

Abstract
Nonalcoholic steatohepatitis (NASH) is the most prevalent cause of chronic liver disease in the Western world. However, an optimum therapy for NASH is yet to be established mandating more in-depth investigation into the molecular pathogenesis of NASH to identify novel regulatory molecules and develop targeted therapies. Here, we unravel a unique function of Astrocyte elevated gene-1/Metadherin (AEG-1/MTDH) in NASH using a transgenic mouse with hepatocyte-specific overexpression of AEG-1 (Alb/AEG-1) and a conditional hepatocyte-specific AEG-1 knockout mouse (AEG-1ΔHEP). Alb/AEG-1 mice developed spontaneous NASH while AEG-1ΔHEP mice were protected from high fat diet (HFD)-induced NASH. Intriguingly, AEG-1 overexpression was observed in livers of NASH patients and WT mice that developed steatosis upon feeding high fat diet. In-depth molecular analysis unraveled that inhibition of PPARα activity resulting in decreased fatty acid β-oxidation, augmentation of translation of fatty acid synthase resulting in de novo lipogenesis, and increased NF-κB-mediated inflammation act in concert to mediate AEG-1-induced NASH. Therapeutically, hepatocyte-specific nanoparticle-delivered AEG-1 siRNA provided marked protection from HFD-induced NASH in wild-type mice.
Conclusion
AEG-1 might be a key molecule regulating initiation and progression of NASH. AEG-1 inhibitory strategies might be developed as a potential therapeutic intervention in NASH patients.
Keywords: AEG-1, NASH, PPAR, fatty acid synthase, inflammation
Introduction
Non-alcoholic fatty liver disease (NAFLD), the most common cause of chronic liver disease in the Western world leading to cirrhosis and hepatocellular carcinoma (HCC), is characterized by initial accumulation of triglycerides (TG) in hepatocytes (hepatic steatosis) with subsequent non-alcoholic steatohepatitis (NASH) (1, 2). The pathogenesis of NASH includes an initial metabolic disturbance that increases the influx of free fatty acids (FFA) and de novo lipogenesis, followed by inflammation, oxidative stress and lipid peroxidation (3). The molecular players regulating these events are gradually being identified leading to evaluation of multiple strategies in clinical trials (1). However, an optimum therapy is yet to be established indicating that more comprehensive understanding of the molecular mechanism of NASH is urgently needed to develop effective therapeutic strategies.
Astrocyte elevated gene-1 (AEG-1), also known as metadherin (MTDH) and LYRIC, is a pleiotropic protein with diverse array of functions (4). AEG-1 is fundamentally required for the activation of NF-κB pathway, a pivotal determinant of inflammation, and AEG-1 regulates NF-κB at multiple levels (5–8). Lipopolysaccharide (LPS)-induced NF-κB activation is markedly abrogated in AEG-1 knockout (AEG-1KO) hepatocytes and macrophages and AEG-1KO mice show resistance to aging-associated inflammation (9).
An essential component of AEG-1 function is its ability to interact with and inhibit the function of retinoid X receptor (RXR) (10). RXR heterodimerizes with nuclear receptors, including Liver X Receptor (LXR), Peroxisome Proliferator Activated Receptor (PPAR) and Farnesoid X Receptor (FXR), and regulate corresponding ligand-dependent gene transcription (11). Cholesterol metabolites, fatty acid derivatives and bile acids serve as endogenous ligands for LXR, PPAR and FXR, respectively, which play an important role in regulating lipid metabolism, hence NASH (12). These transcription factors exert opposing effects, e.g., LXR promotes NASH by augmenting de novo lipogenesis, while PPARα inhibits NASH by augmenting fatty acid β-oxidation (13, 14). Interaction between AEG-1 and RXR in the nucleus blocks co-activator recruitment thereby abrogating ligand-induced gene transcription (10). Interaction of cytoplasmic AEG-1 with RXR blocks RXR nuclear translocation and also induces RXR phosphorylation and hence inactivation (10).
We generated a transgenic mouse with hepatocyte-specific overexpression of AEG-1 (Alb/AEG-1) to analyze its oncogenic function (15). Interestingly we observe that Alb/AEG-1 mice develop spontaneous NASH, and in the present manuscript we further show that a hepatocyte-specific conditional AEG-1 knockout mouse (AEG-1ΔHEP) is resistant to high fat diet-induced NASH, and human NASH patients show overexpression of AEG-1. We demonstrate that AEG-1 promotes NASH by regulating the function of nuclear receptors, promoting translation of lipogenic enzymes and activating inflammation. We also document inhibition of AEG-1 as a potential strategy to protect from NASH. These findings identify AEG-1 as a novel regulator of NASH onset and progression and suggest that AEG-1 inhibition in the liver might serve as an effective interventional therapeutic to halt NASH progression.
Materials and Methods
Mice
Generation and characterization of a hepatocyte-specific AEG-1 transgenic mouse (Alb/AEG-1) in B6/CBA background have been described (15). Floxed AEG-1 mice (AEG-1fl/fl) in C57BL/6 background (9) was crossed with Alb/Cre (Jackson laboratories) mice to generate AEG-1ΔHEPmice. Mice were fed regular chow. For high fat diet (HFD) experiments, 8 weeks old mice were fed a high fat and cholesterol containing diet (Harlan; TD. 88137) for 20–22 weeks. This diet contains 0.2% total cholesterol and 21% total fat by weight which provides 42% kcal (4.5 kcal/gm). All animal studies were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Patient samples
Liver biopsy samples were collected from normal livers (biopsied for ruling out differential diagnosis; n = 32) and NASH patients (n = 21). Stage 1 and 2 NASH patients according to NASH CRN modified Brunt methodology(16) were selected and patients with bridging fibrosis or cirrhosis were excluded (Table S1). The Institutional Review Board (IRB) of New York Methodist Hospital approved these studies.
Tissue culture
Primary mouse hepatocytes were isolated and cultured as described (15). Telomerase immortalized human hepatocytes Hc3716-hTERT are a kind gift from Dr. Tahara (17). HepG3 and QGY-7703 cells were cultured as described (18). Generation and characterization of Hep-pc-4 (control clone) and Hep-AEG-1–14 (a COOH-terminal HA-tagged AEG-1 overexpressing clone) in HepG3 background, and Hep-Consh (HepG3 clone stably expressing control shRNA) and Hep-AEG-1sh (HepG3 clone stably expressing AEG-1 shRNA) have been described (18, 19).
Statistical analysis
Data were represented as the mean ± Standard Error of Mean (S.E.M) and analyzed for statistical significance using one-way analysis of variance (ANOVA) followed by Newman-Keuls test as a post hoc test. A p-value of <0.05 was considered as significant. Overexpression of AEG-1 in NASH patients was analyzed by Pearson’s χ2 test.
Results
Alb/AEG-1 mice develop spontaneous NASH
Analysis of liver sections of 6 months old WT and Alb/AEG-1 littermates showed increased fat droplets in H&E staining, increased fibrosis (assayed by α-smooth muscle actin, Collagen I and picrosirius red staining) and increased inflammation (F4/80 staining for infiltrated macrophages) in Alb/AEG-1 versus WT mice indicating features of NASH (Fig. 1A, S1A–C). Levels of interleukin-6 (IL-6) and vascular endothelial growth factor (VEGF) were significantly higher in Alb/AEG-1 serum than in WT serum in both sexes, except for IL-6 which did not show an increase in female Alb/AEG-1 mice and might be explained by inhibition of IL-6 by estrogen (Fig. 1B) (20). Serum liver enzymes, AST, ALT and Alk Phos, total TG and FFA, and hepatic TG and cholesterol levels were significantly higher in Alb/AEG-1 mice compared to WT (Fig. 1C–E). Total TG level in the conditioned media from an AEG-1-overexpressing clone of human HCC cell line HepG3 (Hep-AEG-1–14) was significantly higher compared to control clone Hep-PC-4 indicating that in vivo observations in mice are maintained in human cell lines (Fig. S1D) (18). Steatosis is often associated with insulin resistance (21). Indeed, basal blood glucose level was significantly increased in Alb/AEG-1 over WT mice (Fig. 1F, top). Glucose tolerance test (GTT) revealed impaired glucose management in Alb/AEG-1 mice in which blood glucose level remained significantly higher 2 h after the initial glucose injection versus WT further indicating reduced insulin sensitivity (Fig. 1F, bottom).
Figure 1.
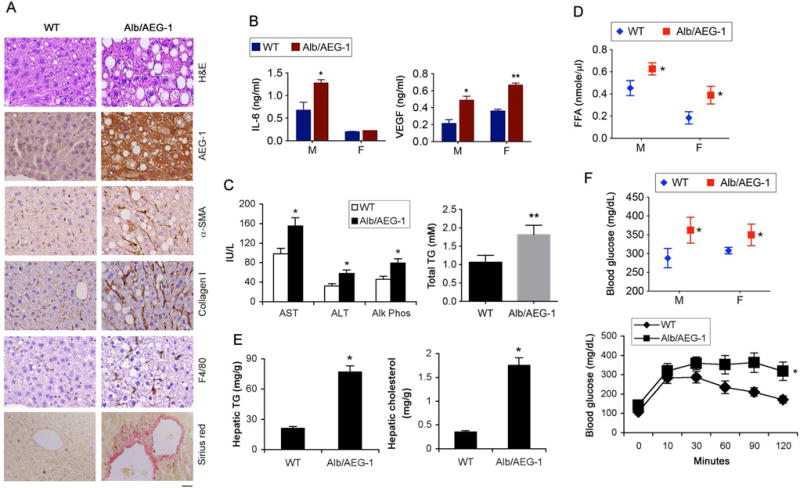
Alb/AEG-1 mice develop NASH. A. H&E, IHC staining with the indicated antibodies and picrosirius red staining in FFPE sections of livers of 6 months old male littermates. Magnification 400×. Scale bar: 20 μm. B–E. IL-6 and VEGF (B), liver enzymes and total TG (C), and free fatty acid (FFA) (D) levels were measured in the sera and hepatic TG and cholesterol levels (E) were analyzed in 6-month old male littermates (n = 6/group). F. Blood glucose level was measured either under basal condition (top) or after glucose tolerance test (bottom). Male mice were used (n = 12/group). For graphs, data represent mean ± SEM. * : p<0.01, ** : p<0.001.
AEG overexpression in the liver is linked with the presence of NASH
To further evaluate the role of AEG-1 in NASH progression, we examined the response of WT and Alb/AEG-1 littermates to 20 weeks of high fat diet (HFD) feeding. A significant increase in liver weight and liver enzymes, and elevated levels of TG, cholesterol and HDL and markedly elevated levels of LDL and VLDL were detected in sera of chow- and HFD-fed Alb/AEG-1 mice vs WT (Fig. 2A–B). Although HFD feeding induced development of steatosis in WT mice, in Alb/AEG-1 mice the steatotic phenotype was more pronounced with large fat droplets, collagen bands (indicated by picrosirius staining) and lobular inflammation (indicated by neutrophil infiltration) (Fig. 2C, S1B and S1E). A significant increase in the levels of mRNA of inflammatory and fibrotic genes was observed in the livers of chow-fed Alb/AEG-1 mice compared to WT which was further augmented upon feeding HFD (Fig. 2D). Liver sections from WT mice that developed HFD-induced steatosis showed a marked increase in AEG-1 expression compared to WT chow-fed controls (Fig. 2E and S1F).
Figure 2.
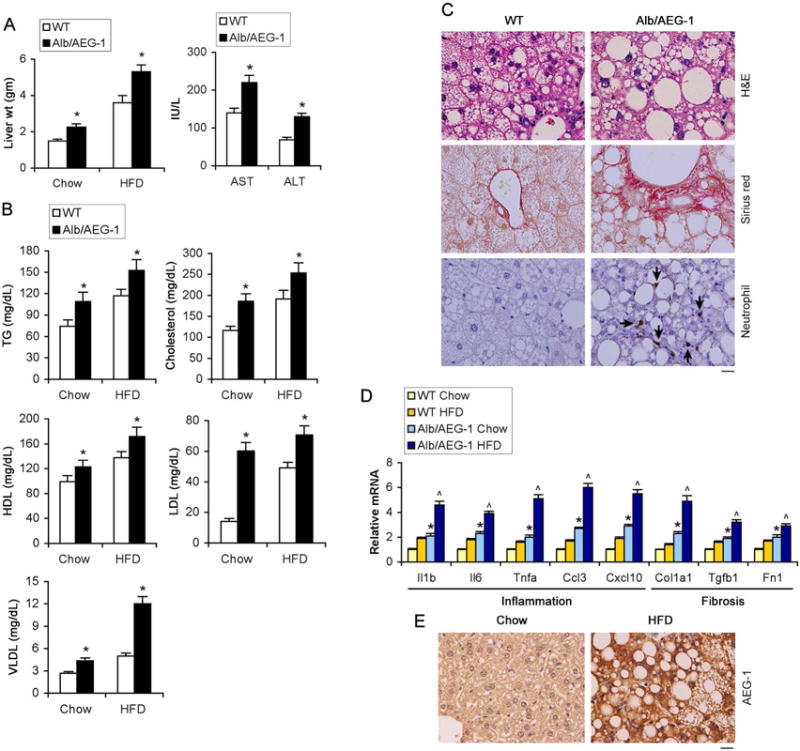
High fat diet (HFD) exacerbates phenotypes of Alb/AEG-1 mice. WT and Alb/AEG-1 littermates were fed HFD for 20 weeks. WT: n = 12; Alb/AEG-1: n = 14. Liver weight (A, left panel), liver enzymes (A, right panel) and plasma lipid profile (B) in chow- and HFD-fed mice at the end of the experiment. Data represent mean ± SEM. * : p<0.01. C. H&E, picrosirius red and neutrophil (arrows) staining of FFPE sections of livers of mice fed HFD. D. Analysis of the mRNA levels of indicated markers of inflammation and fibrosis in the livers of the indicated mice groups (n = 5/group). Data represent mean ± SEM. *: p<0.01 between chow-fed groups; ˄: p<0.01 between HFD-fed groups. E. AEG-1 immunostaining in FFPE sections of livers of WT mice fed chow or HFD. C–D: representative images; magnification 400×; scale bar: 20 μm.
To evaluate the status of AEG-1 in the livers of NASH patients, AEG-1 expression was analyzed by immunohistochemistry in biopsy samples of 32 normal, non-NASH afflicted livers (biopsied for ruling out differential diagnosis) and 21 livers with NASH. Significant overexpression of AEG-1 was detected in NASH patients compared to non-NASH individuals (Pearson’s χ2 test: p<0.0001) (Fig. 3A–B and Table S1).
Figure 3.
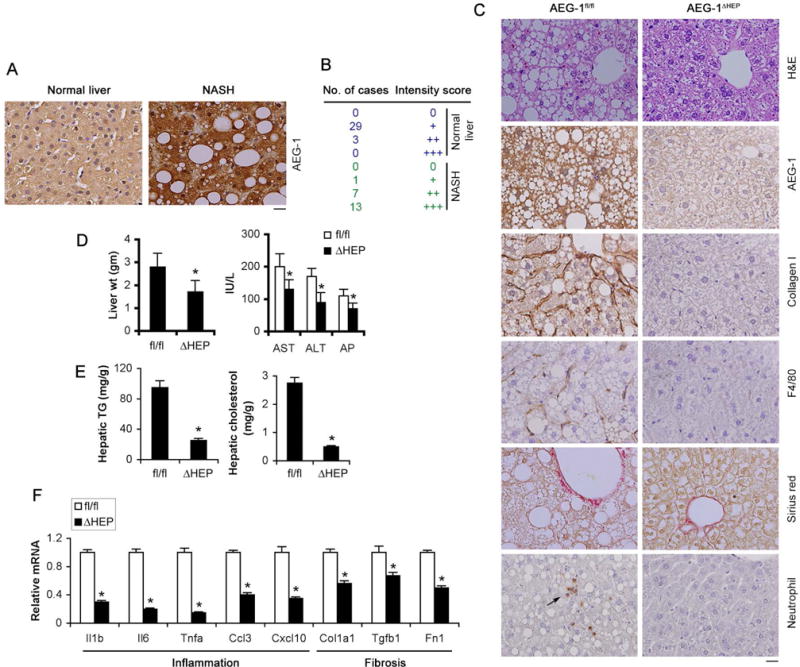
AEG-1 is overexpressed in NASH patients and AEG-1ΔHEP mice are resistant to HFD-induced NASH. A. AEG-1 immunostaining in livers of normal individual and NASH patients. B. Quantification of AEG-1 staining intensity in normal and NASH livers. (Pearson’s χ2 test: p<0.0001). C. H&E, IHC staining with the indicated antibodies and picrosirius red staining in FFPE sections of livers of AEG-1fl/fl and AEG-1ΔHEP littermates fed HFD for 22 wks. D–F. Liver weight (D, left panel), serum levels of liver enzymes (D, right panel), hepatic TG and cholesterol levels (E) and mRNA levels of inflammatory and fibrotic genes in the liver by Q-RT-PCR (F) were measured in HFD-fed AEG-1fl/fl and AEG-1ΔHEP littermates at the end of the study. The indicated mRNA levels were normalized by levels of corresponding chow-fed littermates. AEG-1fl/fl: n = 9. AEG-1ΔHEP: n = 10. A and C: representative images; magnification 400×; scale bar: 20 μm. D–F, data represent mean ± SEM. * : p<0.01.
We next checked which factors induce AEG-1 overexpression in NASH. Treatment with palmitate did not induce AEG-1 (data not shown) in primary mouse hepatocytes and Hc3716 immortalized human hepatocytes (17). However, treatment with TNFα and IL-1β significantly induced AEG-1 mRNA in both cell types which could be inhibited by BMS-3445541, an IκB kinase inhibitor, indicating a potential role of NF-κB in mediating inflammatory cytokine-induced AEG-1 expression (Fig. S1G). Taken together, these findings provide evidence of a positive feedback relationship, wherein NF-κB and AEG-1 regulate each other fostering a chronic inflammatory state.
AEG-1ΔHEP mice are resistant to HFD-induced NASH
To directly investigate whether the presence of AEG-1 in the hepatocyte is essential to NASH initiation, we generated a hepatocyte-specific conditional AEG-1 KO mouse (AEG-1ΔHEP), with complete loss of AEG-1 in the hepatocytes (Fig. S2A–C). Male AEG-1ΔHEP mice showed marked resistance to NASH development following 22 weeks on a HFD (Fig. 3C). Analysis of liver sections revealed decreased fat droplet accumulation, collagen I and F4/80 staining and neutrophil infiltration in AEG-1ΔHEP mice compared to their AEG-1fl/fl littermates (Fig. 3C and S2D) indicating an overall reduction in the phenotypic hallmarks of NASH. Furthermore, liver weight, serum levels of liver enzymes, hepatic TG and cholesterol levels and mRNA levels of inflammatory and fibrotic genes were also dramatically reduced in the livers of HFD-fed AEG-1ΔHEP mice compared to AEG-1fl/fl mice (Fig. 3D–F). These findings indicate that the livers of AEG-1ΔHEP mice are protected from HFD-induced NASH and suggest that AEG-1expression is required for initiation and progression of the disease.
AEG-1 regulates PPARα function
To thoroughly compare the differences in the gene expression profiles between WT and Alb/AEG-1 hepatocytes, an Affymetrix oligonucleotide microarray analysis was performed using WT and Alb/AEG-1 hepatocytes harvested from 12 weeks old mice. Using a false discovery rate (FDR) of 0 (q-value: 0%) as cut-off, 810 genes showed differential change, 366 genes showing upregulation and 444 genes showing downregulation in Alb/AEG-1 hepatocytes compared to WT (a complete list of differentially regulated genes is available in GSE46701). Genes regulating lipid biosynthesis and transport, lipid oxidation, inflammatory cytokines and adipokines were significantly modulated in Alb/AEG-1 hepatocytes consistent with the observed phenotypes (Table 1). Several of these genes were then selected from the data set and independently validated by Q-RT-PCR and Western blotting (Fig. 4A, left panel and 4B, left panel). All differentially changed genes were analyzed using Ingenuity pathway analysis software to identify the upstream regulators the activation or inhibition of which might lead to alterations in downstream genes. A z-score >2 indicates activation and a score of <-2 indicates inhibition. The most robust inhibition in Alb/AEG-1 hepatocytes was observed for genes that are regulated by PPARα, PPARγ and PXR with very significant p-value and a z-score of <-2 (Fig. 4C, top panel). Although inhibition of LXRα, LXRβ and CAR showed significant p-value the z-score was >-2. These findings suggest that there is preferential inhibition of PPAR- and PXR-target genes in Alb/AEG-1 hepatocytes compared to WT hepatocytes. Reduced expression of CAR- and LXR-target genes was also observed. However, overall the magnitude of inhibition and the number of genes were not sufficient to attain a significant z-score. AEG-1 plays a fundamental role in regulating NF-κB and inflammation (6, 9). In concordance, the most robust activation was observed in upstream regulators, such as TLR4, TLR3, IL-1β, TNFα, IL-6 and NF-κB complex, all of which are critical components involved in inflammatory signaling (Fig. 4C, top panel). We performed RNA-Seq analysis on liver samples of chow-fed 12 weeks old AEG-1fl/fl and AEG-1ΔHEP mice. With a p-value cut-off of <0.05, 868 genes showed differential regulation (the complete list is available in GSE85098) a subset of which were then validated (Fig. 4A, right panel and 4B, right panel). Upstream regulator analysis revealed a robust activation of PPARα and PPARγ and inhibition of TNFα downstream events thus corroborating findings from Alb/AEG-1 mice (Fig. 4C, bottom panel). Heat map analysis of PPARα-target genes showed robust clustering in WT and Alb/AEG-1 hepatocytes (Fig. 4D) or AEG-1fl/fl and AEG-1ΔHEP livers (Fig. S3A) further indicating an important role of AEG-1 in regulating PPARα signaling pathway. We therefore focused our studies on AEG-1-mediated regulation of PPARα activity.
Table 1.
Selective deregulated genes related to lipid and inflammatory functions in the hepatocytes of Alb/AEG-1 vs WT mice as identified by microarray analysis. (n = 3 per group)
| Genes | Gene symbol | Fold change |
|---|---|---|
| Lipid biosynthesis and transport | ||
| apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 | Apobec1 | 2.929048 |
| fatty acid binding protein 5, epidermal | Fabp5 | 2.105085 |
| suppressor of cytokine signaling 1 | Socs1 | 1.7178593 |
| apolipoprotein L 9a /// apolipoprotein L 9b | Apol9a /// Apol9b | 1.5561239 |
| apolipoprotein C-II | Apoc2 | 1.531308 |
| elongation of very long chain fatty acids (FEN1/Elo2, SUR4/Elo3, yeast)-like 3 | Elovl3 | 1.4592772 |
| acyl-CoA synthetase medium-chain family member 2 | Acsm2 | 1.606501 |
| sterol regulatory element binding transcription factor 1 | Srebf1 | 0.6958657 |
| phospholipid transfer protein | Pltp | 0.26495898 |
| acetyl-Coenzyme A acetyltransferase 2 | Acat2 | 0.6190338 |
| CD36 antigen | Cd36 | 0.22634816 |
| Lipid oxidation | ||
| peroxisome proliferator activated receptor gamma | Pparg | 0.24888209 |
| isoamyl acetate-hydrolyzing esterase 1 homolog (S. cerevisiae) | Iah1 | 0.5118611 |
| acyl-CoA thioesterase 1 | Acot1 | 0.5617671 |
| peroxisome proliferator activated receptor alpha | Ppara | 0.58591425 |
| acyl-Coenzyme A oxidase 1, palmitoyl | Acox1 | 0.59743994 |
| peroxisomal biogenesis factor 11 alpha | Pex11a | 0.6227678 |
| hydroxysteroid (17-beta) dehydrogenase 10 | Hsd17b10 | 0.6812308 |
| acetyl-Coenzyme A acyltransferase 1A /// 1B | Acaa1a /// Acaa1b | 0.699141 |
| Inflammatory responsive genes in steatosis/obesity | ||
| chemokine (C-C motif) ligand 3 | Ccl3 | 3.694735 |
| chemokine (C-X-C motif) ligand 9 | Cxcl9 | 3.6443808 |
| interleukin 6 | IL6 | 3.4334872 |
| interleukin 33 | Il33 | 2.4694686 |
| secreted phosphoprotein 1 | Spp1 | 2.029259 |
| chemokine (C-C motif) ligand 5 | Ccl5 | 1.9767197 |
| chemokine (C-X-C motif) ligand 2 | Cxcl2 | 1.943742 |
| chemokine (C-C motif) ligand 4 | Ccl4 | 1.9180745 |
| chemokine (C-X-C motif) ligand 10 | Cxcl10 | 1.8839078 |
| matrix metallopeptidase 13 | Mmp13 | 1.8367785 |
| interleukin 10 | Il10 | 1.7319263 |
| interleukin 1 alpha | Il1a | 1.6411645 |
| toll-like receptor 2 | Tlr2 | 1.6023682 |
| vascular endothelial growth factor A | Vegfa | 1.5935463 |
| alpha-2-macroglobulin /// hypothetical protein LOC677369 | A2m /// LOC677369 | 1.4580379 |
| Adipokine related genes | ||
| serine (or cysteine) peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 12 | Serpina12 | 3.493617 |
| ERO1-like (S. cerevisiae) | Ero1l | 2.6987228 |
| serine (or cysteine) peptidase inhibitor, clade A, member 3N | Serpina3n | 1.4795601 |
Figure 4.
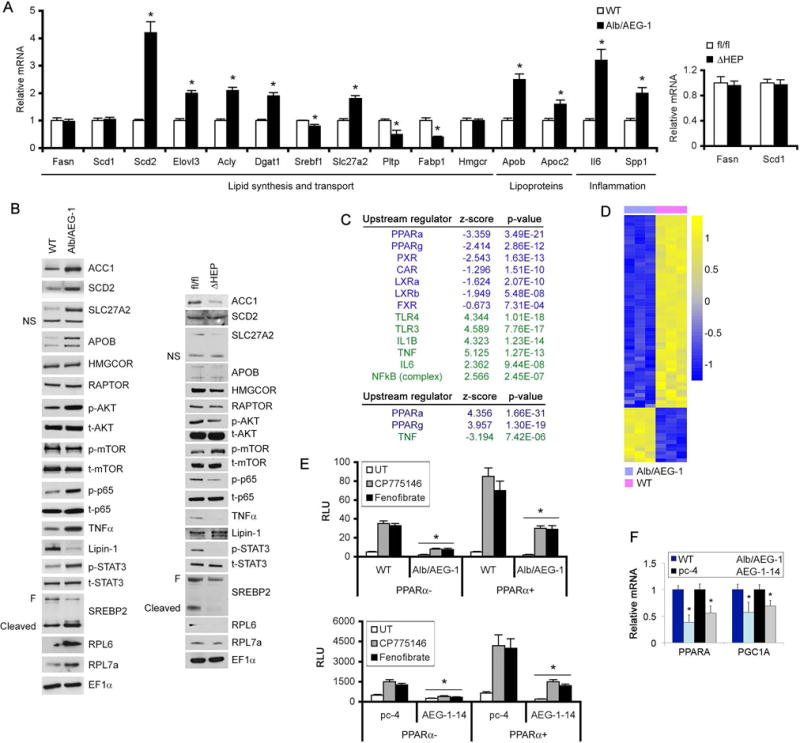
AEG-1 inhibits PPARα function. A. Expression analysis by Q-RT-PCR of the indicated mRNAs in the livers of WT and Alb/AEG-1 mice (left panel, n = 5/group), and AEG-1fl/fl and AEG-1ΔHEP mice (right panel, n = 3/group). Normalized by GAPDH. Expression level in WT and AEG-1fl/fl was considered as 1 for each gene. Data represent mean ± SEM. * : p<0.01. B. Western blotting for the indicated proteins in WT and Alb/AEG-1 livers (left panel), and AEG-1fl/fl and AEG-1ΔHEP livers (right panel). EF1α was used as loading control for each blot and one representative of such control is shown. C. Upstream regulators inhibited or activated in Alb/AEG-1 hepatocytes (top) and chow-fed AEG-1ΔHEP livers (bottom). D. Heat map of expression levels of PPARα target genes in WT and Alb/AEG-1 hepatocytes. E. PPRE luciferase reporter activity in the indicated cells in the presence or absence of PPARα expression plasmid and treated or not with CP775146 (2.5 μM) or Fenofibrate (5 μM). F. Relative PPARα and PGC1α mRNA levels in the indicated cells. Data represent mean ± SEM of at least 3 experiments. *: p<0.01.
We checked PPRE (PPAR response element)-luciferase activity in WT and Alb/AEG-1 hepatocytes as well as Hep-pc-4 and Hep-AEG-1-14 cells. To stimulate PPARα activity cells were treated with synthetic ligands of PPARα, CP775146 and fenofibrate. Both basal and ligand-dependent PPRE-luc activity was significantly inhibited upon AEG-1 overexpression when compared to control in the presence or absence of PPARα expression plasmid (Fig. 4E). Similarly, basal and ligand-dependent PPRE-luc activity was significantly less in QGY-7703 cells when compared to HepG3 cells, expressing high and low levels of endogenous AEG-1, respectively (Fig. S3B). The expression of PPARα and its co-activator PGC1α was downregulated in Alb/AEG-1 hepatocytes versus WT as well as Hep-AEG-1-14 cells versus Hep-pc-4 cells (Fig. 4F and S2C). Downregulation of PPARγ and PPARα co-activator Lipin1 was also observed in Alb/AEG-1 hepatocytes over WT (Fig. S3C). It has been shown that Lipin1 acts as an inducible amplifier of PPARα/PGC1α pathway in hepatocytes (22) and AEG-1-mediated inhibition in Lipin1 would indirectly lead to further abrogation of PPARα signaling.
We next analyzed the expression levels of PPARα target genes that regulate β-oxidation of fatty acids in WT and Alb/AEG-1 hepatocytes. To evaluate the mitochondrial β-oxidation pathway, we analyzed Cpt1a, the rate-limiting enzyme in the carnitine-dependent transport of fatty acids across the mitochondrial membrane, and Acadl and Acadm, which catalyze the initial steps of fatty acid β-oxidation (23). Peroxisomal β-oxidation was evaluated by analyzing Acox1, the first enzyme in this pathway, and Pex11a (23). Both basal and ligand-dependent induction of these genes was blunted in Alb/AEG-1 hepatocytes and augmented in AEG-1−/− hepatocytes compared to controls (Fig. 5A–B). Interestingly, expression of these genes was further suppressed in the livers of HFD-fed Alb/AEG-1 mice when compared to HFD-fed WT mice (Fig. 5C). Levels of β-hydroxybutyrate (3′-HB), a product of FA oxidation, were significantly lower in the sera of Alb/AEG-1 mice versus WT and in Alb/AEG-1 hepatocytes compared to WT hepatocytes (Fig. 5D) further suggesting reduced rate of β-oxidation when AEG-1 is overexpressed. To directly investigate the differences in rates of FA β-oxidation, we measured products of fatty acid β-oxidation in fresh liver homogenates using 14C-palmitate (24). Acetyl-CoA, generated by β-oxidation, enters TCA cycle to generate CO2. 14C-palmitate that does not get oxidized forms acid-soluble metabolites (ASM). Reduced production of these intermediates indicated a significant decrease in the rate of FA β-oxidation in the livers of Alb/AEG-1 versus WT mice (Fig. 5E).
Figure 5.
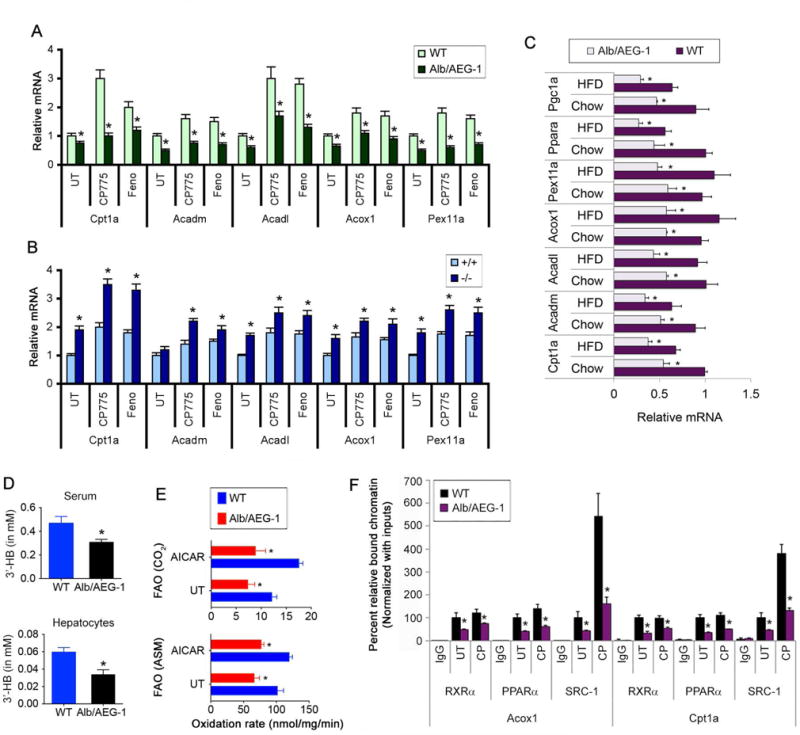
AEG-1 inhibits expression of PPARα target genes and fatty acid β-oxidation. A–C. Analysis of PPARα target genes in WT and Alb/AEG-1 hepatocytes (A), AEG-1+/+ and AEG-1−/− hepatocytes (B) with or without PPARα ligands, and in chow- and HFD-fed WT and Alb/AEG-1 livers (C). Normalized by GAPDH. D. Measurement of 3′-HB level in mice sera and hepatocytes. For A–D, n = 5/group. Data represent mean ± SEM of at least 3 experiments. *: p<0.01. E. Fatty acid β-oxidation rate measured by acid soluble metabolite (ASM) and CO2 in the indicated livers. AICAR is an inducer. Data represent mean ± SEM of at least 3 experiments. *: p<0.01 between WT and Alb/AEG-1. F. ChIP assay to check the binding of the indicated proteins on PPARα targets Acox1 and Cpt1a promoter in WT and Alb/AEG-1 hepatocytes using IgG as control. Data represent mean ± SEM of at least 3 experiments. *: p<0.01.
To directly evaluate the relationship between AEG-1 expression and PPARα activity, we examined recruitment of RXRα/PPARα heterodimer and co-activator SRC-1 on Acox1 and Cpt1a promoter upon CP775146 treatment in WT and Alb/AEG-1 hepatocytes by chromatin immunoprecipitation (ChIP) assay. Previous ChIP-Seq studies have documented binding of PPARα in the promoters of Acox1 and Cpt1a (Fig. S3D) (25). Additionally we generated luciferase reporter vectors under control of Acox1 and Cpt1a promoter amplicons and created mutants of these reporter vectors with point mutations in PPRE. In WT hepatocytes, both the promoter constructs showed increased activity upon co-transfection of PPARα expression construct and treatment with PPARα agonists which was lost in the mutant constructs (Fig. S3E). Upon confirmation that these genes are bona fide PPARα target genes we proceeded with our ChIP assay. A significant decrease in recruitment of RXRα/PPARα heterodimer was observed in Alb/AEG-1 hepatocytes compared to WT hepatocytes (Fig. 5F). This decrease might be attributed to a decrease in total PPARα and decrease in nuclear RXRα in Alb/AEG-1 livers since cytoplasmic AEG-1 can interact with RXR and prevent RXR nuclear translocation (10). However, a marked inhibition of SRC-1 recruitment was observed in Alb/AEG-1 hepatocytes upon ligand treatment confirming that AEG-1 overexpression interferes with co-activator recruitment.
AEG-1 regulates lipogenic enzymes at a translational level
Inhibition of FA β-oxidation might lead to accumulation of TG and FA contributing to steatosis. However, one important component of NASH is de novo lipogenesis and a key enzyme in this pathway is fatty acid synthase (Fasn). LXR transcriptionally regulates Fasn by binding to LXRE elements in its promoter (26). LXR also regulates SREBP-1 which controls transcription of lipogenic genes (27). As described earlier upstream analysis identified a trend involving a decrease in LXR/RXR signaling in Alb/AEG-1 hepatocytes which did not attain a significant z-score. Additionally if LXR function is inhibited there will be a decrease in Fasn resulting in decrease lipogenesis. However, no differences in Fasn mRNA levels were observed either by RNA-Seq analysis of AEG-1+/+ and AEG-1−/− livers or by Affymetrix microarray analysis of Hep-pc4 and Hep-AEG-1-14 cells (9, 18). Fasn mRNA levels were also unchanged between WT and Alb/AEG-1 hepatocytes, and AEG-1fl/fl and AEG-1ΔHEP livers in microarray and RNA-Seq analyses, respectively, as were levels of SCD1, another LXR target gene. This data was confirmed by Q-RT-PCR (Fig. 4A). We also did not observe any change in the total and cleaved forms of SREBP-1 in Alb/AEG-1 and AEG-1ΔHEP livers compared to controls (Fig. 6A). These findings argue that LXR activity may not be affected by AEG-1 status. However, there was a significant increase in Alb/AEG-1 and a decrease in AEG-1ΔHEP livers in FASN protein levels compared to controls (Fig. 6A). Additionally, a dual color 2D gel electrophoresis comparing HepG3 clone stably expressing control shRNA (Hep-Consh) and HepG3 clone stably expressing AEG-1 shRNA (Hep-AEG-1sh) showed a marked decrease in FASN protein levels in Hep-AEG-1sh cells compared to Hep-Consh although Fasn mRNA levels remained unchanged (Fig. S4). To confirm that the increase in the lipogenic enzymes indeed contributes to de novo lipogenesis, 14C-acetate incorporation in fresh liver homogenates was performed followed by analysis of lipid fractions. A significant and robust increase in TG and phospholipid (PL), and small increases in monoglyceride and diglyceride, FFA and cholesterol ester (CE) were observed in Alb/AEG-1 versus WT livers (Fig. 6B).
Figure 6.
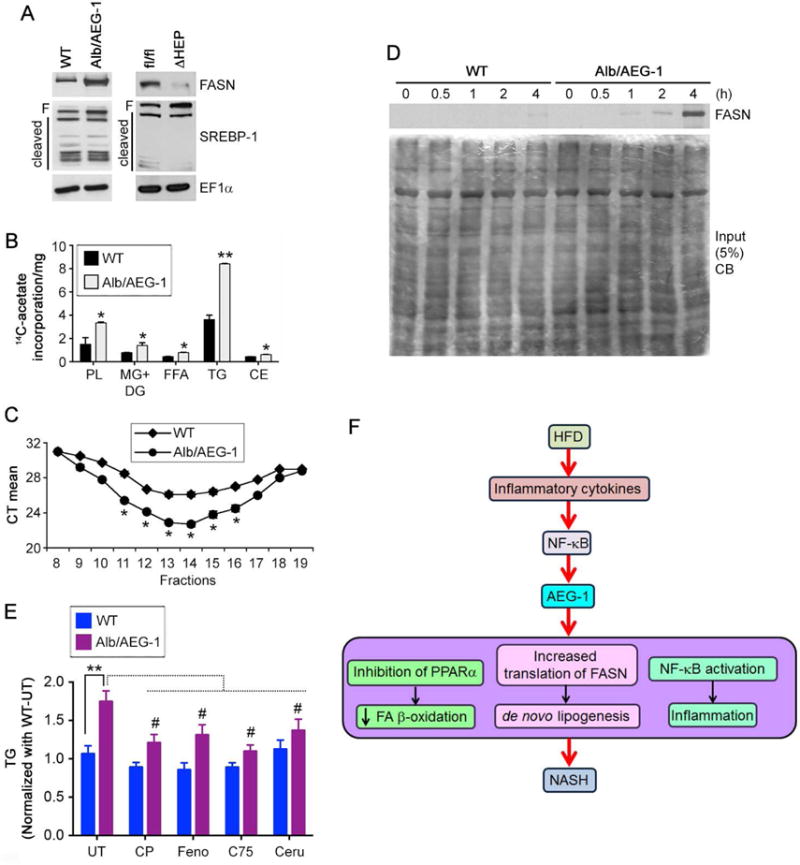
AEG-1 induces de novo lipogenesis. A. Western blotting for the indicated proteins in WT, Alb/AEG-1, AEG-1fl/fl and AEG-1ΔHEP livers. EF1α was used as loading control of each blot and one representative of such control is shown. B. De novo lipogenesis was measured by 14C-acetate incorporation assay. PL: phospholipid; MG: monoglyceride; DG: diglyceride; FFA: free fatty acid; TG: triglyceride; CE: cholesterol ester. Data represent mean ± SEM of three independent experiments. * : p<0.05, ** : p<0.01. C. RNA was extracted from fractions of WT and Alb/AEG-1 hepatocytes centrifuged through a sucrose gradient and subjected to Fasn mRNA level analysis by Q-RT-PCR. Fractions 9–16 typically represent the polysomal fractions. CT: cycle threshold. Data represent mean ± SEM of three independent experiments. *: p<0.05. D. WT and Alb/AEG-1 hepatocytes were labeled with 35S-methionine for the indicated time-points and 500 μg of protein per sample was used to immunoprecipitate FASN. Top, autoradiogram of the immunoprecipitates. Bottom, Coomassie blue (CB) stain of 5% of input. E. WT and Alb/AEG-1 hepatocytes were either untreated or treated with CP775146 (2.5 μM), Fenofibrate (5 μM), C75 (10 μg/ml), or cerulenin (10 μg/ml) and TG level was measured. Data represent mean ± SEM of three independent experiments. **: p<0.001 between UT groups; #: p<0.05 between untreated and treated Alb/AEG-1 hepatocytes. F. Cartoon depicting the molecular mechanism by which AEG-1 is induced in NASH and AEG-1 promotes NASH.
AEG-1 is known to regulate gene expression at the translational level (28). We, therefore, analyzed association of Fasn mRNA to polysomes. Polysomal fractions were collected from WT and Alb/AEG-1 hepatocytes, RNA was extracted from each fraction and an equal amount of RNA from each fraction was subjected to cDNA synthesis and Taqman Q-RT-PCR for Fasn. The mean cycle threshold (CT) value for Fasn amplification was significantly lower in Alb/AEG-1 hepatocytes compared to WT hepatocytes indicating that AEG-1 preferentially helps Fasn mRNA associate with polysomes and thereby facilitates translation (Fig. 6C). To further confirm these findings we analyzed de novo FASN protein synthesis by labeling WT and Alb/AEG-1 hepatocytes with 35S-methionine and then temporally analyzing FASN protein level by immunoprecipitation. In Alb/AEG-1 hepatocytes de novo FASN protein could be detected as early as 1 h after addition of 35S-methionine which continued to increase robustly until 4 h, while in WT hepatocytes it was not detected until 4h at a substantially lower level (Fig. 6D).
To assess the relative contribution of the identified pathways to increased TG level we treated WT and Alb/AEG-1 hepatocytes with PPARα activators, CP775146 and fenofibrate, and FASN inhibitors, C75 and cerulenin. Activation of PPARα and inhibition of FASN significantly reduced TG level in Alb/AEG-1 hepatocytes compared to control (Fig. 6E). Additionally PPARα activation resulted in significant increase in 3′-HB levels in Alb/AEG-1 versus WT hepatocytes (Fig. S3F). Collectively, our findings reveal that inhibition of PPARα, translational upregulation of FASN and NF-κB activation might work in concert to mediate AEG-1-induced NASH (Fig. 6F).
Nanoparticle-delivered AEG-1 siRNA protects from HFD-induced steatosis
We have developed liver-targeted nanoplexes by conjugating polyamidoamine (PAMAM) dendrimers with polyethylene glycol (PEG) and galactose lactobionic acid (PAMAM-PEG-Gal) which were complexed with AEG-1 siRNA (PAMAM-AEG-1si) (Fig. 7A, top) and documented its therapeutic utility in the context of cancer (29). Cationic PAMAM complexes and compacts siRNA, PEG increases stability and increases circulation time and the galactose (lactobionic acid) binds to asialoglycoprotein receptors that are upregulated on liver cells (29). We have now generated PAMAM-AEG-1si specific for mouse AEG-1 and checked its efficacy in inhibiting HFD-induced steatosis. WT C57BL/6 mice (n = 10/group) were fed HFD for a total of 8 wks. After 2 wks of feeding HFD, mice were injected i.v. with PAMAM-siCon (control scrambled siRNA conjugated with PAMAM-PEG-Gal) or PAMAM-AEG-1si (5 μg of siRNA, conjugated with PAMAM-PEG-Gal at a ratio of 10:1) twice weekly for 4 wks (Fig. 7A, bottom). Mice were sacrificed 2 wks after the last injection. PAMAM-AEG-1si treatment resulted in significant knockdown of AEG-1 mRNA only in liver but not in spleen and small intestine confirming efficiency of liver-specific targeting (Fig. 7B) which was also documented by IHC staining of liver sections for AEG-1 (Fig. 7C). Treatment with PAMAM-AEG-1si resulted in profound inhibition of HFD-induced steatosis (detected by H&E and Oil Red O staining), inflammation (detected by F4/80 staining) and fibrosis (detected by Collagen I staining) compared to PAMAM-siCon (Fig. 7C and S5). Liver weight, serum liver enzymes, and hepatic TG and cholesterol levels were significantly less in PAMAM-AEG-1si group vs PAMAM-siCon group indicating efficacy of treatment and protection from HFD-induced liver damage (Fig. 7D). Compared to PAMAM-siCon, PAMAM-AEG-1si treatment resulted in significant increase in PPARα-target genes, and decrease in FASN protein levels and NF-κB activation as indicated by decreased p-p65 levels and Il6 mRNA (Fig. 7E–F). Thus targeting of AEG-1 in the liver might be a useful therapeutic intervention for NASH.
Figure 7.
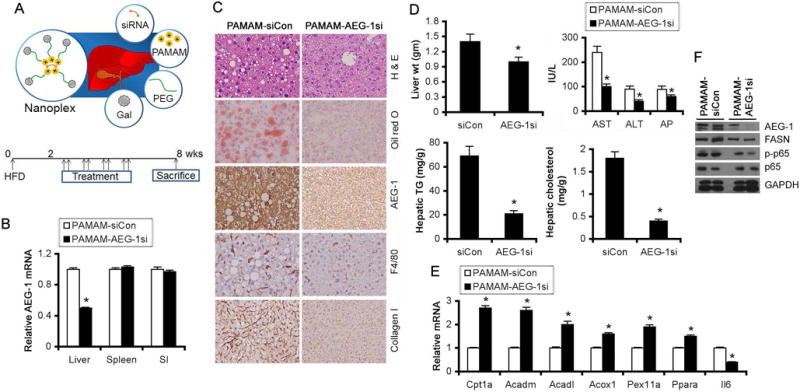
PAMAM-AEG-1si protects from HFD-induced steatosis. A, top. Strategy of targeting siRNA to the liver using PAMAM-PEG-Gal. A, bottom. Treatment protocol of WT C57BL/6 mice fed HFD (n = 10/group). B. AEG-1 mRNA expression by Q-RT-PCR at the end of the study in the indicated organs (normalized by GAPDH). C. H & E and oil red O staining and IHC for AEG-1, F4/80 and Collagen I of liver sections at the end of the study. D. Liver weight, serum liver enzymes, and hepatic TG and cholesterol levels at the end of the study. E. Expression analysis of the indicated mRNAs by Q-RT-PCR in the liver at the end of the study. F. Western blot analysis for the indicated proteins in livers of two independent mice per group at the end of the study. For graphs, data represent mean ± SEM. *: p<0.01.
Discussion
Our present studies reveal that AEG-1 contributes to both arms of the NASH-inducing mechanism, de novo lipogenesis and inflammation. We observe upregulation of AEG-1 in livers of mice chronically fed HFD as well as in NASH patients. Accordingly, there is a vicious cycle where HFD feeding induces AEG-1 and AEG-1 overexpression further augments the consequences of HFD-feeding resulting in aggravation and exacerbation of NASH development and progression. Inflammatory cytokines significantly induced AEG-1 mRNA expression in both mouse and human hepatocytes via NF-κB pathway. Thus, HFD-induced inflammatory events might be both the cause and consequence of AEG-1 overexpression.
We document three potential mechanisms that collaborate in the development of spontaneous NASH in Alb/AEG-1 mice: inhibition of PPARα function; translational regulation of FASN, and induction of inflammation (Fig. 6F). AEG-1 interacts with and inhibits RXR function (10). However, our current findings document that this inhibition of RXR by AEG-1 does not equally influence all RXR heterodimer partners. We observed significant inhibition of PPARα function affecting β-oxidation of fatty acids. However, LXR and FXR functions remained relatively unaffected. The mechanism underlying differential regulation of nuclear receptors by AEG-1 is a question that remains unclear. Co-activators contain multiple ‘LXXLL’ motifs (SRC-1 and PGC1α contain 3) with which they interact with both RXR and its heterodimer partner (30–33). AEG-1 contains only one ‘LXXLL’ motif and our initial yeast two-hybrid assay to identify AEG-1 interacting proteins identified only RXR, but not other nuclear receptors, to interact with AEG-1 (10). The ability of AEG-1 to interact with other nuclear receptors apart from RXR and any potential mechanisms for preferential binding to specific nuclear receptors are essential points that will require further investigation. While SRC-1 functions as a co-activator for a wide range of nuclear receptors, including PPAR and LXR, in the liver PGC1α serves as a co-activator mainly for PPAR (30, 33, 34). It needs to be checked whether AEG-1 exert preferential effect on the recruitment of SRC-1 or PGC1α on nuclear receptors. In addition to inhibition of PPARα/RXR transcriptional activity, a significant decrease in the levels of PPARα and its co-activators PGC1α and Lipin1 was observed in Alb/AEG-1 livers. Thus, there might be additional mechanisms, regulated by AEG-1, that contribute to inhibition of PPARα.
Although AEG-1 can directly activate NF-κB, inhibition of PPARα might also activate NF-κB by relieving the inhibitory effect of PPARα on NF-κB by transrepression (35). Thus AEG-1 effect on NASH mechanistically parallels that of PPARα inhibition. What differentiates AEG-1 is its ability to regulate de novo lipogenesis, the underlying molecular mechanism of this process is complex. We observed neither activation of SREBP-1 nor changes in mRNA of LXR-target genes, particularly Fasn and Scd1, key regulators of lipogenesis. However, we observed activation of SREBP-2 (Fig. 4B), which might induce lipogenic enzymes, such as Acaca, at the mRNA level (36). SREBP-2 promotes cholesterol synthesis and LDL uptake (37). Even though we did not see any difference in HMGcoR (Fig. 4B), a rate-limiting enzyme in cholesterol biosynthesis, at mRNA and protein levels, SREBP-2-mediated increased uptake might explain increased cholesterol, especially the marked increase in LDL, in Alb/AEG-1 mice. However, the molecular mechanism of the differential regulation of SREBP-2 by AEG-1 remains to be determined. Another important point to note is that even though there was robust activation of Akt in Alb/AEG-1 hepatocytes no change was observed for activation of mTOR (Fig. 4B), a master regulator of metabolism and translational control (38). We observed a marked increase in FASN and other lipogenic enzymes at the protein level in Alb/AEG-1 livers. We also document that AEG-1 overexpression facilitated association of Fasn mRNA with polysomes. Moreover, increase in ribosomal proteins (Fig. 4B) imply AEG-1-induced augmentation of translation and protein stability. These observations suggest that translational regulation might be an important mechanism by which AEG-1 promotes de novo lipogenesis. AEG-1 has already been ascribed as a potential RNA-binding protein (39). RNA immunoprecipitation followed by mass spectrometry identified Fasn mRNA to interact with AEG-1 in endometrial cancer cells although the consequence of this interaction was not further analyzed (39). It will be intriguing to further analyze the role of AEG-1 in regulating protein translation and ribosomal biogenesis.
A very important aspect of our study is the profound efficacy of PAMAM-AEG-1si to provide protection from HFD-induced steatosis. To our knowledge, this is the first demonstration that nanoparticle-mediated targeting of a specific gene provides therapeutic benefit in a NASH model. For liver diseases RNAi might be a useful strategy since i.v. injected payload will go directly to the liver thus ensuring high target organ delivery. A Phase I trial employing i.v. administration of lipid nanoparticle (LNP)-conjugated siRNA for vascular endothelial growth factor (VEGF) and kinesin spindle protein (KSP) in cancer patients showed complete regression of metastatic lesion in the liver (40). The success of this clinical trial provides an important precedence for the use of nanoparticles in the treatment of liver diseases and as such suggests that PAMAM-AEG-1si may be a plausible strategy to counteract NASH progression and needs to be further evaluated for potential translational application.
Supplementary Material
Acknowledgments
Financial support
Present study was supported in part by The National Cancer Institute Grant R01 CA138540 and R21 CA183954 and The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant 1R01DK107451-01A1 (DS). CLR is supported by a National Institute of Diabetes And Digestive And Kidney Diseases Grant T32DK007150. Services in support of this project were provided by the VCU Massey Cancer Center Transgenic/Knock-out Mouse Facility supported in part with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Abbreviations
- NASH
Nonalcoholic steatohepatitis
- AEG-1
Astrocyte elevated gene-1
- MTDH
Metadherin
- PPAR
Peroxisome Proliferator Activated Receptor
- FASN
Fatty acid synthase
- FFPE
Formalin-fixed paraffin-embedded
- AST
Aspartate aminotransferase
- ALT
Alanine aminotransferase
- Alk Phos
Alkaline phosphatase
- H & E
Hematoxylin and eosin
- IL-6
Interleukin-6
- VEGF
Vascular endothelial growth factor
- TG
Triglyceride
- HDL
High density lipoprotein
- LDL
Low density lipoprotein
- VLDL
Very low density lipoprotein
- FFA
Free fatty acids
- GTT
Glucose tolerance test
References
- 1.Satapathy SK, Sanyal AJ. Novel treatment modalities for nonalcoholic steatohepatitis. Trends Endocrinol Metab. 2010;21:668–675. doi: 10.1016/j.tem.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarkar D, Fisher PB. AEG-1/MTDH/LYRIC: Clinical Significance. Adv Cancer Res. 2013;120:39–74. doi: 10.1016/B978-0-12-401676-7.00002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emdad L, Sarkar D, Su ZZ, Randolph A, Boukerche H, Valerie K, Fisher PB. Activation of the nuclear factor kappaB pathway by astrocyte elevated gene-1: implications for tumor progression and metastasis. Cancer Res. 2006;66:1509–1516. doi: 10.1158/0008-5472.CAN-05-3029. [DOI] [PubMed] [Google Scholar]
- 6.Sarkar D, Park ES, Emdad L, Lee SG, Su ZZ, Fisher PB. Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008;68:1478–1484. doi: 10.1158/0008-5472.CAN-07-6164. [DOI] [PubMed] [Google Scholar]
- 7.Alexia C, Poalas K, Carvalho G, Zemirli N, Dwyer J, Dubois SM, Hatchi EM, et al. The endoplasmic reticulum acts as a platform for ubiquitylated components of nuclear factor kappaB signaling. Sci Signal. 2013;6:ra79. doi: 10.1126/scisignal.2004496. [DOI] [PubMed] [Google Scholar]
- 8.Krishnan RK, Nolte H, Sun T, Kaur H, Sreenivasan K, Looso M, Offermanns S, et al. Quantitative analysis of the TNF-alpha-induced phosphoproteome reveals AEG-1/MTDH/LYRIC as an IKKbeta substrate. Nat Commun. 2015;6:6658. doi: 10.1038/ncomms7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson CL, Srivastava J, Siddiq A, Gredler R, Emdad L, Rajasekaran D, Akiel M, et al. Genetic deletion of AEG-1 prevents hepatocarcinogenesis. Cancer Res. 2014;74:6184–6193. doi: 10.1158/0008-5472.CAN-14-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava J, Robertson CL, Rajasekaran D, Gredler R, Siddiq A, Emdad L, Mukhopadhyay ND, et al. AEG-1 Regulates Retinoid X Receptor and Inhibits Retinoid Signaling. Cancer Res. 2014;74:4364–4377. doi: 10.1158/0008-5472.CAN-14-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefebvre P, Benomar Y, Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab. 2010;21:676–683. doi: 10.1016/j.tem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53:1023–1034. doi: 10.1002/hep.24148. [DOI] [PubMed] [Google Scholar]
- 13.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem. 2000;275:28918–28928. doi: 10.1074/jbc.M910350199. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava J, Siddiq A, Emdad L, Santhekadur P, Chen D, Gredler R, Shen X-N, et al. Astrocyte elevated gene-1 (AEG-1) promotes hepatocarcinogenesis: novel insights from a mouse model. Hepatology. 2012;56:1782–1791. doi: 10.1002/hep.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 17.Waki K, Anno K, Ono T, Ide T, Chayama K, Tahara H. Establishment of functional telomerase immortalized human hepatocytes and a hepatic stellate cell line for telomere-targeting anticancer drug development. Cancer Sci. 2010;101:1678–1685. doi: 10.1111/j.1349-7006.2010.01576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, Mills AS, et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Invest. 2009;119:465–477. doi: 10.1172/JCI36460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia SK, Pannell L, et al. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocelllular carcinoma. Hepatology. 2011;53:1538–1548. doi: 10.1002/hep.24216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 21.Gruben N, Shiri-Sverdlov R, Koonen DP, Hofker MH. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta. 2014;1842:2329–2343. doi: 10.1016/j.bbadis.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr, et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huynh FK, Green MF, Koves TR, Hirschey MD. Measurement of Fatty Acid oxidation rates in animal tissues and cell lines. Methods Enzymol. 2014;542:391–405. doi: 10.1016/B978-0-12-416618-9.00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, et al. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem. 2002;277:11019–11025. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- 27.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoo BK, Chen D, Su Z-Z, Gredler R, Yoo J, Shah K, Fisher PB, et al. Molecular mechanism of chemoresistance by Astrocyte Elevated Gene-1 (AEG-1) Cancer Res. 2010;70:3249–3258. doi: 10.1158/0008-5472.CAN-09-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajasekaran D, Srivastava J, Ebeid K, Gredler R, Akiel M, Jariwala N, Robertson C, et al. Combination of nanoparticle-delivered siRNA for Astrocyte elevated gene-1 (AEG-1) and all-trans retinoic acid (ATRA): an effective therapeutic strategy for hepatocellular carcinoma (HCC) Bioconjug Chem. 2015;26:1651–1661. doi: 10.1021/acs.bioconjchem.5b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615–630. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rha GB, Wu G, Shoelson SE, Chi YI. Multiple binding modes between HNF4alpha and the LXXLL motifs of PGC-1alpha lead to full activation. J Biol Chem. 2009;284:35165–35176. doi: 10.1074/jbc.M109.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, et al. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12:3357–3368. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 35.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 36.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 39.Meng X, Zhu D, Yang S, Wang X, Xiong Z, Zhang Y, Brachova P, et al. Cytoplasmic Metadherin (MTDH) provides survival advantage under conditions of stress by acting as RNA-binding protein. J Biol Chem. 2012;287:4485–4491. doi: 10.1074/jbc.C111.291518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, Paz-Ares L, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3:406–417. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.