipalmitoyl phosphatidylethanolamine (NBD-PE) and 1.5% N-(lissamine rhodamine B sulfonyl)-1,2-dipalmitoyl phosphatidylethanolamine (rhodamine-PE) were added to a mixture of 55% POPC/15% DOPS/27% POPC before drying down and 0.095 mg of recombinant His6-tagged VAMP2 was added in elution buffer. These mixtures were agitated on a tabletop vortex until the lipids were dissolved (10–15 min). After lipids were dissolved, 2 volumes of reconstitution buffer (25 mm HEPES-KOH, pH 7.8, 100 mm KCl, 10% glycerol, 1 mm DTT) was added dropwise, and the sample was vortexed gently for another 10 min. After 10 min, the sample was transferred to dialysis tubing (10,000 molecular weight cutoff, ThermoScientific) and dialyzed overnight with two dialysis buffer exchanges after 6 h. The dialysis buffer contained 25 mm HEPES-KOH, pH 7.8, 100 mm KCl, 10% glycerol, and 1 mm DTT in two 4-liter volumes. The next morning the samples were retrieved and mixed in equal volumes with a solution of 80% iohexol (Accudenz, Accurate Chemical Co.) in 25 mm HEPES-KOH, pH 7.8, 100 mm KCl, 10% glycerol, and 1 mm DTT. Gradients were assembled in thin-walled centrifuge tubes (#344057, Beckman Coulter) for a SW-55 swinging bucket rotor (Beckman Coulter) with 1.5 ml of the lipid/Accudenz mixture at the bottom, 1.5 ml of 30% Accudenz, and finally 450 μl of 0% Accudenz on the top. Liposomes were floated by centrifugation at 55,000 rpm for 2 h at 4 °C with minimal brake. Liposomes can be visualized as a thin uniform layer of opacity at the 0–30% interface of the gradient. Approximately 0.4 ml of liposomes were removed from each layer by direct puncture with a 27-gauge needle at the 0–30% Accudenz interface. All tubes from each preparation were mixed, aliquoted, and flash-frozen with an ethanol/dry ice bath. Liposomes were stored at −80 °C. Lipid concentrations and recovery rates were quantified using the Beer-Lambert law from NBD-PE absorbance values at 460 nm from liposomes containing NBD-PE that were diluted with dodecyl maltoside to 0.5%. SNARE protein concentrations in liposomes were determined by Coomassie Brilliant Blue R-250 staining of SDS-PAGE gels containing a standard curve of known concentrations of bovine serum albumin (Thermo Scientific) followed by densitometric analysis using the Fiji distribution of ImageJ software (59, 60). SNARE copy number was determined according to previously published methods (47).
Membrane TIRF imaging
Bilayers were prepared from 55% phosphatidylcholine, 15% phosphatidylserine, 29% PE, and 1% DiO liposomes with or without t-SNARE complexes as above except DiO (1%) was added. Coverslips were cleaned by soaking in 2% Hellmanex II solution, sonicated at 50 °C, rinsed in 18 megohms of deionized water and in 100% ethanol and stored in ethanol. For recording, coverslips were rinsed, dried under filtered compressed air, and placed in a microscope-recording chamber. 650 μl of HEPES-KCl was added to 100 μl of proteoliposome mix, and then 750 μl of HEPES-KCl with 10 mm CaCl2 was added. 25 μl of this mixture was placed in the coverslip chamber and sat for 1 h to allow a lipid bilayer to settle on the coverslip. This lipid bilayer was washed with a superfusate of 290 mm HEPES-KCl and 10 mm EGTA titrated with CaCl2 to a final free Ca2+ concentration of 100 nm). The DiO fluorescent bilayer was excited using laser TIRF microscopy under a 60× 1.45 NA lens (Olympus Plan-APO-N). This enabled the correct focal plane to be attained and alignment of the TIRF laser angle to be made. DiO fluorescence was then bleached to extinction by continuous excitation (20 min). Gβγ labeled with Alexa 488 (see above) was pressure-ejected from a pipette over the bilayer to obtain a transient exposure of Gβγ to the lipid bilayer, and the intensity of fluorescence excited with TIRF illumination was then measured with photomultipliers, whereas Gβγ concentrations and subtype were varied in the pipette. The concentration of Gβγ over the bilayer was measured by conventional epifluorescence through the coverslip, and the resulting signal amplitude was compared with known concentrations of labeled Gβγ in the recording chamber. In some experiments fluorescence anisotropy was also recorded to confirm protein-protein interactions occurred. This was achieved by measuring fluorescence through a polarizing beamsplitter with a detector parallel and orthogonal to the plane of the TIRF laser plane polarization angle.
HEK cell culture and transfection
HEK293 cells were maintained in an incubator at 37 °C and 5% CO2 and were passaged every 3–4 days for up to 15 passages. Cells were cultured in Minimum Essential Media (MEM; Life Technologies) supplemented with fetal bovine serum (10%; GE Healthcare), l-glutamine (2 mm; Life Technologies), and penicillin/streptomycin (100 unit ml−1/100 μg ml−1; Mediatech Inc., Manassas, VA). 24 h before transfection, cells were plated in 35-mm dishes according to manufacturer's guidelines for Lipofectamine 2000 (Invitrogen) transfection. Cells were transiently transfected with voltage-gated calcium channel subunits CaV2.2, β2a, an α2δ alongside Gβ1 (either WT, K78A, W332A, or K78A/W332A) and Gγ2 subunits in a 1:1:1:3:3 ratio, respectively. Some control cells were transfected with only the voltage-gated calcium channel subunits. Transfected cells were visually identified by GFP expressed downstream of an IRES (internal ribosome entry site) sequence in the β2a subunit plasmid. Cells were re-plated onto poly-l-lysine–coated coverslips 48–60 h after transfection and left to adhere for 2 h before patch clamp electrophysiology experiments.
Patch clamp electrophysiology experiments
Transfected HEK cells were recorded in the whole cell patch clamp configuration, and all experiments were performed at room temperature. Patch pipette electrodes were pulled from borosilicate glass capillary tubes (World Precision Instruments, Sarasota, FL) using a Sutter P-97 pipette puller (Sutter Instruments, Novato, CA), coated with dental wax (Electron Microscopy Services, Hatfield, PA), and fire-polished using a Narishige MF-830 micro forge (Narishige, Amityville, NY). Pipette resistance was ∼2 megohms when filled with an internal patch pipette solution containing 110 mm CsCl, 4 mm MgCl2·6H2O, 20 mm HEPES, 10 mm EGTA, 4 mm MgATP, 0.35 mm Na2GTP, 14 mm creatine phosphate, pH 7.3, osmolarity ∼305–310 mosmol. Coverslips were placed in a recording bath continually perfused with extracellular solution at a rate of ∼3 ml/min, and cells were viewed on a Nikon TE2000 inverted microscope. Cells were initially washed in a NaCl-based extracellular solution consisting of mm 145 NaCl, 2 mm KCl, 1 mm MgCl2·6H2O, 10 mm glucose, 10 mm HEPES, 2 mm CaCl2, pH 7.3, osmolarity ∼305 mosmol. After obtaining the whole cell recording configuration, the extracellular solution was a tetraethylammonium chloride-based (TEACl) solution containing 145 mm TEACl, 10 mm HEPES, 10 mm glucose, 1 mm MgCl2·6H2O, 5 mm BaCl2, pH 7.3, osmolarity ∼305 mosmol. Transfected HEK cells were voltage-clamped using an Axopatch 200B amplifier, Digidata 1400A interface, and PClamp10 (Clampex) acquisition software (Molecular Devices, Sunnyvale, CA). A double-pulse protocol was used to evoke voltage-gated calcium channel currents (ICa). Cells were stimulated by two identical 20-ms–step depolarizations (P1 and P2) to various potentials (−10, 0, 10, and + 20mV) from a holding potential of −80 mV. The second test pulse (P2; 270 ms after P1) was preceded by a 30-ms–conditioning prepulse to +120 mV. Analog data were filtered at 2 kHz and sampled at 20 kHz. Series resistance was partially compensated (∼60–70%) using the Axopatch circuitry, and ICa were subject to linear capacitance and leak subtraction using P/−8 protocols with leak pulses applied after test pulses. Raw data were analyzed in PClamp (Clampfit) software with ICa amplitude measured 5 ms after P1 or P2 test-pulse onset. Graphing and statistical analysis were performed using OriginPro 7 (Originlab Corp., Northampton, MA) and Prism 5 (GraphPad Software, Inc., La Jolla, CA) software. Statistical significance was determined using ANOVA with Dunnett's multiple pairwise comparison.
Peptide synthesis
Peptide array synthesis was performed using the ResPep SL peptide synthesizer (Intavis AG, Koeln, Germany) according to previously published automated SPOT synthesis methods (30, 61). Peptides correspond to the primary amino acid sequence of the protein of interest on the GenBankTM database for human GNB1, GNGT1, or GNG2.
Alphascreen competition-binding assays
Alphascreen luminescence measurements were performed in an EnSpire multimode plate reader (PerkinElmer Life Sciences) at 27 °C. Biotinylated SNAP-25 was diluted to a 5× concentration of 100 nm in assay buffer (20 mm HEPES, pH 7.0, 10 mm NaCl, 40 mm KCl, 5% glycerol, and 0.01% Triton X-100). An EC80 concentration of 180 nm purified His6-Gβ1γ2 was made in assay buffer. Peptide stocks in DMSO were spotted onto 384-well white OptiPlates (PerkinElmer Life Sciences) at concentration ranges of 1 nm to 100 μm using a Labcyte Echo 555 Omics acoustic liquid handler (Labcyte, Sunnyvale, CA), with DMSO being back-added to a final concentration of 0.1%. 4 μl of Gβ1γ2 solution was incubated with peptide for 5 min while shaking. After 5 min, 1μl of biotinylated SNAP25 was added to a final concentration of 20 nm. Subsequent to incubation while shaking for an additional 5 min, 10 μl of Alphascreen Histidine Detection Kit (nickel chelate) acceptor beads were added to a final concentration of 20 μg/ml in assay buffer. The assay plate was agitated in dim light for 30 min. At that point, Alphascreen Streptavidin Donor Beads were added to a final concentration of 20 μg/ml in dim light. All aqueous solutions in this assay were manipulated by a Velocity 11 Bravo liquid handler (Agilent Automation Solutions, Santa Clara, CA). The final volume in the assay plate was 25 μl. After being spun down briefly to settle all fluid at the bottom of the well, plates were incubated for an additional 1 h at 27 °C before being read in the EnSpire. 20 nm biotinylated recombinant glutathione S-transferase (Schistosoma japonicum) in place of SNAP-25 with Gβ1γ2 were used as a negative control for nonspecific binding in each assay. IC50 concentrations for each peptide were determined by sigmoidal dose-response curve-fitting with variable slope. To have a strong signal in the Alphascreen competition-binding assay that could still be competed with a peptide, we used an EC80 concentration (180 nm) of His6-tagged Gβ1γ2 combined with 20 nm recombinant human SNAP25 biotinylated on primary amine residues with NHS-biotin and increasing concentrations of peptides corresponding to a region on Gβ1 or Gγ1 in the Alphascreen bead assay. As a negative control, peptides were tested for their ability to disrupt a second Alphascreen assay in which donor and acceptor beads were reacted with 50 nm concentrations of a peptide consisting of a biotinylation site and a His-tag peptide (PerkinElmer Life Sciences).
Reconstituted fusion/lipid mixing assay
45 μl of t-SNARE liposomes were reacted with 5 μl of v-SNARE liposomes in a total of 75 μl of assay buffer (25 mm HEPES-KOH, pH 7.8, 100 mm KCl, 10% glycerol, 1 mm DTT, 0.2 mm EGTA) in white 96-well FluoroNunc plates (Thermo Fisher, Waltham, MA). Purified synaptotagmin 1 (10 μm) was added to the buffer along with a concentration-response curve of purified bovine Gβ1γ1 followed by the addition of the t-SNARE liposomes. All components of the fusion reaction, except v-SNARE vesicles, were combined and pre-warmed to 37 C for 15 min. NBD fluorescence (excitation 460 nm/emission 538 nm) was continuously monitored over 80 min in a BioTek Synergy plate reader under continuous agitation, with fluorescence intensity being read every 8 s. After 20 min, CaCl2 was added to a final concentration of 1.2 mm in the assay with 0.2 mm being chelated by EGTA to yield an effective concentration of 1.0 mm. After 80 min, dodecyl maltoside was added to a final concentration of 0.5% to maximally dequench NBD via infinite distance of the NBD:rhodamine FRET pair.
Protein structure visualization
All representatives of protein structure were made using PyMOL.
Statistics
Two-tailed Student's t tests and all concentration-response-curve-fitting sigmoidal dose-response with variable slope were performed using GraphPad Prism v.4.03 for Windows, (GraphPad Software, La Jolla, CA). Sigmoidal dose-response curves were plotted using 1500 line segments. For the purposes of IC50 estimation in sigmoidal concentration-response curves, all IC50 values were calculated using an additional maximal point representing an order of magnitude above the highest concentration tested at the lowest signal value obtained in the study. For two-tailed Student's t tests, p values < 0.05 were considered to be statistically significant. *, p < 0.05; **, p < 0.01.
Author contributions
Z. Z., B. P., M. C. C., R. L. B., C. A. W., K. P. M. C., E. R. C., S. A., and H. E. H. participated in research design. Z. Z., B. P., M. C. C., R. L. B., C. A. W., and S. A. conducted the experiments. Z. Z., C. A. W., A. M. P., K. H., J. A. G., and O. C.-R. contributed new reagents. Z. Z., B. P., M. C. C., R. L. B., C. A. W., K. P. M. C., S. A., and H. E. H. performed the data analysis. Z. Z., K. P. M. C., E. R. C., S. A., and H. E. H. wrote or contributed to the writing of the manuscript.
Acknowledgments
We thank Chantell S. Evans and Huan Bao for expertise in conducting lipid mixing assays as well as Jin Liao and James Gilbert for assistance in protein purification. We thank the following laboratories for providing plasmids: Dr. Aaron Fox (University of Chicago, Chicago, IL); Dr. Roger Colbran (Vanderbilt University, Nashville, TN); Dr. Terry Snutch (University of British Columbia, Vancouver, Canada).
This work was supported, in whole or in part, by National Institutes of Health Grants R01NS052446 (NINDS), R01EY010291 (NEI), and R01MH101679, R01MH061876, and R01MH084874 (National Institute of Mental Health). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G protein–coupled receptor
- Gβγ
- G protein βγ subunit
- SNARE
- soluble N-ethylmaleimide attachment protein receptor
- SNAP-25
- synaptosomal-associated protein of 25 kDa
- VAMP2
- synaptobrevin syt1
- C2AB
- the tandem C2A-C2B domain of synaptotagmin 1
- HEK
- human embryonic kidney cells 293
- TIRF
- total internal reflection microscopy
- PE
- phosphatidylethanolamine
- NBD-PE
- N-(7-nitro-2–1,3-benzoxadiazol-4-yl)-1,2-dipalmitoyl PE
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine)
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- ANOVA
- analysis of variance.
References
- 1. Betke K. M., Wells C. A., and Hamm H. E. (2012) GPCR mediated regulation of synaptic transmission. Prog. Neurobiol. 96, 304–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rizo J., and Xu J. (2015) The synaptic vesicle release machinery. Annu. Rev. Biophys. 44, 339–367 [DOI] [PubMed] [Google Scholar]
- 3. Südhof T. C. (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Augustin I., Rosenmund C., Südhof T. C., and Brose N. (1999) Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461 [DOI] [PubMed] [Google Scholar]
- 5. Jahn R., and Scheller R. H. (2006) SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643 [DOI] [PubMed] [Google Scholar]
- 6. Chapman E. R., and Jahn R. (1994) Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes: autonomous function of a single C2-homologous domain. J. Biol. Chem. 269, 5735–5741 [PubMed] [Google Scholar]
- 7. Perin M. S., Fried V. A., Mignery G. A., Jahn R., and Südhof T. C. (1990) Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature 345, 260–263 [DOI] [PubMed] [Google Scholar]
- 8. Rizo J., and Südhof T. C. (2012) The membrane fusion Enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annu. Rev. Cell Dev. Biol. 28, 279–308 [DOI] [PubMed] [Google Scholar]
- 9. Chapman E. R., Hanson P. I., An S., and Jahn R. (1995) Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J. Biol. Chem. 270, 23667–23671 [DOI] [PubMed] [Google Scholar]
- 10. Zhang X., Kim-Miller M. J., Fukuda M., Kowalchyk J. A., and Martin T. F. (2002) Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron 34, 599–611 [DOI] [PubMed] [Google Scholar]
- 11. Gerona R. R., Larsen E. C., Kowalchyk J. A., and Martin T. F. (2000) The C terminus of SNAP25 is essential for Ca2+-dependent binding of synaptotagmin to SNARE complexes. J. Biol. Chem. 275, 6328–6336 [DOI] [PubMed] [Google Scholar]
- 12. Wang Z., Liu H., Gu Y., and Chapman E. R. (2011) Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J. Cell Biol. 195, 1159–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu H., Bai H., Xue R., Takahashi H., Edwardson J. M., and Chapman E. R. (2014) Linker mutations reveal the complexity of synaptotagmin 1 action during synaptic transmission. Nat. Neurosci. 17, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bai J., Tucker W. C., and Chapman E. R. (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11, 36–44 [DOI] [PubMed] [Google Scholar]
- 15. Oldham W. M., and Hamm H. E. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71 [DOI] [PubMed] [Google Scholar]
- 16. Dunlap K., and Fischbach G. D. (1978) Neurotransmitters decrease the calcium component of sensory neuron action potentials. Nature 276, 837–839 [DOI] [PubMed] [Google Scholar]
- 17. Bean B. P. (1989) Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340, 153–156 [DOI] [PubMed] [Google Scholar]
- 18. Herlitze S., Garcia D. E., Mackie K., Hille B., Scheuer T., and Catterall W. A. (1996) Modulation of Ca2+ channels by G-protein βγ subunits. Nature 380, 258–262 [DOI] [PubMed] [Google Scholar]
- 19. Miller R. J. (1998) Presynaptic receptors. Annu. Rev. Pharmacol. Toxicol. 38, 201–227 [DOI] [PubMed] [Google Scholar]
- 20. Hamid E., Church E., Wells C. A., Zurawski Z., Hamm H. E., and Alford S. (2014) Modulation of neurotransmission by GPCRs is dependent upon the microarchitecture of the primed vesicle complex. J. Neurosci. 34, 260–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isaacson J. S. (1998) GABAB receptor-mediated modulation of presynaptic currents and excitatory transmission at a fast central synapse. J. Neurophysiol. 80, 1571–1576 [DOI] [PubMed] [Google Scholar]
- 22. Takahashi T., Kajikawa Y., and Tsujimoto T. (1998) G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J. Neurosci. 18, 3138–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scanziani M., Capogna M., Gähwiler B. H., and Thompson S. M. (1992) Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron 9, 919–927 [DOI] [PubMed] [Google Scholar]
- 24. Wollheim C. B., Kikuchi M., Renold A. E., and Sharp G. W. (1977) Somatostatin- and epinephrine-induced modifications of 45Ca2+ fluxes and insulin release in rat pancreatic islets maintained in tissue culture. J. Clin. Invest. 60, 1165–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mandarino L., Itoh M., Blanchard W., Patton G., and Gerich J. (1980) Stimulation of insulin release in the absence of extracellular calcium by isobutylmethylxanthine and its inhibition by somatostatin. Endocrinology 106, 430–433 [DOI] [PubMed] [Google Scholar]
- 26. Silinsky E. M. (1984) On the mechanism by which adenosine recptor activation inhibits the release of acetylcholine from motor nerve endings. J. Physiol. 346, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blackmer T., Larsen E. C., Takahashi M., Martin T. F., Alford S., and Hamm H. E. (2001) G protein βγ subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science 292, 293–297 [DOI] [PubMed] [Google Scholar]
- 28. Blackmer T., Larsen E. C., Bartleson C., Kowalchyk J. A., Yoon E. J., Preininger A. M., Alford S., Hamm H. E., and Martin T. F. (2005) G protein βγ directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat. Neurosci. 8, 421–425 [DOI] [PubMed] [Google Scholar]
- 29. Yoon E. J., Gerachshenko T., Spiegelberg B. D., Alford S., and Hamm H. E. (2007) Gβγ interferes with Ca2+-dependent binding of synaptotagmin to the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Mol. Pharmacol. 72, 1210–1219 [DOI] [PubMed] [Google Scholar]
- 30. Wells C. A., Zurawski Z., Betke K. M., Yim Y. Y., Hyde K., Rodriguez S., Alford S., and Hamm H. E. (2012) Gβγ inhibits exocytosis via interaction with critical residues on SNAP-25. Mol. Pharmacol. 82, 1136–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zurawski Z., Rodriguez S., Hyde K., Alford S., and Hamm H. E. (2016) Gβγ binds to the extreme C terminus of SNAP25 to mediate the action of Gi/o-coupled G protein-coupled receptors. Mol. Pharmacol. 89, 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gerachshenko T., Blackmer T., Yoon E. J., Bartleson C., Hamm H. E., and Alford S. (2005) Gβγ acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat. Neurosci. 8, 597–605 [DOI] [PubMed] [Google Scholar]
- 33. Zhou Q., Lai Y., Bacaj T., Zhao M., Lyubimov A. Y., Uervirojnangkoorn M., Zeldin O. B., Brewster A. S., Sauter N. K., Cohen A. E., Soltis S. M., Alonso-Mori R., Chollet M., Lemke H. T., Pfuetzner R. A., et al. (2015) Architecture of the synaptotagmin–SNARE machinery for neuronal exocytosis. Nature 525, 62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brewer K. D., Bacaj T., Cavalli A., Camilloni C., Swarbrick J. D., Liu J., Zhou A., Zhou P., Barlow N., Xu J., Seven A. B., Prinslow E. A., Voleti R., Häussinger D., Bonvin A. M., Tomchick D. R., et al. (2015) Dynamic binding mode of a synaptotagmin-1–SNARE complex in solution. Nat. Struct. Mol. Biol. 22, 555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delaney A. J., Crane J. W., and Sah P. (2007) Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron 56, 880–892 [DOI] [PubMed] [Google Scholar]
- 36. Zhao Y., Fang Q., Straub S. G., Lindau M., and Sharp G. W. (2010) Noradrenaline inhibits exocytosis via the G protein βγ subunit and refilling of the readily releasable granule pool via the αi1/2 subunit. J. Physiol. 588, 3485–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang X.-L., Upreti C., and Stanton P. K. (2016) Gβγ and the C terminus of SNAP-25 are necessary for long-term depression of transmitter release. PLoS ONE 6, e20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Glitsch M. (2006) Selective inhibition of spontaneous but not Ca2+-dependent release machinery by presynaptic group II mGluRs in rat cerebellar slices. J. Neurophysiol. 96, 86–96 [DOI] [PubMed] [Google Scholar]
- 39. Iremonger K. J., and Bains J. S. (2009) Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. J. Neurosci. 29, 7349–7358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoon E. J., Hamm H. E., and Currie K. P. (2008) G protein βγ subunits modulate the number and nature of exocytotic fusion events in adrenal chromaffin cells independent of calcium entry. J. Neurophysiol. 100, 2929–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Hook M. J., Babai N., Zurawski Z., Yim Y. Y., Hamm H. E., and Thoreson W. B. (2017) A presynaptic group III mGluR recruits Gβγ/SNARE interactions to inhibit synaptic transmission by cone photoreceptors in the vertebrate retina. J. Neurosci. 37, 4618–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zamponi G. W., and Currie K. P. (2013) Regulation of Ca(V)2 calcium channels by G protein–coupled receptors. Biochim. Biophys. Acta 1828, 1629–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McDavid S., and Currie K. P. (2006) G-proteins modulate cumulative inactivation of N-type (Cav2.2) calcium channels. J. Neurosci. 26, 13373–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sondek J., Bohm A., Lambright D. G., Hamm H. E., and Sigler P. B. (1996) Crystal structure of a G-protein βγ dimer at 2.1 Å resolution. Nature 379, 369–374 [DOI] [PubMed] [Google Scholar]
- 45. Lambright D. G., Sondek J., Bohm A., Skiba N. P., Hamm H. E., and Sigler P. B. (1996) The 2.0 Å crystal structure of a heterotrimeric G protein. Nature 379, 311–319 [DOI] [PubMed] [Google Scholar]
- 46. Lodowski D. T., Pitcher J. A., Capel W. D., Lefkowitz R. J., and Tesmer J. J. (2003) Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gβγ. Science 300, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 47. Tucker W. C., Weber T., and Chapman E. R. (2004) Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438 [DOI] [PubMed] [Google Scholar]
- 48. Weber T., Zemelman B. V., McNew J. A., Westermann B., Gmachl M., Parlati F., Söllner T. H., and Rothman J. E. (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 [DOI] [PubMed] [Google Scholar]
- 49. Bhalla A., Chicka M. C., Tucker W. C., and Chapman E. R. (2006) Ca2+-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat. Struct. Mol. Biol. 13, 323–330 [DOI] [PubMed] [Google Scholar]
- 50. Gaffaney J. D., Dunning F. M., Wang Z., Hui E., and Chapman E. R. (2008) Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J. Biol. Chem. 283, 31763–31775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ford C. E., Skiba N. P., Bae H., Daaka Y., Reuveny E., Shekter L. R., Rosal R., Weng G., Yang C. S., Iyengar R., Miller R. J., Jan L. Y., Lefkowitz R. J., and Hamm H. E. (1998) Molecular basis for interactions of G protein βγ subunits with effectors. Science 280, 1271–1274 [DOI] [PubMed] [Google Scholar]
- 52. Waldschmidt H. V., Homan K. T., Cruz-Rodríguez O., Cato M. C., Waninger-Saroni J., Larimore K. M., Cannavo A., Song J., Cheung J. Y., Kirchhoff P. D., Koch W. J., Tesmer J. J., and Larsen S. D. (2016) Structure-based design, synthesis, and biological evaluation of highly selective and potent g protein-coupled receptor kinase 2 inhibitors. J. Med. Chem. 59, 3793–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Simonds W. F., Butrynski J. E., Gautam N., Unson C. G., and Spiegel A. M. (1991) G-protein βγ dimers. Membrane targeting requires subunit coexpression and intact γ CAAX domain. J. Biol. Chem. 266, 5363–5366 [PubMed] [Google Scholar]
- 54. Takida S., and Wedegaertner P. B. (2003) Heterotrimer formation, together with isoprenylation, is required for plasma membrane targeting of Gβγ. J. Biol. Chem. 278, 17284–17290 [DOI] [PubMed] [Google Scholar]
- 55. Chicka M. C., Hui E., Liu H., and Chapman E. R. (2008) Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+. Nat. Struct. Mol. Biol. 15, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chapman E. R., Desai R. C., Davis A. F., and Tornehl C. K. (1998) Delineation of the oligomerization, AP-2 binding, and synprint binding region of the C2B domain of synaptotagmin. J. Biol. Chem. 273, 32966–32972 [DOI] [PubMed] [Google Scholar]
- 57. Mazzoni M. R., Malinski J. A., and Hamm H. E. (1991) Structural analysis of rod GTP-binding protein, Gt. limited proteolytic digestion pattern of Gt with four proteases defines monoclonal antibody epitope. J. Biol. Chem. 266, 14072–14081 [PubMed] [Google Scholar]
- 58. Kozasa T., and Gilman A. G. (1995) Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of α12 and inhibition of adenylyl cyclase by αz. J. Biol. Chem. 270, 1734–1741 [DOI] [PubMed] [Google Scholar]
- 59. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schindelin J., Rueden C. T., Hiner M. C., and Eliceiri K. W. (2015) The ImageJ ecosystem: an open platform for biomedical image analysis. Mol. Reprod. Dev. 82, 518–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yim Y. Y., Betke K., and Hamm H. (2015) Using peptide arrays created by the SPOT method for defining protein-protein interactions. In Protein-Protein Interactions: Methods and Applications (Meyerkord C. L., and Fu H., eds.) pp 307–320, Springer New York, New York: [DOI] [PubMed] [Google Scholar]