

Abstract
Cytomegalovirus (CMV) is a major cause of morbidity and mortality in solid-organ transplant recipients. Approximately 60% of adults are CMV seropositive indicating previous exposure. Following resolution of primary infection, CMV remains in a latent state. Reactivation is controlled by memory T cells in healthy individuals; transplant recipients have reduced memory T cell function due to chronic immunosuppressive therapies. In this study, CD8+ T cell responses to CMV polypeptides IE-1 and pp65 were analyzed in sixteen CMV seropositive renal and cardiac transplant recipients longitudinally pre- and post-transplant. All patients received standard of care maintenance immunosuppression, antiviral prophylaxis and CMV viral load monitoring, with approximately half receiving T cell depleting induction therapy. The frequency of CMV-responsive CD8+ T cells, defined by production of effector molecules in response to CMV peptides, increased during the course of a year post-transplant. The increase commenced after the completion of antiviral prophylaxis, and these T cells tended to be terminally differentiated effector cells. Based on this small cohort, these data suggest that even in the absence of disease, antigenic exposure may continually shape the CMV-responsive T cell population post-transplant.
Introduction
Immunosuppression places transplant recipients at an elevated risk of disease associated with viral infections that establish latency, such as CMV. Standard of care to protect recipients from CMV disease consists of frequent monitoring for viremia and treatment with antiviral agents, or antiviral prophylaxis for up to a year post-transplant (1–4). These therapeutic interventions have reduced the incidence of viremia and CMV disease (1, 4), but side effects including leukopenia complicate immunosuppressive management (3, 5). In cardiac and kidney transplant recipients, damage to the allograft contributes to decreased graft function and correlates with increased incidence of rejection (6, 7).
Resolution of primary CMV infection by NK cells, B cells, and T cells (8) is followed by long-term viral latency in CD34+ bone marrow cells and monocytes (8, 9). CMV reactivation is incompletely understood, but is known to occur when latently infected cells differentiate (8, 10). Viral spread is typically restricted by memory T cell responses (8). While reactivation does not lead to disease in healthy individuals, it does result in expansion of CMV-specific memory T cells with age, a phenomenon termed “memory inflation” (11, 12). As much as 40 percent of the CD8+ T cell repertoire may be CMV-reactive in older individuals (13–15).
CMV-specific T cells respond to a variety of CMV gene products by producing cytokines and cytolytic proteins. T cell responses form primarily to the polypeptides immediate early 1 (IE-1), pp50, and pp65 (16, 17), and responses to IE-1 and pp65 are the most thoroughly studied, as reviewed in reference (18). Studies in transplant recipients have largely relied on interferon γ (IFNγ) production to quantify T cell responses to CMV. Research on T cell responses to viral infections, including HIV, HCV, and EBV, indicates that production of multiple inflammatory cytokines and cytolytic proteins is important to viral control (19–21). Cells with multiple functions are called polyfunctional cells. In healthy CMV seropositive individuals, responses to CMV are maintained and the polyfunctional T cell population expands with time (11). Polyfunctionality is elevated in CMV-specific memory T cells, in contrast to HIV (22). Little is known about the role of polyfunctional CMV-responsive T cells in solid organ transplant recipients. T cell polyfunctionality could be more predictive of protection against CMV than production of IFNγ alone. Thus a better understanding of T cell responses in immunosuppressed transplant patients may improve clinical care and outcomes. In these studies, we analyzed polyfunctionality and specificity of CMV-responsive T cells in solid organ transplant recipients pre- and post-transplant.
Materials and Methods
Human subjects
Patients were enrolled at the University of Pennsylvania immediately prior to transplant. Blood samples were collected pre-transplant (day 0) and at intervals of 30, 90, 180, 270 and 360 days post-transplant. This study was subject to IRB approval at the University of Pennsylvania under protocol number 817637 and all patients gave informed consent before participating in the study. All identifiable information was blinded to those performing experiments. De-identified normal donor samples were obtained from the University of Pennsylvania Human Immunology Core and the Stanford Blood Center. All patients were screened for CMV serostatus.
The cohort consisted of renal and cardiac transplant recipients (Table 1). These populations were selected due to their similar immunosuppressive regimens and differing induction therapy. Standard of care immunosuppressive therapy was controlled by the treating physicians. The patients reported in this study are limited to recipients who were CMV seropositive at the time of transplant (Table 1), with a mix of seropositive and seronegative donors (Table 2). One seropositive recipient (#9) had documented CMV viremia two years prior to transplant and was undergoing immunomodulatory treatment for ulcerative colitis at the time of transplant; this subject was excluded from analysis. Another recipient, #17, was excluded from Figures 1–4 and S3–4 due to an episode of CMV viremia (Table S1). After the above exclusions, 16 transplant recipients remained. Day 30 samples were excluded due to low cell numbers.
Table 1.
| Transplant type | Induction | Immunosuppression | Recipient CMV serostatus | # of subjects |
|---|---|---|---|---|
| Renal | Rabbit anti-thymocyte globulin (rATG) | prednisone, mycophenolic acid and tacrolimus | positive | 10 |
| Cardiac | Steroid + basiliximab | prednisone, mycophenolic acid and tacrolimus | positive | 7 |
| Renal + cardiac | Steroid + basiliximab | prednisone, mycophenolic acid and tacrolimus | positive | 1 |
Table 2.
| Transplant | Graft rejection | Previous transplant | Donor CMV status | Recipient CMV status | Induction therapy | Valganciclovir duration | Subject # |
|---|---|---|---|---|---|---|---|
| Renal | No | Renal | − | + | rATG | 4 months | 1 |
| No | No | − | + | rATG | 1 month | 2 | |
| No | No | − | + | rATG | 4 months | 3 | |
| No | No | − | + | rATG | 3 months | 4 | |
| No | No | − | + | rATG | 3 months | 5 | |
| No | No | + | + | rATG | 3 months | 6 | |
| No | Liver | + | + | rATG | 3 months | 7 | |
| No | No | + | + | rATG | 5 months | 8 | |
| No | No | + | + | none | 6 months | 9 | |
| No | No | + | + | rATG | 4 months | 10 | |
| Cardiac | Yes | No | − | + | none | 3 months | 11 |
| No | No | − | + | basiliximab | 5 months | 12 | |
| No | No | − | + | basiliximab | 3 months | 13 | |
| No | concurrent renal | − | + | basiliximab | 5 months | 14 | |
| No | No | − | + | basiliximab | 4 months | 15 | |
| No | No | − | + | none | 3 months | 16 | |
| Yes | No | + | + | basiliximab | 4 months, then from month 7–8 | 17 | |
| No | No | + | + | basiliximab | 3 months | 18 |
Figure 1. Pre-transplant CD8+ T cell responses to CMV polypeptides vary.

A) Time course of blood collection and patient treatment. B) IE-1 stimulated PBMC gated for CD8+ T cells were stained for IFNγ, TNFα, and CD107a. Gates were defined based on an unstimulated control for that sample. Boolean gates were used to identify combinations of expression for the three functional markers. C) % polyfunctional of CD8+ T cells in response to IE-1 (black circles) and pp65 (white squares) in all recipients pre-transplant. Lines represent median and error bars represent interquartile range. n=16.
Figure 4. The post-transplant increase in CMV-responsive T cells is antigen-specific and independent of lymphodepletion.
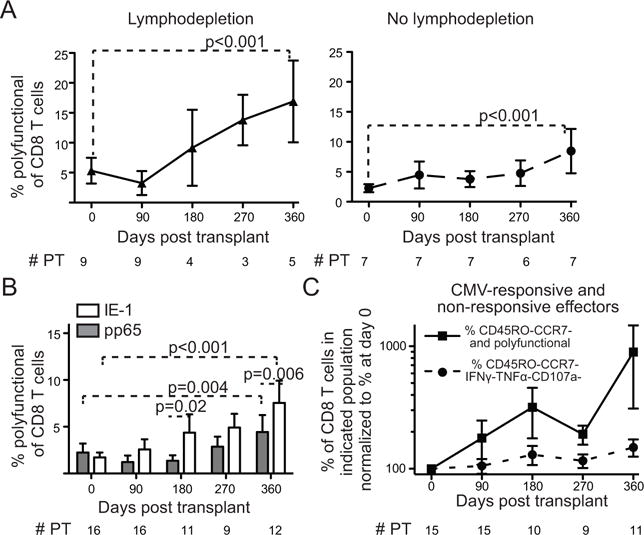
Percent of CD8+ T cells that are polyfunctional in response to IE-1 plus pp65 at each time point. A) Recipients divided between those who received lymphodepletion (left, triangles) and non-lymphodepleting induction (right, circles). B) Percent polyfunctional in response to IE-1 (open bars) or pp65 (gray bars). C) Percent of CD8+ T cells that are CMV-responsive (squares) or CMV non-responsive effectors (circles). For each population, the percentage is normalized to that determined at at day 0. Values represent means and error bars represent SEM. n = # PT as indicated. Trends and pairwise comparisons analyzed using mixed modeling on non-normalized data.
Blood collection and processing
Blood was collected in vacutainer tubes with EDTA (Ethylenediaminetetraacetic acid) as anticoagulant for isolation of peripheral blood mononuclear cells (PBMC) or clot activator for isolation of serum. All transplant recipient samples and a subset of normal donor samples were processed by the University of Pennsylvania Human Immunology Core. The remaining normal donor samples were processed by L.H. using the core protocol. PBMC were isolated from blood collected 1–24 hours previously using a Ficoll gradient and frozen at 5–20×106 cells/mL in FBS (Gemini) with 10% DMSO (Sigma) as previously described (23). Sera were collected by centrifugation at 1000 × g for 15 minutes, supernatant collection, and storage at −80°C.
Viral load monitoring
Viral load monitoring was performed as part of routine care in the University of Pennsylvania Clinical Microbiology laboratory or satellite locations using the COBAS AmpliPrep/COBAS TaqMan CMV Test (Roche), ARUP Laboratories CMV Quantitative PCR, LabCorp CMV Quantitative DNA PCR, or Quest CMV DNA quantitative RT PCR. The COBAS test targets the CMV UL54 DNA polymerase gene, and has a lower limit of 95% detection of 91 IU/mL EDTA plasma. The other tests have lower limits of 390 copies/mL, 200 IU/mL, and 200 IU/mL respectively. Testing was done based on clinical protocol or indication by the treating physician. Timing of viral load testing for each subject is detailed in Table S1. Additional testing on day 360 samples of subjects 1, 9, 13, and 14 was performed on stored sera in the Stanford Clinical Virology Lab using the artus CMV MDx Rotor-Gene kit (Qiagen) targeting the CMV major immediate early (MIE) gene. This test has a lower limit of 95% detection at ~100 IU/mL of serum.
Peptide libraries
Peptide libraries consisting of 15 amino acid peptides with 11 amino acid overlap for the lengths of the polypeptides IE-1 and pp65 were purchased from GenScript (Piscataway, NJ).
Cell stimulation
Cells for normal donor controls and an entire time course of transplant recipient samples were thawed in a single batch as previously described (23) and rested overnight in R10 (RPMI with 10% FBS, 1% Penicillin/Streptomycin (Thermo Fisher, Waltham, MA), and 1% L-glutamine (Thermo Fisher)) with 10 units/mL DNase (Roche Life Science, Indianapolis, IN). Rested cells were counted and re-suspended in 5 mL tubes at 2×106/mL in fresh R10 with 10 units/mL DNase and 3 μL/mL CD28/CD49d costimulatory reagent (BD Biosciences, San Jose, CA). Each sample was divided as follows: IE-1 library (0.8 μg/mL), pp65 library (3.2 μg/mL), PMA (50 ng/mL, Sigma) and ionomycin (1 μg/mL, Sigma), unstimulated. The number of cells per stimulation tube was 1 to 2×106. PMA/ionomycin was used as a positive control. Tubes were placed in an incubator at 37°C and 5% CO2 at an 80° angle. After 1 hour, brefeldin A (2 mg/mL, Life Technologies), monensin (BD Golgistop at 0.7 μL/mL), and antibody to human CD107a (BioLegend, San Diego, CA) were added. Stimulation continued 5–7 hours, and was stopped by addition of 3 mL cold phosphate buffered saline (PBS, Thermo Fisher). Cells were detached from tubes by incubation for 10 minutes in PBS with 2 mM EDTA (Thermo Fisher) at 37°C.
Stain for flow cytometry
Cells were first stained in an antibody to human c-c chemokine receptor (CCR)7 (BioLegend) diluted at 1:20 in PBS for 30 min at 37°C, then in Live/Dead aqua dye (Life Technologies, Carlsbad, CA) diluted at 1:400 in PBS for 20 min at RT. Cells were washed and spun down at 1250 RPM for 10 min, and re-suspended in surface stain prepared in Brilliant Stain Buffer (BD Biosciences) and incubated for 30 min at RT, then washed and spun down. Surface stain included an Fc receptor, CD16, for blocking. Cells were fixed using the BD Cytofix/cytoperm kit and protocol. Fixed cells were stained for 1 h at RT with intracellular antibodies diluted in 1X Perm/Wash buffer from the kit. Cells were washed and spun down, and re-suspended in PBS with 1% paraformaldehyde (Alfa Aesar, Haverhill, MA). Prior to running samples on a flow cytometer, they were re-suspended in PBS. Compensation controls were prepared using unstained and single stained cells or eBioscience Ultracomp eBeads (San Diego, CA).
Antibodies conjugated to fluorescein, phycoerythrin (PE) or four PE conjugates, allophycocyanin (APC) or APC conjugates, Alexa Fluor 700, or Brilliant Violet dyes 421, 570, 605, 650, 711 or 785 were specific for anti-human CD3 (HIT3a), CD4 (S3.5), CD8 (RPA-T8), CD14 (61D3), CD16 (3G8), CD19 (HIB19), CCR7 (G043H7), CD107a (H4A3), CD45RO (UCHL1), IFNγ (B27), and tumor necrosis factor α (TNFα, MAb11) and were purchased from BioLegend, eBioscience, BD, Life Technologies, Abcam (Cambridge, UK), or Beckman-Coulter (Pasadena, CA).
Flow cytometry and data analysis
Samples were analyzed on BD LSRII analyzers (Becton Dickinson, Franklin Lakes, NJ) configured for 18-color analysis at the University of Pennsylvania Flow Cytometry and Cell Sorting Resource Laboratory or the Stanford Shared FACS Facility. CS&T beads (BD Biosciences) were used to standardize analysis between instruments and from day to day.
Gating and data analysis
Flow cytometry data were analyzed using FlowJo version 8.8.7 or 9.9.3 (Treestar, Ashland, OR). Most gates were manual, but Boolean gates were used to measure polyfunctionality. Positive gates for IFNγ, TNFα, and CD107a were defined as shown in Fig. 1 and used as input for FlowJo to make combinatorial Boolean gates for all possible combinations of expression of the 3 markers for a total of 8 gates. Pie charts in Fig. S2 were created using the freely available software package SPICE (NIAID, Bethesda, MD and 24). All other graphs were made in GraphPad Prism 5 (La Jolla, CA).
Statistical analysis
Statistical analyses were completed using GraphPad Prism 5 and Stata Statistical Software version 13.1 (StataCorp, College Station, TX). Equality of variances was measured using Bartlett’s test in Prism (Fig. S3). Due to varying sample number at different time points, generalized linear mixed models assuming a Gaussian distribution and log link were used to analyze trends of frequency of total cells changing over time and identify differences between time points in STATA (Fig. 2–4, S4). 0.05 was the threshold for significance of p-value, with Bonferroni correction used to determine threshold of significance for multiple comparisons.
Figure 2. Polyfunctional CMV-responsive CD8+ T cells tend to be terminally differentiated effectors.
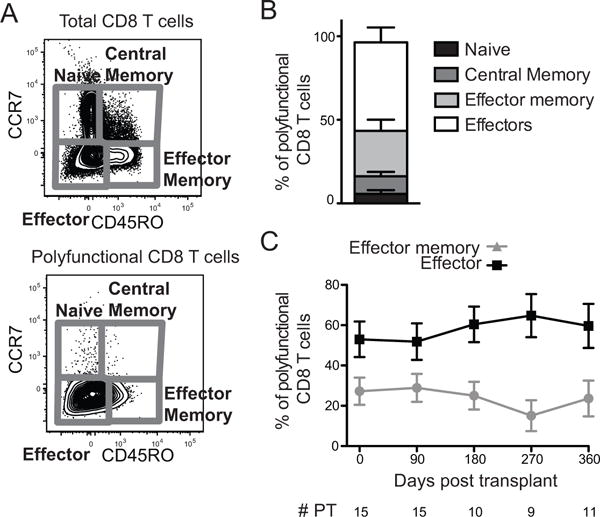
A) Total and polyfunctional CD8+ T cells were gated for CCR7 and CD45RO expression. B) Proportion of polyfunctional CD8+ T cells in each memory/effector subpopulation pre-transplant. Bars stack to a total of 100% of polyfunctional cells. n=15. C) Proportion of polyfunctional CD8+ T cells that are effector (CD45RO−CCR7−, black squares) or effector memory (CD45RO+CCR7−, gray triangles) phenotype in response to IE-1 or pp65. Trends analyzed using mixed-effects generalized linear modeling in STATA. There were no statistically significant differences. Lines represent means and error bars represent SEM. # PT represents the number of patients at each time point. Not all recipients had samples at all time points, resulting in variable n.
Results
Identification of polyfunctional CD8+ T cells
We first developed an in vitro assay using 16-parameter flow cytometry to identify and characterize polyfunctional CD8+ CMV-specific T lymphocytes (Fig. S1). Responses to IE-1 or pp65 (25) were analyzed in CD8+ T cells at the indicated time points pre- and post-transplant (Fig. 1A). Not all recipients had samples collected at every time point. For these studies, we define polyfunctionality as co-expression of two or three of IFNγ, TNFα, and CD107a (Fig. 1B). There was substantial patient-to-patient variation at day 0 in the frequency of polyfunctional IE-1 and pp65 responsive cells (Fig. 1C) and cells expressing any combination of functions (Fig. S2). To determine whether this was due to biological or technical variation, we thawed multiple samples from one normal donor frozen on two occasions. Variation from the mean frequency of polyfunctional cells was lower within the samples from this individual than for the group of all transplant recipients (Fig. S3). This difference in variance was statistically significant (p<0.05) suggesting that differences due to technical variation alone do not account for differences between patients and time points assessed.
Polyfunctional cells tend to be terminally differentiated effectors
In order to determine whether variation in polyfunctional CMV-responsive cells extended to the state of differentiation, we analyzed the memory/effector phenotype on the basis of CCR7 and CD45RO expression (Fig. 2A). Memory T cells can be divided into effector (CD45RO+CCR7−) and central (CD45RO+CCR7+) populations (26). Naïve cells are CCR7+CD45RO−. CCR7−CD45RO− cells comprise a heterogeneous effector population that includes the most terminally differentiated CD8+ T cells. The majority of polyfunctional cells are effectors and effector memory T cells (Fig. 2B). This phenotype was maintained post-transplant. The proportion of polyfunctional CD8+ T cells that were effector or effector memory cells did not change over the time course (Fig. 2C, p>0.05).
The population of CMV-responsive polyfunctional T cells increases in frequency of CD8+ T cells post-transplant
The frequency of CD8+ T cells that are polyfunctional in response to IE-1 and pp65 increased during the course of the year post-transplant (Fig. 3). This increase is statistically significant over the time course (p<0.001), and there was a statistically significant difference between day 0 and day 270 (p=0.02) as well as day 360 (p<0.001). There was no significant difference between any of the shorter time intervals evaluated. Monofunctional cells, in contrast, did not expand post-transplant (Fig. S4, all p>0.05). The increase in CD8+ CMV-responsive cells appears to occur after day 90, correlating with removal of anti-viral prophylaxis between 3 and 4 months in a majority of patients.
Figure 3. The frequency of CMV-responsive T cells increases post-transplant.
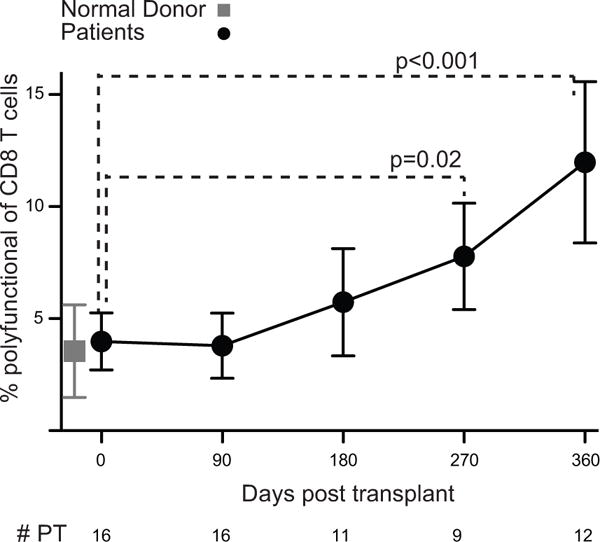
Percent of CD8+ T cells that are polyfunctional in response to IE-1 plus pp65 averaged in all transplant recipients (closed black circles) and normal donor controls (closed gray square). n= # PT as indicated, 7 normal donors. Values represent means and error bars represent SEM. Trends and pairwise comparisons were analyzed using mixed-effects modeling.
We reasoned that changes in abundance, functionality, or specificity of CMV-responsive cells could be due to viral exposure. All PCR testing for CMV DNAemia for these patients was negative except for one PCR for subject #17 (Table S1 and Figure S5). To further investigate evidence of subclinical reactivation, we tested day 360 sera from four recipients for CMV DNAemia. These samples were chosen to represent patients with a range of CMV-responsive cellularity changes. All samples tested negative. Together, these data suggest that the expansion may not require detectable CMV exposure in the blood.
We next investigated other potential etiologies of T cell expansion. Because lymphodepletion is known to lead to rapid re-expansion of surviving lymphocyte pools (27), we hypothesized that the expansion would be restricted to transplant recipients who had received lymphodepleting therapy. As expected, subgroup analysis of PBMC from patients who received rATG as induction therapy showed a significant increase in CMV-responsive CD8+ T cells over the time course (p<0.001) and between day 0 and day 360 (Fig. 4A, p<0.001). Interestingly, CMV-responsive cells also increased during the year following transplantation in those who received non-lymphodepleting induction with anti-IL2R blockade or steroids alone (Fig. 4A, p<0.001). In patients without lymphodepletion, the increase was statistically significant between day 0 with both day 90 (p=0.006) and day 360 (Fig. 4A, p<0.001). There was no statistically significant difference in the rates of CMV-responsive cell expansion among lymphodepleted and non-lymphodepleted recipients (p=0.23). Therefore, lymphodepletion is not necessary for increased relative abundance of CMV-responsive, polyfunctional cells post-transplant.
We next evaluated if the increase in CMV-responsive cells was due to a shift between specificity to IE-1 versus pp65. IE-1 responsive T cells increase in frequency to a greater extent than pp65- responsive T cells (Fig. 4B). Both trajectories were statistically significant over the time course (p<0.01), and there were statistically significant increases between day 0 and day 360 (p<0.01). For IE-1 responsive cells, there was also a statistically significant increase from day 0 to day 180 (p=0.02). The increase initiated by day 90 in IE-1 responsive cells, whereas the kinetics were relatively delayed for pp65-responsive cells. There was a statistically significant difference in the increase of IE-1- and pp65-responsive cells (p<0.0001), specifically at day 180 (p=0.02) and day 360 (p=0.006). These data suggest that there is a shift towards IE-1 specificity post-transplant but that both specificities are affected.
T cell differentiation state, in particular relative proportions of effector and naïve T cells, can be affected by immunosuppression following transplantation. There was no difference in total CD8+ T cells between day 0 and 360: 16.2+/−2.36 versus 18.0+/−2.80 percent of live PBMC (p=0.76). In contrast, the frequency of effector CD8+ T cells increased post-transplant (Fig. 4C). To address whether the increased frequency of polyfunctional CMV-responsive cells resulted from non-specific expansion of the CD8+ effector population, we measured the frequencies of CD8+ T cells that were CMV non-responsive (IFNγ-TNFα-CD107a-) and polyfunctional CMV responsive CD45RO−CCR7− effectors. For CD8+ effector cells that did not respond to IE-1 or pp65, there was no statistically significant increase (Fig. 4C, p=0.05). For CMV-responsive effectors, there were increases from day 0 to days 180 (p<0.001), 270 (p=0.002) and 360 (p<0.001). Comparison of the rate of increase of CMV non-responsive and CMV-responsive effectors showed no association between the trends (p=0.80). Therefore, the increase in effectors cannot fully account for the increase in CMV-responsive cells.
Discussion
In this study, we demonstrate that CMV-responsive CD8+ T cells expand post-transplant. CMV-responsive CD8+ T cells are effectors or effector memory T cells pre- and post-transplant. The frequency of CMV-responsive CD8+ T cells increases post-transplant in 16 patients who, based on standard of care monitoring, lack evidence of CMV viremia or disease. Therefore, overt infection may not be required for antigen-specific changes in immunity in the post-transplant environment.
This study has several major strengths that make it a valuable approach to analyzing T cell immunity to CMV in the context of transplantation. First, the majority of studies of T cell responses to CMV have focused on ELISpot or 4–6 color flow cytometry. The use of 16 color analysis in this study provides substantial information that allows us to concurrently analyze functionality and memory phenotype. Second, studies on responses to IE-1 and pp65 have typically focused on production of IFNγ, and our study extends this to polyfunctional T cells. Third, this study involves longitudinal analysis of changes post-transplant, and demonstrates that these findings apply to recipients of two different organs.
Interpretation of these studies is complicated by several limitations. The small number of patients and heterogeneity of the population complicate our ability to draw conclusions. Viral load monitoring in this study was determined by the treating physician and standard of care; in the absence of weekly viral load monitoring, we cannot formally conclude a lack of viral replication. In addition, these studies utilized T cell function rather than MHC class I tetramer staining. As such, we cannot identify CMV-specific T cells independent of their function. Previous studies indicated that >90% of CMV-specific T cells are also CMV-responsive (28, 29). Therefore we anticipate that our data are consistent with antigen specific populations, but staining with tetramers for a variety of epitopes of IE-1 and/or pp65 would be required to fully analyze this.
This work contributes to understanding of polyfunctionality in the context of CMV-responsive T cells in transplant recipients. Previous work on CMV in transplant recipients has generally focused on IFNγ+ T cells (30–33). Development of CMV disease several years post-transplant is associated with defective CD8+ T cell production of IFNγ and CD107a in response to CMV peptide (34). The presence of polyfunctional CMV-responsive CD8+ T cells after lung transplant correlates with protection from viremia (35). These studies focus on post-transplant polyfunctionality; our study builds on these results by measuring changes in polyfunctionality from pre- to post-transplant. In addition, our results allow comparison of CMV in transplant recipients to studies of polyfunctionality in healthy CMV seropositive individuals (11, 19) and in responses to other latent viral infections (19–21). Consistent with our findings in immunosuppressed transplant recipients, the majority of CMV responsive T cells are polyfunctional in healthy individuals (11, 19). In responses to both HIV and CMV, there is an inverse correlation between viral load and the proportion of the T cell response that is polyfunctional (19). This finding is consistent with antigen-specific expansion of CMV responsive T cells in the absence of viremia. Because only one of the patients in our cohort developed clinical viremia, we cannot conclude that polyfunctionality confers protection. However, in combination with the previously identified correlation between polyfunctional T cells and viral load, our data suggest that expansion of CMV-responsive T cells may protect transplant recipients from viremia.
The lack of detected viral reactivation suggests that controlled and/or subclinical reactivation may also be relevant to the T cell response. Transient CMV viremia controlled by prophylaxis or immunity has been documented (36). Heart, lung, and kidney transplant recipients with CMV viremia had an expansion of T cells producing IFNγ in response to CMV (37). In another study, asymptomatic CMV seropositive renal transplant recipients (6–732 days post-transplant) had a higher frequency of IFNγ+ pp65 responsive CD8+ T cells than seropositive healthy volunteers (38). Thus, IFNγ+ CMV-responsive CD8+ T cells can expand post-transplant in patients with or without detectable viremia. Our findings extend this observation to polyfunctional CD8+ T cells and begin to define the kinetics of this increase.
In contrast to the increase in CMV-responsive CD8+ T cells, only one of our cohort of transplant recipients had CMV viremia detected in standard of care testing or in our subsequent analysis of day 360 samples. There may have been viremia below the threshold of detection, asymptomatic viremia, or virus localized to tissues. Such viral replication could promote T cell expansion without being detectable in peripheral blood. Subclinical viremia may be more likely to occur in patients with seropositive allografts. However, numbers in this study were insufficient to draw conclusions about this possibility. In support of the hypothesis that exposure to virus is the cause, the expansion is first observed at day 180, which is after termination of antiviral prophylaxis in all subjects included in the statistical analyses. The recipient who experienced viremia (#17) had an expansion of polyfunctional cells that was either concurrent with or after their viremic episode. These data are consistent with virus-induced expansion, but we were not able to obtain appropriately timed samples in this patient to confirm this interpretation. Interestingly, in one recipient (#9) with a documented episode of viremia while on immunomodulatory treatment for non-transplant related disease prior to enrollment, approximately 30% of pre-transplant CD8+ T cells were responsive to CMV suggesting that a persistent expansion of CMV-responsive T cells resulted from prolonged exposure to high levels of viral replication.
Another factor suggesting controlled reactivation as the cause of T cell expansion is the differential kinetics of response to IE-1 and pp65 (Fig. 4B). IE-1 is a transcription factor expressed early in the CMV life cycle and required for expression of other CMV genes (39, 40), including pp65 (41). In contrast, pp65 is an important structural component of CMV virions (42), and expressed much later in the viral life cycle. T cell responses to IE-1 form in response to rapidly controlled reactivation that is otherwise undetectable, and can preclude expression of pp65 by reactivated virus (43). Responses to IE-1, and not pp65, have been shown to correlate with protection from CMV disease in heart and lung transplant recipients (30). Differential kinetics are consistent this finding, though our results do not address protection.
Analysis of viral load in healthy individuals demonstrated that CMV DNA is virtually undetectable in the majority of the healthy CMV seropositive population, with the exception of those over the age of 70 (44). Impaired control of CMV in the aged promotes elevated viral load. The over 70 cohort have expanded CMV-responsive T cell populations and elevated CMV-specific antibodies (44). These immune phenotypes are detectable at earlier ages, before viremia is detectable. Consistent findings in mice support this hypothesis: in mice infected with murine CMV defective in replication and spread, production of immunogenic proteins by cells infected during primary infection was sufficient to drive long-term expansion of memory T cells (10). These studies indicate that detectable viremia is not necessary for expansion of T cells in response to CMV.
An alternative potential trigger of the increase in CMV-responsive T cells in our study population is exposure to donor alloantigen. Published research demonstrates that CMV specific T cells can cross-react with alloantigen (45, 46). In addition, memory T cells are resistant to lymphodepletion (47, 48), which can lead to increased levels of alloreactive memory T cells (27). Therefore, a population of CMV-responsive T cells could contain alloreactive cells that expand in response to donor alloantigen. Rejection can be prevented by the immunosuppressive treatment in these recipients.
Another potential cause is non-specific homeostatic expansion. The frequency of CD8+ T cells in the blood is unchanged post-transplant. Therefore, homeostatic expansion does not enlarge the CD8+ T cell niche, and any changes are within the pre-existing niche. Specifically, immunosuppression and lymphodepletion can cause expansion of memory and effector T cells at the expense of other T cell populations (47, 48). We assessed the potential effects of lymphodepletion through comparison of CMV-responsive CD8+ T cells in transplant recipients with and without lymphodepleting induction therapy. The CMV-responsive T cell expansion occurred in both groups of patients. A previous study of T cell responses to CMV found no difference in IFNγ production by ELISpot over the course of a year post-transplant between renal transplant recipients with and without lymphodepleting induction (49). Our data are consistent with this study and extend the finding from IFNγ to polyfunctional CMV responsive T cells. In addition, CMV-responsive effector cells expand to a greater degree than CMV non-responsive effector cells (Figure 4C). If homeostatic proliferation were the cause, there would be no difference in expansion of these populations. In our view, antigen-specific proliferation is a more likely etiology than homeostatic proliferation, suggesting that these T cells are involved in ongoing immune responses.
Our results indicate that CMV seropositive recipients with asymptomatic or no viremia have functional CMV-responsive T cells both pre-transplant and a year post-transplant. Depending on the antigenic stimulus for the expansion, these cells may be protective (CMV-responsive) or pathogenic (alloantigen-responsive). Therefore, larger studies should explore whether this T cell phenotype correlates with protection from CMV. If so, then transplant recipients could be evaluated for T cell responses, and in some cases have immunosuppressive and antiviral doses reduced to allow T cells to control CMV post-transplant. Such therapeutic changes could contribute to improving CMV control and long-term allograft survival (50).
Supplementary Material
Percent of functional CD8+ T cells (positive for at least one of IFNγ, TNFα, CD107a) expressing each possible combination of functional markers in all transplant recipients at day 0. Pie charts of total functional CD8+ cells with each functional population denoted by a different color. Numbers are the same as those in Table 2. Subjects #9 and #17 excluded from other analyses for reasons discussed in the Materials and Methods.
PBMC from one normal donor were frozen in aliquots on two different days, and cells from these aliquots thawed and stimulated on eight separate days. Percent polyfunctional of CD8+ T cells divided by mean for fold difference for patients (PT) at day 0 and normal donor (ND). Bartlett’s test for equal variance was used to compare variance between PT and ND. Lines represent means and error bars represent SEM. n=16.
Percent of CD8+ T cells that are monofunctional in response to IE-1 plus pp65 averaged in all transplant recipients. Values presented separately for IFNγ (left), TNFα (middle), and CD107a (right). n= # PT as indicated. Values represent means and error bars represent SEM. Mixed model measuring differences across time course showed no statistically significant differences.
PBMC were stimulated for 6 hrs with IE-1 or pp65 peptide libraries, and stained for expression of the indicated markers. Cells were sequentially gated on lymphocytes, singlets, live, CD3+ DUMP−, and CD8+CD4−. DUMP consists of CD14, CD16, and CD19. Numbers represent % of cells within the adjacent gate.
Percent of CD8+ T cells that are polyfunctional in response to IE-1 plus pp65 averaged in all transplant recipients without detected viremia (gray circles) and in the viremic patient (#17) individually (red triangles). The viremic episode was seven months post-transplant and coincided with gastrointestinal symptoms. Viremic episode (red arrow) and valganciclovir treatment (blue lines) are indicated for subject 17. n=16 recipients. Values represent means and error bars represent SEM.
PCRs completed as indicated in the Materials and Methods. Of the 18 patients in this cohort (including subjects #9 and #17 excluded as discussed in Materials and Methods), 11 were monitored for CMV DNAemia between 1 and 14 times during the study as part of routine care. The other 7 patients had no CMV viral load testing as part of their post-transplant course and have “N/A” indicated under both viral load and result. If the result is listed as “negative,” there was no CMV above the threshold of detection. *references the one positive viral load result.
Acknowledgments
We thank Dr. Glenn Chertow for suggestions and critical reading of the manuscript, Dr. Holden Maecker for advice on experimental design, and Dr. Maria E. Montez-Rath for advice regarding statistical analyses. This research was supported by grants from the American Heart Association to JSM (13IRG13640042) and the National Institutes of Health to LEH (T32 DK007357-31, T32 AI07290-31).
Abbreviations
- APC
allophycocyanin
- CCR7
c-c chemokine receptor 7
- CMV
cytomegalovirus
- EDTA
Ethylenediaminetetraacetic acid
- IE-1
immediate early 1
- IFNγ
interferon gamma
- TNFα
tumor necrosis factor alpha
- HBSS
Hanks Balanced Salt Solution
- PBMC
Peripheral blood mononuclear cells
- PBS
phosphate buffered saline
- PE
phycoerythrin
- rATG
rabbit anti-thymocyte globulin
- RT
room temperature
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- 1.Khoury JA, Storch GA, Bohl DL, Schuessler RM, Torrence SM, Lockwood M, et al. Prophylactic versus preemptive oral valganciclovir for the management of cytomegalovirus infection in adult renal transplant recipients. Am J Transplant. 2006;6(9):2134–2143. doi: 10.1111/j.1600-6143.2006.01413.x. [DOI] [PubMed] [Google Scholar]
- 2.Preiksaitis JK, Brennan DC, Fishman J, Allen U. Canadian society of transplantation consensus workshop on cytomegalovirus management in solid organ transplantation final report. Am J Transplant. 2005;5(2):218–227. doi: 10.1111/j.1600-6143.2004.00692.x. [DOI] [PubMed] [Google Scholar]
- 3.Razonable RR, Humar A, ASTInfectiousDiseasesCommunityofPractice Cytomegalovirus in solid organ transplantation. Am J Transplant. 2013;13(Supplement 4):93–106. doi: 10.1111/ajt.12103. [DOI] [PubMed] [Google Scholar]
- 4.Kotton CN. CMV: prevention, diagnosis, and therapy. Am J Transplant. 2013;13(Supplement 3):24–40. doi: 10.1111/ajt.12006. [DOI] [PubMed] [Google Scholar]
- 5.Paya C, Humar A, Dominguez E, Washburn K, Blumberg E, Alexander B, et al. Efficacy and safety of valganciclovir vs. oral ganciclovir for prevention of cytomegalovirus disease in solid organ transplant recipients. Am J Transplant. 2004;4(4):611–620. doi: 10.1111/j.1600-6143.2004.00382.x. [DOI] [PubMed] [Google Scholar]
- 6.Fateh-Moghadam S, Bocksch W, Wessely R, Jager G, Hetzer R, Gawaz M. Cytomegalovirus infection status predicts progression of heart-transplant vasculopathy. Transplantation. 2003;76(10):1470–1474. doi: 10.1097/01.TP.0000090163.48433.48. [DOI] [PubMed] [Google Scholar]
- 7.Richardson WP, Colvin RB, Cheeseman SH, Tolkoff-Rubin N, Herrin JT, Cosimi AB, et al. Glomerulopathy associated with cytomegalovirus viremia in renal allografts. N Engl J Med. 1981;305(2):57–63. doi: 10.1056/NEJM198107093050201. [DOI] [PubMed] [Google Scholar]
- 8.Wills MR, Poole E, Lau B, Krishna B, Sinclair JH. The immunology of human cytomegalovirus latency: could latent infection be cleared by novel immunotherapeutic strategies? Cellular and Molecular Immunology. 2015;12(2):128–138. doi: 10.1038/cmi.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodrum FD, Jordan CT, High K, Shenk T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proceedings of the National Academy of Sciences. 2002;99(25):16255–16260. doi: 10.1073/pnas.252630899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snyder CM, Cho KS, Bonnett EL, Allan JE, Hill AB. Sustained CD8+ T cell memory inflation after infection with a single-cycle cytomegalovirus. PLoS Pathog. 2011;7(10):e1002295. doi: 10.1371/journal.ppat.1002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lachmann R, Bajwa M, Vita S, Smith H, Cheek E, Akbar A, et al. Polyfunctional T cells accumulate in large human Cytomegalovirus-specific T cell responses. J Virol. 2012;86(2):1001–1009. doi: 10.1128/JVI.00873-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, et al. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol. 2003;170(4):2022–2029. doi: 10.4049/jimmunol.170.4.2022. [DOI] [PubMed] [Google Scholar]
- 13.Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, et al. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002;169(4):1984–1992. doi: 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- 14.Crough T, Khanna R. Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev. 2009;22(1):76–98. doi: 10.1128/CMR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vescovini R, Biasini C, Fagnoni FF, Telera AR, Zanlari L, Pedrazzoni M, et al. Massive load of functional effector CD4+ and CD8+ T cells against cytomegalovirus in very old subjects. J Immunol. 2007;179(6):4283–4291. doi: 10.4049/jimmunol.179.6.4283. [DOI] [PubMed] [Google Scholar]
- 16.Khan N, Bruton R, Taylor GS, Cobbold M, Jones TR, Rickinson AB, et al. Identification of cytomegalovirus-specific cytotoxic T lymphocytes in vitro is greatly enhanced by the use of recombinant virus lacking the US2 to US11 region or modified vaccinia virus Ankara expressing individual viral genes. J Virol. 2005;79(5):2869–2879. doi: 10.1128/JVI.79.5.2869-2879.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gandhi MK, Khanna R. Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. The Lancet Infectious Diseases. 2004;4(12):725–738. doi: 10.1016/S1473-3099(04)01202-2. [DOI] [PubMed] [Google Scholar]
- 18.Jackson SE, Mason GM, Wills MR. Human cytomegalovirus immunity and immune evasion. Virus Res. 2011;157(2):151–160. doi: 10.1016/j.virusres.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 19.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nature Reviews Immunology. 2008;8(4):247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 20.Smith C, Beagley L, Khanna R. Acquisition of polyfunctionality by Epstein-Barr virus-specific CD8+ T cells correlates with increased resistance to galectin-1-mediated suppression. J Virol. 2009;83(12):6192–6198. doi: 10.1128/JVI.00239-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciuffreda D, Comte D, Cavassini M, Giostra E, Bühler L, Perruchoud M, et al. Polyfunctional HCV-specific T-cell responses are associated with effective control of HCV replication. Eur J Immunol. 2008;38(10):2665–2677. doi: 10.1002/eji.200838336. [DOI] [PubMed] [Google Scholar]
- 22.Riou C, Treurnicht F, Abrahams MR, Mlisana K, Liu MK, Goonetilleke N, et al. Increased memory differentiation is associated with decreased polyfunctionality for HIV but not for cytomegalovirus-specific CD8+ T cells. J Immunol. 2012;189(8):3838–3847. doi: 10.4049/jimmunol.1201488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higdon LE, Lee K, Tang Q, Maltzman JS. Virtual global transplant laboratory standard operating procedures for blood collection, PBMC isolation, and storage. Transplantation Direct. 2016;2(9):e101. doi: 10.1097/TXD.0000000000000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roederer M, Nozzi JL, Nason M. SPICE: Exploration and analysis of post-cytometric complex multivariate datasets. Cytometry Part A. 2011;79A(2):167–174. doi: 10.1002/cyto.a.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clari MA, Aguilar G, Benet I, Belda J, Giménez E, Bravo D, et al. Evaluation of cytomegalovirus (CMV)-specific T-cell immunity for the assessment of the risk of active CMV infection in non-immunosuppressed surgical and trauma intensive care unit patients. J Med Virol. 2013;85(10):1802–1810. doi: 10.1002/jmv.23621. [DOI] [PubMed] [Google Scholar]
- 26.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 27.Tchao NK, Turka LA. Lymphodepletion and homeostatic proliferation: implications for transplantation. Am J Transplant. 2012;12(5):1079–1090. doi: 10.1111/j.1600-6143.2012.04008.x. [DOI] [PubMed] [Google Scholar]
- 28.Maecker HT, Ghanekar SA, Suni MA, He X-S, Picker LJ, Maino VC. Factors affecting the efficiency of CD8+ T cell cross-priming with exogenous antigens. J Immunology. 2001;166:7268–7275. doi: 10.4049/jimmunol.166.12.7268. [DOI] [PubMed] [Google Scholar]
- 29.Maecker HT, Moon J, Bhatia S, Ghanekar SA, Maino VC, Payne JK, et al. Impact of cryopreservation on tetramer, cytokine flow cytometry, and ELISPOT. BMC Immunol. 2005;6:17. doi: 10.1186/1471-2172-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bunde T, Kirchner A, Hoffmeister B, Habedank D, Hetzer R, Cherepnev G, et al. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J Exp Med. 2005;201(7):1031–1036. doi: 10.1084/jem.20042384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar D, Chernenko S, Moussa G, Cobos I, Manuel O, Preiksaitis J, et al. Cell-mediated immunity to predict cytomegalovirus disease in high-risk solid organ transplant recipients. Am J Transplant. 2009;9(5):1214–1222. doi: 10.1111/j.1600-6143.2009.02618.x. [DOI] [PubMed] [Google Scholar]
- 32.Egli A, Binggeli S, Bodaghi S, Dumoulin A, Funk GA, Khanna N, et al. Cytomegalovirus and polyomavirus BK posttransplant. Nephrology Dialysis Transplantation. 2007;22(Supplement 8):viii72–viii82. doi: 10.1093/ndt/gfm648. [DOI] [PubMed] [Google Scholar]
- 33.Egli A, Binet I, Binggeli S, Jäger C, Dumoulin A, Schaub S, et al. Cytomegalovirus-specific T-cell responses and viral replication in kidney transplant recipients. J Transl Med. 2008;6:29. doi: 10.1186/1479-5876-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummins NW, Deziel PJ, Abraham RS, Razonable RR. Deficiency of cytomegalovirus (CMV)-specific CD8+ T cells in patients presenting with late-onsent CMV disease several years after transplantation. Transpl Infect Dis. 2008;11:20–27. doi: 10.1111/j.1399-3062.2008.00344.x. [DOI] [PubMed] [Google Scholar]
- 35.Snyder LD, Chan C, Kwon D, Yi J, Martissa JA, Copeland CAF, et al. Polyfunctional T-cell signatures to predict protection from cytomegalovirus after lung transplantation. Am J Respir Crit Care Med. 2016;193(1):78–85. doi: 10.1164/rccm.201504-0733OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humar A, Paya C, Pescovitz MD, Dominguez E, Washburn K, Blumberg E, et al. Clinical utility of cytomegalovirus viral load testing for predicting CMV disease in D+/R- solid organ transplant recipients. Am J Transplant. 2004;4(4):644–649. doi: 10.1111/j.1600-6143.2004.00391.x. [DOI] [PubMed] [Google Scholar]
- 37.Gerna G, Lilleri D, Fornara C, Comolli G, Lozza L, Campana C, et al. Monitoring of human cytomegalovirus-specific CD4+ and CD8+ T-cell immunity in patients receiving solid organ transplantation. Am J Transplant. 2006;2006(6):2356–2364. doi: 10.1111/j.1600-6143.2006.01488.x. [DOI] [PubMed] [Google Scholar]
- 38.Radha R, Jordan S, Puliyanda D, Bunnapradist S, Petrosyan A, Amet N, et al. Cellular immune responses to cytomegalovirus in renal transplant recipients. Am J Transplant. 2005;5:110–117. doi: 10.1111/j.1600-6143.2003.00647.x. [DOI] [PubMed] [Google Scholar]
- 39.Jean Beltran PM, Cristea IM. The life cycle and pathogenesis of human cytomegalovirus infection: lessons from proteomics. Expert Review of Proteomics. 2014;11(6):697–711. doi: 10.1586/14789450.2014.971116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stinksi MF, Thomsen DR, Stenberg RM, Goldstein LC. Organization and expression of the immediate early genes of human cytomegalovirus. J Virol. 1983;46(1):1–14. doi: 10.1128/jvi.46.1.1-14.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Depto A, Stenberg RM. Regulated expression of the human cytomegalovirus pp65 gene: octamer sequence in the promoter is required for activation by viral gene products. J Virol. 1989;63(3):1232–1238. doi: 10.1128/jvi.63.3.1232-1238.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abate DA, Watanabe S, Mocarski ES. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol. 2004;78(20):10995–11006. doi: 10.1128/JVI.78.20.10995-11006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan N, Best D, Bruton R, Nayak L, Rickinson AB, Moss PAH. T cell recognition patterns of immunodominant cytomegalovirus antigens in primary and persistent infection. J Immunology. 2007;178(7):4455–4465. doi: 10.4049/jimmunol.178.7.4455. [DOI] [PubMed] [Google Scholar]
- 44.Parry HM, Zuo J, Frumento G, Mirajkar N, Inman C, Edwards E, et al. Cytomegalovirus viral load within blood increases markedly in healthy people over the age of 70 years. Immun Ageing. 2016;13 doi: 10.1186/s12979-015-0056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heutinck KM, Yong S, Tonneijck L, van den Heuvel H, van der Weerd NC, van der Pant KA, et al. Virus-specific CD8+ T-cells cross-reactive to donor-alloantigen are transiently present in the circulation of kidney transplant recipients infected with CMV and/or EBV. Am J Transplant. 2016;16(5):1480–1491. doi: 10.1111/ajt.13618. [DOI] [PubMed] [Google Scholar]
- 46.Kaminski H, A FJ. The cell biology of cytomegalovirus: implications for transplantation. Am J Transplant. 2016;16(8):2254–2269. doi: 10.1111/ajt.13791. [DOI] [PubMed] [Google Scholar]
- 47.Rosenblum JM, Kirk AD. Recollective homeostasis and the immune consequences of peritransplant depletional induction therapy. Immunol Rev. 2014;258(1):167–182. doi: 10.1111/imr.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zwang NA, Turka LA. Homeostatic expansion as a barrier to lymphocyte depletion strategies. Current Opinion in Organ Transplantation. 2014;19(4):357–362. doi: 10.1097/MOT.0000000000000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abate D, Saldan A, Fiscon M, Cofano S, Paciolla A, Furian L, et al. Evaluation of cytomegalovirus (CMV)-specific T cell immune reconstitution revealed that baseline antiviral immunity, prophylaxis, or preemptive therapy but not antithymocyte globulin treatment contribute to CMV-specific T cell reconstitution in kidney transplant recipients. J Infect Dis. 2010;202(4):585–594. doi: 10.1086/654931. [DOI] [PubMed] [Google Scholar]
- 50.OPTN & SRTR annual data 2010. Am J Transplant. 2012;12(Supplement s1) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Percent of functional CD8+ T cells (positive for at least one of IFNγ, TNFα, CD107a) expressing each possible combination of functional markers in all transplant recipients at day 0. Pie charts of total functional CD8+ cells with each functional population denoted by a different color. Numbers are the same as those in Table 2. Subjects #9 and #17 excluded from other analyses for reasons discussed in the Materials and Methods.
PBMC from one normal donor were frozen in aliquots on two different days, and cells from these aliquots thawed and stimulated on eight separate days. Percent polyfunctional of CD8+ T cells divided by mean for fold difference for patients (PT) at day 0 and normal donor (ND). Bartlett’s test for equal variance was used to compare variance between PT and ND. Lines represent means and error bars represent SEM. n=16.
Percent of CD8+ T cells that are monofunctional in response to IE-1 plus pp65 averaged in all transplant recipients. Values presented separately for IFNγ (left), TNFα (middle), and CD107a (right). n= # PT as indicated. Values represent means and error bars represent SEM. Mixed model measuring differences across time course showed no statistically significant differences.
PBMC were stimulated for 6 hrs with IE-1 or pp65 peptide libraries, and stained for expression of the indicated markers. Cells were sequentially gated on lymphocytes, singlets, live, CD3+ DUMP−, and CD8+CD4−. DUMP consists of CD14, CD16, and CD19. Numbers represent % of cells within the adjacent gate.
Percent of CD8+ T cells that are polyfunctional in response to IE-1 plus pp65 averaged in all transplant recipients without detected viremia (gray circles) and in the viremic patient (#17) individually (red triangles). The viremic episode was seven months post-transplant and coincided with gastrointestinal symptoms. Viremic episode (red arrow) and valganciclovir treatment (blue lines) are indicated for subject 17. n=16 recipients. Values represent means and error bars represent SEM.
PCRs completed as indicated in the Materials and Methods. Of the 18 patients in this cohort (including subjects #9 and #17 excluded as discussed in Materials and Methods), 11 were monitored for CMV DNAemia between 1 and 14 times during the study as part of routine care. The other 7 patients had no CMV viral load testing as part of their post-transplant course and have “N/A” indicated under both viral load and result. If the result is listed as “negative,” there was no CMV above the threshold of detection. *references the one positive viral load result.