

Abstract
Chronic inflammation may contribute to insulin resistance via molecular cross-talk between pathways for pro-inflammatory and insulin signaling. Interleukin 1 receptor-associated kinase 1 (IRAK-1) mediates pro-inflammatory signaling via IL-1 receptor/Toll-like receptors, which may contribute to insulin resistance, but this hypothesis is untested. Here, we used male Irak1 null (k/o) mice to investigate the metabolic role of IRAK-1. C57BL/6 wild-type (WT) and k/o mice had comparable body weights on low-fat and high-fat diets (LFD and HFD, respectively). After 12 weeks on LFD (but not HFD), k/o mice (versus WT) had substantially improved glucose tolerance (assessed by the intraperitoneal glucose tolerance test (IPGTT)). As assessed with the hyperinsulinemic euglycemic glucose clamp technique, insulin sensitivity was 30% higher in the Irak1 k/o mice on chow diet, but the Irak1 deletion did not affect IPGTT outcomes in mice on HFD, suggesting that the deletion did not overcome the impact of obesity on glucose tolerance. Moreover, insulin-stimulated glucose-disposal rates were higher in the k/o mice, but we detected no significant difference in hepatic glucose production rates (± insulin infusion). Positron emission/computed tomography scans indicated higher insulin-stimulated glucose uptake in muscle, but not liver, in Irak1 k/o mice in vivo. Moreover, insulin-stimulated phosphorylation of Akt was higher in muscle, but not in liver, from Irak1 k/o mice ex vivo. In conclusion, Irak1 deletion improved muscle insulin sensitivity, with the effect being most apparent in LFD mice.
Keywords: Akt PKB, inflammation, insulin resistance, metabolism, muscle, glucose tolerance, IRAK-1
Introduction
Insulin resistance is defined as decreased sensitivity or impaired responsiveness to metabolic actions of insulin (1, 2). In humans, insulin resistance is typically associated with impairments in multiple metabolic processes: insulin-stimulated glucose uptake in skeletal muscle and fat cells, antilipolysis in adipose tissue, and suppression of gluconeogenesis in liver (3–7). Studies have revealed many mechanisms that can induce insulin resistance. These include pro-inflammatory signaling pathways in cells (8–11), animals (12, 13), and humans (14–16). Insulin regulates energy metabolism through a well-defined signaling pathway. As a consequence of binding to the insulin receptor (IR),2 insulin activates the receptor's intrinsic tyrosine kinase, which in turn catalyzes phosphorylation of intracellular substrates including insulin receptor substrate (IRS) proteins (17–19). Phosphotyrosine residues in IRS proteins provide docking sites for SH2 domain–containing signaling molecules including PI3K (20–22) and Grb-2/mSOS (20), leading to activation of PI3K and MAPK pathways. Lipid products of PI3K activate PDK-1, which triggers phosphorylation and activation of downstream serine kinases including Akt and PKC-ζ, enzymes with critical roles in mediating the metabolic actions of insulin (19, 23). Pro-inflammatory cytokines including IL-1, and free fatty acids (FFA), activate innate immune signaling to NF-κB via IL-1– and Toll-like receptors (TLR), which may impair insulin action in metabolic target tissues (24–26). This is mediated, in part, through serine phosphorylation of insulin receptor substrate 1 (IRS-1) by IKKβ, JNK, and IRAK-1, which impairs insulin signaling, thereby inducing insulin resistance (11, 16, 27–33). Understanding the detailed mechanisms of this cross-talk between pro-inflammatory signaling pathways and insulin signaling pathways may help to improve the diagnosis of prediabetic states, and also suggest new strategies to treat obesity and diabetes.
IRAK-1 is homologous to Pelle in Drosophila (34) and a key signaling molecule in mediating the inflammatory responses to IL-1-R and Toll-like receptor. IL-1-R and Toll-like receptors interact with MyD88 to activate IRAK-4, which phosphorylates and activates IRAK-1 (26). Downstream from IRAK-1, TRAF6 interacts with TAK1 to activate the IKK complex leading to phosphorylation of IκBα that then dissociates from NF-κB (35, 36). NF-κB enters the nucleus to promote transcription of many pro-inflammatory genes including interferons and cytokines, such as IL-1 and TNF-α (37–39). IL-1, TLR-2/-4 receptor ligands (e.g. FFA), and TNF-α may cause insulin resistance in metabolic targets via IRS-1 serine phosphorylation catalyzed by IKKβ, IRAK-1, and other innate immune kinases (28, 40–44). Inhibiting IKKβ reduces serine phosphorylation of IRS-1, which in turn ameliorates insulin resistance because of obesity or high-fat diet (16, 32, 45–47). High-activity genetic variants in the human IRAK1 gene are associated with higher circulating C-reactive protein (CRP) (a biomarker for heart disease risk and possibly diabetes) (48). IRAK-1 phosphorylates IRS-1 at Ser24, which impairs binding of IRS-1 to PI3K, thereby impairing metabolic actions of insulin including insulin-stimulated translocation of GLUT4 (28). Interestingly, IRS-2 does not have a corresponding IRAK-1 phosphorylation site (27). Thus, IRAK-1 is not predicted to alter IRS-2 function. Although IRAK-1 may impair metabolic actions of insulin primarily in cells (e.g. muscle) where IRS-1 is the predominant insulin signaling molecule downstream from the insulin receptor, the role of IRAK-1 might be less important in cells such as hepatocytes where IRS-2 plays the major role in mediating insulin actions (49–52).
In this study, we investigated the role of IRAK-1 in regulating glucose metabolism using Irak1 null mice. We showed that Irak1 null mice on low-fat diet (but not on high-fat diet that induces obesity, insulin resistance, and glucose intolerance) have improved glucose tolerance likely mediated by increased insulin sensitivity in skeletal muscle but not in liver. In the context of precision medicine, these data suggest that low IRAK-1 activity in skeletal muscle may have predictive value to identify a subset of insulin-resistant individuals most likely to obtain metabolic benefit from other therapeutic and lifestyle interventions that improve insulin sensitivity.
Results
Irak1 k/o mice display improved glucose tolerance on low-fat, but not on high-fat diet
To investigate whether IRAK-1 deficiency alters glucose metabolism, we studied mice fed for 14 weeks with either low-fat (10% kcal from fat) or high-fat diet (60% kcal from fat). As expected, wild-type mice fed with high-fat diet had higher caloric intake (supplemental Fig. S1, B and D) and also exhibited significantly higher body weight (supplemental Fig. S1, A and C). Although k/o mice ate slightly less food than WT mice on either diet (4.6% less on LFD and 8.9% less on HFD), there were no significant differences in body weight between Irak1 k/o and WT littermates on either diet (supplemental Fig. S1, A and C). These observations suggest that Irak1 k/o mice may have lower energy expenditure than WT littermates.
To investigate whether IRAK-1 deficiency altered glucose metabolism, we performed IPGTTs at baseline and also after 12 weeks of either HFD or LFD. There was no significant difference in baseline IPGTTs on normal chow diet between WT and k/o mice (data not shown). As expected, HFD induced glucose intolerance in both WT and k/o mice (Fig. 1, C and D). Furthermore, there was no significant difference between WT and k/o mice with respect to IPGTTs on the HFD, which induced obesity. By contrast, k/o mice had significantly enhanced glucose tolerance when fed an LFD (Fig. 1, A and B). Therefore, although IRAK-1 deficiency improved glucose tolerance in mice fed with an LFD, it did not protect mice from HFD-induced obesity and impairment in glucose tolerance.
Figure 1.
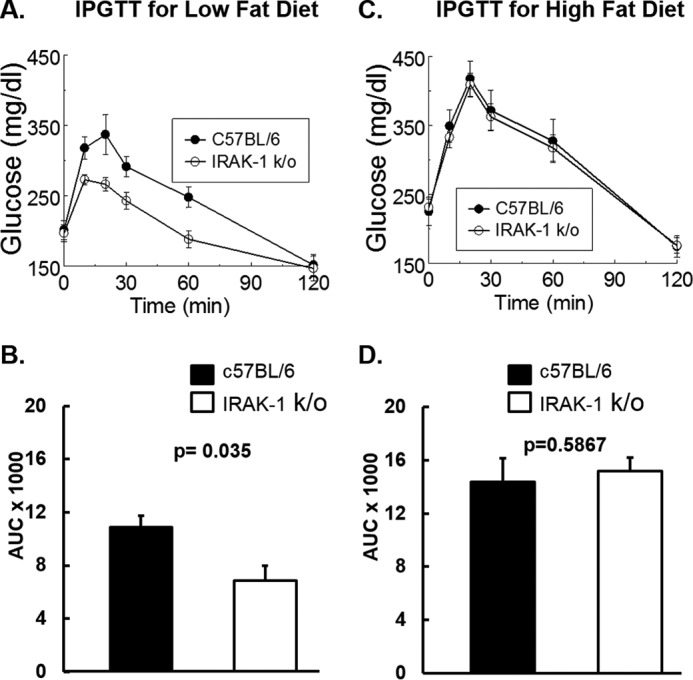
Intraperitoneal glucose tolerance tests (IPGTT) are shown. A–D, 6-week-old wild-type and Irak1 k/o mice were fed with either low-fat (A and B, n = 8) or high-fat diets (C and D, n = 8). After 12 weeks on the designated diets, mice were fasted for 5 h. Glucose (1 g/kg) was injected intraperitoneally and blood glucose was measured at 0, 10, 20, 30, 60, and 120 min after injection. B and D, areas under the curve (AUC) were calculated based on the trapezoidal rule. A, ANOVA for repeated measures, p = 0.004. B, AUCglucose = 10890 ± 840 versus 6850 ± 1100 (mg/dl) × min), p = 0.035. C, ANOVA for repeated measures, p = 0.56. D, AUCglucose = 14370 ± 1800 versus 15180 ± 1040 (mg/dl) × min), p = 0.59. Data are expressed as mean ± S.E.
To investigate whether IRAK-1 deficiency affected insulin sensitivity, we performed insulin tolerance tests (ITT). Based on studies with tissue-selective insulin receptor knock-out mice, the ITT is primarily a reflection of insulin sensitivity in the liver rather than muscle (49, 50). As expected, as judged by responses to ITT, both k/o and WT mice were less insulin sensitive when studied on HFD as compared with LFD (supplemental Fig. S2, A–D). However, there were no significant differences between Irak1 k/o mice and wild-type littermates when compared with each other on either the HFD or the LFD (supplemental Fig. S2, A–D). As demonstrated previously in studies with tissue-specific knock-outs, deletion of insulin receptors in the liver (but not in muscle) impair the ability of insulin to lower plasma glucose levels (49, 50). Thus, the ITT reflects primarily hepatic insulin sensitivity. Taken together, these data suggest that deletion of Irak1 did not protect mice from HFD-induced hepatic insulin resistance characteristic of obesity.
Irak1 k/o mouse displayed improved insulin sensitivity and lowered markers of endothelial dysfunction and insulin resistance
Although ITTs reflect primarily hepatic insulin sensitivity, hyperinsulinemic euglycemic glucose clamp studies are the reference standard for in vivo measurement of insulin sensitivity in extrahepatic tissues including skeletal muscle and fat (1, 53, 54). Because we did not detect effects of Irak1 deletion in mice fed an HFD, we conducted clamp studies in mice fed a normal chow diet. Average glucose was clamped at a steady-state level of 125 mg/dl by adjusting the glucose infusion rate (GIR) appropriately. Irak1 k/o mice required significantly higher glucose infusion rates when compared with WT littermates to maintain steady-state blood glucose levels (Fig. 2, A and B). After normalization for body weight (Fig. 2C) and steady-state blood glucose levels, we calculated an insulin sensitivity index (ISI = GIR/steady-state glucose) (Fig. 2D). The ISI was higher for Irak1 k/o mice than for their WT littermates when fed with a normal chow diet. Thus, deletion of Irak1 improved insulin sensitivity in extrahepatic tissues under these conditions (Fig. 2).
Figure 2.
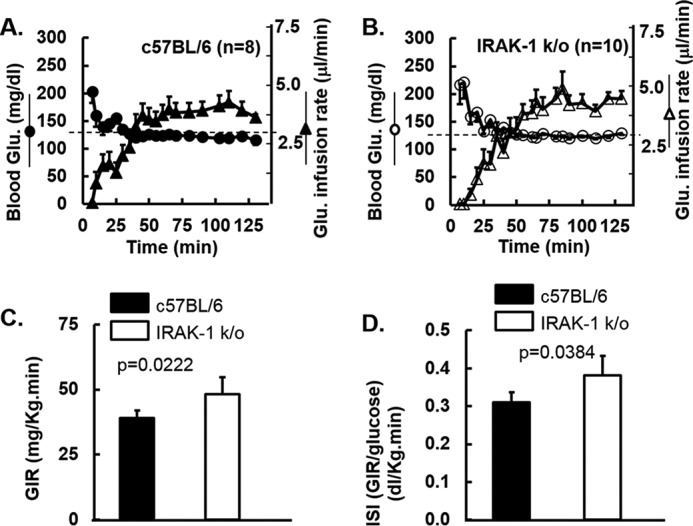
Hyperinsulinemic euglycemic glucose clamp studies in mice fed normal chow diet are shown. A–D, at 28 weeks of age, glucose clamp studies were performed as described under “Experimental Procedures” and previously (94). Mean glucose infusion rates over the 30-min steady-state period at the end of the study were used to estimate the steady-state glucose infusion rate (GIR). Data shown are mean ± S.E. A, for the WT mice (n = 8), blood glucose is indicated with solid circles and GIR with closed triangles. B, for Irak1 k/o mice (n = 10), blood glucose is indicated with open circles and GIR with open triangles. C, GIR normalized for body weight. D, ISI was defined as the GIR (glucose mg/kg/min) divided by the mean glucose level (mg/dl) calculated from the last 30 min of the clamp study when steady-state had been achieved.
IRAK-1 is a key signaling molecule in TLR/IL-1-R/TNFα-R mediated inflammatory responses and may contribute to the pathogenesis of chronic inflammation-associated insulin resistance. To explore this issue, we measured insulin resistance and cardiometabolic and inflammation biomarkers in mice fed for 14 weeks with an LFD and HFD. Fasting (5 h) plasma glucose was significantly lowered in Irak1 k/o mice as compared with wild-type mice fed with LFD (Fig. 3B). Although fasting plasma insulin concentration tended to be lower in Irak1 k/o mice, this did not reach statistical significance (Fig. 3A). Nevertheless, QUICKI, a fasting surrogate index for insulin sensitivity (55, 56), was significantly higher (Fig. 3C), suggesting an improvement of insulin sensitivity for k/o mice when compared with wild-type littermates. There was no difference in adiponectin levels between k/o and wild type littermates (Fig. 3D). Endothelin-1 (ET-1) is a potent vasoconstrictor and a biomarker for endothelial dysfunction associated with insulin resistance (59–61). Interestingly, deletion of Irak1 led to lowered ET-1 levels (Fig. 3E), suggesting an improvement in endothelial function in Irak1 k/o mice consistent with a role for inflammation in endothelial dysfunction related to insulin resistance. TNF-α is a biomarker for inflammation, and is reported to be associated with insulin resistance (57, 58). Plasma levels of TNF-α in our experiments were too low to obtain reliable data with our immunoassay although there was a trend toward decreased levels of TNF-α in k/o mice as compared with WT mice (data not shown), consistent with attenuation of chronic inflammation. These data are consistent with the conclusion that the k/o were more insulin-sensitive than WT mice.
Figure 3.
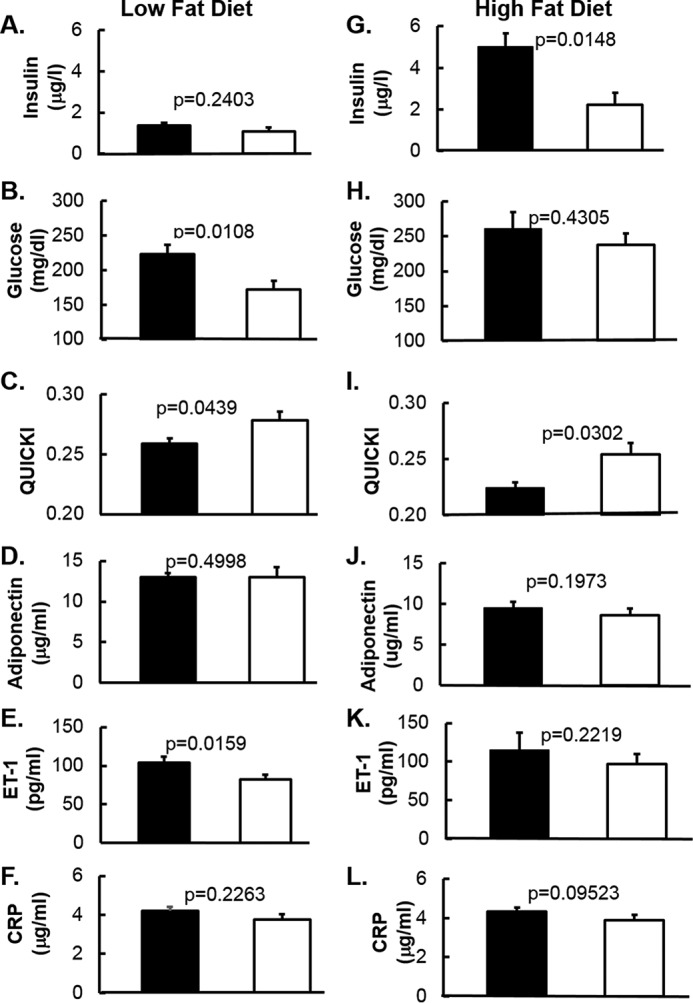
Cardiometabolic biomarkers in mice fed with low-fat diet and high-fat diet are shown. A–L, fasting plasma was obtained after 14 weeks on low-fat diet (A–F) or high-fat diet (G–L). B and H, glucose was measured by YSI 2700 Select Biochemistry Analyzer. Insulin (A and G), adiponectin (D and J), ET-1 (E and K), and CRP (F and L) were measured by ELISA. C and I, QUICKI was calculated as QUICKI = 1/(log (insulin) + log (glucose)) (55, 56, 100). Data are expressed as mean ± S.E.
As expected, when compared with low-fat diet, high-fat diet mice showed higher insulin resistance markers including fasting insulin (Fig. 3G), glucose (Fig. 3H), ET-1 (Fig. 3K), and lower adiponectin (Fig. 3J) and QUICKI (Fig. 3I). This confirmed that 14 weeks of high-fat diet induced obesity (supplemental Fig. S1, C and D) and insulin resistance. However, we did not observe a significant difference in ET-1 (Fig. 3K) between WT and k/o as was seen in the low-fat diet study (Fig. 3E). Adiponectin (Fig. 3J) and CRP (Fig. 3L) also showed no difference between WT and k/o mice. Of note, we observed lower plasma insulin levels in k/o mice (Fig. 3G), and, as a result, higher QUICKI (Fig. 3I).
Improved insulin sensitivity in Irak1 k/o mice may be caused specifically by increased glucose uptake in skeletal muscle
To assess contributions of specific tissues to improvement in glucose metabolism in vivo, we conducted [18F]FDG PET/CT scans to investigate insulin-stimulated [18F]FDG bio-distribution in three insulin-sensitive target tissues: hind limb muscle, abdominal subcutaneous white adipose tissue, and liver (supplemental Fig. S3). We did not observe higher [18F]FDG uptake in the liver post insulin injection in either Irak1 k/o or wild-type mice (Fig. 4, A and B). This suggests that altered liver glucose metabolism did not account for improved insulin sensitivity observed in Irak1 k/o mice. By contrast, in skeletal muscle, [18F]FDG uptake was significantly higher in k/o mice when compared with wild-type littermates. This achieved statistical significance at 20–30 min (i.e. the delayed washout phase of [18F]FDG), but not in the vascular phase (i.e. the 0- to 10-min time frame) (Fig. 4, A and B). These results demonstrated an important role of IRAK-1 in skeletal muscle glucose uptake, and likely underlie the increased insulin sensitivity observed in Irak1 k/o mice on LFD. White adipose tissue of the abdomen also showed a trend toward delayed washout of [18F]FDG after insulin injection in both types of animals, but there was no difference between k/o and wild-type mice (Fig. 4, A and B). Thus, it is unlikely that adipose tissue had an important direct role in mediating the beneficial effect of IRAK-1 deficiency to improve insulin sensitivity.
Figure 4.
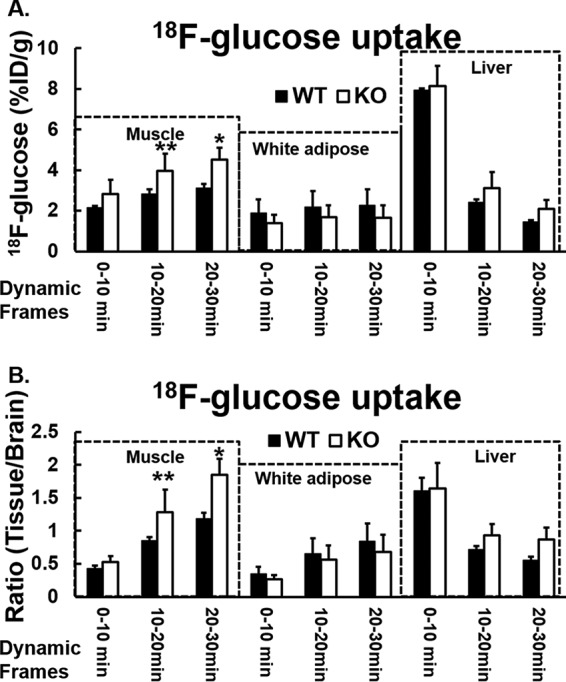
In vivo insulin-stimulated [18F]FDG distribution in mice fed with normal chow diet is shown. A and B, after mice were fasted overnight, [18F]FDG 11.1 MBq (300 Ci) and insulin (1.125 units/kg) in 200 μl of saline were injected via tail vein under anesthesia. Dynamic imaging was acquired for 30 min by Siemens Inveon Small Animal PET/CT Imaging System. The data were obtained by drawing regions of interest (ROI) over target organs of muscle, abdominal subcutaneous white adipose tissue, liver, and brain (98, 99). The [18F]FDG bio-distribution in the target organs/tissues were analyzed at three time points: 10, 20, and 30 min. A, time-dependent distribution percentage of injected dose per gram (%ID/g) of [18F]FDG in the hind limb, liver, and abdominal white fat in Irak1 k/o mice and in wild-type mice. B, relative time-dependent distribution of [18F]FDG in the hind limb, liver, and abdominal white fat (ratio of [18F]FDG:specific tissue/brain) in Irak1 k/o mice and in wild-type mice. WT, n = 3; k/o, n = 3; *, p < 0.04; **, p < 0.03. Data shown are mean ± S.E.
To further support these findings, we performed hyperinsulinemic euglycemic glucose clamp studies with [3-3H]glucose tracer on Irak1 k/o and WT littermates fed with chow diets. The hyperinsulinemic euglycemic clamp with [3-3H]glucose tracer is the reference standard method for assessing insulin sensitivity in muscle, fat, and liver (1, 53, 62). As expected, Irak1 k/o mice required significantly higher glucose infusion rate resulting in a larger insulin sensitivity index as compared with wild-type littermates (Fig. 5, A and B). Glucose appearance rates were comparable at the basal state, but significantly higher in Irak1 k/o mice during insulin infusion (Fig. 5C). When hepatic glucose production was measured, there was no significant difference between Irak1 k/o mice and their WT littermates in either the basal state or the insulin-stimulated state (Fig. 5D). These data are consistent with the PET/CT scan data showing improved insulin sensitivity in skeletal muscle but not liver. This suggests that improvement of insulin sensitivity in Irak1 k/o mice mainly occurs in muscle and/or adipose tissue when compared with wild-type mice.
Figure 5.
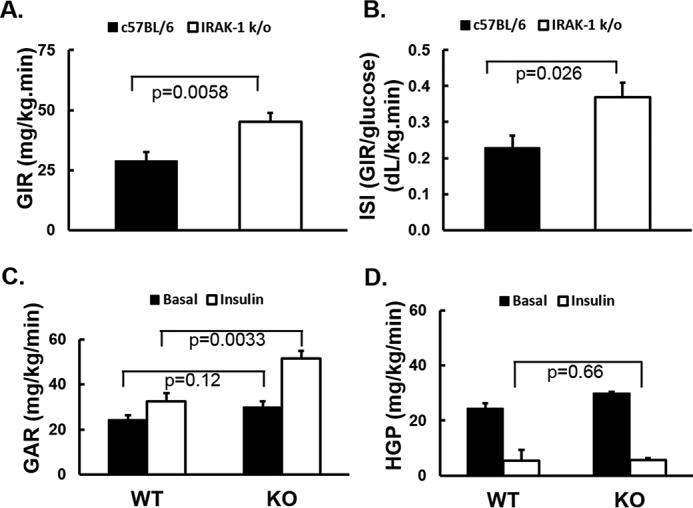
Hyperinsulinemic euglycemic glucose clamp studies with [3-3H]glucose tracer in mice fed with a normal chow diet are shown. A–D, 6 WT and 6 k/o mice underwent glucose clamps with [3H]glucose tracer as described under “Experimental Procedures” and previously (94). At the beginning and the end of each clamp study, blood was collected for calculating insulin-stimulated glucose disposal rates and hepatic glucose production rate. A, GIR normalized for body weight. B, ISI was defined as GIR (mg/kg/min) divided by steady-state glucose level (mg/dl) during the last 30 min of the clamp. C, glucose appearance rate. D, hepatic glucose production rate. Data are expressed as mean ± S.E.
IRAK-1 deficiency is associated with improved intracellular insulin signaling in skeletal muscle and adipose tissue, but not in liver
Cross-talk between the pathways of inflammatory signaling and insulin signaling is likely mediated, at least in part, by serine/threonine phosphorylation of IRS-1, which attenuates insulin signaling, thereby inducing insulin resistance (27). Based on this hypothesis, IRAK-1 deficiency is predicted to alleviate the negative impact of inflammation upon insulin signaling and action. To evaluate this issue, we investigated the effect of IRAK-1 deficiency on the insulin signaling pathways in metabolic insulin target tissues including skeletal muscle, adipose tissue, and liver. First, we measured and compared IRAK-1 protein levels in these tissues from wild-type mice (supplemental Fig. S4A). Quantified by total proteins of tissue lysates, muscle expressed higher IRAK-1 protein (100%) than liver (31%) or fat (42%). Second, we measured IRAK-1 expression in these tissues of WT and k/o mice. As expected, IRAK-1 was markedly lower in all the tissues tested from Irak1 k/o mice (supplemental Fig. S4B). There was a weak band in the Irak1 k/o muscle sample (supplemental Fig. S4B, top panel), but not in fat tissue or liver (supplemental Fig. S4B, middle and lower panels). Although the observed weak band in muscle immunoblots is likely nonspecific, we cannot entirely rule out the possibility that there is less than complete knockout of IRAK-1 in skeletal muscle.
As expected, insulin stimulation showed higher phosphorylation of Akt in all three tissues. Phosphorylation is a proxy for activation of the enzyme by insulin in skeletal muscle, liver, and adipose tissue (Fig. 6). Interestingly, IRAK-1–deficient skeletal muscle showed significantly higher insulin-stimulated phosphorylation of Akt when compared with that of wild-type littermates (Fig. 6, A and B). Whereas fat tissue isolated from Irak1 k/o mice showed a trend toward higher insulin-stimulated Akt kinase activity, this did not reach statistical significance (Fig. 6, E and F). By contrast, liver from Irak1 k/o mice did not show any difference in insulin-stimulated Akt phosphorylation when compared with wild-type littermates (Fig. 6, C and D). This is consistent with in vivo data from both glucose clamp and PET/CT studies showing comparable liver insulin sensitivity in Irak1 k/o and WT mice.
Figure 6.
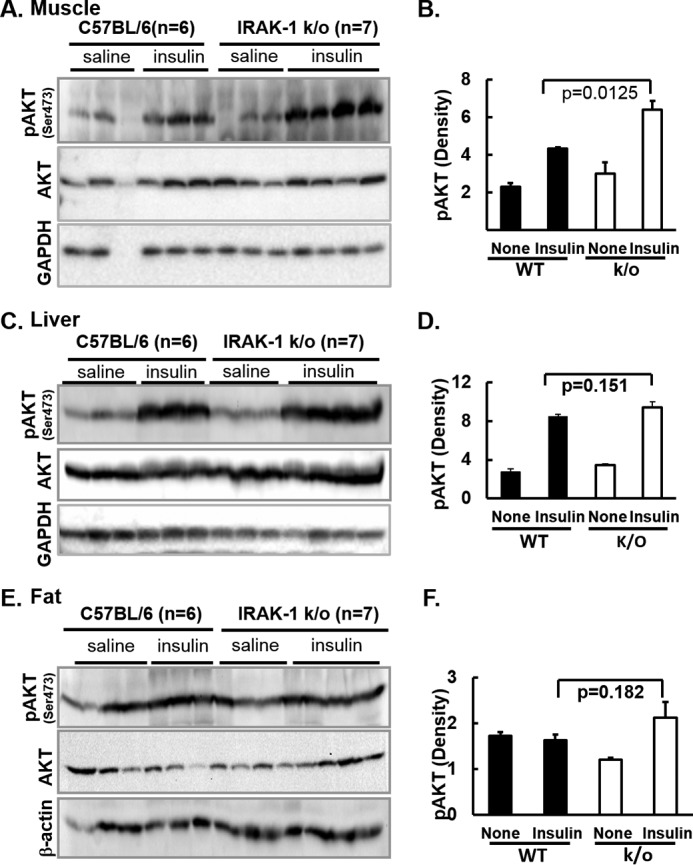
Immunoblotting analysis of insulin signaling pathways in muscle, liver, and abdominal white adipose tissue is shown. A and B, muscle. C and D, liver. E and F, abdominal white adipose tissue. 20-week-old mice fed with chow diet were fasted for 5 h and then given saline (none) or insulin (10 units/kg body weight) via portal vein injection under anesthesia. 5 min after injection, skeletal muscle from hind limb, abdominal fat, and liver were immediately flash frozen and homogenized. These samples were centrifuged and supernatants of tissue lysates were analyzed by immunoblotting with indicated antibodies. A, C, and E, immunoblots with samples from 6 WT and 7 Irak1 k/o mice. B, D, and F, immunoblots for phospho-Akt were quantified by scanning densitometry and normalized for Akt total protein. Data are expressed as mean ± S.E.
Discussion
Protein phosphorylation plays multiple roles related to insulin action and insulin resistance. The insulin receptor possesses intrinsic tyrosine kinase activity, which catalyzes tyrosine phosphorylation of IRS-1, IRS-2, and other endogenous substrates. This in turn triggers signaling pathways that activate downstream serine-specific protein kinases that mediate insulin action (e.g. PDK-1 and Akt). On the other hand, some serine-specific protein kinases participate in multiple biological pathways and are able to serine phosphorylate IRS-1 and IRS-2. For example, some inflammatory signaling pathway kinases (e.g. IKK, JNK, MAPK, and IRAK-1) have been reported to attenuate insulin signaling and induce insulin resistance (27) by promoting serine-specific phosphorylation of components of the insulin signaling pathway (e.g. IRS-1 and/or IRS-2). This has led to the hypothesis that chronic inflammation represents a key mechanism contributing to the pathogenesis of insulin resistance in obesity (63, 64).
Our current studies suggest that suppression of chronic inflammation may be beneficial by ameliorating insulin resistance. IRAK-1 is a critical enzyme in signaling pathways downstream of the IL-1 receptor, TNF-α receptor, and TLRs. In the present study, we demonstrated that deletion of Irak1 improved insulin sensitivity in mice fed an LFD (but not with HFD-induced obesity). Our observations suggest that the improvement in whole body insulin sensitivity is most likely mediated by improved insulin signaling and enhanced insulin sensitivity in skeletal muscle. The significance of this study is that although IRAK-1 is missing in all tissues, it appears that muscle may be the tissue in which IRAK-1 exerts the most important metabolic effects. There may also be secondary metabolic benefits mediated by IRAK-1 absence in white fat and vascular endothelium (28, 65).
Role of IRAK-1 in inflammation-associated insulin resistance
IRAK-1–deficient mice, which have been studied for over a decade, are characterized by a partial impairment in inflammatory response to lipopolysaccharide (LPS) at low dose but normal response to high dose of LPS (66, 67). Despite complete absence of IRAK-1, complete functional impairment has only been observed for a limited number of responses, e.g. interferon-α production (68). Although adverse metabolic effects of LPS in IRAK-1 k/o mice have been studied (69), the metabolic phenotype of Irak1 k/o mice in response to insulin has not been reported previously. Thus, inspired by our previous observation that IRAK-1–mediated phosphorylation exerts a negative impact on IRS-1–mediated insulin signaling (28), the present set of experiments was designed to investigate the role of IRAK-1 in metabolic physiology. Specifically, what is the role of IRAK-1 in the pathogenesis of the insulin-resistant state associated with chronic inflammation? Importantly, deletion of Irak1 did not affect body weight, regardless of the fat content of the diet (i.e. LFD versus HFD). Because body weight was not affected by deletion of Irak1, this eliminated the confounding influence of obesity. Interestingly, deletion of Irak1 was associated with a “healthy phenotype” (i.e. supranormal insulin sensitivity) in mice fed with LFD. This was an unexpected finding as it suggests that IRAK-1 appears to be important under conditions associated with a minimal level of inflammation. By contrast, both genotypes exhibited comparable degrees of insulin resistance when fed a HFD. Taken together, these data suggest that diet-induced obesity essentially eliminated the suprahealthy phenotype associated with Irak1 k/o genotype. This is most likely because there are multiple mechanisms for obesity to cause insulin resistance. Therefore, in the presence of obesity-induced insulin resistance the contribution of IRAK-1 alone is masked.
High-fat diets have been widely used to induce obesity in animal models as well as to study mechanisms of insulin resistance (70). Multiple factors have been reported to contribute to the molecular pathogenesis of obesity-induced insulin resistance. In addition to chronic inflammation (11, 71), other pathogenic factors including hyperlipidemia (72, 73), ectopic lipid accumulation (74), gut hormones (75), oxidative stress (76), ER stress (9, 77), sympathetic nervous system dysfunction (78), and gut microbiota (79) also contribute to insulin resistance in obesity. In this context, it is not surprising that Irak1 deficiency did not protect mice from obesity-induced insulin resistance inasmuch as IRAK-1 is only one of many mechanisms contributing to induction of insulin resistance. Low-fat and normal chow diets are likely associated with minimal low-grade inflammation. Nevertheless, when this low-grade inflammation was suppressed by deleting Irak1, this enhanced insulin sensitivity and glucose tolerance significantly. Our observations in metabolic physiology mirror what has been reported in the context of the physiology of inflammation. Specifically, macrophages from Irak1 k/o mice have impaired production of TNF-α in response to low concentrations of LPS (a ligand for TLR-4 and the TNF-α receptor upstream of IRAK-1). However, this defect can be overcome by high concentrations of LPS, which induce the same quantities of TNF-α in macrophages regardless of Irak1 genotype. Thus, Irak1 deletion only partially impairs the inflammation pathway induced by IL-1/TLR signaling (66, 67). Both IL-1 and TNF-α stimulate increased phosphorylation of IRS-1 (Ser24) in isolated rat adipose cells (28). These previously published data provide a precedent to explain why Irak1 k/o mice exhibit improved insulin sensitivity when fed either an LFD or normal chow diet (i.e. low-level inflammation) whereas the protective effect is overwhelmed on an HFD (i.e. high level of inflammation). Interestingly, although k/o mice have slightly lower food intake (4.5% on LFD and 8.9% on HFD) than that of WT mice, they maintained the same body weight as WT mice. This suggests lower metabolic rate in k/o mice counterbalances the lower food intake. Further study will clarify whether the apparent decrease in energy utilization is mediated by the central nervous system or peripheral tissues.
IRAK-1 is not the only kinase that phosphorylates IRS-1 (Ser24). We and others reported that a number of PKC isoforms also phosphorylate IRS-1 (Ser24) to impair metabolic insulin signaling (80, 81). Therefore, it is possible that IRS-1 Ser24 may be phosphorylated by other kinases in mice where IRAK-1 is missing. However, the fact that we observed a significant and substantial metabolic phenotype in Irak1 k/o mice consistent with abrogation of cross-talk between innate immune signaling and insulin signaling suggests that the contribution of IRAK-1 to regulate whole body glucose homeostasis is important relative to other kinases that also phosphorylate IRS-1 (Ser24).
Interestingly, fasting levels of plasma ET-1, a potent vasoconstrictor, were also significantly lower in k/o mice versus WT (p = 0.016) after 14 weeks of LFD (20-week-old mice). Reduction of plasma ET-1 may be secondary to the reduction of TNF-α, which is a positive regulator of ET-1 expression. ET-1 plays an important role in the pathogenesis of insulin resistance–associated endothelial dysfunction (24, 60, 82). However, there is also likely a direct effect of IRAK-1 absence in endothelial cells to reduce ET-1 synthesis and secretion. In a previous study, we found that siRNA knockdown of TLR-2 (closely upstream of IRAK-1) in primary vascular endothelial cells completely abrogates the effect of FFA (TLR-2 ligand elevated in insulin resistance) to stimulate phosphorylation of IRS-1 (Ser24) (65). Moreover, under conditions of metabolic insulin resistance (i.e. pathway-selective impairment of PI3K-dependent insulin signaling with intact MAPK signaling), the effect of insulin to stimulate increased synthesis and secretion of ET-1 from vascular endothelial cells is greatly enhanced (83–85). Taken together, these data suggest that the ability of IRAK-1 to phosphorylate IRS-1 (Ser24) in endothelium may promote vascular insulin resistance leading to endothelial dysfunction characterized by increased insulin- and FFA-stimulated ET-1 synthesis and secretion. This is relevant because whole body metabolic insulin resistance is characterized by elevated fasting and postprandial insulin and FFA levels as well as pathway-selective impairment in PI3K-dependent insulin signaling and action. We hypothesize that decreased levels of ET-1 mediated directly and indirectly by absence of IRAK-1 may improve endothelial function and increase blood flow to skeletal muscle. This will increase access of insulin and glucose to metabolic tissues and secondarily promote insulin-stimulated glucose uptake and improved glucose tolerance (61).
Role of skeletal muscle in mediating physiological effects of IRAK-1 deficiency
Several lines of evidence suggest that deletion of IRAK-1 increased insulin sensitivity and improved glucose metabolism in skeletal muscle, but not in liver.
Hyperinsulinemic euglycemic glucose clamp studies
Hyperinsulinemic euglycemic glucose clamp studies demonstrated that IRAK-1 deficiency was associated with higher insulin-stimulated glucose disposal rate, but did not alter insulin's ability to suppress the rate of hepatic glucose production. Insulin-stimulated glucose disposal as assessed by clamp studies has been demonstrated to reflect primarily glucose uptake by skeletal muscle. Observations in both mice and humans suggest that insulin tolerance tests primarily reflect hepatic insulin sensitivity. Whereas muscle-specific insulin receptor knock-out mice exhibit normal ITT (49), insulin tolerance was impaired in mice with liver-specific insulin receptor knock-out (50). Similarly, in patients with insulinoma, insulin-driven fasting hypoglycemia is mediated primarily by suppression of hepatic glucose production (86). Thus, the normal ITT observed in Irak1 k/o mice supports the conclusion that the liver responds normally to insulin.
PET/CT studies
In PET/CT studies, muscle and adipose tissue were the principal tissues into which insulin promoted distribution of [18F]FDG. Further, deletion of IRAK-1 selectively increased muscle [18F]FDG uptake. There was no difference in liver [18F]FDG uptake.
Analysis of insulin-stimulated phosphorylation
Deletion of Irak1 enhanced insulin signaling in muscle, but not liver, as assessed by analysis of insulin-stimulated phosphorylation of Akt (Fig. 6) and MAPK (data not shown). There was also a trend toward improved insulin signaling in adipose tissue of k/o mice (although data did not achieve statistical significance).
The PET/CT scan does not provides a direct assessment of glucose uptake, but rather relies on radioactively labeled 2-deoxy-2-fluoro-d-glucose molecule (FDG) uptake as tracer. Because fasting glucose was lower in k/o mice than controls, this may differentially affect FDG tracer uptake in different tissues. As compared with glucose, the 2-hydroxyl group in FDG is replaced by a fluorine atom. Although FDG does undergo phosphorylation on the 6-hydroxyl, it is not a substrate for downstream steps in the glycolytic pathway. Accordingly, the rate of accumulation of FDG is determined by the rate of glucose transport, which is mediated primarily by GLUT-family transporters. FDG uptake by the liver is mediated primarily by GLUT2, which has a Km for glucose of ∼17 mm (101). As shown in Fig. 3B, fasting glucose levels (∼10 mm) are below the Km of GLUT2 to transport glucose, and the majority of GLUT2 molecules would be available to transport FDG with relatively little competition from unlabeled glucose. Accordingly, FDG uptake by the liver would be predicted to be relatively insensitive to ambient glucose levels (for glucose levels substantially below the Km of GLUT2 for glucose). In this context, it is noteworthy that both WT and k/o mice exhibit similar rates of FDG uptake in liver (Fig. 4).
By contrast, the Km for GLUT1 is ∼26 mm and for GLUT4 is ∼4–5 mm (87). Thus, for ambient glucose levels of ∼10 mm, approximately two thirds of GLUT4 molecules would be engaged in transporting glucose whereas the majority of GLUT1 molecules would not be bound to glucose molecules. Although GLUT4 is believed to be the principal glucose transporter in both adipose tissue and skeletal muscle in the presence of insulin, both GLUT1 and GLUT4 are believed to be significant in the absence of insulin. Whereas GLUT1-mediated FDG transport would be predicted to be relatively insensitive to inhibition by glucose levels substantially below the Km = 26 mm, GLUT4-mediated FDG transport would likely be significantly inhibited by glucose levels of ∼10 mm. Thus, it is possible that GLUT1 would be the principal transporter mediating FDG uptake when plasma glucose levels are ∼10 mm in the absence of insulin. In any case, ambient glucose levels would be predicted to exert similar effects on FDG uptake in both adipose tissue and skeletal muscle. In this context, it is noteworthy that rates of FDG uptake in adipose tissue are not significantly different in WT versus IRAK-1 knock-out mice (Fig. 4). In contrast, FDG uptake by skeletal muscle is selective increased in knock-out mice. If the difference observed for skeletal muscle were caused primarily by competitive inhibition at the level of GLUT4, one might have expected to observe a similar difference for adipose tissue, which is not the case.
Taken together, these in vivo and ex vivo analyses suggest that IRAK-1 plays a particularly important role in the metabolic physiology of skeletal muscle, but has little, if any, effect in liver. What are the mechanisms whereby deletion of Irak1 selectively promotes insulin action in muscle rather than liver? Several possibilities deserve consideration. At least in theory, this might be explained by tissue selective expression of IRAK-1. Although our Western blot analyses for muscle, fat, and liver tissues demonstrate that all three tissues express IRAK-1, relative expression of IRAK-1 is higher in muscle than in fat and liver. A second possible explanation is based on tissue-specific roles of insulin receptor substrate molecules. Whereas muscle and adipocytes rely primarily on IRS-1, IRS-2 is more important in liver. As reported previously, IRAK-1 phosphorylates Ser24 on IRS-1. Phosphorylation of Ser24 impairs the signaling function of IRS-1 (28). By contrast, the Ser24 phosphorylation site is not conserved in the IRS-2 molecule (27). Thus, deletion of Irak1 has potential to improve insulin sensitivity in tissues where IRS-1 is important (e.g. skeletal muscle), but is less likely to affect insulin signaling in tissues such as liver where IRS-2 has the dominant role in insulin signaling. Indeed, consistent with this possibility, our observations from clamp studies demonstrated that insulin-stimulated inhibition of hepatic glucose production was comparable among Irak1 k/o and WT mice on normal chow diet or LFD whereas insulin sensitivity in skeletal muscle was improved in the Irak1 k/o mice when compared with WT mice. Furthermore, there is a precedent for other kinases regulating insulin signaling to phosphorylate IRS proteins in an isoform-specific manner because of the presence of a phosphorylation site in one isoform but not another. For example, PKC-ζ phosphorylates IRS-1 but not IRS-2 (88). This IRS isoform-specific phosphorylation may represent a mechanism for determining insulin signaling specificity in different tissues. In immunoblotting studies, we did not detect the presence of phospho-IRS-1 (Ser24) in skeletal muscle in either the absence or presence of insulin stimulation under conditions of our ex vivo insulin signaling studies (in either in wild-type or k/o mice) (data not shown). Although we cannot entirely eliminate the possibility of technical problems, we used a phospho-specific antibody that we previously developed and characterized that detected phospho-IRS-1 (Ser24) in rat fat cells (28) and bovine aortic endothelial cells (65) in vitro. Taken at face value, these data suggest that Ser24 phosphorylation of IRS-1 may not be important in mouse skeletal muscle. Nevertheless, it is also possible that phosphorylation of Ser24 might accelerate degradation of IRS-1, thereby decreasing the level of phospho-IRS-1 (Ser24) to the point where it is not detectable by immunoblotting. On the other hand, our immunoblotting experiments did not detect an alteration in the levels of IRS-1 in IRAK-1 knock-out mice (data not shown). Another possibility is that rapid dephosphorylation of phospho-IRS-1 (Ser24) was occurring in our ex vivo experimental setting such that we were unable to detect a transient but physiologically relevant higher phospho-IRS-1 (Ser24) in skeletal muscle by immunoblotting.
A third possibility to explain why deletion of Irak1 selectively promotes insulin action in muscle rather than liver relates to the cell biology of insulin action in different tissues. In muscle cells, insulin-stimulated glucose uptake is mediated by insulin-stimulated translocation of GLUT4-containing vesicles from an intracellular location to the plasma membrane (89, 90). By contrast, GLUT2-mediated transport of glucose into hepatocytes is not directly regulated by insulin. Moreover, deletion of Irak1 did not alter levels of GLUT4 protein in muscle (supplemental Fig. S4C). Thus, we hypothesize that Irak1 deletion leads to diminished serine phosphorylation of IRS-1, which in turn promotes the activity of the GLUT4 translocation machinery, thereby increasing insulin sensitivity (28, 89). However, as noted above, insulin-stimulated hepatic glucose production was comparable between Irak1 k/o mice and WT mice, making it more likely that it is the predominance of IRS-2 in liver and the lack of an IRAK phosphorylation site on IRS-2 that explains why liver insulin sensitivity did not change in Irak1 k/o mice. Thus, the improvement in glucose tolerance observed in Irak1 k/o mice on normal chow diet or LFD is likely mediated primarily by improved metabolic insulin signaling and action in muscle.
Conclusion: Possible translational implications of the role of IRAK-1 in metabolic physiology
Our studies in a rodent animal model suggest that IRAK-1 deficiency is associated with a healthy phenotype (i.e. supranormal insulin sensitivity), at least in mice fed with either LFD or normal chow diet. Inasmuch as the Irak1 genotype does not affect the degree of insulin resistance in mice fed an HFD, we hypothesize that Irak1 k/o mice will experience a quantitatively greater deterioration of insulin sensitivity when switched from normal chow to HFD. Conversely, Irak1 k/o mice might experience greater metabolic benefit (i.e. a quantitatively greater improvement in insulin sensitivity) if weight loss were induced by shifting the mice from an HFD to an LFD. If our data in this animal model are predictive of human pathophysiology, this prompts the hypothesis that IRAK-1 activity in skeletal muscle may provide a biomarker to predict a beneficial metabolic response to weight loss. If correct, this hypothesis may have significant implications for a precision medicine approach to weight loss. In other words, low levels of skeletal muscle IRAK-1 activity might identify a subset of insulin-resistant patients who are most likely to experience the greatest improvement in insulin sensitivity in response to weight loss.
Experimental procedures
Animal studies
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine. To generate Irak1 null mice and their wild-type control, female homozygotes were first backcrossed to C57BL/6 males for 10 generations. Because the Irak1 null allele was introduced on a genetic background of C57BL/6 mice (Harlan Laboratories), we genotyped for a previously reported mutation in nicotinamide nucleotide transhydrogenase (Nnt), and confirmed that our mice did not carry a mutation in Nnt (91, 92). The Irak1 gene is on the X chromosome (93). Thus, males that are hemizygous for the null allele of Irak1 will be deficient in Irak1 function (k/o) whereas males hemizygous for the functional allele of Irak1 will be wild-type. To simplify breeding and selection of affected mice, we limited our studies to male mice (either Irak1 k/o or Irak1 WT). For studies on different diets, Irak1 k/o versus WT littermate mice were fed with LFD (10% kcal from fat, 70% kcal from carbohydrate) (D12450B, Research Diets) or HFD (60% kcal from fat, 20% kcal from carbohydrate) (D12492, Research Diets) for indicated weeks or chow diet (18% kcal from fat and 58% kcal from carbohydrate) (2018SX, Teklad Global). Body weight and food intake were monitored three times per week.
IPGTT and ITT
After 5 h fasting, either glucose (1 g/kg for IPGTT) or human insulin (0.75 IU/kg for ITT) (Lilly) was injected intraperitoneally (50). Blood glucose was monitored at indicated time points with an Accu-Chek Aviva blood glucometer. Area under the curve (AUC) for IPGTT or area above the curve (AAC) for ITT was calculated based on the trapezoidal rule.
Hyperinsulinemic euglycemic glucose clamp studies
Jugular veins were cannulated 4–5 days before the clamp experiments (53, 94). Mice were acclimated to restrainers three times (3 h each time) before clamp. After 5 h fasting, [3-3H]-glucose was infused intravenously (2.5 μCi bolus followed by constant infusion at 0.05 μCi/min). Blood samples (20 μl) were collected at 90, 100, and 110 min after the initiation of [3H]-glucose infusion for the measurement of [3H]glucose-specific activity, blood glucose concentration, and calculation of basal hepatic glucose production. Then, regular human insulin was infused (60 mU/kg loading bolus followed by constant infusion at 2.5 mU/kg/min), and glucose levels were measured every 10 min. An infusion of 25% dextrose was adjusted to maintain the blood glucose at 120–130 mg/dl. Blood samples were again collected at 100, 110, and 120 min during insulin infusion for calculation of insulin-stimulated glucose disposal rate and hepatic glucose production rate. Glucose infusion rate was calculated as the average glucose infusion rate during the last 30 min of the clamp steady-state period and normalized for body weight. An ISI was defined as GIR divided by the level at which glucose was clamped (GIR/clamp glucose). The rates of glucose appearance and hepatic glucose production were calculated as described previously (94).
Portal vein insulin injection and collection of liver, muscle, and fat tissues
Mice were fasted for 5 h and anesthetized with isoflurane vapor. After making a ventral incision to expose the liver and portal vein, we injected either human insulin (10 units/kg body weight) (Lilly) or saline into the portal vein (95–97). After 5 min, samples of epididymal fat tissue, liver, and hind limb skeletal muscle were collected and flash frozen for future immunoblotting analysis (95–97).
Tissue and cell sample preparation and immunoblotting
Muscle (200 mg) and liver (100 mg) frozen tissues were homogenized at 4 °C in 400 μl lysis buffer (50 mm HEPES, pH 7.4, 2 mm EDTA, 50 mm NaF, 100 mm NaCl, 5 mm DTT, 0.1% Nonidet P-40, 2 mm benzamindine, 0.1 mm Na3VO4, and 5 μl of Roche protein inhibitor mixture set) with a tissue grinder. Tissue lysates were centrifuged at 21,000 × g for 1 h at 4 °C in a table top centrifuge. The supernatants were used for immunoblotting analysis. For fat tissue, 400 mg of frozen tissue was homogenized in 400 μl of fat tissue lysis buffer (10 mm Tris, pH 7.4, 1 mm EDTA, 100 mm NaF, 0.2 mm Na3VO4, and 4 μl of Roche protease inhibitor mixture set). Homogenates were mixed with 900 μl of cold acetone at −80 °C overnight, and then centrifuged at 21,000 × g for 1 h at 4 °C. Supernatants were discarded and the pellets were dried on ice for 5–10 min prior to resuspension in 400 μl of Laemmli sample buffer for immunoblotting analysis. Aliquots (20 μg of total protein) from each tissue sample were separated on a 10% SDS-PAGE gel, transferred to PVDF membranes, and incubated with indicated antibodies as described previously (95–97). Antibodies against IRAK-1, pAkt (S473), Akt, GLUT4, GAPDH, and β-actin were purchased from Cell Signaling Technology Inc. Quantification of images was performed using an imaging device with Image Lab software. Relative pAkt was calculated by dividing pAkt density with that of total Akt. Relative IRAK-1 in muscle, liver, and fat tissues were calculated by dividing all three tissues by muscle IRAK-1 density.
PET/CT scan
3 Irak1 k/o and 3 WT male mice were fasted overnight, and [18F]FDG PET/CT was performed with animals maintained under 2% isoflurane anesthesia on a Siemens Inveon Small Animal PET/CT Imaging System (Siemens Medical Solutions) in the Core for Translational Research in Imaging (CTRIM). [18F]FDG (11.1 MBq; 300 μCi) and insulin (1.125 units/kg body weight) in 200 μl of saline were injected via tail vein of anesthetized mice. Dynamic imaging was acquired for 30 min. Data acquisition, image reconstruction, and region of interest (ROI) analysis were carried out according to methods reported previously (98, 99). The data were obtained by ROI analysis focused on several target organs: hind limb muscle, subcutaneous white adipose tissue (abdominal region), liver, and brain. The quantitative [18F]FDG PET data in the target regions were expressed as percentages of injected dose per gram (%ID/g) at three time points of 0–10, 10–20, and 20–30 min.
ELISA
Adiponectin (Millipore), endothelin-1 (Enzo Life Science), insulin (Concodian), TNF-α (Invitrogen), and CRP (ThermoFisher Scientific) were measured by ELISA according to the manufacturers' instructions.
Statistical analyses
t-tests were performed with Excel software, and analysis of variance (ANOVA) for repeated measures was conducted with Axum 7 software.
Author contributions
X. J. S. designed, performed, and analyzed the experiments and coordinated the study and wrote the paper. S. P. K. performed the animal care, IPGTT and ITT, ELISA measurement, and immunoblotting analysis. D. Z. and H. S. participated in the immunoblotting of animal tissues. Q. C. and X. L. performed and analyzed the experiments shown in supplemental Fig. S3. Z. Y. contributed to the discussion. L. L. contributed to the Irak1 k/o mice. R. R. H. and T. P. C. contributed to the interpretation of data and writing of the manuscript. S. I. T. contributed to the discussion of the project and writing the manuscript. M. J. Q. conceived and coordinated the study, analyzed all data, and wrote and edited the manuscript.
Supplementary Material
This work was supported by American Diabetes Association Grants 1-13-BS-150 (to M. J. Q.) and 1-14-BS-198 (to X. J. S.), NIEHS, National Institutes of Health Grant R01ES024516 (to Z. Y.), U. S. Department of Veterans Affairs Clinical Sciences Research and Development Service Merit Review Award I01CX00635, Baltimore Veterans Affairs Medical Center Geriatric Research Clinical Center (GRECC), and NIDDK, National Institutes of Health Grant 5P30DK072488-12 (to S. I. T.). S. I. T. has worked as a paid consultant for Ionis Pharmaceuticals and Aegerion Pharmaceuticals. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or reflect the position or policy of the Department of Veterans Affairs or the United States government.
This article contains supplemental Figures S1–S4.
- IR
- insulin receptor
- IRS
- insulin receptor substrate
- k/o
- knock-out
- IRAK-1
- interleukin 1 receptor-associated kinase 1
- PI3K
- phosphatidylinositol 3-kinase
- LPS
- lipopolysaccharide
- ET-1
- endothelin-1
- IPGTT
- intraperitoneal glucose tolerance test
- TLR
- Toll-like receptor
- ITT
- insulin tolerance tests
- GIR
- glucose infusion rate
- ISI
- insulin sensitivity index
- QUICKI
- quantitative insulin sensitivity check index
- CRP
- C-reactive protein
- AUC
- area under the curve
- ROI
- region of interest
- ANOVA
- analysis of variance
- LFD
- low-fat diet
- HFD
- high-fat diet.
References
- 1. Muniyappa R., Lee S., Chen H., and Quon M. J. (2008) Current approaches for assessing insulin sensitivity and resistance in vivo: Advantages, limitations, and appropriate usage. Am. J. Physiol. Endocrinol. Metab. 294, E15–E26 [DOI] [PubMed] [Google Scholar]
- 2. Kahn C. R. (1994) Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 43, 1066–1084 [DOI] [PubMed] [Google Scholar]
- 3. Samuel V. T., and Shulman G. I. (2016) The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Invest. 126, 12–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim Y.-B., Kotani K., Ciaraldi T. P., Henry R. R., and Kahn B. B. (2003) Insulin-stimulated protein kinase C λ/ζ activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: reversal with weight reduction. Diabetes 52, 1935–1942 [DOI] [PubMed] [Google Scholar]
- 5. Matsumoto M., Pocai A., Rossetti L., Depinho R. A., and Accili D. (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 6. Coppack S. W., Jensen M. D., and Miles J. M. (1994) In vivo regulation of lipolysis in humans. J. Lipid Res. 35, 177–193 [PubMed] [Google Scholar]
- 7. Wahren J., and Ekberg K. (2007) Splanchnic regulation of glucose production. Annu. Rev. Nutr. 27, 329–345 [DOI] [PubMed] [Google Scholar]
- 8. Jaeschke A., and Davis R. J. (2007) Metabolic stress signaling mediated by mixed-lineage kinases. Mol. Cell 27, 498–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 16, 396–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wellen K. E., Fucho R., Gregor M. F., Furuhashi M., Morgan C., Lindstad T., Vaillancourt E., Gorgun C. Z., Saatcioglu F., and Hotamisligil G. S. (2007) Coordinated regulation of nutrient and inflammatory responses by STAMP2 is essential for metabolic homeostasis. Cell 129, 537–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hotamisligil G. S. (2003) Inflammatory pathways and insulin action. Int. J. Obes. Relat. Metab. Disord. 27, Suppl. 3, S53–S55 [DOI] [PubMed] [Google Scholar]
- 12. Fujimoto Y., Torres T. P., Donahue E. P., and Shiota M. (2006) Glucose toxicity is responsible for the development of impaired regulation of endogenous glucose production and hepatic glucokinase in Zucker diabetic fatty rats. Diabetes 55, 2479–2490 [DOI] [PubMed] [Google Scholar]
- 13. Kewalramani G., Bilan P. J., and Klip A. (2010) Muscle insulin resistance: Assault by lipids, cytokines and local macrophages. Curr. Opin. Clin. Nutr. Metab. Care 13, 382–390 [DOI] [PubMed] [Google Scholar]
- 14. DeFronzo R. A. (2010) Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 53, 1270–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hommelberg P. P., Langen R. C., Schols A. M., Mensink R. P., and Plat J. (2010) Inflammatory signaling in skeletal muscle insulin resistance: Green signal for nutritional intervention? Curr. Opin. Clin. Nutr. Metab. Care 13, 647–655 [DOI] [PubMed] [Google Scholar]
- 16. Shoelson S. E., Lee J., and Goldfine A. B. (2006) Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun X. J., Rothenberg P., Kahn C. R., Backer J. M., Araki E., Wilden P. A., Cahill D. A., Goldstein B. J., and White M. F. (1991) Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 352, 73–77 [DOI] [PubMed] [Google Scholar]
- 18. Sun X. J., Wang L. M., Zhang Y., Yenush L., Myers M. G. Jr., Glasheen E., Lane W. S., Pierce J. H., and White M. F. (1995) Role of IRS-2 in insulin and cytokine signalling. Nature 377, 173–177 [DOI] [PubMed] [Google Scholar]
- 19. Taniguchi C. M., Emanuelli B., and Kahn C. R. (2006) Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96 [DOI] [PubMed] [Google Scholar]
- 20. Sun X. J., Crimmins D. L., Myers M. G. Jr., Miralpeix M., and White M. F. (1993) Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol. Cell. Biol. 13, 7418–7428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Myers M. G. Jr., Backer J. M., Sun X. J., Shoelson S., Hu P., Schlessinger J., Yoakim M., Schaffhausen B., and White M. F. (1992) IRS-1 activates phosphatidylinositol 3′-kinase by associating with src homology 2 domains of p85. Proc. Natl. Acad. Sci. U.S.A. 89, 10350–10354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esposito D. L., Li Y., Cama A., and Quon M. J. (2001) Tyr612 and Tyr632 in human insulin receptor substrate-1 are important for full activation of insulin-stimulated phosphatidylinositol 3-kinase activity and translocation of GLUT4 in adipose cells. Endocrinology 142, 2833–2840 [DOI] [PubMed] [Google Scholar]
- 23. Chen H., Nystrom F. H., Dong L. Q., Li Y., Song S., Liu F., and Quon M. J. (2001) Insulin stimulates increased catalytic activity of phosphoinositide-dependent kinase-1 by a phosphorylation-dependent mechanism. Biochemistry 40, 11851–11859 [DOI] [PubMed] [Google Scholar]
- 24. Muniyappa R., Iantorno M., and Quon M. J. (2008) An integrated view of insulin resistance and endothelial dysfunction. Endocrinol. Metab. Clin. North Am. 37, 685–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dasu M. R., and Jialal I. (2011) Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors. Am. J. Physiol. Endocrinol. Metab. 300, E145–E154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Akira S., and Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 27. Sun X. J., and Liu F. (2009) Phosphorylation of IRS proteins Yin-Yang regulation of insulin signaling. Vitam. Horm. 80, 351–387 [DOI] [PubMed] [Google Scholar]
- 28. Kim J. A., Yeh D. C., Ver M., Li Y., Carranza A., Conrads T. P., Veenstra T. D., Harrington M. A., and Quon M. J. (2005) Phosphorylation of Ser24 in the pleckstrin homology domain of insulin receptor substrate-1 by Mouse Pelle-like kinase/interleukin-1 receptor-associated kinase: Cross-talk between inflammatory signaling and insulin signaling that may contribute to insulin resistance. J. Biol. Chem. 280, 23173–23183 [DOI] [PubMed] [Google Scholar]
- 29. Hotamisligil G. S., Peraldi P., Budavari A., Ellis R., White M. F., and Spiegelman B. M. (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 271, 665–668 [DOI] [PubMed] [Google Scholar]
- 30. Klover P. J., Zimmers T. A., Koniaris L. G., and Mooney R. A. (2003) Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 52, 2784–2789 [DOI] [PubMed] [Google Scholar]
- 31. Klover P. J., Clementi A. H., and Mooney R. A. (2005) Interleukin-6 depletion selectively improves hepatic insulin action in obesity. Endocrinology 146, 3417–3427 [DOI] [PubMed] [Google Scholar]
- 32. Cai D., Yuan M., Frantz D. F., Melendez P. A., Hansen L., Lee J., and Shoelson S. E. (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 11, 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shoelson S. E., Herrero L., and Naaz A. (2007) Obesity, inflammation, and insulin resistance. Gastroenterology 132, 2169–2180 [DOI] [PubMed] [Google Scholar]
- 34. O'Neill L. A., and Greene C. (1998) Signal transduction pathways activated by the IL-1 receptor family: ancient signaling machinery in mammals, insects, and plants. J. Leukocyte Biol. 63, 650–657 [PubMed] [Google Scholar]
- 35. Kenny E. F., and O'Neill L. A. (2008) Signalling adaptors used by Toll-like receptors: An update. Cytokine 43, 342–349 [DOI] [PubMed] [Google Scholar]
- 36. Jenkins K. A., and Mansell A. (2010) TIR-containing adaptors in Toll-like receptor signalling. Cytokine 49, 237–244 [DOI] [PubMed] [Google Scholar]
- 37. Li X., Commane M., Jiang Z., and Stark G. R. (2001) IL-1-induced NFκB and c-Jun N-terminal kinase (JNK) activation diverge at IL-1 receptor-associated kinase (IRAK). Proc. Natl. Acad. Sci. U.S.A. 98, 4461–4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q., Dziarski R., Kirschning C. J., Muzio M., and Gupta D. (2001) Micrococci and peptidoglycan activate TLR2–>MyD88–>IRAK–>TRAF–>NIK–>IKK–>NF-κB signal transduction pathway that induces transcription of interleukin-8. Infect. Immun. 69, 2270–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takaesu G., Ninomiya-Tsuji J., Kishida S., Li X., Stark G. R., and Matsumoto K. (2001) Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol. Cell. Biol. 21, 2475–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yuan M., Konstantopoulos N., Lee J., Hansen L., Li Z. W., Karin M., and Shoelson S. E. (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science 293, 1673–1677 [DOI] [PubMed] [Google Scholar]
- 41. Kim F., Tysseling K. A., Rice J., Pham M., Haji L., Gallis B. M., Baas A. S., Paramsothy P., Giachelli C. M., Corson M. A., and Raines E. W. (2005) Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKβ. Arterioscler. Thromb. Vasc. Biol. 25, 989–994 [DOI] [PubMed] [Google Scholar]
- 42. Kim F., Pham M., Maloney E., Rizzo N. O., Morton G. J., Wisse B. E., Kirk E. A., Chait A., and Schwartz M. W. (2008) Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler. Thromb. Vasc. Biol. 28, 1982–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim J. A., Koh K. K., and Quon M. J. (2005) The union of vascular and metabolic actions of insulin in sickness and in health. Arterioscler. Thromb. Vasc. Biol. 25, 889–891 [DOI] [PubMed] [Google Scholar]
- 44. Dewberry R., Holden H., Crossman D., and Francis S. (2000) Interleukin-1 receptor antagonist expression in human endothelial cells and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 20, 2394–2400 [DOI] [PubMed] [Google Scholar]
- 45. McCarty M. F. (2010) Salsalate may have broad utility in the prevention and treatment of vascular disorders and the metabolic syndrome. Med. Hypotheses 75, 276–281 [DOI] [PubMed] [Google Scholar]
- 46. Goldfine A. B., Fonseca V., Jablonski K. A., Pyle L., Staten M. A., and Shoelson S. E.; TINSAL-T2D (Targeting Inflammation Using Salsalate in Type 2 Diabetes) Study Team (2010) The effects of salsalate on glycemic control in patients with type 2 diabetes: A randomized trial. Ann. Intern. Med. 152, 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fleischman A., Shoelson S. E., Bernier R., and Goldfine A. B. (2008) Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31, 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lakoski S. G., Li L., Langefeld C. D., Liu Y., Howard T. D., Brosnihan K. B., Xu J., Bowden D. W., and Herrington D. M. (2007) The association between innate immunity gene (IRAK1) and C-reactive protein in the Diabetes Heart Study. Exp. Mol. Pathol. 82, 280–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brüning J. C., Michael M. D., Winnay J. N., Hayashi T., Hörsch D., Accili D., Goodyear L. J., and Kahn C. R. (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569 [DOI] [PubMed] [Google Scholar]
- 50. Michael M. D., Kulkarni R. N., Postic C., Previs S. F., Shulman G. I., Magnuson M. A., and Kahn C. R. (2000) Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 6, 87–97 [PubMed] [Google Scholar]
- 51. Moller D. E., Chang P. Y., Yaspelkis B. B. 3rd, Flier J. S., Wallberg-Henriksson H., and Ivy J. L. (1996) Transgenic mice with muscle-specific insulin resistance develop increased adiposity, impaired glucose tolerance, and dyslipidemia. Endocrinology 137, 2397–2405 [DOI] [PubMed] [Google Scholar]
- 52. Kido Y., Burks D. J., Withers D., Bruning J. C., Kahn C. R., White M. F., and Accili D. (2000) Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Clin. Invest. 105, 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ren J. M., Marshall B. A., Mueckler M. M., McCaleb M., Amatruda J. M., and Shulman G. I. (1995) Overexpression of Glut4 protein in muscle increases basal and insulin-stimulated whole body glucose disposal in conscious mice. J. Clin. Invest. 95, 429–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim J. K., Fillmore J. J., Sunshine M. J., Albrecht B., Higashimori T., Kim D. W., Liu Z. X., Soos T. J., Cline G. W., O'Brien W. R., Littman D. R., and Shulman G. I. (2004) PKC-θ knockout mice are protected from fat-induced insulin resistance. J. Clin. Invest. 114, 823–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen H., Sullivan G., and Quon M. J. (2005) Assessing the predictive accuracy of QUICKI as a surrogate index for insulin sensitivity using a calibration model. Diabetes 54, 1914–1925 [DOI] [PubMed] [Google Scholar]
- 56. Quon M. J. (2002) QUICKI is a useful and accurate index of insulin sensitivity. J. Clin. Endocrinol. Metab. 87, 949–951 [DOI] [PubMed] [Google Scholar]
- 57. Hotamisligil G. S. (2000) Molecular mechanisms of insulin resistance and the role of the adipocyte. Int. J. Obes. Relat. Metab. Disord. 24, Suppl. 4, S23–S27 [DOI] [PubMed] [Google Scholar]
- 58. Uysal K. T., Wiesbrock S. M., Marino M. W., and Hotamisligil G. S. (1997) Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610–614 [DOI] [PubMed] [Google Scholar]
- 59. Jiang Z. Y., Zhou Q. L., Chatterjee A., Feener E. P., Myers M. G. Jr., White M. F., and King G. L. (1999) Endothelin-1 modulates insulin signaling through phosphatidylinositol 3-kinase pathway in vascular smooth muscle cells. Diabetes 48, 1120–1130 [DOI] [PubMed] [Google Scholar]
- 60. Potenza M. A., Marasciulo F. L., Chieppa D. M., Brigiani G. S., Formoso G., Quon M. J., and Montagnani M. (2005) Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am. J. Physiol. Heart Circ. Physiol. 289, H813–H822 [DOI] [PubMed] [Google Scholar]
- 61. Kim J. A., Montagnani M., Koh K. K., and Quon M. J. (2006) Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 113, 1888–1904 [DOI] [PubMed] [Google Scholar]
- 62. Ayala J. E., Samuel V. T., Morton G. J., Obici S., Croniger C. M., Shulman G. I., Wasserman D. H., McGuinness O. P.; NIH Mouse Metabolic Phenotyping Center Consortium (2010) Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Models Mech. 3, 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Franceschi C., Bonafè M., Valensin S., Olivieri F., De Luca M., Ottaviani E., and De Benedictis G. (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N.Y. Acad. Sci. 908, 244–254 [DOI] [PubMed] [Google Scholar]
- 64. Minciullo P. L., Catalano A., Mandraffino G., Casciaro M., Crucitti A., Maltese G., Morabito N., Lasco A., Gangemi S., and Basile G. (2016) Inflammaging and anti-inflammaging: The role of cytokines in extreme longevity. Arch. Immunol. Ther. Exp. 64, 111–126 [DOI] [PubMed] [Google Scholar]
- 65. Jang H. J., Kim H. S., Hwang D. H., Quon M. J., and Kim J. A. (2013) Toll-like receptor 2 mediates high-fat diet-induced impairment of vasodilator actions of insulin. Am. J. Physiol. Endocrinol. Metab. 304, E1077–E1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Berglund M., Thomas J. A., Hörnquist E. H., and Hultgren O. H. (2008) Toll-like receptor cross-hyporesponsiveness is functional in interleukin-1-receptor-associated kinase-1 (IRAK-1)-deficient macrophages: Differential role played by IRAK-1 in regulation of tumour necrosis factor and interleukin-10 production. Scand. J. Immunol. 67, 473–479 [DOI] [PubMed] [Google Scholar]
- 67. Swantek J. L., Tsen M. F., Cobb M. H., and Thomas J. A. (2000) IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J. Immunol. 164, 4301–4306 [DOI] [PubMed] [Google Scholar]
- 68. Uematsu S., Sato S., Yamamoto M., Hirotani T., Kato H., Takeshita F., Matsuda M., Coban C., Ishii K. J., Kawai T., Takeuchi O., and Akira S. (2005) Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon- induction. J. Exp. Med. 201, 915–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maitra U., Chang S., Singh N., and Li L. (2009) Molecular mechanism underlying the suppression of lipid oxidation during endotoxemia. Mol. Immunol. 47, 420–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Van Heek M., Compton D. S., France C. F., Tedesco R. P., Fawzi A. B., Graziano M. P., Sybertz E. J., Strader C. D., and Davis H. R. Jr. (1997) Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J. Clin. Invest. 99, 385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sell H., Habich C., and Eckel J. (2012) Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 8, 709–716 [DOI] [PubMed] [Google Scholar]
- 72. Kim Y. B., Shulman G. I., and Kahn B. B. (2002) Fatty acid infusion selectively impairs insulin action on Akt1 and protein kinase C λ/ζ but not on glycogen synthase kinase-3. J. Biol. Chem. 277, 32915–32922 [DOI] [PubMed] [Google Scholar]
- 73. Yu C., Chen Y., Cline G. W., Zhang D., Zong H., Wang Y., Bergeron R., Kim J. K., Cushman S. W., Cooney G. J., Atcheson B., White M. F., Kraegen E. W., and Shulman G. I. (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277, 50230–50236 [DOI] [PubMed] [Google Scholar]
- 74. Eckardt K., Taube A., and Eckel J. (2011) Obesity-associated insulin resistance in skeletal muscle: role of lipid accumulation and physical inactivity. Rev. Endocr. Metab. Disord. 12, 163–172 [DOI] [PubMed] [Google Scholar]
- 75. Mishra A. K., Dubey V., and Ghosh A. R. (2016) Obesity: An overview of possible role(s) of gut hormones, lipid sensing and gut microbiota. Metabolism 65, 48–65 [DOI] [PubMed] [Google Scholar]
- 76. Talior I., Yarkoni M., Bashan N., and Eldar-Finkelman H. (2003) Increased glucose uptake promotes oxidative stress and PKC-δ activation in adipocytes of obese, insulin-resistant mice. Am. J. Physiol. Endocrinol. Metab. 285, E295–E302 [DOI] [PubMed] [Google Scholar]
- 77. Kim O. K., Jun W., and Lee J. (2015) Mechanism of ER stress and inflammation for hepatic insulin resistance in obesity. Ann. Nutr. Metab. 67, 218–227 [DOI] [PubMed] [Google Scholar]
- 78. Lambert G. W., Straznicky N. E., Lambert E. A., Dixon J. B., and Schlaich M. P. (2010) Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol. Ther. 126, 159–172 [DOI] [PubMed] [Google Scholar]
- 79. Esteve E., Ricart W., and Fernández-Real J. M. (2011) Gut microbiota interactions with obesity, insulin resistance and type 2 diabetes: Did gut microbiote co-evolve with insulin resistance? Curr. Opin. Clin. Nutr. Metab. Care 14, 483–490 [DOI] [PubMed] [Google Scholar]
- 80. Nawaratne R., Gray A., Jørgensen C. H., Downes C. P., Siddle K., and Sethi J. K. (2006) Regulation of insulin receptor substrate 1 pleckstrin homology domain by protein kinase C: Role of serine 24 phosphorylation. Mol. Endocrinol. 20, 1838–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Greene M. W., Ruhoff M. S., Roth R. A., Kim J. A., Quon M. J., and Krause J. A. (2006) PKCδ-mediated IRS-1 Ser24 phosphorylation negatively regulates IRS-1 function. Biochem. Biophys. Res. Commun. 349, 976–986 [DOI] [PubMed] [Google Scholar]
- 82. Han S. H., Quon M. J., and Koh K. K. (2007) Reciprocal relationships between abnormal metabolic parameters and endothelial dysfunction. Curr. Opin. Lipidol. 18, 58–65 [DOI] [PubMed] [Google Scholar]
- 83. Chen H., Lin A. S., Li Y., Reiter C. E., Ver M. R., and Quon M. J. (2008) Dehydroepiandrosterone stimulates phosphorylation of FoxO1 in vascular endothelial cells via phosphatidylinositol 3-kinase- and protein kinase A-dependent signaling pathways to regulate ET-1 synthesis and secretion. J. Biol. Chem. 283, 29228–29238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Formoso G., Chen H., Kim J. A., Montagnani M., Consoli A., and Quon M. J. (2006) Dehydroepiandrosterone mimics acute actions of insulin to stimulate production of both nitric oxide and endothelin 1 via distinct phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-dependent pathways in vascular endothelium. Mol. Endocrinol. 20, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 85. Muniyappa R., Montagnani M., Koh K. K., and Quon M. J. (2007) Cardiovascular actions of insulin. Endocr. Rev. 28, 463–491 [DOI] [PubMed] [Google Scholar]
- 86. Rizza R. A., Haymond M. W., Verdonk C. A., Mandarino L. J., Miles J. M., Service F. J., and Gerich J. E. (1981) Pathogenesis of hypoglycemia in insulinoma patients: Suppression of hepatic glucose production by insulin. Diabetes 30, 377–381 [DOI] [PubMed] [Google Scholar]
- 87. Nishimura H., Pallardo F. V., Seidner G. A., Vannucci S., Simpson I. A., and Birnbaum M. J. (1993) Kinetics of GLUT1 and GLUT4 glucose transporters expressed in Xenopus oocytes. J. Biol. Chem. 268, 8514–8520 [PubMed] [Google Scholar]
- 88. Lee S., Lynn E. G., Kim J. A., and Quon M. J. (2008) Protein kinase C-ζ phosphorylates insulin receptor substrate-1, -3, and -4 but not -2: Isoform specific determinants of specificity in insulin signaling. Endocrinology 149, 2451–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cong L. N., Chen H., Li Y., Zhou L., McGibbon M. A., Taylor S. I., and Quon M. J. (1997) Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol. Endocrinol. 11, 1881–1890 [DOI] [PubMed] [Google Scholar]
- 90. Fasshauer M., Klein J., Ueki K., Kriauciunas K. M., Benito M., White M. F., and Kahn C. R. (2000) Essential role of insulin receptor substrate-2 in insulin stimulation of Glut4 translocation and glucose uptake in brown adipocytes. J. Biol. Chem. 275, 25494–25501 [DOI] [PubMed] [Google Scholar]
- 91. Toye A. A., Lippiat J. D., Proks P., Shimomura K., Bentley L., Hugill A., Mijat V., Goldsworthy M., Moir L., Haynes A., Quarterman J., Freeman H. C., Ashcroft F. M., and Cox R. D. (2005) A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48, 675–686 [DOI] [PubMed] [Google Scholar]
- 92. Freeman H., Shimomura K., Horner E., Cox R. D., and Ashcroft F. M. (2006) Nicotinamide nucleotide transhydrogenase: a key role in insulin secretion. Cell Metab. 3, 35–45 [DOI] [PubMed] [Google Scholar]
- 93. Sperry J. L., Zolin S., Zuckerbraun B. S., Vodovotz Y., Namas R., Neal M. D., Ferrell R. E., Rosengart M. R., Peitzman A. B., and Billiar T. R. (2014) X chromosome-linked IRAK-1 polymorphism is a strong predictor of multiple organ failure and mortality postinjury. Ann. Surg. 260, 698–703; discussion 703–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. He L., Sabet A., Djedjos S., Miller R., Sun X., Hussain M. A., Radovick S., and Wondisford F. E. (2009) Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 137, 635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Folli F., Saad M. J., Backer J. M., and Kahn C. R. (1992) Insulin stimulation of phosphatidylinositol 3-kinase activity and association with insulin receptor substrate 1 in liver and muscle of the intact rat. J. Biol. Chem. 267, 22171–22177 [PubMed] [Google Scholar]
- 96. Saad M. J., Araki E., Miralpeix M., Rothenberg P. L., White M. F., and Kahn C. R. (1992) Regulation of insulin receptor substrate-1 in liver and muscle of animal models of insulin resistance. J. Clin. Invest. 90, 1839–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Saad M. J., Folli F., Kahn J. A., and Kahn C. R. (1993) Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J. Clin. Invest. 92, 2065–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mirbolooki M. R., Upadhyay S. K., Constantinescu C. C., Pan M. L., and Mukherjee J. (2014) Adrenergic pathway activation enhances brown adipose tissue metabolism: A [18F]FDG PET/CT study in mice. Nucl. Med. Biol. 41, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Thorn S. L., deKemp R. A., Dumouchel T., Klein R., Renaud J. M., Wells R. G., Gollob M. H., Beanlands R. S., and DaSilva J. N. (2013) Repeatable noninvasive measurement of mouse myocardial glucose uptake with 18F-FDG: Evaluation of tracer kinetics in a type 1 diabetes model. J. Nucl. Med. 54, 1637–1644 [DOI] [PubMed] [Google Scholar]
- 100. Chen H., Sullivan G., Yue L. Q., Katz A., and Quon M. J. (2003) QUICKI is a useful index of insulin sensitivity in subjects with hypertension. Am. J. Physiol. Endocrinol. Metab. 284, E804–E812 [DOI] [PubMed] [Google Scholar]
- 101. Burant C. F., and Bell G. I. (1992) Mammalian facilitative glucose transporters: Evidence for similar substrate recognition sites in functionally monomeric proteins. Biochemistry 31, 10414–10420 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.