

Abstract
Subcellular mislocalization of the microtubule-associated protein Tau is a hallmark of Alzheimer disease (AD) and other tauopathies. Six Tau isoforms, differentiated by the presence or absence of a second repeat or of N-terminal inserts, exist in the human CNS, but their physiological and pathological differences have long remained elusive. Here, we investigated the properties and distributions of human and rodent Tau isoforms in primary forebrain rodent neurons. We found that the Tau diffusion barrier (TDB), located within the axon initial segment (AIS), controls retrograde (axon-to-soma) and anterograde (soma-to-axon) traffic of Tau. Tau isoforms without the N-terminal inserts were sorted efficiently into the axon. However, the longest isoform (2N4R-Tau) was partially retained in cell bodies and dendrites, where it accelerated spine and dendrite growth. The TDB (located within the AIS) was impaired when AIS components (ankyrin G, EB1) were knocked down or when glycogen synthase kinase-3β (GSK3β; an AD-associated kinase tethered to the AIS) was overexpressed. Using superresolution nanoscopy and live-cell imaging, we observed that microtubules within the AIS appeared highly dynamic, a feature essential for the TDB. Pathomechanistically, amyloid-β insult caused cofilin activation and F-actin remodeling and decreased microtubule dynamics in the AIS. Concomitantly with these amyloid-β–induced disruptions, the AIS/TDB sorting function failed, causing AD-like Tau missorting. In summary, we provide evidence that the human and rodent Tau isoforms differ in axodendritic sorting and amyloid-β–induced missorting and that the axodendritic distribution of Tau depends on AIS integrity.
Keywords: Alzheimer disease, amyloid-beta (Aβ), cell polarity, microtubule, neurodegeneration, Tau protein (Tau), tauopathy, AIS
Introduction
Tau is one of the major microtubule-associated proteins (MAPs)4 in neurons and promotes axonal microtubule (MT) assembly and stabilization (1, 2). In the human CNS, Tau occurs in 6 isoforms, which are characterized by the presence or absence of the second repeat (3R or 4R-Tau) or the presence or absence of N-terminal inserts (0N-, 1N-, or 2N-Tau; see Fig. 1A for details) (3, 4). In adult humans, the two 2N isoforms (2N3R and 2N4R) are underrepresented, only 3–4% each, compared with 16.7% each if all six isoforms were equally expressed. In adult mice, there is no 3R isoform, but the three 4R isoforms are more evenly represented (varying between 20 and 40% each). During development, the isoform ratio of human Tau shifts from predominantly 3R-Tau isoforms to a roughly even ratio of 3R and 4R (Table 1) (4–7). Tau is generally considered an axonal protein in mature neurons (8), but in Alzheimer disease (AD) and other tauopathies, missorting of Tau protein into the somatodendritic compartment is an early pathological event (9, 10). In cell culture and mouse models of AD and frontotemporal lobar degeneration with tauopathy (FTLD-Tau), missorting of Tau coincides with synapse loss and functional impairments (11–13).
Figure 1.

The second repeat and inserts synergistically prevent Tau from retrograde passage through the TDB in the AIS, whereas mutations cause retrograde leakage into the soma. A, bar diagrams of the studied isoforms of Tau. Red, alternatively spliced N-terminal inserts (N1 and N2); green, repeat domain; yellow, repeat 2 (R2). Mutated 2N4R-Tau species are displayed in the bottom half, and mutations are represented by red vertical lines. B, photoconversion of primary rat cortical neurons transfected with control 2N4R-TauD2 (top) or 2N4R-TauD2-KXGE mutant (bottom). 1 and 5, reference image of unconverted protein before photoconversion to identify soma and axon in the green channel. 2 and 6, before photoconversion, there is no fluorescence in the red channel. 3 and 4a, after photoconversion in the region of interest (dotted box, red), cells transfected with 2N4R-TauD2 (top) show movement of TauD2 beyond the region of interest but not into the soma (red triangle) and dendrites, indicating a retrograde barrier in the initial part of the axon. 7 and 8a, in contrast, cells transfected with 2N4R-TauD2-KXGE mutant show penetration of photoconverted protein into the somatodendritic compartment. Pictograms in 4b and 8b are a graphical representation of 2N4R-TauD2 unable to cross the diffusion barrier within the AIS (4b, cell body remains dark) and 4KXGE-TauD2, which crosses the AIS and reaches the cell soma (8b, cell body lights up). C, level of Tau leaking through the barrier into the soma after photoconversion shown for several Tau isoforms (blue) and Tau mutations (red). Isoforms lacking both the second repeat (R2) and the second insert (N2) penetrate into the somatodendritic compartment. Pseudophosphorylated Tau (KXGE) or FTLD-Tau related mutations (A152T and ΔK280) also cause incomplete axonal retention. Note that the tightness of the barrier does not depend on the propensity of Tau for β-structure that controls aggregation (compare ΔK280 and ΔK280PP). D, decay of a selection of photoconverted Dendra2 and TauD2 species in the photoconverted area of the axon. Axonal diffusion rates correlate with the MT binding potential of the species involved in some cases (e.g. Dendra2 → low and 8-repeat Tau → high), but not in case of FTLD-related Tau mutations. Note that the half-life of 2N4R-Tau is roughly intermediate between those of 8-repeat Tau and KXGE-Tau (indicated by dotted colored lines). a.u., arbitrary units. E, quantification of the half-lives in the photoconverted area of the axon (distal to the AIS) of all Tau isoforms, including 2N4R-Tau (§), Dendra2 alone (†), 8-repeat Tau (‡), KXGE-Tau (●), A152T-Tau, Tau ΔK280, and Tau ΔK280PP. Bars marked as Dendra2 alone (†), 8-repeat Tau (‡), 2N4R-Tau (§), and KXGE-Tau (●) are the quantifications of the decay curves displayed in D. *, p < 0.05. Error bars, S.E.
Table 1.
mRNA and protein levels of the different isoforms of Tau in human and mouse brain
| Tau clone names (3) | Analogous Tau isoform (N, inserts; R, repeats) | Isoform fraction adult human mRNA (4) | Isoform fraction adult human protein (cortex)a (5) | Isoform fraction adult mouse mRNA (6) | Isoform fraction adult mouse protein (6) |
|---|---|---|---|---|---|
| hTau40 | 2N4R | 3 | 8 ± 7 | 38 | 38 |
| hTau34 | 1N4R | 24 | 23 | 30 | 41 |
| hTau24 | 0N4R | 23 | 13 | 32 | 21 |
| hTau39 | 2N3R | 4 | 3 | NAb | NA |
| hTau37 | 1N3R | 25 | 26 | NA | NA |
| hTau23 | 0N3R | 21 | 19 | <1 | <1 |
a Note that the values in column 4 do not add up to 100%, probably due to sample variation in Ref. 5.
b NA, not applicable.
Abnormal splicing that only affects the isoform ratios of Tau without altering the amino acid sequence is sufficient to cause FTLD-Tau (4, 14, 15). Tauopathies can be classified depending on the isoform of Tau that appears in pathological paired helical filaments, neurofibrillary tangles, or thioflavin-negative accumulations of Tau (16–18). Pick disease is a 3R tauopathy, corticobasal degeneration and progressive supranuclear palsy (PSP) are 4R tauopathies, AD is a 3R+4R tauopathy, and familial cases of FTLD-Tau with mutations of Tau can be either 3R or 4R or 3R+4R tauopathies (18). Also, the interactome is different for the different isoforms of Tau (19). Thus, Tau isoform distribution and function may be crucial for disease development, but these aspects are understudied.
The mechanisms proposed for the polarized distribution of Tau in adult neurons include (i) preferential binding to MTs in the axon, (ii) preferential degradation in the somatodendritic compartment (20), (iii) axonal transport of Tau mRNA (21), (iv) preferred protein translation in axons (22), and (v) preferred retention by an axonal barrier (23). Because most protein synthesis takes place in the cell body, the bulk of Tau must transit from the cell body to the axon through the axon initial segment (AIS), which roughly coincides with a retrograde Tau barrier. The AIS is enriched with cytoskeletal scaffolding proteins ankyrin G and βIV spectrin, which together cluster voltage-gated sodium and potassium channels (Nav and Kv, respectively) and link them to the actin cytoskeleton (24). Functionally, the AIS plays a role in action potential initiation and modulation as well as neuronal polarity (25). Several studies describe the barrier function of the AIS as being based on lipids, membranes, F-actin, or MTs, depending on the components studied (23, 26–30). The internal structure of the AIS and its relationship to barrier functions has remained elusive since its discovery (31), but recent studies have revealed a dense, fibrillar/globular submembranous coat containing the classical AIS proteins (e.g. ankyrin G and βIV spectrin), which covers MT bundles (32).
The AIS and neuronal polarity are established early in development in vivo and can be mirrored in vitro. With the emergence of the axon, Tau becomes gradually enriched in the axon concurrent with AIS establishment (33, 34). Similarly, Tau isoforms change from the fetal to the adult distribution, due to alternative splicing (Table 1). The initially high Tau phosphorylation decreases with neuronal maturation, but in neurodegeneration Tau becomes “hyperphosphorylated,” concomitant with missorting and aggregation. Hyperphosphorlyation of Tau in turn acts on the AIS localization within the axon, thereby decreasing neuronal excitability (35). A major kinase acting on Tau is GSK3β, which phosphorylates many of the Ser-Pro or Thr-Pro motifs and is implicated in neurodegeneration (36). GSK3β is important for AIS function because it is an integral part of the AIS architecture, where it induces local tethering of β-catenin and ankyrin G (37).
We recently discovered a barrier that blocks the retrograde passage of Tau from the axon into the soma and dendrites (termed the Tau diffusion barrier (TDB), located at the AIS) (23). Here, we investigate the relationship between the TDB and the AIS and their effects on different isoforms of Tau. We found that the TDB has different retention efficiencies for different isoforms of Tau. The TDB functions not only in a retrograde fashion but also anterogradely; it partially retains the largest isoform of Tau in the dendrites, where it enhances dendritic growth and spine formation. Knockdown of structural components of the AIS decreases the Tau barrier efficiency. The Tau and AIS kinase GSK3β induces missorting of endogenous and exogenous Tau, but independently of Tau phosphorylation. STED nanoscopy and live-cell imaging show that MTs are major constituents of the TDB in the AIS, with unusual dynamics. Exposure of neurons to amyloid-β oligomers results in impairment of the F-actin–based filter in the AIS and decreased MT dynamics, resulting in failure of proper axodendritic Tau sorting.
Results
The Tau diffusion barrier is isoform-selective
Previous studies have shown that there is a retrograde diffusion barrier for Tau at the AIS that retards the back-diffusion of Tau from the axon once it has entered it (23). These results were obtained using a Tau fusion protein with the photoconvertible GFP derivative Dendra2 (38), linked to the N terminus of full-length Tau (TauD2). This allowed us to observe the movement of axonal Tau after local UV irradiation. Phosphorylated forms of TauD2 were able to penetrate the barrier to some extent. In the earlier study, we focused on the Tau barrier with regard to full-length Tau (2N4R) only and its modification by phosphorylation. However, Tau contains several domains that may also modulate its functions.
In the present study, we tested all six isoforms of Tau (see Table 1 for comparisons and nomenclature of the isoforms and Fig. 1A for a diagram). Transfected species of TauD2 were distributed throughout the cell (Fig. 1B, 1 and 5, green emission of unconverted TauD2). Then ∼15 μm of the proximal axon (∼50 μm away from the cell body) were irradiated with UV light, causing the Dendra2 to convert from green to red fluorescence. This was then imaged for up to 1 h, and signals were measured in the axon or the cell body (indicated by red rectangles and red triangles, respectively) (Fig. 1B).
The reference values are represented by Dendra2 alone (barrier fully open, unrestricted diffusion from axon into cell body, Fig. 1C, fluorescence (F) ∼100 in arbitrary units (a.u., see “Materials and methods”)) and the 8-repeat construct of Tau (barrier fully closed, F ∼2.5 (background), almost no detectable diffusion into cell body). For the 4R isoforms, the barrier is relatively tight (F ∼5–10, teal bars), and for two of the 3R isoforms, there is intermediate leakiness (F ∼25, blue bars). For 2N4R-Tau mutants, the leakiness is intermediate to high (F ∼25–70, red bars). This is particularly apparent for the 4KXGE mutant of 2N4R-Tau (bar 9), where the target sites of the kinase MARK, the four KXGS motifs in the repeats, were pseudophosphorylated to KXGE, causing loss of MT affinity of this Tau construct (39).
Tau carrying mutations derived from FTLD or PSP also shows decreased MT affinity (40). Because the TDB correlates with Tau–MT interaction, we tested the TDB for (i) 2N4R-Tau mutated at sites causing FTLD (pro-aggregant Tau with deletion of Lys-280), (ii) a control version of this mutation that cannot aggregate due to the insertion of two prolines (anti-aggregant Tau ΔK280PP), and (iii) the mutant A152T of 2N4R-Tau implicated in PSP (41). All of these mutations increased the leakiness of the TDB for retrograde diffusion back into the cell soma (Fig. 1C, red bars). The example of A152T-Tau is remarkable, because a single point mutation outside the conventional MT-binding region (amino acids ∼200–370) can enhance the mobility of Tau in neurons. One exception to the above classification is 2N3R-Tau, where the barrier is almost tight (as in 2N4R), indicating that the second N-terminal insert (amino acids 74–102, exon 3) contributes strongly to the functional retention by the Tau barrier, although it lies outside the MT-binding domain. Some of these variations correlate with the affinity of Tau for MTs, but not all, so that we conclude that MT affinity alone is not sufficient to explain Tau retention in the axon.
The diffusion rates of the Tau species can be assessed by the fluorescence increase in the cell body (as shown above), but also by the decay in the photoconverted area, which reflects the diffusion of Tau both toward the cell body (retrogradely) and away (anterogradely toward the axon terminal) (Fig. 1, D and E). The reference values are set by Dendra2 alone (rapid decay because of fast diffusion, short half-life of ∼2 min) and 8R-Tau (very slow diffusion because of tight MT binding, long half-life of ∼46 min). Overall, tight MT binding correlates with slow diffusion within the axon (e.g. half-times Dendra2 < 4KXGE-Tau < 2N4R-Tau < 8-repeat Tau).
Exogenous Tau is efficiently sorted into axons of wild-type and TauKO neurons except the largest isoform, 2N4R
In the experiments described so far, we overexpressed the isoforms of TauD2 at high levels (∼20-fold of endogenous Tau) to be able to investigate the barrier. In this setting, using primary neurons at an early stage of polarization (8–9 days in vitro (DIV)), Tau is initially present in all compartments. Subsequently, we photoconverted a subset of molecules in the axon to observe their propagation. As an alternative approach, to confirm the functioning of the TDB at low Tau levels, we used a protocol that limits Tau expression to 1–3-fold of endogenous Tau (estimated at ∼1 μm (42)), which required antibody-mediated amplification of the transfected constructs. We also waited longer (4–6 days) to allow the transfected versions of Tau to reach their destiny compartment. We also used more mature well-polarized neurons (13 DIV) with longer processes from TauKO and wild-type mice. We found that the shorter human 4R isoforms (0N4R and 1N4R) and the 3R isoforms were highly enriched in axons, whereas the longest isoform (2N4R) was in part excluded from axons (Fig. 2 (A and B), supplemental Figs. S1 and S2 and supplemental Table S1).
Figure 2.

2N4R-Tau is retained in the somatodendritic compartment in wild-type neurons, where it induces accelerated spine and dendrite growth, whereas 0N4R-Tau can enter axons. Different isoforms of TauD2 were cotransfected with tdTomato (volume marker) for 6 days into primary neurons (7 DIV) derived from TauKO or wild-type mice. A and B, TauD2 transfected into TauKO cells (A) and wild-type cells (B). Transfected Tau was stained with an antibody against human Tau (CP27). Arrowheads, axon; arrows, dendrites. A, 0N4R-Tau shows strong enrichment in axons (for more constructs, see supplemental Fig. S1). B, wild-type neurons expressing endogenous Tau in addition to transfected exogenous TauD2 (stained with CP27). Exogenous and endogenous Tau are stained with a pan-Tau antibody (K9JA), and dendrites are stained with an antibody against MAP2. Columns 1 and 2, 0N4R-Tau is enriched in the axon. As a result, axonal Tau concentration is higher than in neighboring untransfected axons where only endogenous Tau is present, but it can barely be detected in dendrites with a pan-Tau antibody. Column 2 (bottom) shows magnified images from the boxed areas, and axon is indicated by arrowheads. Columns 3 and 4, 2N4R-Tau is not enriched in axons. Column 5, 2N4R-Tau pseudophosphorylated at the KXGS motifs (KXGS motifs mutated to KXGE) shows strong axonal enrichment. C1, quantification of axonal enrichment of different TauD2 isoforms and 2N4R-TauD2 mutations reveals lower axonal presence of the longest isoform of Tau (2N4R) compared with the other isoforms. The exception is the human-specific isoform 1N3R, not present in rodents. The mutations 4KXGE, A152T, and ΔK280PP of 2N4R-TauD2 increase the axonal sorting, whereas the mutation ΔK280 has no effect. C2, graphical sketch of the different distributions of the representative isoforms 0N4R (strong axonal enrichment) and 2N4R (weak axonal enrichment). Left panels, Tau distribution depicted in green. Right panels, Tau distribution in green presented relative to a volume marker with unbiased distribution in red, resulting in a ratiometric image. D and E, wild-type neurons with tdTomato as an unbiased volume/morphology marker. D, TauD2 transfection induces spine formation and maturation in young (10 DIV; 1–6) neurons, and Tau knockdown abolishes spine maturation in old (21 DIV; 7 and 8) neurons. 1, neurons only transfected with a control vector (Dendra2) show no spine formation and very few filopodia, representative of neurons on the brink of (but before) spine development. 2, transfection of 0N3R-Tau results in elongated filopodia-like protrusions up to 10 μm. 3, by contrast, 0N4R-Tau has no effect on dendritic spine/filopodia formation. 4, 2N4R-Tau-transfected cells show dendritic processes with mushroom heads (arrowheads), indicative of mature spines. 5 and 6, mutations of the KXGS motifs in the repeat domain to KXGE (5) or KXGA (6) of 2N4R-Tau prevent formation of protrusions and acceleration of spine maturation. For a comprehensive list of effects of all isoforms and several mutations, see supplemental Table S1. 7, aged control neurons display normal mature spines. 8, aged neurons transfected with shRNA against Tau display impaired spines. E1, Tau transfection induces dendritic outgrowth. Left, neuron transfected with a control vector (Dendra2). Right, neuron transfected with the 2N4R-Tau. The longest dendrites are indicated by red dashes. E2, quantification of the longest dendrite of cells transfected as indicated. F, graphical sketch of the dendritic effects of the representative Tau isoforms 0N3R (filopodia spines), 0N4R (no dendritic effects), and 2N4R (elongated dendrites and premature development of spines). *, p < 0.05. Error bars, S.E.
To quantify effects, we used an enrichment factor (e.f.), which is calculated by the normalized ratio of axonal presence versus dendritic presence of transfected Tau (see “Materials and methods”). Quantitatively, axonal e.f. was ∼75, 85, and 95 for 2N4R, 1N4R, and 0N4R, respectively. The 3R versions of Tau without or two inserts (i.e. 0N3R and 2N3R) showed axonal enrichment but to different extents (e.f. of ∼90 and 95), whereas the 1N3R isoform was, with an e.f. of 70, less efficiently sorted (supplemental Table S1 and Fig. 2C). With the exception of 2N4R-TauD2, Tau isoforms and mutants that were able to partially penetrate the TDB retrogradely were also axonally enriched in neurons transfected with a specific isoform. For example, 4KXGE-2N4R-TauD2 showed retrograde penetration of the TDB (Fig. 1, B and C) (23) and also axonal enrichment (i.e. anterograde penetration of the TDB), in contrast to other mutations (ΔK280, ΔK280PP, A152T, and all 2N4R-TauD2), which showed only slight retrograde penetration of the TDB and correspondingly only limited axonal sorting (Fig. 2 (A–C), supplemental Figs. S1 and S2, and supplemental Table S1). Because 2N4R-Tau has a much higher affinity with MTs than the tested mutations, this confirms that strong MT binding can only partly explain axonal sorting of Tau. Surprisingly, we found no differences for TauKO (KO of all isoforms (43)) and wild-type cells (for full comparisons, see supplemental Figs. S1 and S2), indicating that even when Tau is absent during development and after establishment of polarity, Tau can be as efficiently sorted as in wild-type conditions.
Dendritic Tau accelerates spine formation and dendritic elongation
In rodents, 3R-Tau is mainly expressed during the first week of neuronal development and decreases during the second week, whereas 4R-Tau is mainly expressed after 2 weeks. Because mice overexpressing the 2N4R isoform of Tau show enhanced LTP early in life (44) and throughout life (45), we hypothesized that Tau has a dendritic function in synapse formation. Because dendritic synapses sit on spines, we chose to express the Tau isoforms in young neurons, before spines are formed (7 DIV). We found that some isoforms of TauD2 induce accelerated spine formation, but with strikingly different phenotypes (Fig. 2D). For example, 0N3R induced elongated spine necks and mature spine heads, unlike filopodia (Fig. 2D, 2). 0N4R had almost no effect, whereas 2N4R-Tau induced mature spine heads (Fig. 2D and supplemental Table S1). Expression of the other native isoforms of Tau showed intermediate effects on spine development, but all of them induced an increase in spine number and either an increase in spine maturity or spine length. In detail, 1N4R-Tau showed intermediate effect on spine maturation, number, and length, positioning it right between 0N4R and 2N4R-Tau (supplemental Table S1). 2N3R-Tau showed intermediate and 1N3R-Tau showed slight increase in all parameters investigated (spine number, length, and maturity), positioning them between 0N4R and 2N4R. The mutations tested (ΔK280, ΔK280PP, A152T, KXGE, and KXGA, expressed on the basis of 2N4R-TauD2) resulted in more spines of variable phenotype, indicating that these mutations might affect the spine maturation and also spine/filopodia dynamics (Fig. 2D and supplemental Table S1). Because overexpression of Tau results in enhanced spine maturation, we tested whether there was an effect of acute knockdown of Tau via shRNA on spine maturation. When applied during the period of spine maturation (14–21 DIV, which coincides with the transition from 3R- to 4R-Tau), knockdown of all Tau isoforms via shRNA results in impaired spine formation (consistent with Ref. 46) (Fig. 2D, 7 and 8). However, we found no differences between wild-type and TauKO neurons in terms of aberrant spine formation after transfection of individual Tau isoforms. This indicates that forced expression of different isoforms (thereby also neglecting the physiological mixture of isoforms) results in aberrant spine development independent of the presence or absence of endogenous Tau. Not surprisingly, expression of Tau induced elongation of dendrites, but to different extents (Fig. 2E; graphical summary of Fig. 2, D and E, in Fig. 2F). Elongation of dendrites required the presence of N-terminal inserts and the presence of intact KXGS motifs but were largely unaltered in the case of the tested mutations (ΔK280, ΔK280PP, and A152T; all expressed on the basis of 2N4R-TauD2). Thus, elongation of dendrites correlates roughly with increased dendritic presence for some of the native Tau isoforms (high dendritic presence for N-containing isoforms results in elongated dendrites, whereas 0N-Tau isoforms do not elongate dendrites), except for the Tau mutations tested and the 2N3R isoform (intermediate dendritic presence). This indicates that the presence of the two N-terminal inserts is sufficient to induce dendrite elongation.
Structural components of the axon initial segment are required for the tightness of the anterograde and retrograde Tau diffusion barrier
Because the position of the TDB coincides roughly with that of the AIS, we determined which of the major known components of the AIS are essential for TDB function. Interestingly, knockdown of any of the AIS components by shRNA, such as neuronal cell adhesion molecules (NrCAM), Nav, or scaffold proteins (βIV spectrin or AnkG) (47), led to slight leakage of 2N4R-TauD2 from the axon into the soma (4–6-fold above background levels (supplemental Fig. S3, A and B)). This leakage is similar to the leakiness of one of the shorter isoforms of Tau (e.g. 0N3R; Fig. 1C). In addition, knockdown of the MT end-binding proteins EB1 and EB3, which are enriched at the AIS (48), led to similar enhanced retrograde leakage of TauD2 (supplemental Fig. S3, A and B). Thus, each of these AIS components contribute to the maintenance of the retrograde TDB.
We next tested whether knockdown of the different structural components of the AIS would also lead to enhanced anterograde movement of Tau. We thus expressed the longest isoform of Tau (2N4R-TauD2), which shows little anterograde axonal propagation compared with the other isoforms (Fig. 2 (A and B) and supplemental Table S1), in combination with knockdown of the AIS components (i.e. NrCAM, NaV, βIV spectrin, AnkG, EB1, and EB3; supplemental Fig. S3 (C and D), and see supplemental Fig. S4 (A, D, and E) for controls). In control conditions (empty shRNA vector, unrelated protein (Spast)), 2N4R-TauD2 is present slightly more in axons (∼100 μm from the cell body) than in the cell body or dendrites, as shown above, and MAP2 signals are low in axons (∼3-fold lower than in the cell body and 5-fold lower than in dendrites; supplemental Fig. S3, C and D). Knockdown of the AIS components AnkG and EB1 results in an increase of 2N4R-TauD2 in the axon by ∼50%, whereas the other components (NrCAM, Nav, βIV spectrin, and EB3) show no measurable effect. Knockdown of any component of the AIS caused invasion of MAP2 into the axon (supplemental Fig. S3D). Interestingly, none of the knockdowns resulted in impaired axonal sorting of endogenous rat Tau (supplemental Fig. S4). Together, these results indicate that the AIS structural components are important for both the regulation of anterograde and retrograde passage of exogenous Tau but do not hinder anterograde passage of endogenous Tau.
The protein kinase GSK3β is a structural component tethered to the AIS, affects the activity of AIS-based sodium channels (37), and is crucial for cell polarity and pathological development of tauopathies, including Alzheimer's (49, 50). We thus tested the effect of GSK3β on Tau distribution. Overexpression of GSK3β induces pronounced missorting even of endogenous Tau, in contrast to other structural components of the AIS mentioned above. Missorted Tau is phosphorylated at the KXGS motifs (supplemental Fig. S5A), despite the fact that GSK3β cannot phosphorylate Tau directly at these motifs. This is consistent with the hypothesis that Tau becomes phosphorylated by other kinases (e.g. MARK), after it has transited into the somatodendritic compartment (51). Missorting of Tau is recognized as a pathological sign and can be induced by exposure to Aβ (e.g. see supplemental Fig. S5A (1), arrowheads) and other forms of molecular stress (51). However, exposing GSK3β-transfected neurons to Aβ did not further increase phosphorylation or missorting of Tau (supplemental Fig. S5A). This indicates that either Aβ exposure and GSK3β activity activate the same pathway, or GSK3β activity changes the cell in a different fashion so that Aβ cannot exert its toxicity anymore. We also tested the state of GSK3β activity after exposure of primary neurons to Aβ, using phosphorylation at Ser-9 as a readout. Time slicing up to 24 h showed that there is a brief 30-min period of phosphorylation of GSK3β (inactivation), which then returns to baseline within 3 h (supplemental Fig. S5B). This indicates that there is no activation of GSK3β after exposure to Aβ.
We previously found that Aβ-induced Tau missorting requires loss of dendritic MTs (12). Here, we find that missorting of Tau is induced by overexpression of GSK3β (wild-type and constitutively active GSK3β-S9A), but not by inactive GSK3β-S9E, and thereby induced missorting of Tau does not lead to MT breakdown (supplemental Fig. S6A). Hence, MT loss cannot account for Tau missorting induced by GSK3β. We thus tested whether missorting of Tau induced by GSK3β is due to a change of Tau or a change of the TDB. Cotransfection of 2N4R-TauD2 and GSK3β results in enhanced retrograde propagation of axonal TauD2 into the cell body, indicative of a dysfunctional barrier, whereas inactive GSK3β (S9E mutant) has no effect (Fig. 3, A and B). GSK3β can phosphorylate Tau at many sites, possibly changing the diffusion capability of Tau or its interaction with the TDB. To determine the effect of GSK3β on the TDB independently of Tau phosphorylation, we cotransfected GSK3β with non-phosphorylatable TauD2 (17AP-TauD2). 17AP-TauD2 contains 17 proline-directed serines (putative GSK3β phosphorylation sites) mutated to alanine (17AP-TauD2). 17AP-TauD2 still results in enhanced retrograde propagation to the soma when cotransfected with GSK3β, but not when expressed alone or co-expressed with a control (CFP; Fig. 3, A and B). Diffusion rates within axons are the same for 2N4R-TauD2 irrespective of GSK3β activity (Fig. 3C). However, 17AP-TauD2 shows reduced diffusion by 50% already after 10 min within the axon (Fig. 3C, purple). This points toward an underestimation of the TDB dysfunction because there is less Tau available to cross the TDB. Taken together, this indicates that GSK3β influences Tau sorting via modifying either the TDB or some other factors maintaining the polarized distribution of Tau (e.g. inhibiting the Tau kinase MARK), but not by phosphorylating Tau.
Figure 3.
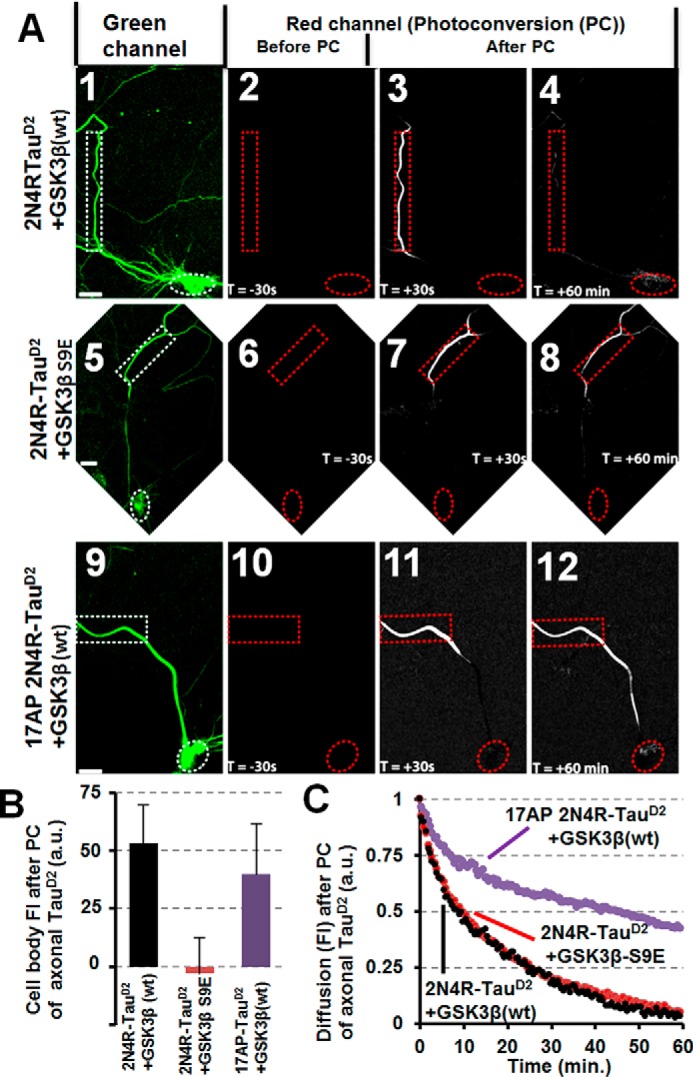
GSK3β induces missorting of Tau independently of phosphorylation of the SP/TP motifs of Tau. Primary cortical rat neurons (7 DIV) were cotransfected for 3 days with different versions of 2N4R-TauD2 and GSK3β (tagged with CFP). A, panels 1, 5, and 9 show cells in the green channel before photoconversion (PC); panels 2, 6, and 10 show cells in the red channel before PC; and panels 3, 4, 7, 8, 11, and 12 show cells in the red channel after photoconversion at the indicated times. Scale bars, 20 μm. A (1–4), primary rat cortical neurons (7 DIV) cotransfected with TauD2 and GSK3β(WT) show leakage of the Tau diffusion barrier and retrograde propagation of Tau back into the cell body (quantified in B (black bar)). A (5–8), inactive GSK3β-S9E does not cause leakage because TauD2 is retained in the axon and does not move into the cell body (quantified in B, red bar). A (9–12), mutating the GSK3β target sites of Tau to alanine (17 SP/TP motifs mutated to AP, AP17-TauD2) results in a leaky barrier so that AP17-TauD2 still shows retrograde propagation back into the cell body (quantified in B, purple bar). B, quantification of A as indicated. C, unable to be phosphorylated at its SP/TP motifs, AP17-Tau is revealed to have a very slow diffusion rate (purple trace). Penetration of the TDB can be seen, nonetheless, as was shown in panels 9–12 of A and the purple bar in B. Error bars, S.E.
AIS structure contributes to Tau diffusion barrier
Sequence motifs in the 5′- or 3′-UTRs of endogenous Tau mRNA have been implicated in the polarized trafficking and translation of Tau (21, 22). Our exogenously expressed Tau lacks these motifs, yet the protein is also sorted in a polarized fashion. This suggests that the mRNA-based mechanisms cannot account alone for the sorting of Tau protein. Because known structural elements of the AIS are necessary to maintain the tightness of the Tau diffusion barrier (supplemental Fig. S3), we hypothesized that there may be additional structural elements of the TDB not integrated into the AIS structure that determine the isoform-specific blockage of Tau. We previously showed that the efficiency of the TDB is decreased when MT dynamics are disturbed by the MT destabilizer nocodazole (i.e. loss of MTs made the barrier leaky) (23).
To examine MT-related properties, the beginning of the AIS was defined as the point where an AIS marker (NrCam) reaches 50% of its level within the AIS, typically 17 μm away from the edge of the cell body. This coincides roughly with the point where MAP2 (a somatodendritic marker) decreases to ∼50% of its value in the axon hillock (Fig. 4, A and B). Acetylated tubulin (a marker for stable MTs) starts low and increases slightly within the AIS, and polyglutamylated tubulin (stable MTs and a potential target for MT-severing enzymes) remains low throughout the AIS (Fig. 4 (A and C) and supplemental Fig. S7). Tyrosinated tubulin (a marker for dynamic MTs) continuously decreases from the beginning of the AIS onward to the axon, consistent with mainly dynamic MTs in the cell body and mainly stable MTs in the axon (Fig. 4C; for volume-normalized calculations and pictograms, see supplemental Fig. S7). We next used fixation protocols that either facilitate the visualization of polymerized tubulin (MTs) or free tubulin. This revealed high levels of tubulin in the AIS compared with the axon or the cell soma (∼2.5-fold higher) but only a low fraction of stainable MTs (Fig. 4D). The ratio of free tubulin versus MTs is ∼2:1 in the AIS and ∼1.5:1 in the cell body, but only 1:1 in the axon (Fig. 4D). This indicates a predominance of unstable MTs or depolymerized tubulin in the AIS.
Figure 4.

Posttranslational modifications of MTs and STED nanoscopy demonstrate highly dynamic MTs within the AIS. Primary cortical neurons were fixed at 9–12 DIV and stained as indicated. The AIS is marked by dotted lines. A, examples of images of cells highlighting axon initial segments. Arrowheads, axons; arrows, dendrites. Asterisks, staining artifacts. A1 and A2, double staining with MAP2 (blue) as a dendritic marker and NrCam (red) as an AIS marker allows identification of the AIS. Acetylated tubulin (green) was used as an axonal marker, highlighting stable MTs. Insets, graphical sketches. A3, magnification of the boxed area of A2. Note that there is no signal for acetylated tubulin within the AIS. B–D, quantification of immunofluorescence intensities from the middle of the cell body over the AIS to the proximal axon. Red vertical line, beginning of the AIS. The right sections of graphs indicate distal axons. On average, the AIS begins about 15–20 μm from the center of the cell body and extends for about 20–30 μm as the AIS markers gradually change toward their values in the proximal axon. B, the beginning of the AIS is characterized by a decrease of MAP2 (blue) and an increase of NrCAM (red). C, levels of tyrosinated tubulin (corresponding to dynamic MTs; red) are lower in the AIS than in the cell body but higher than in the distal axons. Levels of acetylated tubulin are low at the proximal AIS. D, different fixation and extraction procedures were used for the preferred observation of unpolymerized tubulin (green) or MTs (brown) (see “Materials and methods”). Levels of free tubulin are high within the AIS, yet levels of MTs are low, indicating high levels of unpolymerized tubulin. E–G, STED nanoscopy of MTs within the AIS. E and F, normal fixation/extraction method to favor stable MTs. E1 and F1, there are no MTs stainable with an antibody against α-tubulin (E1) and very few fragmented MTs with an antibody against tyrosinated (dynamic) MTs (F1) within the AIS (space between dotted lines). E2 and F2, NrCam was used for identification of the AIS. G, extraction and fixation were conducted in buffers containing the MT stabilizer taxol and the molecular densifier PEG. Left panels, NrCam and MAP2 were used to identify the AIS. Arrow, point of increasing NrCam signal and decreasing MAP2 signal, hence the beginning of the AIS. Right, high density of MTs within the AIS stained by an antibody against tyrosinated (dynamic) MTs; arrow at the same position as in the left panels.
We next attempted to visualize MTs in the AIS with STED nanoscopy. Within the AIS, no MTs are observed when using monoclonal antibodies against unmodified α-tubulin (Fig. 4E) or acetylated tubulin, and very few MTs are visible with an antibody against tyrosinated tubulin (Fig. 4F) with normal fixation procedures (i.e. no application of MT-modifying substances or permeabilization prior to fixation). This indicates that MTs within the AIS are too dynamic to be visualized with standard staining techniques. Only coapplication of the molecular densifier PEG, which creates high osmotic pressure allowing the fixatives to fix MT before they fall apart, together with the MT stabilizer taxol prior to fixation allowed us to observe MTs within the AIS, emphasizing the transient nature of these MTs (Fig. 4G).
Next, we examined MT dynamics directly by imaging of transfected EB3-GFP within the AIS. Due to the high density of dynamic MTs in the AIS (compared with the low density of dynamic MTs in axons, which are easier to image), a specialized imaging procedure was required to visualize EB3 comets within the dendrites and the AIS, using high-frame rate imaging combined with noise reduction (see “Materials and methods”). This revealed that within the AIS, there is a high concentration of transfected EB3 and a high density of EB3 comets, compared with axons and dendrites (Fig. 5, A and B). This again illustrates that MTs are highly dynamic and short-lived within the AIS. All comets observed within the AIS moved anterogradely, indicating a homogeneous polarity of MTs with plus ends pointing into the axon (in contrast to dendrites that contain MTs with mixed orientations (52)). The high concentration of EB3 comets hints at a hub or organization center for MTs at the AIS, probably affecting MT-based transport. Costaining of EB3 in fixed cells with MAP2 and mitochondria illustrates that the EB3 hub lies within the AIS (Fig. 5, C and D).
Figure 5.
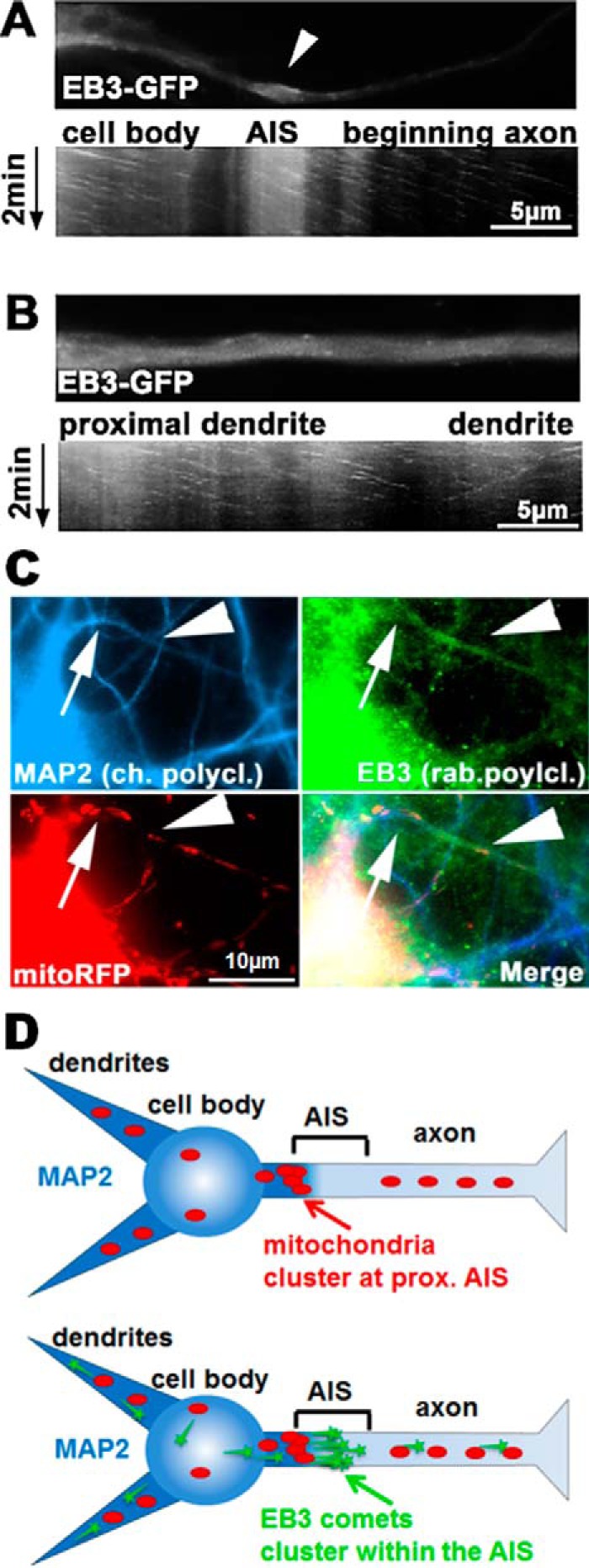
EB3 clusters in the proximal region of the AIS. Primary cortical neurons aged 9 DIV were transfected with EB3-GFP for 2 days. A and B, live imaging of EB3 and time-resolved kymographs of the AIS. A, EB3 comets are enriched and dynamic within the AIS. Top, first frame of time-lapse imaging of the AIS and the proximal axon. Arrow, thickening of the AIS typically present over 5–10 μm within the AIS. Bottom, kymograph of the EB3 comets show unidirectional movement only anterogradely, typical of the axon, and comets within the AIS thickening. B, analogous region as in A, but in a proximal dendrite. Note that EB3 comets move in both directions, and there is no thickening or area of enrichment of EB3. C, cells were fixed 2 days after transfection with mitoRFP and stained for MAP2 and EB3 for identification of the AIS. Arrow, beginning of AIS (end of MAP2 staining) and mitochondrial agglomeration proximal to the AIS; arrowhead, thickening of the AIS and enrichment with EB3 distal to the mitochondrial cluster. D, graphical sketch of mitochondrial and EB3 positioning at the AIS. Mitochondria cluster at the proximal end of the AIS (where MAP2 diminishes), whereas EB3 staining is localized within the AIS.
Exposure of neurons to Aβ results in activation of cofilin, loss of F-actin, and decreased MT growth within the AIS
Missorting of Tau induced by Aβ oligomers is not caused by breakdown of the retrograde barrier, but by failure of newly synthesized Tau to sort into the axon (51). This could be due to either a tightening of the anterograde barrier or loss of sorting-enabling structures. We thus tested whether Aβ exposure connects pathological missorting of Tau with defective elements of the AIS/TDB.
We first tested whether Aβ binds to the AIS. Aβ localizes to dendrites (primarily dendritic spines), but less to axons (90% less binding, judged by immunofluorescence). In contrast, Aβ localizes fairly well to the AIS (25% of dendritic localization; supplemental Fig. S8A). As a result of Aβ exposure, ankyrin G throughout the AIS and F-actin within the AIS decrease, indicative of a direct effect of Aβ on the AIS (supplemental Fig. S8B).
An F-actin meshwork represents a cytoplasmic barrier, which hinders non-axonal proteins and larger beads to enter the axon (27, 28, 53). We previously found that in dendritic spines, Aβ exposure leads to a rapid loss of F-actin (51). Testing F-actin content in the AIS both by live-cell imaging and phalloidin-based stainings reveals that F-actin is lost and remodeled rapidly after exposure to Aβ (i.e. within 15 min), but not, for example, in the dendritic shaft, where F-actin accumulates (Fig. 6, A and B). As cause for this actin remodeling, we found that cofilin, an F-actin–severing enzyme, becomes activated (dephosphorylated) in the AIS with a time course comparable with that of decreasing F-actin (Fig. 6). Thus, activation of cofilin could result in loss or remodeling of F-actin. Live imaging with an F-actin–sensitive probe (f-tractin) confirmed that the area covered by F-actin within the AIS increased, but only after 1.5 h of treatment with Aβ (Fig. 7A), whereas in control conditions, there was no change. This timing is in agreement with the increase in missorting of Tau (51) and the decrease of cofilin activity after initial activation by Aβ (Fig. 6). We next tested whether loss of F-actin alone would lead to missorting of Tau, due to simple loss of its cytoplasmic filter function. Treatment of primary neurons with various concentrations (0.5–2.5 μm) of the F-actin–destabilizing drug latrunculin A for various periods (5 min to 24 h) in no case lead to missorting of Tau, despite >90% reduction of F-actin staining upon treatment. Instead, co-treatment of primary neurons with latrunculin A and Aβ led to an attenuation (50%) of Aβ-induced missorting. Conversely, treatment of cells with the F-actin overstabilizer jasplakinolide (1 μm) was sufficient to induce missorting of Tau, even independently of Aβ (Fig. 7C).
Figure 6.

Aβ exposure results in cofilin activation and loss of F-actin within the AIS. Primary neurons 21 DIV were treated with 1 μm Aβ as indicated and then fixed and stained as indicated. A, co-staining of the AIS with phospho-cofilin (p-Cofilin) (via a phosphorylation-dependent antibody) and F-actin (stained via phalloidin) shows positive stainings in control conditions (A1) but reveals a loss of phospho-cofilin (dephosphorylation results in activation of cofilin and severing of F-actin) and loss of F-actin signal after 15 min of Aβ exposure (A2). B, quantification of phospho-cofilin signals (B1) and F-actin signals (B2). Error bars, S.E.
Figure 7.

Aβ disrupts the Tau diffusion barrier via impairment of MT dynamics in the AIS. A, kymograph (A1) of the AIS of an f-tractinCitrine-transfected primary cortical neuron (12 DIV) for 4 days after treatment with Aβ. The F-actin-labeling protein f-tractinCitrine accumulates within the AIS, indicating an increase in area covered by F-actin. Boxed areas, regions of higher F-actin concentrations within the AIS. Arrows, increasing F-actin (1–1.5 h). Quantification (A2) reveals a constant increase in fluorescence intensity (FI) of f-tractineCitrine after 0.5-h exposure to Aβ. B, kymographs (B1) of the AIS of EB3-tdTomato-transfected primary cortical neurons 11 DIV untreated (top) or treated with Aβ (1 μm) for 1.5 h. Quantification (B2) reveals decreased speed of EB3 comets only within the AIS. C, treatment of primary neurons (20 DIV) with jasplakinolide (Jaspl.) and Aβ, but not latrunculin-A (LatA), results in missorting of endogenous Tau. Neurons were treated for 3 h as indicated and stained with K9JA for total Tau (green). C1, control neurons show little Tau presence in the somatodendritic compartment (left), whereas treatment with jasplakinolide (1 μm) results in increased Tau presence in the somatodendritic compartment. cb, cell body; d, dendrite. C2, quantification of missorting of Tau, as indicated. D, 2N4R-TauD2 and 0N4R-TauD2 as well as Dendra2 were transfected into primary rat cortical neurons (8 DIV) for 3 days. D1, images depict neurons 10 min post-photoconversion in the soma; insets show cells before photoconversion and distribution of the unconverted protein in green. Left, 0N4R-TauD2-transfected neurons show fast anterior movement of photoconverted 0N4R-TauD2. Right, treatment with 1 μm Aβ for 1 h reduces axonal sorting of 0N4R-TauD2. D2, quantification of experiments as shown in D1. Cells transfected with Dendra2 alone or 0N4R-TauD2 show anterograde penetration of the TDB, whereas 2N4R-TauD2 shows reduced anterograde movement into the axon in control conditions (black bars). Treatment with Aβ for 1 h reduces retention of Dendra2 and 2N4R-TauD2 but shows a trend for enhanced retention of 0N4R-TauD2. E, axonal sorting of the 0N4R-Tau is disrupted by Aβ treatment. Primary cortical neurons 16 DIV were transfected with the different rodent isoforms of Tau tagged with citrine (mTauCit; green) and the volume marker tdTomato (red) for 5 days. E1, 2N4R-mTauCit shows little enrichement in the axon (left panels), whereas the 0N4R-mTauCit isoform is enriched in the axon (middle panels, arrow; green), but this enrichment is lost after Aβ treatment (right panels). E2, quantification of the axonal enrichment of all rodent Tau isoforms tagged with citrine (mTauCit) with and without treatment with 1 μm Aβ for 3 h. The shorter 0N isoforms are enriched in the axon (arrow), but this enrichment is partially lost after Aβ treatment. *, p < 0.05; **, p < 0.01. Error bars, S.E.
Thus, only after cell stress ceases is cofilin activity attenuated, and actin in the AIS repolymerizes. Tau entry into the axon is then blocked by aberrant repolymerization of F-actin in the AIS. This finding would be in agreement with earlier data that indicate that Aβ-oligomers lose their toxicity after a brief time (1–3 h) in cell culture and that the peak of missorting of Tau also appears only after the Aβ-oligomers lose their toxicity (51, 54). How can the aberrantly polymerized F-actin block Tau entry? EB3-live imaging of the AIS revealed that 1.5 h after Aβ insult, the EB3 comet speed was reduced by ∼20% (0.1 μm/s) (Fig. 7B). This indicates that repolymerizing actin might exert its effect on Tau sorting via strangulating MT polymerization within the AIS.
Aβ exposure disrupts Tau isoform–specific axodendritic sorting
Previously, we showed that Aβ causes impaired translocation of Tau into axons rather than retrograde leakage of Tau from the axon to the soma (51). The effect of Aβ on the TDB has not been determined so far. We transfected neurons with 2N4R-TauD2 (strong dendritic presence) as well as 0N4R-TauD2 (strong axonal presence) and measured axonal sorting in the presence and absence of 1 μm Aβ. Axonal translocation was determined by photoconversion of TauD2 in the soma and imaging of axon. 2N4R-TauD2 is poorly sorted into the axon, whereas 0N4R-TauD2 is translocated to the axon and past the AIS (100 μm from the axon hillock) within 10 min. Aβ treatment decreases the axonal sorting of 0N4R-TauD2 (Fig. 7D). Conversely, 2N4R-TauD2 and Dendra2 show increased axonal penetration after Aβ treatment (Fig. 7D, 2). This hints at an alteration of the AIS structure as a result of Aβ, probably related to the restructuring of F-actin in the AIS.
To test whether the sorting of rodent (mouse) Tau (mTau) isoforms is different from that of the human isoforms, we cloned the four rodent Tau isoforms (0N3R-mTau, 0N4R-mTau, 1N4R-mTau, and 2N4R-mTau) tagged with citrine (mTauCit). We found that mTau isoforms behave slightly differently from human Tau isoforms; differences in axodendritic sorting among the isoforms are less pronounced, and only 0N4R-mTauCit is more sorted into the axon (axonal e.f. of ∼100 versus ∼85 of the other isoforms (Fig. 7E)). This is in contrast to the human Tau isoforms, which show a wider variation between the isoforms (e.f. ranging from ∼70 to ∼95). We next tested the contribution of the isoforms to missorting by transfecting individual isoforms into neurons and then exposed cells to Aβ. This revealed that the axonal sorting of 0N4R-mTauCit is reduced (∼30%) after Aβ insult, whereas the other rodent isoforms (0N3R, 1N4R, and 2N4R) show no change (Fig. 7E). In summary, we argue that missorting of Tau is due to somatodendritic accumulation of the normally axonally targeted 0N4R-Tau. Aβ-caused missorting can be induced at developmental stages where only 0NxR isoforms are present, but 0N3R-Tau distribution is unchanged after Aβ insult. Thus, missorted Tau must consist mainly of 0N4R-Tau.
Discussion
The Tau diffusion barrier functions both anterogradely and retrogradely, and its tightness is isoform-specific
In the present study, we investigated the TDB and the function of the AIS with respect to physiological sorting and pathological missorting of Tau. We find that longer isoforms of Tau, 2N4R and 1N4R, show almost no retrograde passage from the axon into the soma, whereas all of the shorter splice variants of Tau are able to overcome the retrograde barrier (Fig. 1). The same short isoforms of Tau also show enhanced axonal enrichment (i.e. anterograde sorting) within 4–6 days when expressed at nearly endogenous levels (Fig. 2). This finding was confirmed by photoconversion of Dendra2-tagged Tau in the soma, which did propagate more slowly into the axon in the case of the longest Tau isoform (2N4R) compared with a medium-sized, well-axonally sorted Tau (0N4R; Fig. 7D). This indicates that the TDB can function in both directions. Interestingly, isoform-specific sorting of Tau occurs both in TauKO and in wild-type neurons, even when exogenous Tau is transfected after the establishment of polarity. Thus, either the Tau sorting machinery is not Tau-specific and develops normally in the absence of Tau, or the cell can rapidly establish Tau sorting even after the process of neuronal polarization has been accomplished. The isoform-specific differences of anterograde sorting (i.e. axonal enrichment of somatically produced Tau) or retrograde passage from the axon to the soma are independent of MT affinity (55) or axonal diffusion rates (Figs. 1 and 2). In the case of 3R-Tau, the second insert appears to decrease the ability of Tau to pass the TDB. Considering that 2N3R-Tau is bigger than 0N4R-Tau, also increased size would correlate with inability to cross the TDB. This idea would be in agreement with a previously proposed nonspecific size-dependent filter (27, 28). However, mutants of the longest isoform of Tau, derived from FTLD or PSP (i.e. ΔK280 and A152T), and also a pseudophosphorylation mutant (4KXGE; all four KXGS motifs mutated to KXGE) show enhanced retrograde and anterograde passage of Tau, indicating that size alone is not the sole determinant.
Tau increases spine development and dendritic growth in an isoform-dependent manner
The longest isoform of Tau (2N4R-Tau) along with the other 4R isoforms is expressed only after the initial polarization has taken place (i.e. not during embryonal development), both in rodents and humans. When expressed in low endogenous-like levels, some Tau isoforms (e.g. 3R-Tau species, especially 0N3R) induce filopodia-like spine growth. Other isoforms (0N4R and 1N4R) had no or little effect on promoting spine development. 2N4R-Tau, however, induces striking maturation of spines and elongation of dendrites (Fig. 2, D–F). This is in line with the appearance of specific Tau isoforms during development; the short (3R) isoforms are expressed early in development, driving filopodia spine formation, whereas later in life, the longer isoforms (e.g. 2N4R) are expressed (6), which concurs with spine maturation. Accordingly, knockdown of Tau at the time of spine formation and maturation (14 DIV) results in loss of spine formation. This points toward a bona fide role for Tau in dendritic development. Future studies (e.g. by knocking down individual isoforms of Tau while maintaining the physiological ratio of the remaining isoforms) will have to elucidate which isoform of Tau during which state of development is responsible for or contributes to normal spine development.
Sorting of Tau can be achieved independently of regulatory translation but is modulated by phosphorylation of Tau and AIS structural integrity
We studied the mechanistic/structural manner in which the TDB is able to confine the Tau protein to the axonal compartment. Previously, Tau mRNA was shown to be translocated to the axon by a 3′-UTR axonal targeting signal, implying local translation of Tau (21). However, the exogenous Tau constructs that we used to investigate the TDB and the axonal enrichment do not carry the 3′-UTR. Thus, in our model, Tau mRNA does not contribute to the sorting of Tau.
Phosphorylation of Tau probably has a function in sorting of Tau; pseudophosphorylation at the KXGS motifs enhances anterograde and retrograde propagation of Tau in the axon (23, 56). Activation of the kinase MARK (primarily responsible for phosphorylation of these epitopes) lags behind Aβ-induced missorting of Tau, indicative of a role in resorting of Tau. Here we show that Tau pseudophosphorylated at the KXGS motifs shows enhanced axonal enrichment compared with wild-type Tau (Fig. 2 and supplemental Figs. S1 and S2), indicating the importance of this phospho-site for sorting. Another prominent Tau kinase, GSK3β, is a major player within the AIS, where it is anchored (37), but it does not phosphorylate the KXGS motifs of Tau. Testing the influence of GSK3β on the TDB revealed that GSKβ induces accumulation of endogenous and exogenous Tau in the somatodendritic compartment (Fig. 3 and supplemental Fig. S5). In the case of retrograde missorting of exogenous Tau, this effect is independent of direct phosphorylation of Tau by GSK3β, because even non-phosphorylatable Tau shows the same retrograde propagation from the axon into the soma (Fig. 3). Thus, the interplay of GSK3β with components of the AIS/TDB is essential for sorting and missorting of Tau in our model. Knockdown of other structural components of the AIS results in weakening of the TDB. Tau transit through the TDB is facilitated when classical AIS components (e.g. AnkG and EB1) are knocked down via shRNA. All knockdowns also induce increased invasion of the dendritic marker MAP2 into the proximal part of the axon. This indicates that the classical AIS components contribute to the TDB, but also to the nonspecific filter function of the AIS.
Structures around the TDB: Highly dynamic MTs in the AIS
MT destabilization by nocodazol results in missorting and breakdown of the TDB, whereas MT stabilization by taxol prevents missorting after Aβ-insult (23, 51). We thus hypothesized that the structure of MTs within the AIS holds the key to the TDB. Surprisingly, MTs within the AIS are extremely dynamic. Markers of stable MTs (acetylation and polyglutamylation) are absent in the AIS. MTs could not be stained in the AIS without the use of the molecular densifier PEG and taxol, emphasizing the transient nature of these MTs (Fig. 4), similarly to previous studies (32). EB3-based live imaging of MT dynamics confirmed the dynamic nature of AIS MTs and revealed a hub of EB3 comet spawning at the central part of the AIS (Fig. 5), consistent with observations of enrichment of EB proteins in the AIS (29).
Of note, EB3 comet speed (and thus MT growth rates) is unchanged in the different parts of the AIS and also in the axon (constant at ∼0.5 μm/s for the proximal axon, the AIS itself, and the axon hillock); only after an AD-like insult with Aβ oligomers does the EB3 comet speed decrease by ∼20% (Fig. 7B).
AD-like insult by Aβ causes axodendritic missorting of Tau due to activation of cofilin and loss of MT growth within the AIS
Aβ oligomers bind preferentially to dendritic spines. However, within axons, the AIS is a preferential target for Aβ (supplemental Fig. S8). Because the AIS is composed of a multitude of regulatory elements and receptors (57), Aβ binding may impair the function of the AIS. This is indeed the case for the TDB, which lies within the AIS. Aβ exposure causes activation of cofilin, resulting in increased severing, loss, and subsequent remodeling of F-actin. Whereas this leads to increased amounts of F-actin in the dendritic shaft probably due to redistribution from shriveling spines, the first effect in the AIS is a decrease of F-actin. Thus, the observed activation of cofilin and consequent loss of F-actin, particularly within the AIS, perturb the F-actin meshwork in the AIS. This meshwork acts as a filter to retain non-axonal proteins in the soma. The structural organization appears different, depending on the techniques employed, but probably consists of spectrin IV-bound regularly spaced F-actin rings; still, a large portion of the actin cytoskeleton in the AIS is thought to be dynamic (58, 59). In the case of the TDB, breakdown of F-actin results in anterograde penetration of the “dendritic” 2N4R-Tau, but also failure of proper sorting into the axon of the stronger axonally sorted 0N4R-Tau (Figs. 6 and 7). Thus, the failure of the TDB results in axodendritic missorting of Tau that is isoform-specific.
Aβ-induced loss of F-actin within the AIS may also result in facilitated diffusion of other proteins; Dendra2, for example, shows ∼2 times faster diffusion into the axon. Aβ-induced disruption of F-actin within the AIS would explain the increased anterograde propagation of the longest isoform of Tau, but why is the normally strongly axonally targeted major isoform of Tau (0N4R) missorted after Aβ insult (Fig. 7)? First, because Aβ also disrupts the anterograde polymerization of MTs within the AIS, this would decrease the pumping mechanism that transports the Tau–tubulin complex into the axon (56). Second, because the peak of missorting of Tau (3–6 h (51)) lags substantially behind the loss of F-actin and activation of cofilin (0.1–1 h; Fig. 6), probably the subsequent (aberrant) remodeling of F-actin is responsible for missorting, rather than the loss of F-actin per se. Consistently, the F-actin stabilizer jasplikanolide induces missorting, but not the F-actin destabilizer latrunculin.
Remodeling of the F-actin cytoskeleton within the AIS would result also in the impairment of MT polymerization within the center of the AIS, although it is currently unclear whether there is a direct causal link and whether there is an effector protein. Anyhow, the remodeling of F-actin within the AIS does not occur prior to 1 h after Aβ exposure. Thus, strangulation of MT growth by aberrant F-actin repolymerization within the AIS would start after Aβ has lost its toxicity and F-actin starts to repolymerize. This we indeed observed (Figs. 6 and 7A). Tau is then unable to traffic along MTs, decreasing its ability to pass the anterograde TDB, resulting in somatodendritic accumulation (classical missorting). The cause-effect relationship, however, is far from obvious, because it was shown recently that Tau stabilizes MT growth, judged by EB1 and EB3 comet number and comet path length (60). Thus, the reduced entry of Tau into the AIS (due to aberrant F-actin remodeling) may also contribute to reduced MT growth.
Conclusion
We show here that the barrier function of the TDB within the AIS can operate both in the anterograde and retrograde direction. For 90–97% of Tau molecules, where the second insert is absent, Tau passes through the TDB anterogradely and is retained in the axon. In the case of the longest isoform, 2N4R (∼3–10% of Tau), Tau molecules are partially retained in the somatodendritic compartment, where they enhance dendritic outgrowth and spine formation. Passage of the TDB barrier in both directions is facilitated when Tau itself is mutated (i.e. in the case of pathological mutations, phosphorylation, knockdown of AIS components, or overexpression of GSK3β (an AIS and AD kinase)). Pathomechanistically, we show that AD-like Aβ insult leads to cofilin activation and remodeling of F-actin within the AIS, resulting in decreased MT mobility. This leads to somatodendritic accumulation of 0N4R-Tau, hence classical missorting, but also enhanced axonal penetration of 2N4R-Tau. In summary, we provide evidence for physiological and pathomechanistic differences of the different Tau isoforms and their role in sorting and missorting and demonstrate their functional importance for MT and actin dynamics within the AIS for the maintenance of the TDB.
Materials and methods
Cell culture and transfection
Dissociated cortical neurons were prepared from embryonic day 18 Wistar rats or embryonic day 16 C57BL6JRj wild-type or TauKO mice according to Ref. 61 and plated on a poly-d-lysine (50 μg/ml)-coated coverglass. After 4 DIV, 0.3 μm cytosine arabinoside (Sigma, Munich, Germany) was added to the cultures to reduce glial growth. Cultures were maintained in NeuroBasal medium with B27 (Invitrogen) or NS21 (PanBiotech), l-glutamine (PAA), and penicillin/streptomycin (PAA/PanBiotech) at 37 °C, 5% CO2. Lipofectamine-based transfection for experiments for retrograde Tau movement was conducted as described in Ref. 23 (high protein expression). Transfection for experiments investigating the Tau distribution after 4–6 days of low expression was conducted as described in Ref. 62, making use of standard Lipofectamine 2000 (L2K; Invitrogen) reagent, but not OptiMEM. Briefly, for low protein expression, the DNA amount was limited to 1.2 μg/construct/well of a 6-well multiwell plate or to 0.6 μg/construct/well of 12- or 24-well plates. Instead of OptiMEM, pure unsupplemented Neurobasal medium (Gibco) was used. L2K/DNA mix was prepared as recommended (5 min + 30 min of incubation). Next, cell culture medium already in the multiwell plates (conditioned medium) was carefully removed, leaving a small amount of conditioned medium in the well (1 ml for 6-well plates or 0.5 ml for 12- and 24-well plates). The removed conditioned medium was saved. Cells were then incubated for 6 h for cells older than 14 DIV or for 3 h for cells between 7 and 10 DIV) with the L2K transfection mix. Next, cells were washed twice in the multiwell plates with preheated and carboxylated unsupplemented neurobasal medium, taking great care that cells never fell dry (critically important for older cells). Next, the previously removed conditioned medium was used to allow expression of transfected constructs for 4–6 days. Exposure of cells to reagents and media other than conditioned medium was kept to an absolute minimum to avoid cell degeneration (see also Ref. 62 for critical steps and modulation of expression).
Plasmid construction
The cloning of Dendra2-hTau40 was reported previously (23). Cloning of various isoforms of Tau was done either by PCR-based amplification or subcloning of restriction-digested fragments into the respective restriction sites. PS-CFP2 vector was purchased from Evrogen, and cDNA of human full-length Tau was subcloned into the C terminus of PS-CFP2. Tagging of Tau was tested with several tags (PS-CFP2, Dendra2, CFP, and HA) and without tags, without noticeable differences in cellular distribution (supplemental Fig. S9). Cloning of various shRNAs was performed in pShuttleCMV/mRFP/siRNA/H1RNA Promotor plasmid. The sequences of shRNAs used were as follows: Nav1.x, 5′-GTTCGACCCTGACGCCACT-3′; ankG, 5′-GCCGTCAGTACCATCTTCT-3′; βIV spectrin, 5′-CACTGGATAGCCGAGAAGG-3′; NrCAM, 5′-CAATAATCCTCCGAAGTG-3′; EB1 5′-TCTGACAAAGATAGAACAG-3′; EB3 5′-ACTATGATGGAAAGGATTAC-3′; and spastin, 5′-GGAAUGGUAUAAGAAAGGUAUCGAA-3′. All plasmids were verified by restriction analysis and DNA sequencing. Successful knockdown was demonstrated in Refs. 63–66. Adenovirus production and transfection were as described (67).
Imaging, photoconversion, and analysis
A Fluoview1000 confocal microscope (Olympus, Hamburg, Germany) equipped with a SIM scanner (which allows for simultaneous stimulation and imaging) and a ×60 objective live-cell imaging chamber and ZDC system for z-drift compensation was used for photoconversion experiments and image acquisition. For time-lapse live-cell imaging, cells were plated on glass-bottom dishes and were kept in the imaging chamber (37 °C, 65% humidity and supplied with 5% CO2). Images were taken every 30 s for all applications. Before photoconversion, green fluorescence of Dendra2 was acquired by excitation at 488 nm and detection over the range of 500–550 nm. Photoconversion was performed during the imaging interval by rapidly illuminating the selected area with a 405-nm laser scanner (1–2 exposures of 1 s each, 405-nm laser power 15%). Thereafter, red fluorescence was acquired by excitation at 561 nm and detection over the range 570–670 nm. In the case of PS-CFP2 protein, cyan fluorescence was acquired by excitation at 405 nm with 0.2% laser power and detection over the range of 420–510 nm. Photoconversion was performed similar to Dendra2, and green fluorescence of PS-CFP2 was acquired by excitation at 488 nm and detection over the range of 500–550 nm. For comparison of Dendra2 and PSCFP, fluorescence intensities were normalized using Dendra2 or PS-CFP2 alone and hTau40 tagged with the different fluorescent proteins as controls; fluorescent photoswitchable protein (PSCFP2 and Dendra2) cell body intensities after axonal conversion were set to 100, and baseline levels were normalized by noise subtraction. For knockdown of the different AIS components, PSCFP2 was used because knockdown vectors contained mRFP, which had to be used to identify cells expressing the shRNA. Fluorescence intensity profiles and values according to the indicated time points and area were determined by the microscopy software (Olympus FV1000 Software). Microscopic settings as well as the selection of the regions of interest were kept similar to allow comparison between different experiments, as described previously (23). STED imaging was conducted as recommended by the manufacturer (Leica) with a gSTED system on TCS SP8 secondary antibodies conjugated with CF488 (VWR) or Alexa488 (Dianova) as chromophores and AntifadeGold mounting medium (Invitrogen). Images were analyzed using manufacturer software (Leica LAS AF Lite). Standard immunofluorescence was conducted essentially as described before (12, 51), and see supplemental Table S2 for a list of antibodies used. Axons were identified by the absence of MAP2 and presence of Tau staining, branching pattern of ∼90°, recurrence to cell body and crossing of processes of the same cell, length > 300 μm, and constant diameter. Dendrites were identified by positive MAP2 stain, the presence of dendritic spines, branching pattern of ∼60°, constantly decreasing diameter, and length < 300 μm. Cell bodies, dendrites, and axons have very different diameters, which is impossible to judge by light microscopy. We therefore used a volume marker (tdTomato) and compared the intensities of transfected constructs (i.e. TauD2 stained with human Tau-specific antibody CP27) with the intensities of the volume marker. The volume marker was assumed to be distributed throughout the cell in an unbiased manner, thus making comparisons of the different Tau constructs possible. Values given under “Results” and in the figures were calculated by dividing the normalized (i.e. CP27 fluorescence intensities (Tau) versus tdTomato (Tom) fluorescence intensities) axonal values (AV) versus dendritic values (DV). The formula, therefore, is (TauAV/TomAV)/(TauDV/TomDV); for illustration purposes, values were multiplied by a factor of 50 (to attain a maximal value of 100).
For the evaluation of spine development after transfection of different Tau isoforms and mutations leading to Fig. 2D and supplemental Table S1, at least three cultures from each genotype of wild-type and TauKO hippocampal neurons and several cultures from rat cortical and hippocampal neurons were included.
MT stabilization during extraction was conducted using 3-min extraction at room temperature in MT-stabilizing buffer (PEM buffer: 2% polyethylene glycol (Mr 10,000), 100 mm PIPES-KOH, pH 6.9, 1 mm MgCl2, 1 mm EGTA) containing 1% Triton X-100 and 2 μm taxol (all Sigma or VWR) after a quick rinse in PBS (PAA/Sigma). Cells were then fixed with 3.7% formaldehyde and 0.5% glutaraldehyde in PEM buffer for 20 min. Mitochondria and EB3 live imaging was conducted at 2-s intervals with an MM-based Leica DM6000, processed, and analyzed with Metamorph software.
Other procedures
Western blotting, ATP and LDH measurements, and statistical analysis (*, p < 0.05; **, p < 0.01 versus control if not indicated otherwise) were done as described before (12, 51). Dephosphorylation of endogenous rat Tau was done without phosphatase inhibitors but with protease inhibitors, making use of alkaline phosphate (FastAP, ThermoFisher Scientific) for 16 h. Oligomeric amyloid-β was prepared as described before (51). Briefly, Aβ40 and Aβ42 (both 21st peptide) were mixed in a 7:3 ratio and oligomerized via sequential dissolution in 40 mm NaOH to a 2 mm Aβ solution and consequently diluted in PBS to a 100 μm solution. This solution was then incubated for 1 h at 37 °C. For complete characterization and differences from unmixed preparations, see Ref. 51.
Author contributions
Y. K., F. D., E.-M. M., E. M., and H. Z. conceived the study, and H. Z., E. M., F. D., and E.-M. M. wrote the paper. Y. K. conducted the experiments leading to Fig. 1 and supplemental Fig. S3, A and B. Data were processed and illustrated by F. D., and H. Z., F. D. and H. Z. jointly designed and conducted experiments leading to Figs. 3 and 7D. J. B. designed genetic constructs and provided critical help with molecular biology. All other experiments were conceived, conducted, processed, and illustrated by J. L. and H. Z.
Supplementary Material
Acknowledgments
We thank Sabrina Hübschmann for excellent technical assistance and Hana Dawson (Duke University) for TauKO mice. STED nanoscopy was done at the Light Microscopy Facility at DZNE (Bonn, Germany).
This work was supported by the German Center for Neurodegenerative Diseases (DZNE), Max-Planck Society (MPG), Metlife Foundation, and Tau Consortium. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2 and Figs. S1–S9.
- MAP
- microtubule-associated protein
- STED
- stimulated emission depletion
- MT
- microtubule
- AD
- Alzheimer disease
- FTLD
- frontotemporal lobar degeneration
- FTLD-Tau
- FTLD with tauopathy
- PSP
- progressive supranuclear palsy
- AIS
- axon initial segment
- TDB
- Tau diffusion barrier
- FI
- fluorescence intensity
- e.f.
- enrichment factor
- NrCAM
- neuronal cell adhesion molecules
- Nav and Kv
- voltage-gated sodium and potassium channels, respectively
- mTau
- mouse Tau
- a.u.
- arbitrary units.
References
- 1. Morris M., Maeda S., Vossel K., and Mucke L. (2011) The many faces of tau. Neuron 70, 410–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang Y., and Mandelkow E. (2016) Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21 [DOI] [PubMed] [Google Scholar]
- 3. Goedert M., Spillantini M. G., Potier M. C., Ulrich J., and Crowther R. A. (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andreadis A. (2012) Tau splicing and the intricacies of dementia. J. Cell. Physiol. 227, 1220–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trabzuni D., Wray S., Vandrovcova J., Ramasamy A., Walker R., Smith C., Luk C., Gibbs J. R., Dillman A., Hernandez D. G., Arepalli S., Singleton A. B., Cookson M. R., Pittman A. M., de Silva R., et al. (2012) MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum. Mol. Genet. 21, 4094–4103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bullmann T., Holzer M., Mori H., and Arendt T. (2009) Pattern of tau isoforms expression during development in vivo. Int. J. Dev. Neurosci. 27, 591–597 [DOI] [PubMed] [Google Scholar]
- 7. Takuma H., Arawaka S., and Mori H. (2003) Isoforms changes of tau protein during development in various species. Brain Res. Dev. Brain Res. 142, 121–127 [DOI] [PubMed] [Google Scholar]
- 8. Binder L. I., Frankfurter A., and Rebhun L. I. (1985) The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braak H., and Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 [DOI] [PubMed] [Google Scholar]
- 10. Ballatore C., Lee V. M., and Trojanowski J. Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 [DOI] [PubMed] [Google Scholar]
- 11. Hoover B. R., Reed M. N., Su J., Penrod R. D., Kotilinek L. A., Grant M. K., Pitstick R., Carlson G. A., Lanier L. M., Yuan L. L., Ashe K. H., and Liao D. (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zempel H., Thies E., Mandelkow E., and Mandelkow E. M. (2010) Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van der Jeugd A., Hochgrafe K., Ahmed T., Decker J. M., Sydow A., Hofmann A., Wu D., Messing L., Balschun D., D'Hooge R., and Mandelkow E. M. (2012) Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 123, 787–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hara M., Hirokawa K., Kamei S., and Uchihara T. (2013) Isoform transition from four-repeat to three-repeat tau underlies dendrosomatic and regional progression of neurofibrillary pathology. Acta Neuropathol. 125, 565–579 [DOI] [PubMed] [Google Scholar]
- 15. Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R. C., Stevens M., et al. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 [DOI] [PubMed] [Google Scholar]
- 16. Delacourte A., Sergeant N., Wattez A., Gauvreau D., and Robitaille Y. (1998) Vulnerable neuronal subsets in Alzheimer's and Pick's disease are distinguished by their tau isoform distribution and phosphorylation. Ann. Neurol. 43, 193–204 [DOI] [PubMed] [Google Scholar]
- 17. Sergeant N., Wattez A., and Delacourte A. (1999) Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J. Neurochem. 72, 1243–1249 [DOI] [PubMed] [Google Scholar]
- 18. Dickson D. W., Kouri N., Murray M. E., and Josephs K. A. (2011) Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J. Mol. Neurosci. 45, 384–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu C., Song X., Nisbet R., and Götz J. (2016) Co-immunoprecipitation with Tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J. Biol. Chem. 291, 8173–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hirokawa N., Funakoshi T., Sato-Harada R., and Kanai Y. (1996) Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J. Cell Biol. 132, 667–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aronov S., Aranda G., Behar L., and Ginzburg I. (2001) Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J. Neurosci. 21, 6577–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morita T., and Sobue K. (2009) Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J. Biol. Chem. 284, 27734–27745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li X., Kumar Y., Zempel H., Mandelkow E. M., Biernat J., and Mandelkow E. (2011) Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J. 30, 4825–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bender K. J., and Trussell L. O. (2012) The physiology of the axon initial segment. Annu. Rev. Neurosci. 35, 249–265 [DOI] [PubMed] [Google Scholar]
- 25. Buffington S. A., and Rasband M. N. (2011) The axon initial segment in nervous system disease and injury. Eur. J. Neurosci. 34, 1609–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi T., Storrie B., Simons K., and Dotti C. G. (1992) A functional barrier to movement of lipids in polarized neurons. Nature 359, 647–650 [DOI] [PubMed] [Google Scholar]
- 27. Winckler B., Forscher P., and Mellman I. (1999) A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature 397, 698–701 [DOI] [PubMed] [Google Scholar]
- 28. Song A. H., Wang D., Chen G., Li Y., Luo J., Duan S., and Poo M. M. (2009) A selective filter for cytoplasmic transport at the axon initial segment. Cell 136, 1148–1160 [DOI] [PubMed] [Google Scholar]
- 29. Nakata T., and Hirokawa N. (2003) Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol. 162, 1045–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sohn P. D., Tracy T. E., Son H. I., Zhou Y., Leite R. E., Miller B. L., Seeley W. W., Grinberg L. T., and Gan L. (2016) Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol. Neurodegener. 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palay S. L., Sotelo C., Peters A., and Orkand P. M. (1968) The axon hillock and the initial segment. J. Cell Biol. 38, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones S. L., Korobova F., and Svitkina T. (2014) Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J. Cell Biol. 205, 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dotti C. G., Sullivan C. A., and Banker G. A. (1988) The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 8, 1454–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zempel H., and Mandelkow E. (2014) Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 37, 721–732 [DOI] [PubMed] [Google Scholar]
- 35. Hatch R. J., Wei Y., Xia D., and Götz J. (2017) Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 133, 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Terwel D., Muyllaert D., Dewachter I., Borghgraef P., Croes S., Devijver H., and Van Leuven F. (2008) Amyloid activates GSK-3β to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 172, 786–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tapia M., Del Puerto A., Puime A., Sánchez-Ponce D., Fronzaroli-Molinieres L., Pallas-Bazarra N., Carlier E., Giraud P., Debanne D., Wandosell F., and Garrido J. J. (2013) GSK3 and β-catenin determines functional expression of sodium channels at the axon initial segment. Cell. Mol. Life Sci. 70, 105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gurskaya N. G., Verkhusha V. V., Shcheglov A. S., Staroverov D. B., Chepurnykh T. V., Fradkov A. F., Lukyanov S., and Lukyanov K. A. (2006) Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 24, 461–465 [DOI] [PubMed] [Google Scholar]
- 39. Drewes G., Ebneth A., Preuss U., Mandelkow E. M., and Mandelkow E. (1997) MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 89, 297–308 [DOI] [PubMed] [Google Scholar]
- 40. Goedert M., Ghetti B., and Spillantini M. G. (2012) Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coppola G., Chinnathambi S., Lee J. J., Dombroski B. A., Baker M. C., Soto-Ortolaza A. I., Lee S. E., Klein E., Huang A. Y., Sears R., Lane J. R., Karydas A. M., Kenet R. O., Biernat J., Wang L. S., et al. (2012) Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Human molecular genetics 21, 3500–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cleveland D. W., Hwo S. Y., and Kirschner M. W. (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 116, 227–247 [DOI] [PubMed] [Google Scholar]
- 43. Dawson H. N., Ferreira A., Eyster M. V., Ghoshal N., Binder L. I., and Vitek M. P. (2001) Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 44. Kremer A., Maurin H., Demedts D., Devijver H., Borghgraef P., and Van Leuven F. (2011) Early improved and late defective cognition is reflected by dendritic spines in Tau.P301L mice. J. Neurosci. 31, 18036–18047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sydow A., Van der Jeugd A., Zheng F., Ahmed T., Balschun D., Petrova O., Drexler D., Zhou L., Rune G., Mandelkow E., D'Hooge R., Alzheimer C., and Mandelkow E. M. (2011) Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 31, 2511–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen Q., Zhou Z., Zhang L., Wang Y., Zhang Y. W., Zhong M., Xu S. C., Chen C. H., Li L., and Yu Z. P. (2012) Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem. Int. 60, 233–242 [DOI] [PubMed] [Google Scholar]
- 47. Rasband M. N. (2010) The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 11, 552–562 [DOI] [PubMed] [Google Scholar]
- 48. Leterrier C., Vacher H., Fache M. P., d'Ortoli S. A., Castets F., Autillo-Touati A., and Dargent B. (2011) End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc. Natl. Acad. Sci. U.S.A 108, 8826–8831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Medina M., and Avila J. (2014) New insights into the role of glycogen synthase kinase-3 in Alzheimer's disease. Expert Opin. Ther. Targets 18, 69–77 [DOI] [PubMed] [Google Scholar]
- 50. Kim Y. T., Hur E. M., Snider W. D., and Zhou F. Q. (2011) Role of GSK3 signaling in neuronal morphogenesis. Front. Mol. Neurosci. 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zempel H., Luedtke J., Kumar Y., Biernat J., Dawson H., Mandelkow E., and Mandelkow E. M. (2013) Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 32, 2920–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baas P. W., and Lin S. (2011) Hooks and comets: the story of microtubule polarity orientation in the neuron. Dev. Neurobiol. 71, 403–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cruz D. A., Lovallo E. M., Stockton S., Rasband M., and Lewis D. A. (2009) Postnatal development of synaptic structure proteins in pyramidal neuron axon initial segments in monkey prefrontal cortex. J. Comp. Neurol. 514, 353–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuperstein I., Broersen K., Benilova I., Rozenski J., Jonckheere W., Debulpaep M., Vandersteen A., Segers-Nolten I., Van Der Werf K., Subramaniam V., Braeken D., Callewaert G., Bartic C., D'Hooge R., Martins I. C., et al. (2010) Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goode B. L., Chau M., Denis P. E., and Feinstein S. C. (2000) Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. J. Biol. Chem. 275, 38182–38189 [DOI] [PubMed] [Google Scholar]
- 56. Konzack S., Thies E., Marx A., Mandelkow E. M., and Mandelkow E. (2007) Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J. Neurosci. 27, 9916–9927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kam T. I., Gwon Y., and Jung Y. K. (2014) Amyloid β receptors responsible for neurotoxicity and cellular defects in Alzheimer's disease. Cell. Mol. Life Sci. 71, 4803–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leterrier C. (2016) The axon initial segment, 50 years later: a nexus for neuronal organization and function. Curr. Topics Membr. 77, 185–233 [DOI] [PubMed] [Google Scholar]
- 59. Jones S. L., and Svitkina T. M. (2016) Axon initial segment cytoskeleton: architecture, development, and role in neuron polarity. Neural Plast. 2016, 6808293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sayas C. L., Tortosa E., Bollati F., Ramírez-Ríos S., Arnal I., and Avila J. (2015) Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J. Neurochem. 133, 653–667 [DOI] [PubMed] [Google Scholar]
- 61. Zempel H., and Mandelkow E. M. (2017) Tracking Tau in neurons: how to grow, fix, and stain primary neurons for the investigation of Tau in all developmental stages. Methods Mol. Biol. 1523, 327–334 [DOI] [PubMed] [Google Scholar]
- 62. Zempel H., Luedtke J., and Mandelkow E. M. (2017) Tracking Tau in neurons: how to transfect and track exogenous Tau into primary neurons. Methods Mol. Biol. 1523, 335–340 [DOI] [PubMed] [Google Scholar]
- 63. Hedstrom K. L., Xu X., Ogawa Y., Frischknecht R., Seidenbecher C. I., Shrager P., and Rasband M. N. (2007) Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J. Cell Biol. 178, 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Komarova Y., Lansbergen G., Galjart N., Grosveld F., Borisy G. G., and Akhmanova A. (2005) EB1 and EB3 control CLIP dissociation from the ends of growing microtubules. Mol. Biol. Cell 16, 5334–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Straube A., and Merdes A. (2007) EB3 regulates microtubule dynamics at the cell cortex and is required for myoblast elongation and fusion. Curr. Biol. 17, 1318–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Riano E., Martignoni M., Mancuso G., Cartelli D., Crippa F., Toldo I., Siciliano G., Di Bella D., Taroni F., Bassi M. T., Cappelletti G., and Rugarli E. I. (2009) Pleiotropic effects of spastin on neurite growth depending on expression levels. J. Neurochem. 108, 1277–1288 [DOI] [PubMed] [Google Scholar]
- 67. Thies E., and Mandelkow E. M. (2007) Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 27, 2896–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.