

Abstract
We performed orthogonal technology comparisons of concurrent peripheral blood and biopsy tissue samples from 69 kidney transplant recipients, who underwent a comprehensive algorithm-driven clinical phenotyping. The sample cohort included patients with normal protocol biopsies and stable transplant function (TX, n=25), subclinical acute rejection (subAR, n=23), and clinical acute rejection (cAR, n=21). Comparisons between microarray and RNA sequencing (RNA-seq) signatures were performed, demonstrating a strong correlation between the blood and tissue compartments for both technology platforms. A number of shared differentially expressed genes and pathways between subAR and cAR in both platforms strongly suggest that these two clinical phenotypes form a continuum of alloimmune activation. SubAR is associated with fewer or less expressed genes than cAR in blood, whereas in biopsy tissues, this clinical phenotype demonstrates a more robust molecular signature for both platforms. The discovery work done in this study confirms a clear ability to detect gene expression profiles for TX, subAR and cAR in both blood and biopsy tissue, yielding equivalent predictive performance, that is agnostic to either technology or platform. Our data also provide strong biologic insights into the molecular mechanisms underlying these signatures, underscoring their logistical potential as molecular diagnostics to improve clinical outcomes following kidney transplantation.
Introduction
The survival benefits of solid organ transplants in the United States are well documented (1), but little impact on long-term kidney graft survival despite significant improvements in short-term outcomes (2–4). In the OPTN/SRTR 2013 Annual Data Report, kidney graft failure rates for deceased donor transplants were 2.7% at 6 months, 3.5% at 1 year for transplants in 2012–2013, 11.8% at 3 years for transplants in 2010–2011, 23.8% at 5 years for transplants in 2008–2009, and 49.4% at 10 years for transplants in 2002–2003 (5). Moreover, after the first year, the accumulation of late acute rejection episodes doubles the total incidence from 11–15% to 25% by 5 years (5).
There is a pressing clinical need for more sensitive and specific objective surrogate markers to predict allo-immune graft injury to potentially inform management and improve outcomes (6, 7). Serum creatinine is an insensitive and nonspecific marker of allo-immune kidney injury (8). While over-immunosuppression increases multiple drug-related risks (9), under-immunosuppression increases risks of clinical acute rejection (cAR), potentially actionable subclinical acute rejection (subAR) defined by histologically-determined acute rejection in the absence of renal dysfunction, and ultimately chronic rejection (CR), the leading immunological cause of long term graft loss (10–12). A molecular signature to detect subAR would substantially improve our ability to monitor patients following transplantation, to intervene early at a stage of immune-mediated rejection with minimal tissue injury also monitoring response to this intervention, potentially reducing the development of interstitial fibrosis and tubular atrophy (IFTA). The development of IFTA leads to lower GFR (13) and graft failure (14). IFTA can develop surprisingly early post-transplant with approximately 15% incidence at three months to 30% at one year and 40% at two years (15–18). Studies revealed that treatment of subAR was beneficial and led to improved short (12) and long-term renal function (19), although no randomized clinical trial has tested this premise, primarily because molecular tests that can detect subAR are only now emerging. Consistent with our view of the importance of maintaining effective immunosuppression we recently mapped gene expression signatures from histologically and clinically phenotyped kidney biopsies (20). Biopsies of cAR and CR shared > 85% of known immune/inflammatory pathways validated in the literature for cell-mediated rejection revealing an arc of immune-mediated transplant injury. In parallel, subAR, found only by doing protocol biopsies is associated with the development of chronic injury/IFTA and worse graft function and survival (3, 10, 13, 16–18, 21–26).
We have previously demonstrated, using microarray analyses, that peripheral blood whole genome expression profiling in kidney transplant recipients can accurately distinguish patients with stable function and normal histology (TX), clinical and histologic cAR, and acute dysfunction with no rejection (ADNR) (27). Affymetrix DNA microarrays represent an established, FDA-approved diagnostic testing technology. Costs for DNA microarrays have dropped significantly over the past decade and with improved workflows and analytical tools are now comparable to other methods used routinely in commercial diagnostic laboratories. In parallel, rapid advances in next generation sequencing (NGS) technologies provide a cost-effective high throughput RNA sequencing (RNA-seq) approach and a clear potential to enable even lower cost gene expression profiling and faster workflows than can be obtained today with DNA microarrays (28, 29). Two of the most widely used NGS platforms; Illumina and IonTorrent have FDA-approved clinical diagnostic workflows.
As molecular technology platforms rapidly evolve, concerns have been raised that the ability to detect diagnostic and predictive gene signatures, discovered and validated on one technology or platform may not necessarily carry over to others. This concern is particularly relevant as molecular biomarkers begin their journey towards becoming commercial, especially given differences between technologies in logistics such as cost, turnaround time, point of service, and the use of commercially available kits. There is a paucity of technical studies comparing DNA microarray and RNASeq technologies, and even fewer studies comparing their relative capabilities as diagnostic tools. Technical studies comparing gene expression detection by microarrays and NGS technologies have shown good correlations (30–33). A recent study showed that microarray-based models and RNA-seq performed similarly in clinical endpoint prediction in ~ 500 biopsies of primary human neuroblastomas (34). To our knowledge, there are no studies comparing these orthogonal technologies in biopsy-documented, precisely phenotyped kidney transplant recipients.
We hypothesized that the ability to detect molecular signatures of cAR and subAR in both peripheral blood and biopsy tissue would be agnostic to the technology platform used to assess gene expression. To test this hypothesis, we profiled peripheral blood and biopsy tissue derived from 69 precisely phenotyped kidney transplant recipients (TX=25, subAR=23, cAR=21) selected from Northwestern University’s Comprehensive Transplant Center’s Biorepository. After removing technical outliers (GAPDH ratios >4 for microarrays and RNA-seq samples with total reads < 1 million), gene expression profiles were obtained by Affymetrix DNA microarray (peripheral blood n=69, biopsies n=65; total n=134). In parallel, RNA-seq was done on the biopsies of the same patient cohort using the Ion Torrent Proton sequencing platform (n=65) and the matching peripheral blood on the Illumina NextSeq platform (n=45). Thus, a total of 244 global gene expression profiles were obtained on three different commercial platforms and their approved workflows. Our data support the ability to detect signatures of TX, subAR and cAR in both blood and tissue compartments, yielding equivalent predictive performance, agnostic to the technology or platform used.
Materials and Methods
Patients and Samples
The Northwestern University Comprehensive Transplant Center houses a large repository of samples (NU biorepository) from transplant recipients. Kidney transplant recipients at NU undergo surveillance biopsies at 3, 12 and 24 months post-transplantation or for-cause biopsies in response to renal dysfunction. All patients who undergo biopsies are approached to provide informed consent (NU IRB #STU00025946) to enroll in our biorepository. In addition to blood samples (2.5ml PAXgene tube), a biopsy core was obtained and stored in RNAlater (Thermo Fisher, Waltham, MA). Biopsy slides were read by local pathologists, and also by a central pathologist in a blinded fashion using Banff 2007 criteria (35), and all pathology slides are digitally archived (Aperio Digital Scanner, Buffalo Grove, IL). The repository has the stored clinical and laboratory data for all patients. All samples for gene expression were derived from recipients who had clinical and laboratory data available, as well as a histologic assessment of their biopsies. All patients who participated in this study were >18 years of age, and recipients of a primary or subsequent kidney transplant alone. Recipients of multi-organ or prior non-renal transplants and patients with HIV were excluded. Standard immunosuppression at Northwestern consists of alemtuzumab induction with tacrolimus and mycophenolate maintenance (prednisone-free).
Definitions and algorithm used for precision clinical phenotyping of study subjects
Stable renal function: serum creatinine <2.3 mg/dL and <20% variability in the last 3 measurements. Acute renal dysfunction: >20% increase in serum creatinine compared to the previous 3 measurements. Normal histology: no rejection (no T-cell mediated or antibody mediated rejection of borderline or higher) or other abnormal histology (Banff fibrosis score ci=0 or 1 and ct=0 or 1, and both i and t scores =0) on a surveillance biopsy from a patient with stable renal function. Central reads were used for the clinical phenotyping algorithm. TX (n=25): 1) surveillance biopsy, 2) stable renal function, and 3) normal histology; subAR (n=23): 1) surveillance biopsy, 2) stable renal function, and 3) histology showing rejection (16 borderline and 7 Banff grade 1A); cAR (n=21): 1) for-cause biopsy prompted by 2) acute renal dysfunction, and 3) histology showing acute rejection (7 borderline, 6 Banff 1A, 6 1B, 1 2A, 1 2B - 5/21 (23%) has positive C4d staining). All clinical phenotypes were reviewed and blinded by JJF and MMA prior to submitting samples for molecular studies.
Gene expression profiling and statistical analysis
RNA was extracted using the PAXgene Blood RNA system (Qiagen, Valencia, CA) and Ambion GLOBINclear (Life Technologies, Carlsbad, CA). Regardless of technology used, total RNA from biopsies was extracted using the AllPrep DNA\RNA|Protein extraction kits (Qiagen). Ribosomal clearance was performed on all samples with the GeneRead rRNA Depletion Kit (Qiagen).
DNA Microarrays
Affymetrix Human Genome U133 Plus 2.0 GeneChips (plate format) were used for gene expression in blood and biopsies. Biotinylated cRNA was prepared with Ambion MessageAmp Biotin II kit (Ambion) and all samples run on the Affymetrix GeneTitan MC instrument. The primary transcript detection method is hybridization and detection of fluorescently labeled cRNA to custom 23mer DNA probes.
Ion Torrent Sequencing
RNA-seq of biopsies was done using the Ion Torrent Proton II (Thermo Fisher). Briefly, 50ng RNA was used to generate sequencing libraries. RNA concentration was assessed using the Qubit RNA BR Assay kit (Bio-Rad, Hercules, CA). Sequencing libraries were generated using the Ion AmpliSeq™ Transcriptome Human Gene Expression Kit (Thermo Fisher), and barcoded using Ion Express barcodes. cDNA library quality was assessed using the Agilent® 2200 TapeStation System and the High Sensitivity D1000 ScreenTape System (Agilent Technologies, Santa Clara, CA). Templates were run on the Ion PI™ v3 chips using the Ion Chef system and Hi-Q™ Chef kits. Sequencing depth per sample was 12 million genome-aligned reads. The primary transcript detection strategy depends on using Ion Torrents custom designed primers targeting multiple exons for all known protein coding genes.
Illumina Sequencing
All blood samples for RNA-seq were profiled using an Illumina NextSeq instrument (Illumina Inc. San Diego, CA). Total RNA was converted to cDNA using Ovation RNA-seq kits (NuGEN, San Carlos, CA) and S1 endonuclease digestion (Promega, Madison, WI) (36). Digested cDNA libraries were end-repaired, A-tailed and indexed adapter ligated. Ligation product was purified on Agencourt AMPure XP beads (Beckman Coulter Genomics, Carlsbad, CA) followed by 2% agarose size selection. Purified product was amplified for 15 PCR cycles and size selection repeated. Libraries were assessed on an Agilent Bioanalyzer and quantitated by Quant-iT ds DNA BR Assay kits (Invitrogen) and a Qubit Fluorimeter (Invitrogen). Cluster generation and 100bp single-end read sequencing was done following manufacturer’s instructions. Sequencing depth per sample was 15 million genome-aligned reads. The primary transcript detection strategy is capture of the cDNAs clusters on a plastic surface followed by brief PCR amplification and incorporation of fluorescently tagged dNTPs guided by transcript sequences. All expression data have been deposited to the NIH GEO repository.
Statistical methodologies
Microarray signals were normalized with frozen robust multi-array analysis (fRMA)(37). Predictions were done using the SVM algorithm implemented in R using the caret package (38). Diagnostic performance was based on retrospective prediction of known clinical phenotypes. Predictive accuracy was calculated using the formula (TP+TN)/(TP+FP+FN+TN); (TP–True Positive, TN–True Negative, FP–False Positive, FN–False Negative). Diagnostic metrics included sensitivity, specificity, and area under the curve (AUC). Clinical study parameters were tested by multivariate logistic regression with an adjusted (Wald test) p-value and false discovery rate (FDR) calculation. Results were classified at three levels of statistical confidence: False discovery rate (FDR) <10%, p value <0.005, and <0.05. Pathways were mapped using Ingenuity Pathway Analysis (IPA; Qiagen).
Results
Clinical Phenotypes and other patient characteristics
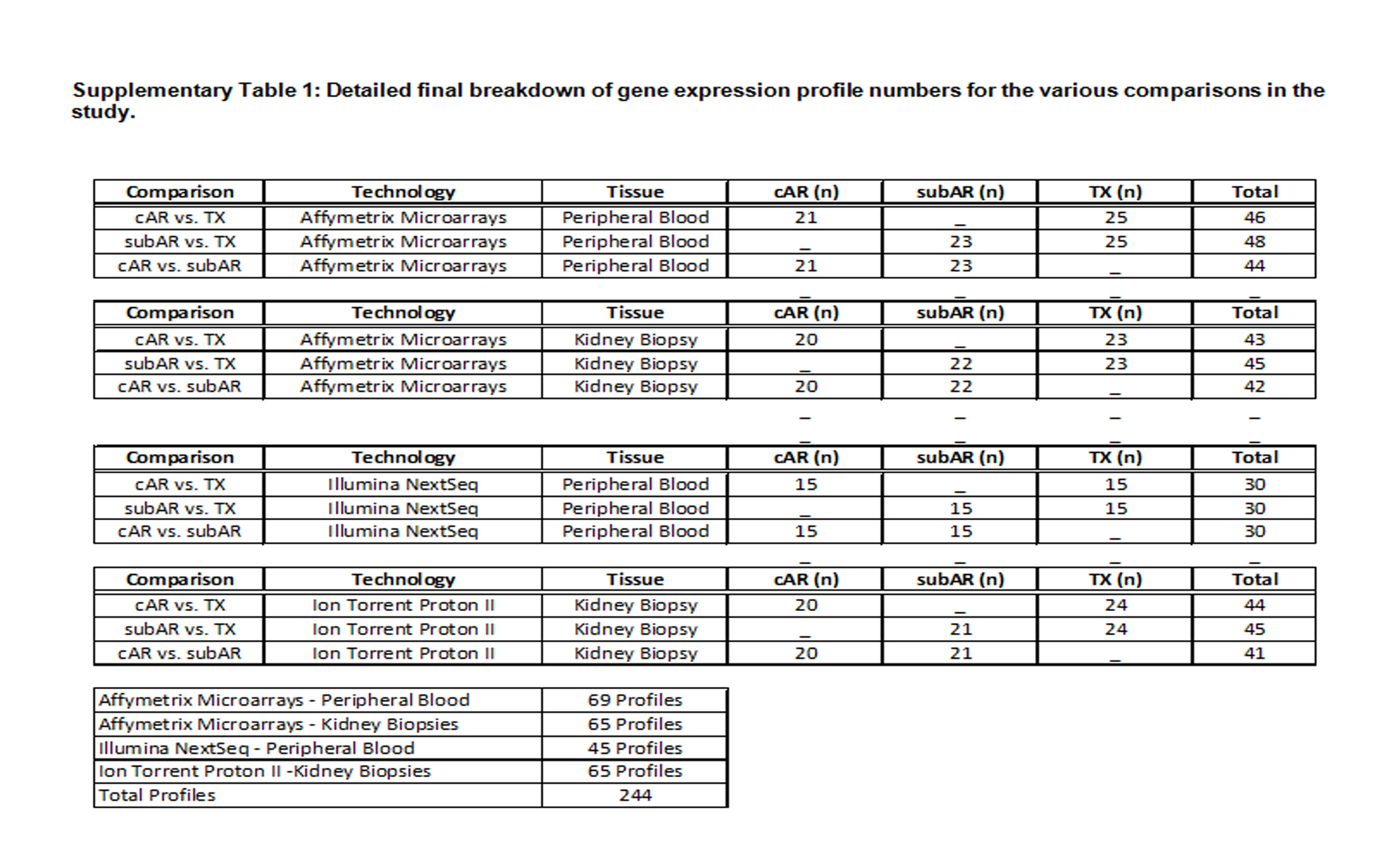
Peripheral blood and biopsy tissue samples were analyzed for gene expression from 69 kidney transplant recipients enrolled at the time of biopsy into the NU biorepository, who had undergone algorithm-driven precise clinical phenotyping (TX, n=25; subAR, n=23; cAR, n=21). The mean age was 49.3 years (range 22–71 years); 35% were female; 52% were deceased donor recipients. Blood was collected at the time of biopsy in all cases. The mean number of days post-transplant to the time of biopsy was 288 days (range 8–748) for subAR, 268 days (range 84–2228) for TX, and 921 days (range 15–2876) for cAR. All clinical characteristics of the study population are given in Table 1. Given various technical sample issues, the final breakdowns of samples used in this study are in Table S1.
Table 1.
Clinical characteristics of all study subjects
| Phenotype | AR | subAR | TX | Significance |
|---|---|---|---|---|
| Sample (n) | 21 | 23 | 25 | NS |
| Recipient Age (±SD) | 46.3 ± 12.6 | 47.3 ± 13.3 | 53.8 ± 8.3 | NS |
| Recipient Sex (Female %) | 23.8 | 34.7 | 44.0 | NS |
| Recipient Ethnicity (AA %) | 14.2 | 26.1 | 32 | NS |
| Time to Biopsy (Days) | 921.2 ± 936.5 | 287.6 ± 170.2 | 267.2 ± 426.1 | AR vs. TX - p = 0.01 subAR vs. TX - p = 0.01 |
| Donor Age (±SD) | 40.4 ± 14.5 | 40.01 ± 13.4 | 41.1 ± 13.7 | NS |
| Donor Sex (Female %) | 10.0 | 10.0 | 15.0 | NS |
| Donor Ethnicity (AA %) | 4.0 | 6.0 | 6.0 | NS |
| HLA Mismatches | 4.1 ± 1.6 | 3.8 ± 1.9 | 3.9 ± 1.7 | NS |
| PRA (> 20%) | 7.0 | 12.0 | 14.0 | NS |
| Donor Type (Deceased Donor %) | 9.0 | 9.0 | 10.0 | NS |
| Pre-Tx Diabetes (Type II %) | 3.0 | 6.0 | 5.0 | NS |
| DGF (%) | 4.0 | 6.0 | 6.0 | NS |
| Induction (alemtuzumab %) | 89.4 | 91.3 | 92.1 | NS |
| Immunosuppression (CNI %) | 90.4 | 95.6 | 96.0 | NS |
| Creatinine (±SD) | 3.4 ± 2.1 | 1.5 ± 0.5 | 1.4 ± 0.4 | AR vs. subAR p = 0.0003 subAR vs. TX p = 0.0005 |
| Steroids (%) | 19 | 30 | 32 | NS |
| C4d Staining positive (%) | 23 | 8.0 | 8.6 | NS |
SD - Standard Deviation; AA - African American; PRA - Panel Reactive Antibodies; DGF - Delayed Graft Function
Classifiers and predictive performance
Analyses of two-class (phenotype) comparisons were performed at three levels of statistical confidence: 1) stringent False Discovery Rate (FDR) <10%, 2) conservative p value <0.005, and 3) relaxed p value <0.05. Less stringent statistical limits are justified for smaller data sets for early stage discovery studies such as ours (69 subjects; 244 total gene expression profiles). Table 2 shows the differentially expressed genes in blood and biopsies comparing transcript detection by microarrays and RNA-seq. Statistically significant numbers of differentially expressed genes are observed between cAR vs. TX and subAR vs. cAR in both blood and biopsies. However, fewer differentially expressed genes were observed for the subAR vs. TX in the peripheral blood by either microarrays or RNA-seq. In contrast significant and equivalent numbers of genes are detected for the subAR vs. TX in the biopsies.
Table 2.
Differentially expressed in blood and biopsy by microarrays and RNA-seq using statistical confidence levels of FDR<10% and p<0.005 and p< 0.05
| Microarray Blood | DE Genes FDR<10% | p<0.005 | p<0.05 |
|---|---|---|---|
| cAR vs. TX | 1326 | 855 | 2286 |
| subAR vs. TX | 0 | 33 | 720 |
| Microarray Biopsy | DE Genes FDR<10% | p<0.005 | p<0.05 |
| cAR vs. TX | 7529 | 5192 | 7377 |
| subAR vs. TX | 804 | 796 | 2932 |
| RNA-seq Blood | DE Genes FDR<10% | p<0.005 | p<0.05 |
| cAR vs. TX | 1198 | 832 | 2566 |
| subAR vs. TX | 3 | 294 | 1647 |
| NGS Biopsy | DE Genes FDR<10% | p<0.005 | p<0.05 |
| cAR vs. TX | 8481 | 5316 | 8921 |
| subAR vs. TX | 604 | 749 | 2564 |
cAR, clinical acute rejection; DE, differential expression; FDR, false discovery rate; RNA-seq, RNA sequencing; subAR, subclinical acute rejection; TX, transplant excellence.
To measure the correlation of differential expression, blood vs. biopsies and microarrays vs. RNA-seq, M (log ratios) and A (mean/average) (MA) scale plots were created (Figures 1a-b). Principal Components Analysis (PCA) (Figure 2) shows that the differentially expressed genes in the biopsies, by virtue of their larger numbers and robust statistical significances, separate phenotypes more efficiently than blood for both platforms.
Figure 1.

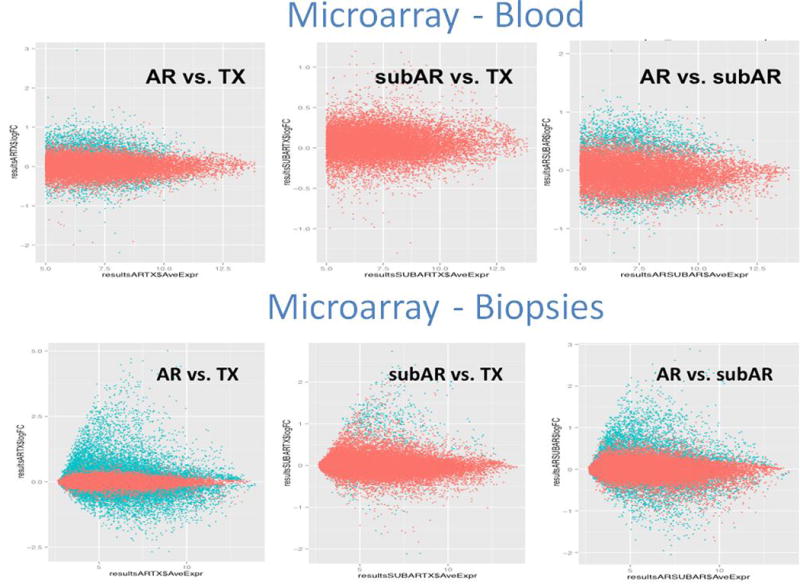
Correlation between the blood findings and the biopsy findings comparing the two analytical methodologies (microarrays and RNA-seq). The similarity between the technologies was also reflected in the consistently higher number of differentially expressed genes in the biopsy compared to the blood. The M (log ratios) and A (average) scale (MA) plots for all comparisons are shown in Figures 1a for RNAseq (NGS) and b for the microarrays.
Figure 2.

Supervised Principal Components Analysis (PCA) plots for the two tissues and technologies showing the separation of the phenotypes using the maximum set classifiers.
Next the performance of optimal classifiers based on the differentially expressed genes to predict the precision phenotypes was tested. Classifiers were selected using a preselected set of features (probesets) picked by the SVM algorithm in an unbiased fashion and run using both cross validation and bootstrapping. A number of methods for are available, but we used two common methods for internal validation using data-splitting 5-fold: cross validation (Table 3a), that splits the data into smaller cohorts and the more rigorous bootstrapping (Table 3b) that leverages the whole dataset. Classifiers were selected using a preselected set of features (probesets) picked by the SVM algorithm in an unbiased fashion and run using both cross validation and bootstrapping.
Table 3.
| a: Predictive performance and comparison of clinical phenotypes in blood and biopsy by microarrays and RNA-seq using 5-fold cross validation. | ||||||
|---|---|---|---|---|---|---|
| Microarray Blood | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 200 | 0.857 | 0.848 | 0.825 | 0.876 | 0.957 |
| subAR vs. TX | 100 | 0.652 | 0.792 | 0.742 | 0.712 | 0.954 |
| Microarray Biopsy | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 150 | 1.000 | 0.920 | 0.913 | 1.000 | 1.000 |
| subAR vs. TX | 250 | 0.918 | 0.872 | 0.863 | 0.923 | 0.993 |
| RNA-seq Blood | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 30 | 1.000 | 0.933 | 0.937 | 1.000 | 1.000 |
| subAR vs. TX | 250 | 0.867 | 0.933 | 0.928 | 0.875 | 0.962 |
| NGS Biopsy | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 100 | 0.990 | 0.925 | 0.916 | 0.991 | 0.990 |
| subAR vs. TX | 300 | 0.714 | 0.866 | 0.824 | 0.776 | 0.926 |
| b: Predictive performance and comparison of clinical phenotypes in blood and biopsy by microarrays and RNA-seq using bootstrap methodology | ||||||
|---|---|---|---|---|---|---|
| Microarray Blood | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 300 | 0.941 | 0.904 | 0.888 | 0.950 | 0.963 |
| subAR vs. TX | 20 | 0.714 | 0.861 | 0.833 | 0.756 | 0.817 |
| Microarray Biopsy | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 200 | 0.918 | 0.869 | 0.850 | 0.930 | 1.000 |
| subAR vs. TX | 350 | 0.916 | 0.705 | 0.687 | 0.923 | 0.951 |
| RNA-seq Blood | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 250 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| subAR vs. TX | 150 | 0.937 | 1.000 | 1.000 | 0.909 | 0.877 |
| NGS Biopsy | # Genes* | Sensitivity | Specificity | PPV | NPV | AUC |
| cAR vs. TX | 30 | 0.974 | 0.976 | 0.974 | 0.976 | 0.999 |
| subAR vs. TX | 200 | 0.767 | 0.902 | 0.891 | 0.787 | 0.778 |
Best classifier set; AUC, area under the curve; cAR, clinical acute rejection; NPV, negative predictive value; PPV, positive predictive value; subAR, subclinical acute rejection; TX, transplant excellence.
The best microarray classifier sets for peripheral blood using 5-fold cross-validation predicted cAR vs. TX and subAR vs. TX with AUCs of 0.95, respectively. By the rigorous bootstrapping the AUCs were 0.96 and 0.82. The best microarray classifier sets for the biopsies using 5-fold cross-validation predicted cAR vs. TX and subAR vs. TX with AUCs 1.00 and 0.99 respectively. By bootstrapping the AUCs were 1.00 and 0.95.
The best RNA-seq classifier sets for peripheral blood using 5-fold cross-validation predicted cAR vs. TX and subAR vs. TX with AUCs 1.00 and 0.96, respectively. By bootstrapping the AUCs were 1.00 and 0.87. The best RNA-seq classifier sets for the biopsies using 5-fold cross-validation predicted cAR vs. TX and subAR vs. TX with AUCs of 0.99 and 0.92 respectively and by bootstrapping the AUCs were 0.99 and 0.77.
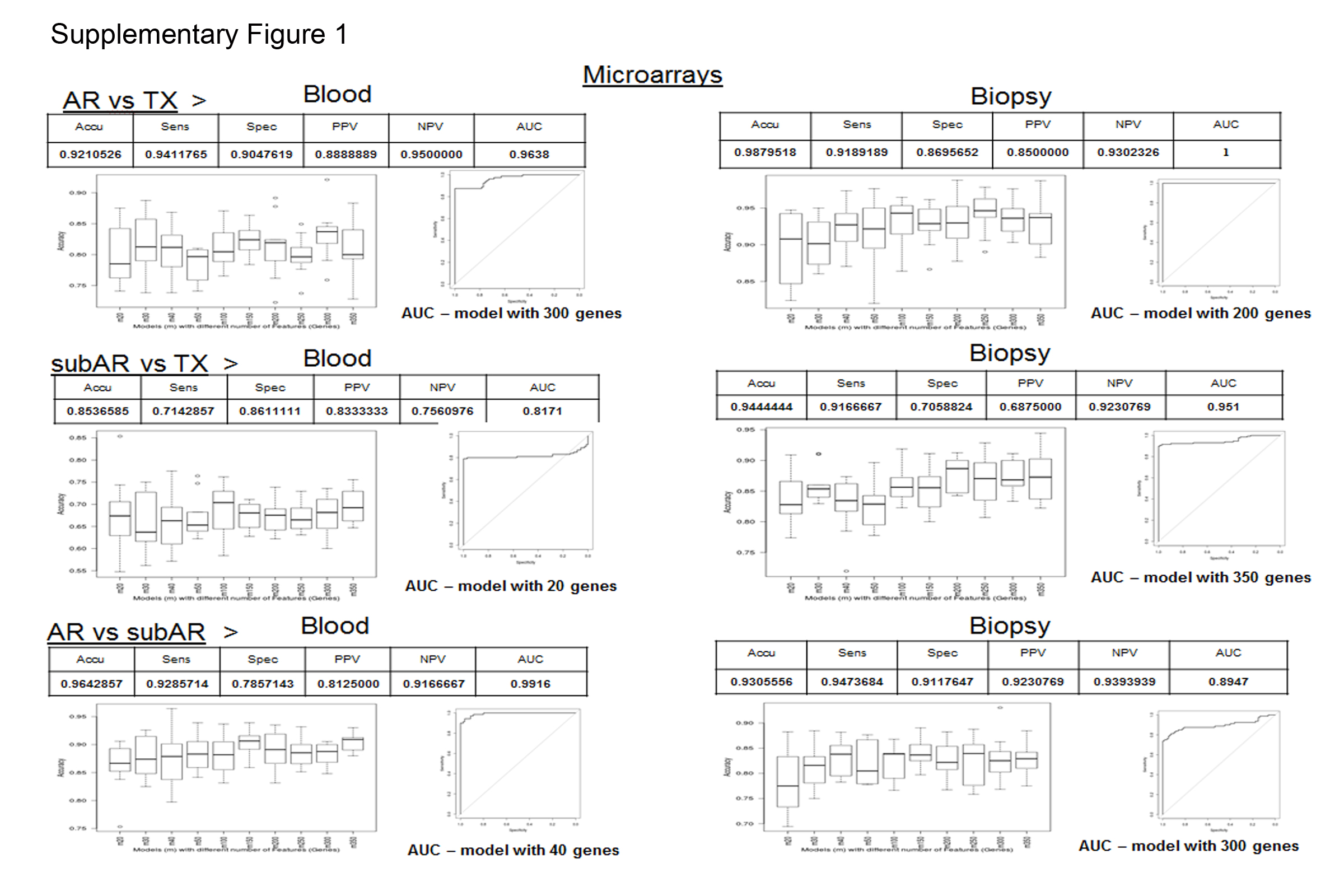
Thus, this first analysis comparing DNA microarrays to RNA-seq suggests that RNA-seq may have a slight performance advantage that must be tested with multiple independent external cohorts as data over-fitting is always possible with internal validation methods. Receiver Operating Curves (ROCs) using the best performing classifiers for the microarray classifier sets by bootstrapping are shown in Figure S1 and RNA-seq classifiers in Figure S2.
Differentially expressed genes present in both the blood and biopsies regardless of technology platform (shared genes)
The differentially expressed shared genes for blood and biopsies by microarrays and RNA-seq are shown in Table 4. The cAR vs. TX and subAR vs. TX comparisons were significant at p <0.05. cAR vs. TX was also significant at a more stringent cut-off of FDR <10%. Even though genes are shared between blood and biopsy the directionality of change (up or down regulation in a specific comparison) may not be the same. Thus, the simple observation of a shared gene in two compartments does not mean their function is biologically the same. Indeed, half the shared genes showed the opposite fold change direction for all comparisons suggesting that the blood and the biopsy are truly independent immunological compartments with distinctly different biological processes (Table 5).
Table 4.
Shared and differentially expressed genes blood and biopsy by microarrays and RNA-seq using statistical confidence levels of FDR<10%, p<0.005 and p<0.05
| Microarray Blood vs. Biopsy FDR<10% |
Blood (DE genes) |
Biopsy (DE genes) |
Overlap | Blood Unique |
Biopsy Unique |
p-value |
|---|---|---|---|---|---|---|
| cAR vs. TX | 1326 | 7530 | 937 | 389 | 6593 | 0.0001 |
| subAR vs. TX | 0 | 804 | 0 | 0 | 804 | NA |
| Microarray Blood vs. Biopsy p<0.005 | Blood (DE genes) | Biopsy (DE genes) | Overlap | Blood Unique | Biopsy Unique | p-value |
| cAR vs. TX | 856 | 5192 | 425 | 431 | 4767 | 0.0001 |
| subAR vs. TX | 33 | 796 | 1 | 32 | 795 | 0.132 |
| Microarray Blood vs. Biopsy p<0.05 | Blood (DE genes) | Biopsy (DE genes) | Overlap | Blood Unique | Biopsy Unique | p-value |
| cAR vs. TX | 2287 | 7376 | 456 | 1831 | 8466 | 0.0001 |
| subAR vs. TX | 720 | 2931 | 180 | 540 | 2751 | 0.0001 |
| RNA-seq Blood vs. Biopsy FDR<10% | Blood (DE genes) | Biopsy (DE genes) | Overlap | Blood Unique | Biopsy Unique | p-value |
| cAR vs. TX | 1198 | 8482 | 236 | 962 | 8246 | 0.0001 |
| subAR vs. TX | 3 | 604 | 0 | 3 | 604 | NA |
| RNA-seq Blood vs. Biopsy p<0.005 | Blood (DE genes) | Biopsy (DE genes) | Overlap | Blood Unique | Biopsy Unique | p-value |
| cAR vs. TX | 833 | 5317 | 94 | 739 | 5223 | 0.0001 |
| subAR vs. TX | 295 | 750 | 4 | 291 | 746 | 0.336 |
| RNA-seq Blood vs. Biopsy p<0.05 | Blood (DE genes) | Biopsy (DE genes) | Overlap | Blood Unique | Biopsy Unique | p-value |
| cAR vs. TX | 2256 | 8922 | 514 | 2052 | 8408 | 0.0001 |
| subAR vs. TX | 1647 | 2565 | 97 | 1550 | 2468 | 0.003 |
cAR, clinical acute rejection; DE, differential expression; FDR, false discovery rate; NGS, next generation sequencing; subAR, subclinical acute rejection; TX, transplant excellence.
Table 5.
Comparison of the directionality of fold-changes among the shared genes between blood and biopsies by microarrays and RNA-seq.
| Comparison | Fold change (same direction) % of genes |
Fold change (opposite direction) % of genes |
|---|---|---|
| Microarray cAR vs. TX | 54.3 | 45.7 |
| Microarray subAR vs. TX | 47.2 | 52.8 |
| NGS cAR vs. TX | 46.5 | 53.5 |
| NGS subAR vs. TX | 48.5 | 51.5 |
We tested the hypothesis that subAR is a state of allo-immune/inflammatory rejection at which point the amount of tissue injury has not exceeded the threshold to cause clinical transplant dysfunction (i.e. acute renal dysfunction; decreased eGFR). If this were true, one prediction would be that subAR is a milder form of cAR manifesting relatively lower gene expression levels for genes upregulated in cAR and higher expression of genes downregulated in cAR.
Therefore, we plotted the subset of shared genes between the cAR vs. TX and the subAR vs. TX comparisons in both blood and biopsies. As shown in Figures 3a-d, the majority of the shared cAR vs. TX genes (green dots) that had the same fold-change directionality (up or down regulated), also had a greater fold change than the same genes in the subAR vs. TX.
Figure 3.

Scatter plots of the fold changes for cAR vs TX (x-axes) and subAR vs. TX (y-axes) to demonstrate that that cAR fold changes are of greater magnitude than subAR changes. 3a, Microarrays - Blood 3b, RNA-seq - Blood 3c, Microarray - Biopsies 3d RNA-seq - Biopsies. Green dots denote greater cAR vs TX fold changes and blue dots denote greater subAR vs. TX fold changes. The table shows the number of genes in each comparison and the overlapping genes that were plotted to create figures 3a-d.
Pathway mapping of shared and unique genes between blood and biopsies
Pathway mapping of blood vs. biopsy genes was done using IPA. A p<0.05 and >10 molecules that were differentially expressed in each pathway filter was used. We combined the microarray and RNA-seq data using the following criteria: 1) genes for mapping were comprised of shared genes; 2) shared genes had the same fold change direction. For the cAR vs. TX comparison there were 109 shared pathways between blood and biopsy. The top 10 shared pathways ranked by p-value were Mitochondrial Dysfunction, B Cell Receptor, Oxidative Phosphorylation, Protein Ubiquitination, NFAT, Tec Kinase, HGF, CD28, PI3K/AKT and Glucocorticoid Receptor Signaling. In contrast there were only 8 shared pathways between blood and biopsy for the subAR vs. TX comparison. These were B Cell Receptor, T Lymphocyte Apoptosis, CD28, CTLA4 in Cytotoxic T Lymphocytes, Leukocyte Extravasation, Phospholipase C, NFAT Regulation of the Immune Response and T Cell Receptor Signaling (Table 6).
Table 6.
Overlapping pathways between blood vs. biopsy genes for the cAR vs. TX and subAR vs. TX comparisons.
| Pathway | cAR vs. TX - Peripheral Blood Molecules (n) |
cAR vs. TX - Biopsy Pathways Molecules (n) |
|---|---|---|
| 14-3-3-mediated Signaling | 34 | 67 |
| Actin Nucleation by ARP-WASP Complex | 15 | 33 |
| Acute Myeloid Leukemia Signaling | 23 | 52 |
| Acute Phase Response Signaling | 35 | 82 |
| Aldosterone Signaling in Epithelial Cells | 40 | 77 |
| Androgen Signaling | 31 | 57 |
| Antiproliferative Role of Somatostatin Receptor 2 | 20 | 38 |
| Apoptosis Signaling | 23 | 51 |
| Assembly of RNA Polymerase II Complex | 16 | 23 |
| Axonal Guidance Signaling | 91 | 176 |
| B Cell Receptor Signaling | 40 | 110 |
| Breast Cancer Regulation by Stathmin1 | 51 | 98 |
| Cardiac Hypertrophy Signaling | 55 | 103 |
| CCR3 Signaling in Eosinophils | 30 | 62 |
| CCR5 Signaling in Macrophages | 20 | 37 |
| CD28 Signaling in T Helper Cells | 33 | 78 |
| Cholecystokinin/Gastrin-mediated Signaling | 25 | 53 |
| Clathrin-mediated Endocytosis Signaling | 45 | 90 |
| Colorectal Cancer Metastasis Signaling | 52 | 112 |
| CREB Signaling in Neurons | 45 | 83 |
| CTLA4 Signaling in Cytotoxic T Lymphocytes | 24 | 45 |
| CXCR4 Signaling | 41 | 86 |
| Docosahexaenoic Acid (DHA) Signaling | 16 | 26 |
| EGF Signaling | 20 | 44 |
| Endometrial Cancer Signaling | 17 | 37 |
| Endothelin-1 Signaling | 40 | 81 |
| Ephrin Receptor Signaling | 36 | 89 |
| ErbB Signaling | 24 | 51 |
| ErbB4 Signaling | 18 | 35 |
| ERK/MAPK Signaling | 42 | 96 |
| Erythropoietin Signaling | 22 | 47 |
| Estrogen Receptor Signaling | 35 | 65 |
| Fc Epsilon RI Signaling | 26 | 60 |
| Fcγ Receptor-mediated Phagocytosis in Macrophages and | 22 | 52 |
| fMLP Signaling in Neutrophils | 34 | 71 |
| G Beta Gamma Signaling | 23 | 46 |
| Gap Junction Signaling | 41 | 76 |
| GDNF Family Ligand-Receptor Interactions | 21 | 43 |
| Germ Cell-Sertoli Cell Junction Signaling | 35 | 90 |
| Gαq Signaling | 39 | 81 |
| Glioblastoma Multiforme Signaling | 38 | 73 |
| Glioma Signaling | 30 | 53 |
| Glucocorticoid Receptor Signaling | 70 | 141 |
| GM-CSF Signaling | 19 | 43 |
| GNRH Signaling | 32 | 63 |
| HER-2 Signaling in Breast Cancer | 21 | 51 |
| HGF Signaling | 31 | 72 |
| Huntington’s Disease Signaling | 54 | 117 |
| α-Adrenergic Signaling | 28 | 40 |
| iCOS-iCOSL Signaling in T Helper Cells | 30 | 69 |
| IGF-1 Signaling | 32 | 62 |
| IL-1 Signaling | 22 | 52 |
| IL-12 Signaling and Production in Macrophages | 38 | 68 |
| IL-17 Signaling | 20 | 47 |
| IL-2 Signaling | 19 | 42 |
| IL-3 Signaling | 26 | 48 |
| IL-8 Signaling | 49 | 102 |
| Insulin Receptor Signaling | 33 | 66 |
| Integrin Signaling | 50 | 107 |
| Interferon Signaling | 11 | 21 |
| JAK/Stat Signaling | 25 | 51 |
| LPS-stimulated MAPK Signaling | 20 | 51 |
| Melanocyte Development and Pigmentation Signaling | 23 | 43 |
| Melanoma Signaling | 15 | 33 |
| Mitochondrial Dysfunction | 43 | 110 |
| Molecular Mechanisms of Cancer | 72 | 176 |
| mTOR Signaling | 42 | 97 |
| Myc Mediated Apoptosis Signaling | 18 | 43 |
| NGF Signaling | 28 | 62 |
| Non-Small Cell Lung Cancer Signaling | 21 | 49 |
| NRF2-mediated Oxidative Stress Response | 47 | 98 |
| Ovarian Cancer Signaling | 30 | 63 |
| Oxidative Phosphorylation | 36 | 74 |
| P2Y Purigenic Receptor Signaling Pathway | 37 | 61 |
| p53 Signaling | 30 | 63 |
| p70S6K Signaling | 39 | 64 |
| Pancreatic Adenocarcinoma Signaling | 27 | 69 |
| PDGF Signaling | 21 | 52 |
| phagosome maturation | 31 | 74 |
| Phospholipase C Signaling | 48 | 115 |
| PI3K Signaling in B Lymphocytes | 31 | 71 |
| PI3K/AKT Signaling | 32 | 73 |
| PKCθ Signaling in T Lymphocytes | 29 | 74 |
| Production of Nitric Oxide and Reactive Oxygen Species in Ma | 49 | 98 |
| Prolactin Signaling | 24 | 49 |
| Prostate Cancer Signaling | 24 | 55 |
| Protein Kinase A Signaling | 73 | 165 |
| Protein Ubiquitination Pathway | 61 | 138 |
| Pyridoxal 5’-phosphate Salvage Pathway | 16 | 32 |
| RANK Signaling in Osteoclasts | 24 | 52 |
| RAR Activation | 44 | 89 |
| Regulation of eIF4 and p70S6K Signaling | 33 | 86 |
| Regulation of IL-2 Expression in Activated and Anergic T Lym | 19 | 51 |
| Renal Cell Carcinoma Signaling | 23 | 43 |
| Renin-Angiotensin Signaling | 32 | 65 |
| Role of NFAT in Cardiac Hypertrophy | 46 | 93 |
| Role of NFAT in Regulation of the Immune Response | 45 | 105 |
| Superpathway of D-myo-inositol (1,4,5)-trisphosphate Metabol | 10 | 14 |
| Systemic Lupus Erythematosus Signaling | 51 | 98 |
| T Cell Receptor Signaling | 26 | 64 |
| Tec Kinase Signaling | 39 | 97 |
| Telomerase Signaling | 25 | 62 |
| Thrombin Signaling | 45 | 98 |
| Thrombopoietin Signaling | 20 | 37 |
| Type II Diabetes Mellitus Signaling | 28 | 62 |
| UVB-Induced MAPK Signaling | 18 | 33 |
| VEGF Family Ligand-Receptor Interactions | 22 | 40 |
| VEGF Signaling | 29 | 50 |
| Xenobiotic Metabolism Signaling | 55 | 122 |
| Pathway | subAR vs. TX - Peripheral Blood Molecules (n) |
subAR vs. TX - Biopsy Pathways Molecules (n) |
|---|---|---|
| B Cell Receptor Signaling | 28 | 22 |
| Calcium-induced T Lymphocyte Apoptosis | 14 | 10 |
| CD28 Signaling in T Helper Cells | 21 | 21 |
| CTLA4 Signaling in Cytotoxic T Lymphocytes | 20 | 15 |
| Leukocyte Extravasation Signaling | 36 | 23 |
| Phospholipase C Signaling | 35 | 24 |
| Role of NFAT in Regulation of the Immune Response | 29 | 26 |
| T Cell Receptor Signaling | 25 | 16 |
Pathways highlighted in red signify shared pathways
An overall analysis of the cAR vs. TX and subAR vs TX in both blood and biopsies revealed 6 key shared pathways (CD28 in T Helper Cells, CTLA4 in Cytotoxic T Lymphocytes, Leukocyte Extravasation, Phospholipase C, NFAT in Regulation of the Immune Response and T Cell Receptor Signaling). Even though these pathways are shared, there were molecules that mapped to the same pathways but were not identical. For example, in the CD28 signaling pathway, the CD28 molecule was only differentially expressed in the blood but PIK3CA was seen in both tissues (Supplementary Table 2).
Discussion
We performed orthogonal technology comparisons of gene expression from both peripheral blood and biopsy tissue compartments concurrently obtained from 69 algorithm-driven clinically phenotyped kidney transplant recipients. We used two high-throughput global gene expression profiling technologies: microarray analysis using Affymetrix Gene Arrays and RNA-seq using both the Ion Torrent and Illumina platforms.
The results of the study demonstrate: 1) diagnostic performance based on the ability to retrospectively predict known clinical phenotypes using SVM were equivalent across technologies and platforms (Affymetrix DNA microarrays, Ion Torrent and Illumina RNA-seq), 2) optimal classifiers selected using the SVM algorithm were different reflecting the technical differences inherent in the chemistries for detection and quantification of gene expression across platforms 3) despite significant overlap in the differentially expressed genes between the blood and biopsies, directionality of these changes differed considerably between compartments, suggesting that these represent distinct immune compartments, and 4) allo-immune/inflammatory pathway mapping to compare differential gene expression in blood and biopsies revealed a number of different genes in each of these two compartments, but these mapped to similar pathways across all technologies. Thus, our ability to detect signatures for TX, subAR and cAR in both compartments appears to be agnostic to the technology or platform used, and equivalent predictive performance metrics were obtained. These data suggest that the ability to develop signatures on one platform should result in a similar ability to develop an equivalent performing signature on another, in response to potential logistical and commercial advantages of new and evolving technologies. To our knowledge, this is the first study to compare orthogonal technologies for molecular diagnostics using mRNA-based differential gene expression in kidney transplant recipients.
We addressed important and often overlooked aspects of biomarker discovery. To avoid over-training, we used both 5-fold cross validation as well as a full leave-one-out bootstrapping methodologies. Of interest, the actual optimal classifiers were different for each platform, and therefore workflows will need to be developed for different technology platforms for introduction into clinical practice. Nonetheless, we show that they each give equivalent predictive performances on TX, cAR and subAR. Thus, decisions made by clinical laboratories regarding which commercial test to use will likely be based on issues other than performance, such as test costs, details of workflow, cost of equipment and its maintenance, and the choice of data analyses pipelines to report results to a clinician. This outcome provides a strong comfort level for the use of microarrays or RNA-seq, as well as that of specific RNA-seq platforms to develop predictive molecular biomarkers to diagnose and predict rejection.
It also appears that the peripheral blood changes may not be as robust as the changes at the tissue site of injury. The subAR signature in the peripheral blood appears to signal a milder form of cAR, consistent with previous data showing similar cellular processes between subAR and cAR (44). Our data show a high level of sharing of differentially expressed genes and pathways between cAR and subAR . SubAR is represented by fewer and less expressed genes than cAR in blood. This is further supported by our analysis that 65–70% of subAR cases are detected by using a signature that was discovered for the diagnosis of cAR in blood (Affymetrix DNA microarrays - data not shown).
Traditional invasive biopsy-based diagnoses are vulnerable to the challenge of sampling errors and differences between the interpretations of individual pathologists (39) (40). The 2007 Banff group previously defined and subsequently refined the pathologic criteria for post-implantation biopsies using the same criteria for implantation biopsies (35).The inter-observer concordance that measures reproducibility between pathologists showed that graft correlates such as acute tubular injury, inflammation in non-scarred areas (Banff i score), and interstitial inflammation had the poorest concordance rates. The Banff working group on pre-implantation biopsies recently concluded that significant limitations remain (41). Despite concerns about histology being the ‘gold standard’, as well as the ongoing debate about the virtues of the local versus central histology reads, we chose to use the central read for the histology component of our clinical phenotype algorithms. We believe that we we may be able to demonstrate a better correlation between the molecular phenotype of the biopsy with long-term outcome compared to either the local or central histology reads. The pros and cons of the currently used platforms to profile rejection phenotypes and build predictive models have been very nicely discussed in a review of genomics in understanding and prediction of Clinical Renal Transplant Injury(42). In this study the slightly better performance of the RNA-seq classifiers was attributed to low signal-to-noise ratio compared to microarrays, and a larger dynamic range of expression levels over which transcripts can be reliably detected as shown in other studies (43, 44). Similarly, the sequencing depth in this study was lower than the generally agreed consensus of ~ 30–40 million reads (43) that is suited to detect major splice isoforms which was not our focus. We tested the power of the detected genes to discriminate the clinical phenotypes and to compare how well the signatures perform.
We also believe that it is important to map the gene networks to show that our analyses are not simply statistical but also founded in the biology of rejection and tissue injury. Despite shared genes, the comparison of the blood and biopsy expression profiles also shows clear differences. Our results demonstrate that the tissue injury and damage begins in the graft and that the blood reveals genes, unique to that compartment, but are nevertheless also informing the prediction of clinical phenotypes consistent with our previous work and that of others (20, 27, 45–49).
While classifiers cannot be locked across technologies or platforms, the predictive performance appears to be equivalent and agnostic to the platform suggesting the ability to develop signatures across platforms with relatively little technical variation and estimation of expression levels of RNA transcripts (50), as these become more robust and cost-effective (31).
This study has some limitations: the sample size was small, and only appropriate for a discovery study as intended, but we demonstrate with the bootstrapping methodology which is the closest approximation to real error estimates in a simulation of independent cohorts; no appreciable deterioration of the predictive metrics for our signatures. While we provided internal validation through widely accepted data-splitting cross-validation bootstrapping methods, we did not perform external independent validation of the signatures described. This was not the intended goal of this study. In fact, these studies are ongoing in the NIH-funded CTOT study of 300 transplant recipients followed serially for two years. Instead, our primary objective to assess whether different technology platforms could be used to detect differential gene expression profiles that signal immune activation in both peripheral blood and biopsy tissue. We also acknowledge that our population had no cases of pure antibody-mediated rejection (AMR), although 23% of the cAR biopsy samples showed C4d positive staining. Also, the majority of patients were Caucasian. However, we have successfully validated our biopsy molecular phenotypes in a Brazilian cohort of 94 patients of significantly different racial and ethnic backgrounds (48) suggesting strong unifying immune mechanisms despite differences in racial, ethnic and genetic backgrounds.
In conclusion, we present a proof-of-concept discovery study that evaluates the predictive performance and profiling capabilities of two complementary techniques of global gene expression profiling for discrimination of three post-transplant phenotypes. This is also the first study to our knowledge of kidney transplant rejection that has performed peripheral blood and biopsy profiling and compared them.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
NIH U19 AI063603 (DRS, SMK, RT, NR, TG, FH); Mendez National Institutes of Transplantation (RT, EV); Northwestern University Internal Funding (MMA, JJF) and Transplant Genomics Inc. (RMF, SR).
Abbreviations
- ADNR
acute dysfunction with no rejection by biopsy histology
- AUC
area under the curve
- cAR
clinical acute rejection
- AMR
antibody mediated rejection
- CMV
cytomegalovirus
- DE
differential expression
- FDR
false discovery rate
- FN
false negative
- FP
false positive
- fRMA
frozen robust multi-array analysis
- IA
immune activation
- IFTA
interstitial fibrosis/tubular atrophy
- IQ
immune quiescence
- NGS
next generation sequencing
- SubAR
subclinical acute rejection
- TGI
Transplant Genomics Inc.
- TN
true negative
- TP
true positive
- TX
transplant with excellent function
Footnotes
Disclosure
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. DRS, SMK, JJF and MMA are founding scientists and have ownership stock in TGI. RMF and SR are full-time employees at TGI. The other authors have no conflicts of interest to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Table S1: Detailed final breakdown of gene expression profile numbers for the various comparisons in the study.
Table S2 Title: List of cAR vs. TX and subAR vs. TX pathways and their constituent molecules in the peripheral blood and biopsies with shared genes (blood and biopsy) highlighted in red.
Figure S1: Performance metrics for microarray based classifiers. Table on top for each comparison shows accuracy, sensitivity, specificity, positive and negative predictive values. Figure on the left for each comparison shows the box and whisker plots for the accuracies for different classifiers based on the number of probesets used to develop the classifier. Figure on right for each comparison shows the Receiver Operating Characteristic curve for the best classifier. The three comparisons on the left are for peripheral blood and the three comparisons on the right are for the biopsies.
Figure S2: Performance metrics for RNA-seq based classifiers. Table on top for each comparison shows accuracy, sensitivity, specificity, positive and negative predictive values. Figure on the left for each comparison shows the box and whisker plots for the accuracies for different classifiers based on the number of probesets used to develop the classifier. Figure on right for each comparison shows the Receiver Operating Characteristic curve for the best classifier. The three comparisons on the left are for peripheral blood and the three comparisons on the right are for the biopsies.
References
- 1.Rana A, Gruessner A, Agopian VG, Khalpey Z, Riaz IB, Kaplan B, et al. Survival Benefit of Solid-Organ Transplant in the United States. Jama Surgery. 2015;150(3):252–259. doi: 10.1001/jamasurg.2014.2038. [DOI] [PubMed] [Google Scholar]
- 2.Lamb KE, Lodhi S, Meier-Kriesche HU. Long-Term Renal Allograft Survival in the United States: A Critical Reappraisal. American Journal of Transplantation. 2011;11(3):450–462. doi: 10.1111/j.1600-6143.2010.03283.x. [DOI] [PubMed] [Google Scholar]
- 3.Matas AJ, Gillingham KJ, Humar A, Kandaswamy R, Sutherland DER, Payne WD, et al. 2202 kidney transplant recipients with 10 years of graft function: What happens next? American Journal of Transplantation. 2008;8(11):2410–2419. doi: 10.1111/j.1600-6143.2008.02414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montgomery RA. One kidney for life. American Journal of Transplantation. 2014;14(7):1473–1474. doi: 10.1111/ajt.12772. [DOI] [PubMed] [Google Scholar]
- 5.Matas AJ, Smith JM, Skeans MA, Thompson B, Gustafson SK, Stewart DE, et al. OPTN/SRTR 2013 Annual Data Report: kidney. American Journal of Transplantation. 2015;(15 Suppl 2):1–34. doi: 10.1111/ajt.13195. [DOI] [PubMed] [Google Scholar]
- 6.Lo DJ, Kaplan B, Kirk AD. Biomarkers for kidney transplant rejection. Nat Rev Nephrol. 2014;10(4):215–225. doi: 10.1038/nrneph.2013.281. [DOI] [PubMed] [Google Scholar]
- 7.Willis JC, Lord GM. Immune biomarkers: the promises and pitfalls of personalized medicine. Nat Rev Immunol. 2015;15(5):323–329. doi: 10.1038/nri3820. [DOI] [PubMed] [Google Scholar]
- 8.Yilmaz S, Isik I, Afrouzian M, Monroy M, Sar A, Benediktsson H, et al. Evaluating the accuracy of functional biomarkers for detecting histological changes in chronic allograft nephropathy. Transplant international : official journal of the European Society for Organ Transplantation. 2007;20(7):608–615. doi: 10.1111/j.1432-2277.2007.00494.x. [DOI] [PubMed] [Google Scholar]
- 9.Malvezzi P, Rostaing L. The safety of calcineurin inhibitors for kidney-transplant patients. Expert opinion on drug safety. 2015;14(10):1531–1546. doi: 10.1517/14740338.2015.1083974. [DOI] [PubMed] [Google Scholar]
- 10.Nankivell BJ, Chapman JR. The significance of subclinical rejection and the value of protocol biopsies. American Journal of Transplantation. 2006;6(9):2006–2012. doi: 10.1111/j.1600-6143.2006.01436.x. [DOI] [PubMed] [Google Scholar]
- 11.Racusen LC, Solez K, Colvin RB, Bonsib SM, Castro MC, Cavallo T, et al. The Banff 97 working classification of renal allograft pathology. Kidney international. 1999;55(2):713–723. doi: 10.1046/j.1523-1755.1999.00299.x. [DOI] [PubMed] [Google Scholar]
- 12.Rush D, Nickerson P, Gough J, McKenna R, Grimm P, Cheang M, et al. Beneficial effects of treatment of early subclinical rejection: a randomized study. Journal of the American Society of Nephrology : JASN. 1998;9(11):2129–2134. doi: 10.1681/ASN.V9112129. [DOI] [PubMed] [Google Scholar]
- 13.Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Allen RD, Chapman JR. Natural history, risk factors, and impact of subclinical rejection in kidney transplantation. Transplantation. 2004;78(2):242–249. doi: 10.1097/01.tp.0000128167.60172.cc. [DOI] [PubMed] [Google Scholar]
- 14.d’Ardenne AJ, Dunnill MS, Thompson JF, McWhinnie D, Wood RF, Morris PJ. Cyclosporin and renal graft histology. Journal of clinical pathology. 1986;39(2):145–151. doi: 10.1136/jcp.39.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heilman RL, Smith ML, Kurian SM, Huskey J, Batra RK, Chakkera HA, et al. Transplanting Kidneys from Deceased Donors With Severe Acute Kidney Injury. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15(8):2143–2151. doi: 10.1111/ajt.13260. [DOI] [PubMed] [Google Scholar]
- 16.Kirk AD, Jacobson LM, Heisey DM, Radke NF, Pirsch JD, Sollinger HW. Clinically stable human renal allografts contain histological and RNA-based findings that correlate with deteriorating graft function. Transplantation. 1999;68(10):1578–1582. doi: 10.1097/00007890-199911270-00024. [DOI] [PubMed] [Google Scholar]
- 17.Nankivell BJ, Fenton-Lee CA, Kuypers DR, Cheung E, Allen RD, O’Connell PJ, et al. Effect of histological damage on long-term kidney transplant outcome. Transplantation. 2001;71(4):515–523. doi: 10.1097/00007890-200102270-00006. [DOI] [PubMed] [Google Scholar]
- 18.Shishido S, Asanuma H, Nakai H, Mori Y, Satoh H, Kamimaki I, et al. The impact of repeated subclinical acute rejection on the progression of chronic allograft nephropathy. Journal of the American Society of Nephrology : JASN. 2003;14(4):1046–1052. doi: 10.1097/01.asn.0000056189.02819.32. [DOI] [PubMed] [Google Scholar]
- 19.Loupy A, Vernerey D, Tinel C, Aubert O, Duong van Huyen JP, Rabant M, et al. Subclinical Rejection Phenotypes at 1 Year Post-Transplant and Outcome of Kidney Allografts. Journal of the American Society of Nephrology : JASN. 2015;26(7):1721–1731. doi: 10.1681/ASN.2014040399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Modena BD, Kurian SM, Gaber LW, Waalen J, Su AI, Gelbart T, et al. Gene Expression in Biopsies of Acute Rejection and Interstitial Fibrosis/Tubular Atrophy Reveals Highly Shared Mechanisms That Correlate With Worse Long-Term Outcomes. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2016;16(7):1982–1998. doi: 10.1111/ajt.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi BS, Shin MJ, Shin SJ, Kim YS, Choi YJ, Moon IS, et al. Clinical significance of an early protocol biopsy in living-donor renal transplantation: ten-year experience at a single center. American Journal of Transplantation. 2005;5(6):1354–1360. doi: 10.1111/j.1600-6143.2005.00830.x. [DOI] [PubMed] [Google Scholar]
- 22.Heilman RL, Devarapalli Y, Chakkera HA, Mekeel KL, Moss AA, Mulligan DC, et al. Impact of subclinical inflammation on the development of interstitial fibrosis and tubular atrophy in kidney transplant recipients. American Journal of Transplantation. 2010;10(3):563–570. doi: 10.1111/j.1600-6143.2009.02966.x. [DOI] [PubMed] [Google Scholar]
- 23.Kee TY, Chapman JR, O’Connell PJ, Fung CL, Allen RD, Kable K, et al. Treatment of subclinical rejection diagnosed by protocol biopsy of kidney transplants. Transplantation. 2006;82(1):36–42. doi: 10.1097/01.tp.0000225783.86950.c2. [DOI] [PubMed] [Google Scholar]
- 24.Moreso F, Ibernon M, Goma M, Carrera M, Fulladosa X, Hueso M, et al. Subclinical rejection associated with chronic allograft nephropathy in protocol biopsies as a risk factor for late graft loss. American Journal of Transplantation. 2006;6(4):747–752. doi: 10.1111/j.1600-6143.2005.01230.x. [DOI] [PubMed] [Google Scholar]
- 25.Rush DN, Henry SF, Jeffery JR, Schroeder TJ, Gough J. Histological findings in early routine biopsies of stable renal allograft recipients. Transplantation. 1994;57(2):208–211. doi: 10.1097/00007890-199401001-00009. [DOI] [PubMed] [Google Scholar]
- 26.Seron D, Moreso F. Protocol biopsies in renal transplantation: prognostic value of structural monitoring. Kidney international. 2007;72(6):690–697. doi: 10.1038/sj.ki.5002396. [DOI] [PubMed] [Google Scholar]
- 27.Kurian SM, Williams AN, Gelbart T, Campbell D, Mondala TS, Head SR, et al. Molecular classifiers for acute kidney transplant rejection in peripheral blood by whole genome gene expression profiling. American Journal of Transplantation. 2014;14(5):1164–1172. doi: 10.1111/ajt.12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kircher M, Kelso J. High-throughput DNA sequencing--concepts and limitations. BioEssays: news and reviews in molecular, cellular and developmental biology. 2010;32(6):524–536. doi: 10.1002/bies.200900181. [DOI] [PubMed] [Google Scholar]
- 29.Wetterstrand KA. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP) 2016 Available from: https://www.genome.gov/27565109/the-cost-of-sequencing-a-human-genome/
- 30.Bradford JR, Hey Y, Yates T, Li Y, Pepper SD, Miller CJ. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC genomics. 2010;11:282. doi: 10.1186/1471-2164-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 2008;18(9):1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trost B, Moir CA, Gillespie ZE, Kusalik A, Mitchell JA, Eskiw CH. Concordance between RNA-sequencing data and DNA microarray data in transcriptome analysis of proliferative and quiescent fibroblasts. Royal Society open science. 2015;2(9):150402. doi: 10.1098/rsos.150402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PloS one. 2014;9(1):e78644. doi: 10.1371/journal.pone.0078644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W, Yu Y, Hertwig F, Thierry-Mieg J, Thierry-Mieg D, Wang J, et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome biology. 2015;16:133. doi: 10.1186/s13059-015-0694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M, et al. Banff 07 classification of renal allograft pathology: updates and future directions. American Journal of Transplantation. 2008;8(4):753–760. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 36.Head SR, Komori HK, Hart GT, Shimashita J, Schaffer L, Salomon DR, et al. Method for improved Illumina sequencing library preparation using NuGEN Ovation RNA-Seq System. Biotechniques. 2011;50(3):177–180. doi: 10.2144/000113613. [DOI] [PubMed] [Google Scholar]
- 37.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11(2):242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuhn M. Building Predictive Models in R Using the caret Package. J Stat Softw. 2008;28(5):1–26. [Google Scholar]
- 39.Azancot MA, Moreso F, Salcedo M, Cantarell C, Perello M, Torres IB, et al. The reproducibility and predictive value on outcome of renal biopsies from expanded criteria donors. Kidney international. 2014;85(5):1161–1168. doi: 10.1038/ki.2013.461. [DOI] [PubMed] [Google Scholar]
- 40.Haas M, Segev DL, Racusen LC, Bagnasco SM, Melancon JK, Tan M, et al. Arteriosclerosis in kidneys from healthy live donors: comparison of wedge and needle core perioperative biopsies. Archives of pathology & laboratory medicine. 2008;132(1):37–42. doi: 10.5858/2008-132-37-AIKFHL. [DOI] [PubMed] [Google Scholar]
- 41.Liapis H, Gaut JP, Klein C, Bagnasco S, Kraus E, Farris AB, 3rd, et al. Banff Histopathological Consensus Criteria for Preimplantation Kidney Biopsies. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2016 doi: 10.1111/ajt.13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menon MC, Keung KL, Murphy B, O’Connell PJ. The Use of Genomics and Pathway Analysis in Our Understanding and Prediction of Clinical Renal Transplant Injury. Transplantation. 2016;100(7):1405–1414. doi: 10.1097/TP.0000000000000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 44.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flechner SM, Kurian SM, Head SR, Sharp SM, Whisenant TC, Zhang J, et al. Kidney transplant rejection and tissue injury by gene profiling of biopsies and peripheral blood lymphocytes. American Journal of Transplantation. 2004;4(9):1475–1489. doi: 10.1111/j.1600-6143.2004.00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halloran PF, Famulski K, Reeve J. The molecular phenotypes of rejection in kidney transplant biopsies. Current opinion in organ transplantation. 2015;20(3):359–367. doi: 10.1097/MOT.0000000000000193. [DOI] [PubMed] [Google Scholar]
- 47.Kurian SM, Heilman R, Mondala TS, Nakorchevsky A, Hewel JA, Campbell D, et al. Biomarkers for early and late stage chronic allograft nephropathy by proteogenomic profiling of peripheral blood. PloS one. 2009;4(7):e6212. doi: 10.1371/journal.pone.0006212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Khatri P, Sigdel TK, Tran T, Ying L, Vitalone MJ, et al. A peripheral blood diagnostic test for acute rejection in renal transplantation. American Journal of Transplantation. 2012;12(10):2710–2718. doi: 10.1111/j.1600-6143.2012.04253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roedder S, Sigdel T, Salomonis N, Hsieh S, Dai H, Bestard O, et al. The kSORT assay to detect renal transplant patients at high risk for acute rejection: results of the multicenter AART study. PLoS medicine. 2014;11(11):e1001759. doi: 10.1371/journal.pmed.1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu X, Fu N, Guo S, Yan Z, Xu Y, Hu H, et al. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC genomics. 2009;10:161. doi: 10.1186/1471-2164-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.