

Abstract
Leishmaniasis is one of the leading globally neglected diseases, affecting millions of people worldwide. Leishmania infection depends on the ability of insect-transmitted metacyclic promastigotes to invade mammalian hosts, differentiate into amastigotes, and replicate inside macrophages. To counter the hostile oxidative environment inside macrophages, these protozoans contain anti-oxidant systems that include iron-dependent superoxide dismutases (SODs) in mitochondria and glycosomes. Increasing evidence suggests that in addition to this protective role, Leishmania mitochondrial SOD may also initiate H2O2-mediated redox signaling that regulates gene expression and metabolic changes associated with differentiation into virulent forms. To investigate this hypothesis, we examined the specific role of SODA, the mitochondrial SOD isoform in Leishmania amazonensis. Our inability to generate L. amazonensis SODA null mutants and the lethal phenotype observed following RNAi-mediated silencing of the Trypanosoma brucei SODA ortholog suggests that SODA is essential for trypanosomatid survival. L. amazonensis metacyclic promastigotes lacking one SODA allele failed to replicate in macrophages and were severely attenuated in their ability to generate cutaneous lesions in mice. Reduced expression of SODA also resulted in mitochondrial oxidative damage and failure of SODA/ΔsodA promastigotes to differentiate into axenic amastigotes. SODA expression above a critical threshold was also required for the development of metacyclic promastigotes, as SODA/ΔsodA cultures were strongly depleted in this infective form and more susceptible to reactive oxygen species (ROS)-induced stress. Collectively, our data suggest that SODA promotes Leishmania virulence by protecting the parasites against mitochondrion-generated oxidative stress and by initiating ROS-mediated signaling mechanisms required for the differentiation of infective forms.
Keywords: differentiation, Leishmania, mitochondria, redox signaling, superoxide dismutase (SOD), iron superoxide dismutase
Introduction
Leishmaniasis affects an estimated 12 million people worldwide, with an estimated 350 million at risk of infection (1). Depending on the Leishmania species, symptoms vary from self-healing skin lesions to a visceralizing form that can be lethal. The absence of efficacious and cost-effective drugs, combined with the emergence of drug resistance, accentuates the critical need for new therapeutic targets.
The life cycle of Leishmania spp. alternates between non-virulent promastigotes inside the insect vectors and virulent amastigotes inside mammalian hosts. Transmission of the parasites to mammals occurs through sand fly bites (2). Amastigotes are adapted to survive and replicate inside acidic parasitophorous vacuoles of macrophages. After ingestion by sand flies during a blood meal, amastigotes transform into promastigotes, which replicate in the fly's digestive tract. As nutrients become depleted, promastigotes cease to replicate and move up the sand fly gut toward the proboscis, where they mature into infective metacyclic forms. Metacyclics are reintroduced into new hosts during the next feeding cycle and enter host macrophages where they transform into amastigotes. To adapt to the rapidly changing environmental conditions during its life cycle, Leishmania undergoes extensive morphological and metabolic changes orchestrated at the post-transcriptional and post-translational levels (3–5). Despite significant progress in characterizing these life cycle-associated developmental changes, the molecular pathways that initiate differentiation in Leishmania are still poorly understood.
Over the last 2 decades, a role for reactive oxygen species (ROS)2 as regulators of physiological and biological responses (redox biology) has emerged (6–8). Two of the most abundant ROS generated as by-products of mitochondrial respiration or as end products of metabolic reactions, O2˙̄ and H2O2, have been linked to cell fate determination. Subtle increases in intracellular O2˙̄ levels promote cell proliferation, whereas low-level accumulation of H2O2 can inhibit cell growth and initiate differentiation (9–11). Recent studies specifically implicate the mitochondrion-generated ROS in intracellular signaling (12–14). O2˙̄, the highly toxic ROS resulting from reduction of O2 by complexes I, II, and III of the mitochondrial electron transport chain (ETC) (15), is rapidly converted by cytosolic or mitochondrial dismutases (SOD) into H2O2. Because of its higher stability, membrane diffusibility, and ability to promote target-specific thiol modifications, H2O2 is considered a primary intracellular signaling ROS molecule (7, 10).
Recent evidence indicates that H2O2 generated within mitochondria through the action of SOD plays a central role in regulating differentiation of L. amazonensis promastigotes into infective amastigotes (16, 17). Given that SOD enzymes in trypanosomatid protozoa exclusively utilize iron as an essential co-factor (18), these enzymes have emerged as important links between iron and ROS-regulated differentiation pathways in L. amazonensis. Notably, the role of H2O2 in triggering promastigote to amastigote differentiation was directly demonstrated during studies of iron uptake in L. amazonensis promastigotes (16). Subsequent studies with L. amazonensis lines defective in mitochondrial iron import (LMIT1/Δlmit1) implicated mitochondrial iron-dependent SOD (SODA) and mitochondrion-generated ROS in the generation of virulent forms (17).
Further supporting a role for SODA and H2O2 in the development of virulence in L. amazonensis, two cues known to trigger in vitro amastigote differentiation, low pH and high temperature (19), are also effective triggers of ROS generation (20–22). Exposure to elevated temperature is thought to lead to hyper-polarization of mitochondria, increased respiratory rate, and an ROS surge that is not only tolerated by Leishmania but is actually used as a differentiation signal. Stationary phase promastigotes show increased resistance to oxidative stress and enhanced SOD activity as they differentiate into amastigotes. In contrast, low SOD activity induces logarithmic phase promastigotes to accumulate higher levels of ROS and to undergo apoptosis, a process that can be reversed by overexpression of mitochondrial SODA (21).
Here, we confirm the prediction that iron import into mitochondria is required for activity of L. amazonensis SODA. Furthermore, by targeting the SODA gene for allelic knock-out and examining the phenotype of mutant lines, we conclude that SODA expression is required for maintaining mitochondrial redox balance and also for the development of parasite virulent forms.
Results
Amastigote differentiation is associated with mitochondrial iron import and activation of mitochondrial SODA
In earlier work, we identified LMIT1, an L. amazonensis mitochondrial iron transporter, and we suggested that the reduced ability of LMIT1/Δlmit1 promastigotes to import iron into mitochondria and activate the iron-dependent SODA might be responsible for their failure to differentiate into amastigotes (17). This hypothesis was largely based on the reduced SOD activity observed in total parasite extracts and on morphological evidence for mitochondrial oxidative damage in LMIT1/Δlmit1 parasites subjected to the low pH/high temperature protocol for axenic amastigote differentiation. To directly investigate the specific role of SODA in the signaling process leading to amastigote differentiation, we developed antibodies capable of distinguishing the mitochondrial SODA from glycosomal SODB, and we compared SOD activity in mitochondrial fractions of wild-type and LMIT1/Δlmit1 parasites undergoing low pH/high temperature-induced axenic differentiation.
Specific polyclonal antibodies against mitochondrial SODA and SODB were generated using purified recombinant proteins (supplemental Fig. S1). The immunofluorescence localization of anti-SODA antibodies was identical to the staining pattern of MitoTracker Red, a mitochondrion-specific dye (Fig. 1A). This result demonstrates the mitochondrial localization of endogenous SODA, as proposed earlier based on overexpression of GFP-tagged SODA (23).
Figure 1.
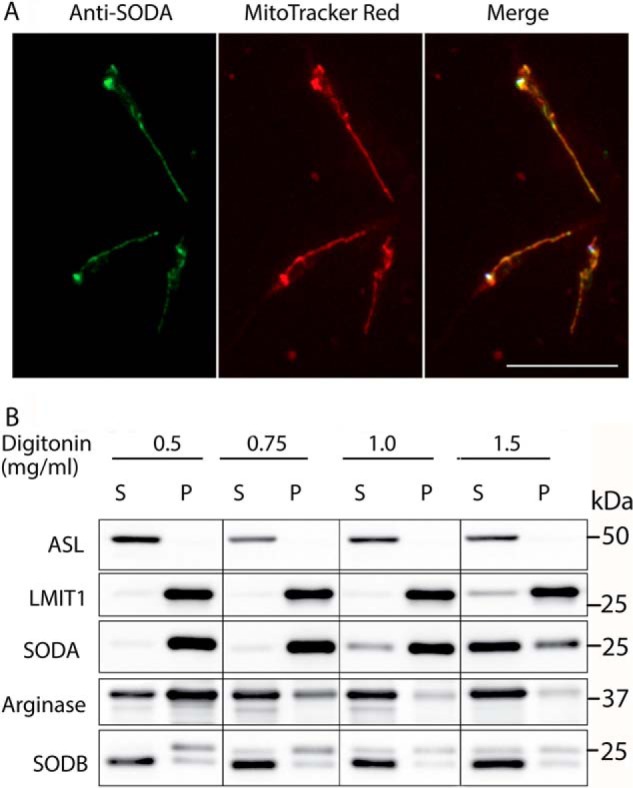
SODA localizes to mitochondria. A, immunolocalization of SODA in L. amazonensis promastigotes was performed using polyclonal antibodies against SODA (green), and mitochondria were stained with MitoTracker Red (red). Merging the two images (merge) confirmed the mitochondrial localization of SODA. Bar, 4 μm. B, SODA is enriched in mitochondrial fractions. Subcellular fractionation of L. amazonensis promastigotes expressing FLAG-tagged LMIT1 was performed using increasing concentrations of digitonin as indicated. Proteins in the supernatant (S) and pellet (P) fractions were detected by Western blotting using antibodies against cytoplasmic adenosuccinate lyase, mitochondrial FLAG-tagged LMIT1, and glycosomal arginase. SODA and SODB were detected with specific polyclonal antisera and LMIT1–3×FLAG with an anti-FLAG monoclonal antibody.
To obtain mitochondrion-enriched fractions free of glycosomal contaminants, subcellular fractionation of promastigotes expressing a 3×FLAG-tagged form of the LMIT1 mitochondrial iron transporter was carried out with increasing concentrations of digitonin, as described previously (17). Western blotting detection of organelle-specific markers (adenosuccinate lyase for cytosol (24), LMIT1-FLAG for mitochondria (17), and arginase for glycosomes (25)) showed that pellets obtained after 1 mg/ml digitonin treatment contained ∼98% of the total LMIT1-FLAG and less than 5% of the total cellular arginase, reflecting mitochondrial enrichment in fractions mostly devoid of glycosomal contaminants (Fig. 1B). Immunoblot with the specific antibodies against SODA or SODB confirmed that >90% of the endogenous SODA co-fractionated with LMIT1 in mitochondrion-enriched fractions, whereas >95% of endogenous SODB was found in the soluble fraction (Fig. 1B).
SOD activity was then quantified in mitochondrion-enriched fractions prepared from wild-type (WT) and LMIT1/Δlmit1 promastigotes. These two lines were subjected to the low pH/high temperature conditions previously shown to trigger amastigote differentiation in WT but not in LMIT1/Δlmit1 (17). WT parasites showed a gradual increase in mitochondrion-associated SOD activity and SODA protein levels during differentiation (Fig. 2, A–C), consistent with the SOD activity increase we reported earlier for whole-cell extracts (17). In contrast, although SOD activity in LMIT1/Δlmit1 cells was slightly higher during early differentiation (6 h), it was ∼2.5-fold lower than WT at the 48-h time point when most WT parasites had already assumed the rounded morphology typical of amastigotes (Fig. 2A). Western blot analysis showed comparable amounts of SODA protein and only trace levels of SODB in both WT and LMIT1/Δlmit1 mitochondrial fractions (Fig. 2, B and C), indicating that the reduced SOD activity in LMIT1/Δlmit1 cells is likely to reflect SODA inactivation as a result of impaired LMIT1-mediated iron import into mitochondria. Importantly, we also observed down-regulation (of about 2-fold) in the expression of ascorbate-dependent peroxidase (APX) in LMIT1/Δlmit1 mitochondrial lysates, when compared with WT (Fig. 2, B and C). Expression of APX, a mitochondrial protein required for the enzymatic breakdown of H2O2 in Leishmania, is known to be up-regulated in response to H2O2 accumulation (26). Collectively, these results demonstrate that differentiation of L. amazonensis promastigotes into infective amastigotes is associated with activation of the mitochondrial iron-dependent SODA and accumulation of H2O2, a ROS previously shown to directly trigger differentiation (16).
Figure 2.

LMIT1/Δlmit1 promastigotes undergoing axenic differentiation show similar SODA protein levels but reduced mitochondrial SOD activity. A, log phase (∼2 × 107/ml) wild-type (WT) and single knock-out (LMIT1/Δlmit1) promastigotes were induced to differentiate into axenic amastigotes by shifting to pH 4.5/32 °C culture conditions and incubated for 48 h. Mitochondrion-enriched pellet fractions prepared by digitonin (1 mg/ml) treatment of parasites were collected at the indicated time points and assayed for SOD activity. The data represent the mean ± S.D. of triplicate determinations and are representative of four independent experiments (Student's t test compared with WT: **, p = 0.008). B, mitochondrial extracts used for the SOD assay were resolved by 12% SDS-PAGE (10 μg protein/lane), and Western blottings were probed with specific antibodies against SODA, SODB, and APX. Anti-tubulin antibodies were used was used as loading controls. C, SODA protein levels relative to the tubulin in mitochondrial extracts, estimated by quantification of the respective band intensities on Western blottings.
SODA is an essential gene in L. amazonensis and in Trypanosoma brucei procyclics
To further understand how mitochondrial SODA regulates the ROS signaling pathway leading to amastigote differentiation, we proceeded to generate SODA null mutants. Knock-out constructs carrying drug resistance gene cassettes flanked by 5′- and 3′-UTR regions of the SODA gene were generated and used for gene replacement through homologous recombination (supplemental Fig. S2A). Replacement of a single SODA allele with HYG or PHLEO drug resistance gene cassettes was possible, and allelic integration into the desired locus was confirmed in each case by PCR and Southern blot analysis (supplemental Fig. S2B). SODA/ΔsodA promastigotes showed ∼53% reduction in SODA protein levels as the parasites reached early stationary growth, when compared with WT (Fig. 3). During the log phase of growth, both SODA and SODB were expressed at lower levels, and no significant difference was observed between WT and SODA/ΔsodA (Fig. 3, A and B). Repeated attempts to generate a SODA null line by replacing the second allele were unsuccessful. Similar to our previous experience with LMIT1, a SODA ORF was consistently detected in lines resistant to both phleomycin and hygromycin, even when the intended in situ integration of the targeting drug resistance markers was confirmed. Thus, SODA appears to be essential for the viability of L. amazonensis promastigotes.
Figure 3.

Deletion of a single SODA allele reduces the amount of SODA protein in promastigotes entering stationary phase. A, protein samples (10 μg of protein/lane) prepared from whole-cell lysates of wild-type (WT) or SODA/ΔsodA promastigotes at log phase (day 2) or early stationary phase (day 4) of growth were analyzed by Western blotting using antibodies specific for SODA or SODB. Tubulin expression levels in the lysates were used as loading controls. B, SODA and SODB protein levels relative to tubulin estimated by quantification of the band intensities on Western blots. The data represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments (Student's t test, **, p = 0.006).
Providing further evidence that SODA is an essential gene in trypanosomatid parasites, tetracycline-inducible RNAi-mediated ablation of SODA in T. brucei procyclic forms (supplemental Fig. S3A) markedly impaired the parasites' ability to sustain replication after 72 h in culture (supplemental Fig. S3B). T. brucei procyclics were reported to be partially dependent on mitochondrial metabolism (27, 28), suggesting that the phenotype we observed may be related to gradual mitochondrial damage as accumulated ROS reach toxic levels in the absence of SODA.
SODA deficiency severely impairs the ability of Leishmania promastigotes to differentiate into virulent forms
Given our inability to generate SODA null mutants, we compared the properties of single knock-out SODA/ΔsodA (hyg) and WT promastigotes. When promastigote cultures were shifted to axenic amastigote growth conditions (pH 4.5, 32 °C), after an initial lag period of 48 h WT parasites replicated steadily throughout the monitored period (144 h) (Fig. 4A). In contrast, parasites lacking one SODA allele showed very poor growth and a gradual loss in viability, as indicated by propidium iodide staining (data not shown). Marked differences were also observed in parasite morphology 48 h after the low pH/high temperature shift. About 95% of the WT population showed the expected change into the rounded/aflagellated amastigote form, whereas the majority (∼55%) of the viable SODA/ΔsodA parasites retained long flagella and showed marked morphological abnormalities (Fig. 4, B and C). Transmission electron microscopy (TEM) analysis revealed extensive mitochondrial alterations in SODA/ΔsodA parasites (Fig. 4D) that were reminiscent of what we previously observed in LMIT1/Δlmit1 promastigotes (17). Mitochondria were enlarged and deformed, with an overall reduction in electron density and accumulation of dense aggregates in the matrix. Normal mitochondrial morphology and a normal ability to differentiate and replicate as amastigotes were restored when SODA/ΔsodA parasites were complemented with episomally expressed SODA (SODA/ΔsodA + SODA) (Fig. 4, A–C).
Figure 4.

SODA/ΔsodA parasites fail to transform into amastigotes following the low pH/high temperature differentiation cue. Wild-type (WT), SODA single knock-out (SODA/ΔsodA), and complemented SODA single knock-out (SODA/ΔsodA + SODA) promastigote cultures were shifted to low pH (4.5)/high temperature (32 °C) culture conditions (0 h). Samples were taken at the indicated time intervals, assessed for viability by FDA staining, and for differentiation into the amastigote form by light microscopy morphological examination (loss of long flagella). A, number of viable cells in WT, SODA/ΔsodA, and SODA/ΔsodA +SODA cultures estimated at different time intervals following induction of low pH/high temperature mediated differentiation. B, percentage of viable rounded forms with a shortened flagellum in WT, SODA/ΔsodA, and SODA/ΔsodA + SODA cultured for 48 h in amastigote growth conditions. At least 200 FDA-stained parasites were counted in each sample. The data represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments (**, p ≤ 0.009). C, SEM analysis of parasite morphology in WT, SODA/ΔsodA, and SODA/ΔsodA + SODA cultures. Bars, 10 μm. D, TEM micrographs of WT, SODA/ΔsodA, and SODA/ΔsodA + SODA parasite sections. m = mitochondria; k = kinetoplast, and n = nucleus. Bars, 1 μm.
In addition to the low pH/high temperature stimuli, in previous work we showed that up-regulation of the iron uptake machinery in response to iron deprivation also triggers differentiation of L. amazonensis promastigotes into amastigotes in a process dependent on mitochondrial H2O2, the product of SODA (16, 17). Thus, we also examined the effects of iron deprivation on the growth and differentiation of SODA/ΔsodA promastigotes. When cultured in iron-deficient medium, both WT and SODA-deficient parasites were initially able to replicate but reached a maximum cell density (∼3 × 107/ml) that was lower than what is normally observed in complete growth medium (Fig. 5A, compare with WT in Fig. 6A). However, in contrast to WT, the SODA/ΔsodA promastigote count steadily declined after reaching the peak density (Fig. 5A), resembling the “population crash” we previously reported for Δlit1/Δlit1 (lacking the ferrous iron transporter LIT1) and LMIT1/Δlmit1 (partially deficient in the mitochondrial iron importer LMIT1) parasites grown in iron-depleted medium (16, 17). Microscopic analysis of the cultures showed that on day 5 of iron deprivation >55% of WT parasites lacked a visible flagellum and had assumed the rounded amastigote-like morphology, whereas only 20% of viable SODA/ΔsodA cells were able to undergo this transformation (Fig. 5B). Complementation with episomally expressed SODA (SODA/ΔsodA + SODA) partially restored both the growth pattern and the parasite's ability to transform into amastigote-like forms (Fig. 5, A and B). Taken together, the results described in this section show that mitochondrial SODA is important for the differentiation of L. amazonensis promastigotes into amastigotes after both types of stimulation, low pH/high temperature and iron deprivation.
Figure 5.

SODA/ΔsodA promastigotes under iron deprivation fail to transform into non-flagellated forms and show an altered growth pattern. Wild-type (WT), SODA single knock-out (SODA/ΔsodA), or complemented SODA single knock-out (SODA/ΔsodA + SODA) promastigotes grown to mid-log phase (1–2 × 107/ml) in regular growth media were transferred to iron-depleted media. A, viability and growth were monitored over the indicated time period by microscopic counting of FDA-positive cells. B, percentage of WT SODA/ΔsodA or SODA/ΔsodA + SODA parasites in iron-deficient cultures that exhibit a rounded morphology and shortened flagellum at the indicated time points. The data represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments (Student's t test, ***, p = 0.001, and **, p = 0.008).
Figure 6.

SODA/ΔsodA promastigotes are more susceptible to ROS stress. A, growth curves for wild-type (WT), SODA single knock-out (SODA/ΔsodA), or SODA single knock-out complemented (SODA/ΔsodA + SODA) parasites in regular promastigote growth medium. B, effect of menadione on the survival of (WT, SODA/ΔsodA, or SODA/ΔsodA + SODA promastigotes. The parasites were cultured in the presence of increasing concentrations of menadione for 48 h followed by estimation of viable cell numbers by FDA staining. The data expressed as percentage of the number of viable cells in cultures without menadione represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments.
Stationary phase SODA/ΔsodA promastigotes are more susceptible to ROS stress and are impaired in metacyclogenesis
We next examined whether a single copy of SODA was sufficient to sustain growth and development during the promastigote stage. When grown in complete promastigote culture medium (containing iron and 10% FBS), SODA/ΔsodA promastigotes grew at a similar rate as WT during the early logarithmic phase of growth but entered stationary phase earlier at a density of 5–6 × 107 cells/ml (day 5). In contrast, WT parasites kept growing until reaching stationary phase at 7–8 × 107 cells/ml (day 7) (Fig. 6A). Moreover, after day 5, the SODA/ΔsodA parasite population showed a steady decline, a process that was partially reverted in the complemented SODA/ΔsodA + SODA line. These results suggest that SODA activity is also important for promastigote viability during the stationary phase of growth.
To investigate whether an impaired ability to detoxify mitochondrial ROS products due to SODA deficiency might cause promastigote death, we exposed WT, SODA/ΔsodA, and SODA/ΔsodA + SODA mid-log promastigotes to increasing concentrations of menadione, a drug that induces O2˙̄ generation in mitochondria. SODA/ΔsodA parasites showed markedly higher sensitivity to the drug, with a drop in viability from >60% in WT to <30% in SODA/ΔsodA after exposure to 4 μm menadione. As expected, complemented SODA/ΔsodA + SODA promastigotes were more resistant to menadione toxicity when compared with SODA/ΔsodA parasites (Fig. 6B). Collectively, our data suggest that normal levels of SODA expression are required for protection against the accumulation of mitochondrion-generated ROS during the late-log and stationary phases of promastigote growth.
Environmental stress (e.g. nutrient and oxygen deprivation) experienced by Leishmania promastigotes attached to the sand fly midgut is thought to be important for their transformation into infective metacyclic forms (29). However, very little is known about the actual signaling pathway involved in Leishmania metacyclogenesis in vivo. Considering our evidence for a role of SODA-mediated redox signaling in the promastigote-amastigote transition, and the increased SODA protein levels observed in stationary phase promastigotes in culture, we investigated whether SODA was also required for metacyclic promastigote development. The number of metacyclic forms was quantified on day 6 WT, SODA/ΔsodA, and SODA/ΔsodA +SODA stationary promastigote cultures after selective agglutination with an L. amazonensis promastigote-specific antibody. SODA/ΔsodA cultures showed ∼3-fold reduction in the yield of metacyclics compared with WT, a phenotype partially restored by SODA complementation (SODA/ΔsodA + SODA) (Fig. 7A). Scanning electron microscopy (SEM) analysis revealed a normal promastigote morphology for all three lines during the log phase of growth (day 3), but during stationary phase (day 7) the elongated and slender forms characteristic of metacyclic forms were only observed in WT and SODA/ΔsodA + SODA (Fig. 7B). Thus, the SODA/ΔsodA stationary phase promastigote population was largely devoid of slender metacyclic forms and contained a large number of cells with abnormal morphology.
Figure 7.

Loss of a single SODA allele impairs metacyclogenesis. A, infective metacyclic forms were quantified microscopically from stationary (day 7) WT, SODA/ΔsodA, or SODA/ΔsodA + SODA cultures after agglutinating promastigotes with the m3A.1 monoclonal antibody. The data represent the mean ± S.D. of the percentage of metacyclics in triplicate determinations and represent data obtained from four independent experiments (**, p = 0.003; *, p = 0.01). B, SEM images of representative parasite morphologies observed in day 7 cultures of wild-type (WT), SODA single knock-out (SODA/ΔsodA), or SODA single knock-out complemented (SODA/ΔsodA + SODA) promastigotes. Arrowheads point to metacyclic forms. Bars, 4 μm. C, mitochondrial membrane potential (ψm) WT, SODA/ΔsodA, or SODA/ΔsodA + SODA parasites during different phases of growth determined by quantification of JC-1 uptake. The data represent the mean ± S.D. of three independent experiments. (Student's t test, **, p ≤ 0.01).
To assess mitochondrial function, we treated promastigotes with JC-1, a lipophilic cationic dye whose rate of accumulation inside mitochondria is directly dependent on the maintenance of an active mitochondrial membrane potential (Δψm) (17, 30). The shift of JC-1 from a monomeric form under low concentrations to an aggregated form inside mitochondria is detected as a shift from green (emission 530 nm) to red (emission 590 nm) fluorescence. Thus, the 590 nm/530 nm ratio in this assay provides an accurate quantification of the ΔΨm-dependent amount of dye imported into mitochondria. Increase in the 590 nm/530 nm fluorescence ratio was observed for all three lines on days 2 and 3 of culture, indicating healthy mitochondrial activity during the logarithmic phase of growth. However, SODA/ΔsodA parasites showed a significant reduction in the 590 nm/530 nm fluorescence ratio on day 7, an effect reversed by SODA complementation (Fig. 7C). This result suggests that SODA expression from a single allele is not sufficient to maintain normal mitochondrial function as the parasites enter the stationary phase of growth, when differentiation into virulent metacyclic forms is initiated.
Deletion of one SODA allele reduces mitochondrial SOD activity in stationary phase promastigotes
Whole-cell extracts from wild type, SODA/ΔsodA, and SODA/ΔsodA + SODA were prepared at different phases of promastigote growth and assayed for SOD activity. A reduction in the total SOD activity was observed in SODA/ΔsodA parasites in late-log (day 5) and stationary phase (day 7) (Fig. 8A). Because this biochemical activity assay does not distinguish between the mitochondrial SODA and the glycosomal SODB, we analyzed the same parasite extracts by immunoblotting with antibodies specific for each of the enzymes. As observed for the total SOD activity, SODA protein expression in the WT and SODA/ΔsodA + SODA parasites was similar and increased gradually as the cells entered stationary phase, whereas significantly less SODA was detected in SODA/ΔsodA extracts at the same time points (days 5 and 7) (Fig. 8B). In contrast, the levels of SODB protein were more stable and comparable between the three lines, showing only slightly elevated levels on days 5 and 7 (Fig. 8B). These results suggest that the abnormal mitochondrial function observed in SODA/ΔsodA promastigotes is likely to be a consequence of reduced SODA expression during the stationary phase of growth. This finding is in agreement with the mitochondrial localization of SODA (Fig. 1A) and with a lack of involvement of the glycosomal SODB isoform.
Figure 8.

Reduced SOD activity in SODA/ΔsodA promastigotes in stationary phase. A, whole-cell lysates were prepared from wild-type (WT), SODA single knock-out (SODA/ΔsodA), or SODA single knock-out complemented (SODA/ΔsodA + SOD) promastigotes collected at indicated time points during growth and assayed for SOD activity. The data represent the mean ± S.D. of triplicate determinations and are representative of four independent experiments. (Student's t test compared with WT: day 5, **, p = 0.04; day 7, ***, p = 0.008, and **, p = 0.03.) B, Western blot analysis (with 10 μg protein/lane) showing the relative levels of SODA and SODB proteins in the whole-cell extracts of WT, SODA SODA/ΔsodA, or SODA/ΔsodA + SOD promastigotes used for the SOD activity assay in A. The ratio of SODA relative to tubulin protein in each sample is indicated. Tubulin expression levels were was used as loading controls.
Consistent with this view, progression into the stationary phase of growth was associated with a reduction in both the total SOD activity and the amount of SODA protein in mitochondrial fractions isolated from SODA/ΔsodA parasites, when compared with WT (Fig. 9, A and B). As expected, only trace amounts of SODB were detected in the same mitochondrial fractions. Slightly elevated levels of SOD activity were observed in mitochondrial extracts from SODA/ΔsodA on day 3, possibly as a response to elevated ROS stress in SODA-deficient parasites during the logarithmic phase of growth.
Figure 9.

SODA protein levels and SOD activity are reduced in mitochondrial fractions of SODA/ΔsodA stationary phase promastigotes. Lysates of mitochondrion-enriched fractions of wild-type (WT) or SODA single knock-out (SODA/ΔsodA) promastigotes were prepared from cells collected at different times of growth. A, SOD activity assay was performed with the mitochondrial lysates. The data represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments. (Student's t test compared with WT: day 5, **, p = 0.03; day 7, **, p = 0.008.) B, Western blottings of mitochondrial lysates (10 μg of protein/lane) were used to compare the amounts of SODA, APX, and SODB protein in WT and SODA/ΔsodA promastigotes. The ratio of SODA and APX relative to tubulin protein in each sample is indicated. The low levels of SODB detected indicate that there was minimal contamination of the mitochondrial fractions with glycosomes. Tubulin expression levels were used as loading controls.
Mitochondrial ROS generation is required for development of virulent forms
The data discussed above suggested that the inability of stationary phase SODA/ΔsodA promastigotes to efficiently differentiate into metacyclic forms might be due to inadequate generation of H2O2, which is known to function as an amastigote differentiation signaling molecule in L. amazonensis (16). However, an alternative explanation is that the differentiation defect of SODA/ΔsodA promastigotes was merely a result of mitochondrial dysfunction, caused by O2˙̄-induced damage to Fe-S cluster ETC proteins. To distinguish between these possibilities, wild-type and SODA/ΔsodA promastigotes were treated with MitoTempo, a mitochondrially targeted anti-oxidant that acts as a SOD mimetic, converting available O2˙̄ to H2O2 (31–33). Importantly, MitoTempo also reduces the overall leakage of electrons from the mitochondrial respiratory chain, thereby inhibiting production of all ROS, including O2˙̄, H2O2, and peroxynitrite (31). Accordingly, we found that treatment of both WT and SODA/ΔsodA promastigotes with MitoTempo improved parasite survival during the stationary phase of growth (Fig. 10A). MitoTempo also restored the Δψm of the SODA/ΔsodA stationary phase promastigotes to wild-type levels (Fig. 10B). These effects on promastigote survival and mitochondrial membrane potential were observed with both low (15 μm) and high (50 μm) concentrations of MitoTempo. Interestingly, when we examined the yield of metacyclic forms, an improvement was observed after treatment of SODA/ΔsodA parasites with 15 μm but not with 50 μm MitoTempo (Fig. 10C). We observed a similar concentration-dependent effect in the expression of the H2O2 reporter protein APX; 15 μm MitoTempo restored APX expression to WT levels, but such an effect was not seen after treatment with 50 μm. In WT parasites, both concentrations of MitoTempo caused >2-fold reduction in both the yield of purified metacyclics (Fig. 10C) and in APX protein levels (Fig. 10D). Thus, although mitochondrial function was restored in SODA/ΔsodA promastigotes treated with 50 μm MitoTempo, this treatment did not increase the yield of metacyclic stage differentiation, suggesting that mitochondrial dysfunction is not the primary cause for the inability to differentiate SODA-deficient parasites. In WT parasites, where a normal complement of SODA may reduce the availability of O2˙̄, MitoTempo may not stimulate H2O2 production, as indicated by the lower levels of APX expression under these conditions (Fig. 10D). Collectively, the results of these experiments reinforce the view that SODA-mediated H2O2 production plays a central role in the development of Leishmania virulent forms.
Figure 10.

MitoTempo restores survival, mitochondrial potential (ψm), and metacyclogenesis of SODA single knock-out (SODA/ΔsodA) promastigotes to wild-type (WT) levels. A, growth curves of WT or SODA/ΔsodA promastigote cultures in the absence or presence of varying concentrations of MitoTempo (as indicated). B, mitochondrial membrane potential (ψm) in stationary phase (day 7) of WT or SODA/ΔsodA parasites cultured in the absence (0) or presence of 15 μm (15) and 50 μm (50) MitoTempo were determined by quantifying JC-1 uptake. The data represent the mean ± S.D. of three independent experiments (Student's t test, **, p = 0.008). C, infective metacyclic forms were quantified microscopically from stationary (day 7) of WT or SODA/ΔsodA cultures grown in the absence or presence of 15 or 50 μm MitoTempo (as indicated) following agglutination of promastigotes with the m3A.1 monoclonal antibody. The data represent the mean ± S.D. of the percentage of metacyclics in triplicate determinations and are representative of three independent experiments (**, p ≤ 0.005). D, Western blot analysis showing the relative levels of APX protein in extracts of stationary phase (day 7) WT or SODA/ΔsodA promastigotes that were cultured in absence or presence of MitoTempo (concentrations as indicated) and used for purification of metacyclic forms (C). Tubulin expression levels were used as loading control. The ratio of APX relative to tubulin in each sample is indicated.
Deletion of one SODA allele results in strong loss of virulence
Metacyclic promastigotes from WT, SODA/ΔsodA, and SODA/ΔsodA + SODA promastigotes were purified from day 5 stationary cultures, and their viability was assessed with the dye FDA and compared for their ability to establish infections in mouse bone marrow macrophages (BMMs). Because of the reduced ability of SODA/ΔsodA to undergo metacyclogenesis, larger culture volumes were used to obtain comparable numbers of viable metacyclic forms. BMM infection with WT parasites progressed normally, and after the typical 24-h lag period the parasites replicated intracellularly as amastigotes, progressively increasing in number (Fig. 11A). In contrast, a steep decline in the number of intracellular parasites was observed with the SODA/ΔsodA line between 3 and 24 h after BMM infection, with only very few intact parasites being detectable after 72 h. Episomal expression of SODA enhanced the ability of the parasites to survive intracellularly, as indicated by a markedly increased number of parasites detected after infection of BMM with the SODA/ΔsodA + SODA line. The SODA complemented parasites were also able to replicate intracellularly, albeit at a slower rate when compared with the WT line (Fig. 11A). These results indicate that normal levels of SODA expression are necessary for L. amazonensis to establish successful infections in host macrophages.
Figure 11.

Deletion of a single SODA allele results in a marked loss of virulence that is partially restored by complementation with episomally expressed SODA. The ability of metacyclic promastigotes purified from wild-type (WT), SODA single knock-out (SODA/ΔsodA), or SODA single knock-out complemented (SODA/ΔsodA + SODA) stationary phase cultures to infect macrophages (BMM) and generate cutaneous lesions in mice was compared. A, BMMs isolated from C57BL/6 mice were infected with metacyclics for 3 h, fixed immediately (3 h), or after further incubation for 24, 48, or 72 h, and the number of intracellular parasites was determined microscopically. The data represent the mean ± S.D. of triplicate determinations and are representative of more than three independent experiments. B, female C57BL/6 mice were inoculated in their left hind footpad with viable WT, SODA/ΔsodA, or SODA/ΔsodA + SODA metacyclics, and lesion development was determined by weekly caliper measurements. The data correspond to the mean ± S.D. of values obtained from five individual mice in each group. C, parasite loads in the infected footpad tissues were determined 9 weeks after infection (n = 5), **, p = 0.013; *, p = 0.132.
We also examined the role of SODA in the parasite's ability to induce cutaneous lesions in mice. Purified viable metacyclic forms were injected into the footpads of C57BL/6 mice, and the progression of cutaneous lesions was quantified over a period of 9 weeks (Fig. 11B). A steady growth in lesion size was recorded for WT L. amazonensis up to week 9. In contrast, mice infected with SODA/ΔsodA metacyclics showed no evidence of lesion formation. Quantification of the parasite tissue load after 9 weeks showed a >104-fold higher number of parasites in the footpad tissues of mice injected with WT parasites, when compared with the group infected with the SODA/ΔsodA line. This phenotype was also partially restored in the complemented SODA/ΔsodA + SODA line (Fig. 11C). The inability to fully restore virulence was not unexpected, considering that lack of robust complementation is commonly observed in transgenic Leishmania (17, 34–38). These findings demonstrate that L. amazonensis must maintain normal levels of expression of the mitochondrial enzyme SODA to replicate inside host macrophages and establish cutaneous infections in vivo.
Discussion
Leishmania spp. are among the few organisms that can survive and replicate in the hostile environment of macrophage phagolysosomes, where degradative enzymes and ROS function as effective mechanisms of protection against pathogens. O2˙̄ and •NO are two key ROS molecules generated by macrophages to neutralize invading microorganisms (39). To successfully evade this onslaught, Leishmania species have developed multiple adaptive features that include an antioxidant defense repertoire that includes trypanthione/trypanthione reductase, peroxidases, and three iron-dependent SODs (39–41). The Leishmania glycosomal iron-dependent SOD isoforms SODB1 and SODB2 are developmentally regulated, as indicated by reports of elevated SODB1 transcripts in amastigotes and of SODB2 in promastigotes, respectively (42, 43). Leishmania chagasi SODB1 null mutants are not viable, and parasite lines lacking one SODB1 allele have markedly reduced viability inside macrophages (42). The mitochondrial SOD isoform SODA was previously proposed to protect Leishmania mitochondria from oxidative stress (21, 44), but prior to this study its physiological function had not been investigated. Here, we investigated the physiological role of L. amazonensis SODA in light of recent evidence implicating this iron-dependent enzyme in ROS generation inside mitochondria, a process proposed to play a key role in the development of the virulent life cycle stages of Leishmania (17).
We previously identified and functionally characterized the L. amazonensis mitochondrial iron importer LMIT1 (17). Similar to what we report here for SODA, LMIT1 null mutants are not viable, consistent with the importance of iron for the assembly of Fe-S cluster proteins and ETC function. We also found that LMIT1/Δlmit1 promastigotes partially impaired in mitochondrial iron import are markedly defective in the ability to differentiate into infective amastigote forms. LMIT1/Δlmit1 parasites also showed a significant drop in mitochondrial iron content, a decrease in aconitase (a Fe-S cluster protein) and SOD activity, and extensive damage to mitochondria following oxidative stress. Based on these findings, we hypothesized that mitochondria is the major site where O2˙̄ is generated and then converted by the iron-dependent SODA to H2O2, previously shown to act as a signal for differentiation (17). However, our initial studies of the role of SOD in L. amazonensis differentiation did not distinguish between the mitochondrial SODA and the glycosomal SODB1 and SODB2 isoforms. In this study, by measuring SOD activity in SODA-enriched mitochondrial fractions with little or no contaminating SODB, we clearly demonstrate that the >50% reduction in SOD activity observed in LMIT1/Δlmit1 L. amazonensis promastigotes in response to a differentiation stimuli can be attributed to SODA.
Our inability to generate SODA null mutants suggests that SODA is also essential for the long-term survival of L. amazonensis. This conclusion is consistent with the presence of functioning mitochondria in both promastigote and amastigote forms, and the well-established role of SOD in detoxifying ROS generated through electron leakage from the respiratory chain (6, 8). The critical role played by SODA in protecting mitochondria from endogenous ROS accumulation is further evident from the lethal phenotype we observed following RNAi-mediated knockdown of the SODA ortholog Tb927.5.3350 in procyclic forms of T. brucei. No phenotype was observed following RNAi-mediated silencing of the T. brucei SODA ortholog Tb927.5.3350 in bloodstream forms (18), in agreement with the dependence on active mitochondrial metabolism of T. brucei procyclics but not bloodstream forms, which possess only rudimentary mitochondria (27, 28).
SODA expression in L. amazonensis promastigotes increased progressively during culture reaching maximal levels on the stationary phase, presumably as a direct consequence of build up in oxidative stress. Such priming of the antioxidant defense system was proposed to facilitate virulence development, by preparing Leishmania parasites for the invasion of host macrophages (17, 26, 42). The ∼50% reduction in both SODA activity and SODA protein levels following entry into stationary phase is probably responsible for the sudden decline in cell viability we observed in SODA/ΔsodA promastigote cultures. In addition, the slight but reproducible increase in SOD activity observed in early log phase cultures of SODA/ΔsodA parasites is consistent with a putative surge in O2˙̄ levels during the initial phase of promastigote growth. In this scenario, impaired conversion of O2˙̄ into H2O2 by SODA might explain the faster growth rate observed for SODA/ΔsodA promastigotes during the log phase, given the known role of H2O2 in arresting cell growth and promoting cell differentiation in Leishmania and other organisms (9, 16, 26, 45, 46).
Metacyclic promastigotes derived from single allele deletion mutants of SODA showed a drastically reduced ability to establish infections in macrophages or to induce cutaneous lesions in mice. Because the parasite's ability to invade host cells was apparently not compromised, this loss of virulence may have resulted from enhanced susceptibility to macrophage-generated toxic oxygen radicals or from an inability to generate sufficient amounts of the amastigote differentiation signal, H2O2 (17). The latter possibility is strengthened by the failure of SODA/ΔsodA promastigotes to differentiate axenically into amastigotes when exposed to two different differentiation stimuli: low pH/high temperature or iron deprivation. Promastigote to amastigote differentiation involves a major shift in metabolism characterized by increased dependence on the tricarboxylic acid cycle and mitochondrial respiration for energy production and less dependence on glycolysis (47). In addition, to maintain a neutral intracellular pH while replicating in the acidic parasitophorous vacuole environment, intracellular amastigotes have to establish a strong intracellular proton gradient and actively pump H+ out of the cells (4, 19). Both processes require up-regulation of electron transport, which is a major source of O2˙̄ generation. Thus, the high mortality rate we observed as SODA/ΔsodA promastigotes failed to differentiate into amastigotes is consistent with a requirement for a threshold level of active SODA to counter the O2˙̄ surge and initiate H2O2-mediated signaling. This view is in agreement with our earlier characterization of the LMIT1/Δlmit1 line that is deficient in mitochondrial iron import (17).
An additional finding that emerged from this study is the importance of SODA and its product H2O2 in the generation of infective metacyclic promastigotes (a process that occurs in nature within the digestive tract of sand fly vectors (48)). Although it was possible to obtain sufficient viable metacyclic promastigotes to perform virulence assays, we observed a sharp decline in the yield of these infective stages, a phenotype that was reversed by episomal expression of SODA and by treatment with SOD mimetic MitoTempo. This finding significantly expands the evidence supporting the important role of mitochondrion-generated ROS as a regulator of virulence in Leishmania parasites.
Experimental procedures
Leishmania culture
L. amazonensis (IFLA/BR/67/PH8) was a kind gift from Dr. David Sacks (Laboratory of Parasitic Diseases, NIAID, National Institutes of Health). Promastigote forms were cultured in vitro at 26 °C in M199 media (pH 7.4) supplemented with 10% heat-inactivated FBS, 0.1% hemin (Frontier Scientific; 25 mg/ml in 50% triethanolamine), 10 mm adenine (pH 7.5), 5 mm l-glutamine and 5% penicillin/streptomycin (36). To induce axenic differentiation into amastigote forms, promastigote cultures (∼2–4 × 107/ml) were mixed with equal volumes of acidic amastigote media (M199 containing 0.25% glucose, 0.5% trypticase, and 40 mm sodium succinate (pH 4.5)) and incubated at 32 °C. Differentiated amastigotes were maintained in amastigote media at 32 °C. Parasite viability was determined by fluorescence microscopy following FDA (Sigma) and propidium iodide (Sigma) staining as described previously (16). Ability to differentiate was quantitated as the percentage of promastigotes with long flagella (undifferentiated) versus rounded forms with short flagella (differentiated) parasites via phase-contrast microscopy. At least 200 viable cells per sample were scored.
Estimation of growth and differentiation of L. amazonensis promastigotes in iron-depleted growth media were done as described earlier (16, 17). Briefly, mid-log phase L. amazonensis promastigotes (∼2 × 107/ml) were harvested and resuspended in iron-depleted media at a concentration of 2 × 106/ml. Cell growth was quantitated by microscopic counting of FDA-stained cells at different times, as indicated.
Menadione sensitivity was determined by seeding promastigotes in log-phase culture (∼2 × 107/ml) at 4 × 105/ml with or without increasing concentrations of menadione. Following 48 h of incubation at 26 °C, parasite viability was determined by fluorescence microscopy following FDA staining. To assess the effect of the mitochondrial SOD mimetic MitoTempo (Sigma) on L. amazonensis growth and differentiation, the antioxidant was added daily to promastigote cultures at the indicated concentrations for the whole duration of the experiment.
T. brucei culture and RNAi
T. brucei procyclics from the 29-13 strain (49) that stably expressed T7 polymerase and Tet repressor were cultured at 27 °C in SM9 media containing 15 μg/ml G418 (Gibco) and 50 μg/ml hygromycin (Invitrogen) and supplemented with 10% tetracycline-free FBS (Atlanta Biological) (50). Growth of T. brucei cultures was monitored by counting using a Beckman Coulter counter.
Generation of RNAi cell lines
A 427-bp gene sequence targeting the T. brucei SODA gene (Tb927.5.3350) for RNAi-mediated knockdown was identified using RNAit software (51) and amplified from T. brucei genomic DNA using the following oligonucleotides: TbSODA-HindIII forward (FD), GAAAGCTTGTTGGAGCTGCACTACACGA, and TbSodA-XbaI reverse (RV), GATCTAGACCCACACATCCACTGTGAAG (introduced HindIII and XbaI restriction sites are indicated as italicized and underlined nucleotides). The amplified gene fragment was then cloned into p2T7-LMIT1 (17) by replacing the LMIT1 gene using corresponding restriction sites to create the RNAi construct pTbSODA-KD. The resulting plasmid linearized with NotI was electroporated into T. brucei, and stable transfectants (2T7/SODA) were obtained after limiting dilution in 96-well plates with phleomycin (2.5 μg/ml) and selection as described (50).
RNAi-mediated knockdown
dsRNA synthesis was induced by the addition of 1 μg/ml tetracycline to cultures of clonal cell lines at 1 × 106/ml starting concentration. Cells growing in the presence or absence of tetracycline were counted daily using a hemocytometer and diluted to the initial starting concentrations. SODA knockdown was confirmed by performing Western blot analyses of whole-cell lysates at different time points following tetracycline addition, using anti-SODA rabbit polyclonal antibodies raised against Leishmania SODA.
Generation of L. amazonensis SODA single knock-out cell lines
The L. amazonensis SODA ORF was genetically targeted for replacement with gene deletion constructs containing the hygromycin resistance gene or neomycin phosphotransferase through homologous recombination, as described earlier (17, 52). Sequences upstream and downstream of the SODA ORF were cloned using the following primers containing SfiI restriction enzyme sites (underlined): LamSODA 5′SfiI-A, FD (GAGGCCACCTAGGCCCGAAGAGGGAGTTGTG), and LamSODA 5′SfiI B, RV (GAGGCCACGCAGGCCGAGTAGTGAGGTGCTTT), to amplify the 5′ sequence; LamSODA 3′SfiI-C FD, GAGGCCTCTGTGGCCTGCTTGGTGCCAACGCG, and LamSODA 3′SfiI-D RV, and GAGGCCTGACTGGCCGCTGACAACTGCACG for 3′ UTR. Four-part ligation using the PCR-amplified 5′- and 3′-flanking sequences, drug resistance cassettes, and the plasmid backbone was carried out. Positive clones were identified by analyzing SfiI restriction digests of plasmid DNA samples and confirmed by sequencing with specific primers as described (52). The targeting fragment used to transfect L. amazonensis promastigotes was released by PacI digestion, gel-purified, and used for electroporation. Leishmania clones with a single SODA allele deletion (SODA/ΔsodA) were isolated based on the ability of transformants to grow on agar plates containing hygromycin (100 μg/ml) or neomycin (50 μg/ml) and analyzed by Southern blotting and PCR to verify integration of the drug cassette in the desired location.
For generation of a rescue plasmid expressing Leishmania SODA with C-terminal hemagglutinin tag, a two-step PCR amplification strategy was employed. In the first round, a 693-bp fragment of the SODA ORF was amplified with primers SODA HA, FD, AACCCGGGACATATGTTCCGCCGTGTCTCGATG (SmaI site underlined), and SODA HA, RV, CTGGGACGTCGTATGGGTAAAGCTTCTTCGTGGC, that allowed for removal of the endogenous stop codon and introduction of an in-frame HA tag. The PCR product was used as template in a second round of amplification using SODA-HA, FD, as sense and HA TAG2, RV, TTGGATCCTTAAGCGTAGTCTGGGACGTCGTATGG (BamHI site underlined), as antisense primers. The final PCR products were digested with BamHI and SmaI and cloned into pXG-SAT (courtesy of Prof. S. Beverley, Washington University). Transfected Leishmania clones were selected in plates containing 50 μg/ml nourseothricin (Jena Biosciences), and expression of HA-tagged SODA was confirmed by Western blotting.
Expression of recombinant SODA, SODB, and APX proteins and generation of antibodies
To produce recombinant histidine-tagged SOD proteins, the genes encoding SODA, SODB, and APX were PCR-amplified from L. amazonensis genomic DNA using primers SODAexp, FD (GGCATATGTTCCGCCGTGTCTCG), and SODAexp RV, (GGAAGCTTCTTCGTGGCCTTTTC) for SODA; SODBexp FD (AACATATGCCGTTCGCTGTTCAGCCGCT), and SODBexp RV (TTAAGCTTCAGATCACTGTTGACGTAGTG) for SODB; and APXexp FD (CCACACATGTTCGGCACCTCGCGG) and APXexp RV (TTCAAAGCTTGCTCCCCGACGCGG) for APX. Forward primers for SODA and SODB contained an NdeI restriction site, and the APX gene contained a PciI site, contiguous with the start codon. Reverse primers were engineered to include a HindIII restriction site that removed the endogenous stop codon and allowed for synthesis of a six-histidine tag. The resulting PCR products were cloned in the pET28b(+) expression plasmid (Novagen) using NdeI and HindIII restriction sites, and the resulting plasmids were used to transform Escherichia coli BL21(DE3)pLysS (Novagen). Expression of His6-tagged SODA, SODB, and APX protein in soluble form was achieved by inducing transformed E. coli strains with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside (overnight at room temperature in media supplemented with 2% ethanol). His6-tagged SODA, SODB, or APX proteins were purified from bacterial cell extracts by nickel column chromatography with His-60 nickel superflow resin (Clontech) followed by elution with 0.3 m imidazole buffer, according to the manufacturer's protocol. Homogeneity of the purified proteins was ascertained by SDS-PAGE.
Polyclonal antibodies were raised against purified SODA, SODB, and APX proteins by periodic injection of rabbits with purified protein samples (the Pocono Rabbit Farm and Laboratory). Specificity of the antisera was assessed by Western blotting.
Isolation of mitochondrial fractions
Mitochondrial enrichment was performed as described previously (17) using 5 × 108 promastigotes from stationary phase cultures. The cells were washed three times with MES buffer (20 mm MOPS (pH 7.0), 250 mm sucrose, and 3 mm EDTA) and resuspended in 500 μl of MES supplemented with 1 mg/ml digitonin and protease inhibitor mixture (Roche Applied Science). Following a 5-min incubation at room temperature, the cell suspensions were centrifuged for 5 min (10,000 × g at 4 °C), and the supernatant was collected as the cytoplasmic fraction. The pellet was washed once with MES buffer and used for further analysis as the mitochondrial fraction or stored at −80 °C until further use.
Determination of SOD activity in whole-cell and mitochondrial extracts
SOD activity was determined in whole-cell extracts as described earlier (16, 17). To estimate mitochondrial SOD activity, mitochondrial fractions obtained following subcellular fractionation as described above were resuspended in buffer A and lysed by sonication. SOD activity in the lysates was measured using the SOD Assay Kit-WST (Dojindo Molecular Technologies, Inc.) according to the manufacturer's protocol. Standard curves were generated using known concentrations of horseradish SOD (Sigma). Protein content was determined using BCATM protein assay kit (Thermo Fisher Scientific).
Localization of SODA by immunofluorescence microscopy
Immunolocalization of SODA was performed as described previously (17). To confirm the mitochondrial localization of SODA, promastigotes were incubated with MitoTracker Red CMXRos (Invitrogen) followed by fixation with 4% paraformaldehyde and attachment to poly-l-lysine-coated slides (multitest 8-well; MP Biomedicals). Following treatment with 50 mm NH4Cl, the cells were permeabilized with 0.1% Triton X-100 in PBS, blocked with PBS 5% horse serum and 1% bovine serum albumin (BSA) for 1 h at room temperature, and incubated with anti-SODA rabbit polyclonal antibodies (1:10,000 dilution in PBS 1% BSA) for 1 h followed by anti-rabbit IgG AlexaFluor 488 (Invitrogen) 1:5000 dilution in PBS 1% BSA for 1 h, and staining with 2 μg/ml DAPI for 1 h. Slides were mounted with ProLong Gold antifade reagent (Invitrogen); images were acquired through a Deltavision Elite Deconvolution microscope (GE Healthcare) and processed using Volocity Suite (PerkinElmer Life Sciences).
Assays for mitochondrial activity
Mitochondrial membrane potential (Δψm) was estimated using the MitoProbe JC-1 assay kit (Invitrogen). 1 × 107 promastigote cells were incubated with 10 μm JC-1 for 15 min at 27 °C, washed, and resuspended in PBS. Fluorescence measured at 530 and 590 nm using a SpectraMaxM5e microtiter plate reader (Molecular Devices) was used determine the Δψm (530:590 ratio).
To visualize the mitochondrial staining pattern, promastigotes were placed in glass-bottom dishes (MatTek Corp.) for live imaging on a Nikon Eclipse Ti inverted microscope with a ×100 NA 1.4 objective (Nikon) equipped with a Hamamatsu C9100-50 camera and mCherry and FITC filters. Acquired images were analyzed with the Volocity Software Suite (PerkinElmer Life Sciences).
Electron microscopy
For scanning EM, parasites fixed in 2.5% (v/v) glutaraldehyde in 0.1 m sodium cacodylate buffer (pH 7.4) for 60 min and attached to poly-l-lysine-coated coverslips were rinsed briefly with PBS, fixed with 0.1 m cacodylate buffer (pH 7.4), treated with osmium tetroxide for 1 h, acetone-dehydrated, and critical point dried from CO2. After sputter coating with Au/Pd, the preparations were imaged in an Amray 1820D scanning electron microscope. For transmission EM, fixed parasite cells were post-fixed with osmium tetroxide, and cell sections were prepared as described before (38). Final images were obtained using a Zeiss EM10CA electron microscope.
Quantification of Leishmania intracellular growth in macrophages
Macrophage infection assays were carried out as described previously (16, 17, 37, 38). A total of 1 × 106 BMMs from C57/BL6 mice (Charles River Laboratories), plated on glass coverslips in 3-cm dishes 24 h prior to the experiment, were infected with at a 1:5 multiplicity of infection with metacyclic forms purified from stationary phase promastigote cultures (5-day-old) using the m3A1 monoclonal antibody (53). After allowing 3 h for invasion, BMMs were washed three times in PBS and incubated for the indicated times at 34 °C. Coverslips were retrieved after 3 (initial infection) 24, 48, and 72 h of incubation, fixed in 4% PFA, permeabilized with 0.1% Triton X-100 for 10 min, and stained with 10 μg/ml DAPI for 1 h. The total number of macrophages and the total number of intracellular parasites per microscopic field (100× N.A. 1.3 oil immersion objective, Nikon E200 epifluorescence microscope) were determined, and the results expressed as intracellular parasites per 100 macrophages. At least 300 host cells, in triplicate, were analyzed for each time point. The data were analyzed for statistical significance using an unpaired Student's t test (p < 0.05 was considered significant).
In vivo virulence and parasite load estimation
A total of 1 × 106 infective metacyclics purified from WT, SODA/ΔsodA, or SODA/ΔsodA + SODA stationary phase promastigote cultures and resuspended in a volume of 50 μl of PBS were used to inoculate 6-week-old female C57BL/6 mice (n = 5 per group) in the left hind footpad. Progression of footpad lesion development was monitored through weekly measurements with a caliper (Mitutoyo Corp., Japan), quantitating the difference between the left and right hind footpads. The parasite load was estimated in infected tissue collected from footpads of mice sacrificed 11 weeks post-infection using a limiting dilution assay (54).
Author contributions
B. M. and N. W. A. conceived and designed the research. B. M., M. F. L. S., D. C. M., and J. P. B. M., performed the experiments; B. M., M. F. L. S., D. C. M., and J. P. B. M. analyzed the data; B. M. and N. W. A. prepared the manuscript.
Supplementary Material
Acknowledgments
We thank Jason Hauzel and Jennifer Jensen for providing excellent technical support. We also thank Drs. P. Yates (Oregon Health and Science University) and S. M. Beverley (Washington University) for their generous gifts of plasmids; Drs. B. Ullman and J. Boitz (Oregon Health and Science University) for antibodies; Dr. K. Hill (UCLA) for the T. brucei strains and plasmids, and T. Maugel (Laboratory for Biological Ultrastructure; University of Maryland) for EM technical assistance. We are also thankful to Drs. I. Hamza and A. Flannery for stimulating discussions and advice.
This work was supported by National Institutes of Health Grant R01 AI067979 (to N. W. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S3.
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- ASL
- adenosuccinate lyase
- TEM
- transmission electron microscopy
- SEM
- scanning electron microscopy
- BMM
- bone marrow-derived macrophage
- FDA
- fluorescein diacetate
- ETC
- electron transport chain
- FD
- forward
- RV
- reverse.
References
- 1. World Health Organization (2010) Control of the leishmaniases. World Health Organ. Tech. Rep. Ser., 2010, xii–xiii, 1–186 [PubMed] [Google Scholar]
- 2. Bates P. A. (2008) Leishmania sand fly interaction: progress and challenges. Curr. Opin. Microbiol. 11, 340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenzweig D., Smith D., Opperdoes F., Stern S., Olafson R. W., and Zilberstein D. (2008) Retooling Leishmania metabolism: from sand fly gut to human macrophage. FASEB J. 22, 590–602 [DOI] [PubMed] [Google Scholar]
- 4. Tsigankov P., Gherardini P. F., Helmer-Citterich M., and Zilberstein D. (2012) What has proteomics taught us about Leishmania development? Parasitology 139, 1146–1157 [DOI] [PubMed] [Google Scholar]
- 5. Lahav T., Sivam D., Volpin H., Ronen M., Tsigankov P., Green A., Holland N., Kuzyk M., Borchers C., Zilberstein D., and Myler P. J. (2011) Multiple levels of gene regulation mediate differentiation of the intracellular pathogen Leishmania. FASEB J. 25, 515–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reczek C. R., and Chandel N. S. (2015) ROS-dependent signal transduction. Curr. Opin. Cell Biol. 33, 8–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schieber M., and Chandel N. S. (2014) ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sena L. A., and Chandel N. S. (2012) Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsukagoshi H., Busch W., and Benfey P. N. (2010) Transcriptional regulation of ROS controls transition from proliferation to differentiation in the root. Cell 143, 606–616 [DOI] [PubMed] [Google Scholar]
- 10. Lennicke C., Rahn J., Lichtenfels R., Wessjohann L. A., and Seliger B. (2015) Hydrogen peroxide–production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 13, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee S., Tak E., Lee J., Rashid M. A., Murphy M. P., Ha J., and Kim S. S. (2011) Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 21, 817–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Porporato P. E., Payen V. L., Pérez-Escuredo J., De Saedeleer C. J., Danhier P., Copetti T., Dhup S., Tardy M., Vazeille T., Bouzin C., Feron O., Michiels C., Gallez B., and Sonveaux P. (2014) A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766 [DOI] [PubMed] [Google Scholar]
- 13. Chandel N. S. (2015) Evolution of mitochondria as signaling organelles. Cell Metab. 22, 204–206 [DOI] [PubMed] [Google Scholar]
- 14. Diebold L., and Chandel N. S. (2016) Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 100, 86–93 [DOI] [PubMed] [Google Scholar]
- 15. Quinlan C. L., Perevoshchikova I. V., Hey-Mogensen M., Orr A. L., and Brand M. D. (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mittra B., Cortez M., Haydock A., Ramasamy G., Myler P. J., and Andrews N. W. (2013) Iron uptake controls the generation of Leishmania infective forms through regulation of ROS levels. J. Exp. Med. 210, 401–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mittra B., Laranjeira-Silva M. F., Perrone Bezerra de Menezes J., Jensen J., Michailowsky V., and Andrews N. W. (2016) A trypanosomatid iron transporter that regulates mitochondrial function is required for Leishmania amazonensis virulence. PLoS Pathog. 12, e1005340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor M. C., and Kelly J. M. (2010) Iron metabolism in trypanosomatids, and its crucial role in infection. Parasitology 137, 899–917 [DOI] [PubMed] [Google Scholar]
- 19. Zilberstein D., and Shapira M. (1994) The role of pH and temperature in the development of Leishmania parasites. Annu. Rev. Microbiol. 48, 449–470 [DOI] [PubMed] [Google Scholar]
- 20. Wilson M. E., Andersen K. A., and Britigan B. E. (1994) Response of Leishmania chagasi promastigotes to oxidant stress. Infect. Immun. 62, 5133–5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alzate J. F., Arias A. A., Moreno-Mateos D., Alvarez-Barrientos A., and Jiménez-Ruiz A. (2007) Mitochondrial superoxide mediates heat-induced apoptotic-like death in Leishmania infantum. Mol. Biochem. Parasitol. 152, 192–202 [DOI] [PubMed] [Google Scholar]
- 22. Riemann A., Schneider B., Ihling A., Nowak M., Sauvant C., Thews O., and Gekle M. (2011) Acidic environment leads to ROS-induced MAPK signaling in cancer cells. PLoS ONE 6, e22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Getachew F., and Gedamu L. (2007) Leishmania donovani iron superoxide dismutase A is targeted to the mitochondria by its N-terminal positively charged amino acids. Mol. Biochem. Parasitol. 154, 62–69 [DOI] [PubMed] [Google Scholar]
- 24. Boitz J. M., Strasser R., Yates P. A., Jardim A., and Ullman B. (2013) Adenylosuccinate synthetase and adenylosuccinate lyase deficiencies trigger growth and infectivity deficits in Leishmania donovani. J. Biol. Chem. 288, 8977–8990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. da Silva M. F., Zampieri R. A., Muxel S. M., Beverley S. M., and Floeter-Winter L. M. (2012) Leishmania amazonensis arginase compartmentalization in the glycosome is important for parasite infectivity. PLoS ONE 7, e34022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pal S., Dolai S., Yadav R. K., and Adak S. (2010) Ascorbate peroxidase from Leishmania major controls the virulence of infective stage of promastigotes by regulating oxidative stress. PLoS ONE 5, e11271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Weelden S. W., Fast B., Vogt A., van der Meer P., Saas J., van Hellemond J. J., Tielens A. G., and Boshart M. (2003) Procyclic Trypanosoma brucei do not use Krebs cycle activity for energy generation. J. Biol. Chem. 278, 12854–12863 [DOI] [PubMed] [Google Scholar]
- 28. Bringaud F., Rivière L., and Coustou V. (2006) Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol. Biochem. Parasitol. 149, 1–9 [DOI] [PubMed] [Google Scholar]
- 29. Bates P. A. (2007) Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int. J. Parasitol. 37, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mehta A., and Shaha C. (2004) Apoptotic death in Leishmania donovani promastigotes in response to respiratory chain inhibition: complex II inhibition results in increased pentamidine cytotoxicity. J. Biol. Chem. 279, 11798–11813 [DOI] [PubMed] [Google Scholar]
- 31. Dikalov S. (2011) Cross-talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nazarewicz R. R., Dikalova A., Bikineyeva A., Ivanov S., Kirilyuk I. A., Grigor'ev I. A., and Dikalov S. I. (2013) Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid. Redox Signal. 19, 344–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murphy M. P., and Smith R. A. (2007) Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 47, 629–656 [DOI] [PubMed] [Google Scholar]
- 34. Gaur U., Showalter M., Hickerson S., Dalvi R., Turco S. J., Wilson M. E., and Beverley S. M. (2009) Leishmania donovani lacking the Golgi GDP-Man transporter LPG2 exhibit attenuated virulence in mammalian hosts. Exp. Parasitol. 122, 182–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oyola S. O., Evans K. J., Smith T. K., Smith B. A., Hilley J. D., Mottram J. C., Kaye P. M., and Smith D. F. (2012) Functional analysis of Leishmania cyclopropane fatty acid synthetase. PLoS ONE 7, e51300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huynh C., Sacks D. L., and Andrews N. W. (2006) A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J. Exp. Med. 203, 2363–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Flannery A. R., Huynh C., Mittra B., Mortara R. A., and Andrews N. W. (2011) LFR1 ferric iron reductase of Leishmania amazonensis is essential for the generation of infective parasite forms. J. Biol. Chem. 286, 23266–23279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miguel D. C., Flannery A. R., Mittra B., and Andrews N. W. (2013) Heme uptake mediated by LHR1 is essential for Leishmania amazonensis virulence. Infect. Immun. 81, 3620–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Assche T., Deschacht M., da Luz R. A., Maes L., and Cos P. (2011) Leishmania-macrophage interactions: insights into the redox biology. Free Radic. Biol. Med. 51, 337–351 [DOI] [PubMed] [Google Scholar]
- 40. Kima P. E. (2014) Leishmania molecules that mediate intracellular pathogenesis. Microbes Infect. 16, 721–726 [DOI] [PubMed] [Google Scholar]
- 41. Adak S., and Pal S. (2013) Ascorbate peroxidase acts as a novel determiner of redox homeostasis in Leishmania. Antioxid. Redox Signal. 19, 746–754 [DOI] [PubMed] [Google Scholar]
- 42. Plewes K. A., Barr S. D., and Gedamu L. (2003) Iron superoxide dismutases targeted to the glycosomes of Leishmania chagasi are important for survival. Infect. Immun. 71, 5910–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ghosh S., Goswami S., and Adhya S. (2003) Role of superoxide dismutase in survival of Leishmania within the macrophage. Biochem. J. 369, 447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Getachew F., and Gedamu L. (2012) Leishmania donovani mitochondrial iron superoxide dismutase A is released into the cytosol during miltefosine induced programmed cell death. Mol. Biochem. Parasitol. 183, 42–51 [DOI] [PubMed] [Google Scholar]
- 45. Owusu-Ansah E., and Banerjee U. (2009) Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 461, 537–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Groeger G., Quiney C., and Cotter T. G. (2009) Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal. 11, 2655–2671 [DOI] [PubMed] [Google Scholar]
- 47. Naderer T., Ellis M. A., Sernee M. F., De Souza D. P., Curtis J., Handman E., and McConville M. J. (2006) Virulence of Leishmania major in macrophages and mice requires the gluconeogenic enzyme fructose-1,6-bisphosphatase. Proc. Natl. Acad. Sci. U.S.A. 103, 5502–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turco S. J., and Sacks D. L. (2003) Control of Leishmania-sand fly interactions by polymorphisms in lipophosphoglycan structure. Methods Enzymol. 363, 377–381 [DOI] [PubMed] [Google Scholar]
- 49. Wirtz E., Leal S., Ochatt C., and Cross G. A. (1999) A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 [DOI] [PubMed] [Google Scholar]
- 50. Oberholzer M., Lopez M. A., Ralston K. S., and Hill K. L. (2009) Approaches for functional analysis of flagellar proteins in African trypanosomes. Methods Cell Biol. 93, 21–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Redmond S., Vadivelu J., and Field M. C. (2003) RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol. Biochem. Parasitol. 128, 115–118 [DOI] [PubMed] [Google Scholar]
- 52. Fulwiler A. L., Soysa D. R., Ullman B., and Yates P. A. (2011) A rapid, efficient and economical method for generating leishmanial gene targeting constructs. Mol. Biochem. Parasitol. 175, 209–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pinto-da-Silva L. H., Fampa P., Soares D. C., Oliveira S. M., Souto-Padron T., and Saraiva E. M. (2005) The 3A1-La monoclonal antibody reveals key features of Leishmania (L) amazonensis metacyclic promastigotes and inhibits procyclics attachment to the sand fly midgut. Int. J. Parasitol. 35, 757–764 [DOI] [PubMed] [Google Scholar]
- 54. Tabbara K. S., Peters N. C., Afrin F., Mendez S., Bertholet S., Belkaid Y., and Sacks D. L. (2005) Conditions influencing the efficacy of vaccination with live organisms against Leishmania major infection. Infect. Immun. 73, 4714–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.