

Abstract
Reduced expression of the Indy (‘I am Not Dead, Yet’) gene in lower organisms promotes longevity in a manner akin to caloric restriction. Deletion of the mammalian homolog of Indy (mIndy, Slc13a5) encoding for a plasma membrane associated citrate transporter expressed highly in the liver, protects mice from high-fat diet and aging-induced obesity and hepatic fat accumulation through a mechanism resembling caloric restriction. We aimed to study a possible role of mIndy in human hepatic fat metabolism. In obese, insulin resistant patients with NAFLD, hepatic mIndy expression was increased and mIndy expression was also independently associated with hepatic steatosis. In non-human primates, a two year high fat, high sucrose diet increased hepatic mIndy expression. Liver microarray analysis showed that high mIndy expression was associated with pathways involved in hepatic lipid metabolism and immunological processes. Interleukin-6 (IL-6) was identified as a regulator of mIndy by binding to its cognate receptor. Studies in human primary hepatocytes confirmed that IL-6 markedly induced mIndy transcription via the IL-6-receptor (IL-6R) and activation of the transcription factor Stat3 and a putative start site of the human mIndy promoter was determined. Activation of the IL-6-Stat3 pathway stimulated mIndy expression, enhanced cytoplasmic citrate influx and augmented hepatic lipogenesis in vivo. In contrast, deletion of mIndy completely prevented the stimulating effect of IL-6 on citrate uptake and reduced hepatic lipogenesis. These data show that mIndy is increased in liver of obese humans and non-human primates with NALFD. Moreover, our data identify mIndy as a target gene of IL-6 and determine novel functions of IL-6 via mINDY. Targeting human mINDY may have therapeutic potential in obese patients with NAFLD.
Keywords: Indy, Liver, NAFLD, Insulin Resistance, IL-6
Introduction
Reduced Indy (I am Not Dead, Yet) gene expression in D. melanogaster and C. elegans promotes longevity in a manner akin to caloric restriction in most studies (1–4). Long-lived flies with lower Indy expression have decreased whole body fat stores, lower expression of insulin-like proteins, and increased mitochondrial number (3, 5). The Indy gene product is a cation-independent, electroneutral tricarboxylate carrier (6, 7), able to transport citrate across the plasma membrane as its preferred substrate. Indy is highly expressed in organs involved in energy homeostasis in flies (8). In mammals, the gene product of Slc13a5 (mIndy) encodes the sodium-coupled citrate transporter NaCT (mINDY); it shares the highest sequence and functional similarity with D. melanogaster INDY (9) and is highly expressed in liver tissue (6, 7, 10–12). In mammals, mINDY mediates an electrogenic cotransport of a various di- and tricarboxylates, with the highest affinity for citrate (6). Citrate and succinate are mINDY substrates with the highest plasma concentrations (11). The amino acid sequence of the N-terminal sodium- and the carboxy-binding pocket are highly conserved between many species, from bacteria to mice to human (13).
Recently, our lab translated findings from Drosophila into mammals by showing that mIndy deletion (mINDY−/−) mimics important aspects of caloric restriction in mice (7), without reducing caloric intake. mINDY−/− mice gain less weight on a high-fat diet and during the aging process together with lower liver fat content and reduced insulin resistance (7). Furthermore, we showed that liver specific mIndy knockdown using anti-sense oligonucleotides (ASOs) in adult rats reduced hepatic lipid storage and enhanced hepatic insulin sensitivity upon feeding a HFD (14). Finally, a competitive, stereo sensitive small molecule inhibitor of the mINDY transporter offered complete protection from diet-induced glucose intolerance in mice and ameliorated diet-induced fatty liver disease as shown by an independent research group (15).
The cytokine interleukin-6 (IL-6) is a pleiotropic cytokine with a complex role in inflammation and metabolic disease. IL-6 binding to its cell surface receptor with subsequent activation of janus family kinases and phosphorylation of signal transducer and activator of transcription 3 (STAT3), promoting its nuclear translocation, DNA binding, and subsequent target gene expression, including important genes of the acute phase reaction. Moreover, PI-3 phosphorylation through IL-6 results in Akt activation. IL-6 also activates the pro-oncogenic Ras/Raf/MAPK 1/2 signaling pathway. In obesity and the metabolic syndrome, cytokines, such as IL-6, are highly secreted from adipose tissue and have been proposed to promote accompanying metabolic diseases, i.e. insulin resistance, type 2 diabetes and non-alcoholic fatty liver disease (NAFLD) (16–19). However, anti-inflammatory and beneficial metabolic actions of IL-6 have been reported as well (20–22). Thus, the exact role of IL-6 in metabolic regulation remains highly controversial (23).
Given the therapeutic potential of mINDY inhibition in metabolic disease and aging, our aim was to investigate the role of the human mIndy homolog in the pathophysiology of obesity and NAFLD, and the regulation of its expression under conditions associated with obesity. We observed that mIndy expression was increased in obese patients with NAFLD and that the cytokine IL-6 is a potent regulator of the human mIndy homolog.
RESULTS
Human mIndy Tissue Distribution
To determine mIndy distribution in human subjects, semi-quantitative PCR measurements of mIndy expression were performed in different tissues. mIndy expression was markedly higher in liver tissue compared to all other tissues studied (Figure 1A), with skeletal muscle and colon showing minimal mIndy expression levels confirming previous reports in mice (7), rats (12, 14) and human subjects (11, 24).
Figure 1. Human mIndy tissue distribution and functional characteristics.
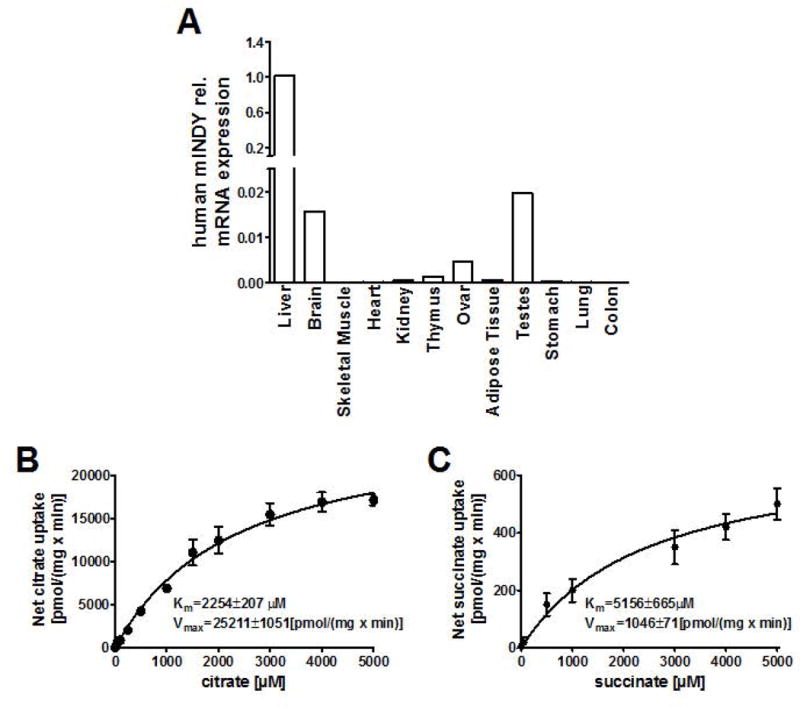
A) Human mIndy mRNA tissue expression in different tissues. B and C) mINDY transport kinetics for citrate (B) and succinate (C) were carried out in HEK293 cells transfected with either the pIndy-human.31 plasmid or empty expression vector pcDNA3.1(+). The net uptake is expressed as the difference between the uptake of substrates into HEK293 cells overexpressing human mINDY and pcDNA-transfected controls (n=3–6 for each concentration). Km values were determined by fitting the data to a non-linear regression curve fit.
Transport kinetics of human mINDY
Human mIndy (SLC13A5) cDNA was cloned, transfected into HEK293 cells, and the uptake capacities of mINDY for citrate and succinate were analyzed. mINDY proved to be a high capacity transporter for citrate over a broad range of concentrations (0–5000 μM), which encompasses physiological concentrations (25) (Km=2254±207 μM, Vmax=25117±1051 [pmol/mg·min−1]) and a succinate transporter with an intermediate capacity (Km=5156±665 μM, Vmax=1046±71 [pmol/mg•min−1]) (Figure 1B and 1C).
Human hepatic mINDY mRNA expression is increased in insulin resistant patients with obesity and nonalcoholic fatty liver disease
Forty-nine liver samples were collected from patients undergoing hepatic surgery and processed for the quantitative determination of mIndy mRNA levels. With respect to current diagnostic criteria (26) all subjects presenting with ≥5% steatotic hepatocytes in our histopathological analysis were considered as having NAFLD. Accordingly, 37% of our study subjects had no sign of any pathological condition in the respective liver tissue, compared to 63% suffering from NAFLD. Liver samples were collected before the blood supply to the liver was clamped (Pringel maneuver) to minimize cold ischemia. Characteristics of patients are given in Table 1. mIndy expression was positively correlated to BMI (r=0.361, P=0.011, Figure 2A), waist circumference (r=0.418, P=0.003, Figure 2B), body fat (r=0.441, P=0.020, Figure 2C), and nominally for insulin resistance as assessed by HOMA-IR (r=0.333, P=0.019), and hepatic insulin resistance index (r=0.316, P=0.027). Of note, the strongest correlation was present with histology determined steatosis (r=0.688, P<0.001, Figure 2D). To determine whether mIndy expression was independently associated with hepatic steatosis, we adjusted for several potential confounders, including age, sex, waist circumference and insulin resistance, by means of multivariate linear regression analysis. The degree of liver histology-determined steatosis remained significantly associated with mIndy expression (Model in Table 1B), suggesting that mIndy independently predicts human hepatic steatosis. Total RNA microarrays from liver samples of patients with low (1±0.1 AU) and high mINDY expression (3.4±0.4 AU), matched for BMI and other metabolic parameters, except for hepatic fat content (34±5% versus 16±2 %, for high versus low expressed mIndy, respectively, P=0.02, n=3) revealed biological processes involved in hepatic lipid metabolism and the acute phase response being activated in patients with high mIndy expression in gene ontology enrichment analysis (Table 2).
Table 1.
Human patient characteristics and model calculation
| n (male) | 49 (23) |
| age (years) | 59±2 |
| BMI (kg/m2) | 26.7±0.9 |
| body fat (%) | 32.9±1.4 |
| waist circumference (cm) | 96.1±2.3a |
| SBP (mm Hg) | 133±3 |
| DBP (mm Hg) | 73±2 |
| AST/ALT | 1.26±0.10 |
| HOMA-IR | 2.84±0.45 |
| NAS (0–8) | 1.72±0.24a |
| Liver steatosis | 16.66±3.22 |
| steatosis score (0–3) | 0.92±0.13a |
| ethanol intake (drinks/week) | 1.4±0.3 |
| Model Dependent variable: Hepatic steatosis |
|
|---|---|
| Independent variable |
Beta coefficient (P-value) R = 0.760; R2 = 0.577, adj. R2 = 0.496, P < 0.001 |
| Sex | 0.106 (0.45) |
| Age | 0.123 (0.38) |
| Waist | 0.279 (0.100) |
| Insulin Resistance (HOMA-IR) |
0.285 (0.046) |
| Hepatic mINDY expression | 0.442 (0.008) |
Significant correlations are shown in bold.
Figure 2. mIndy expression in the liver of patients with different BMI and liver fat contents.
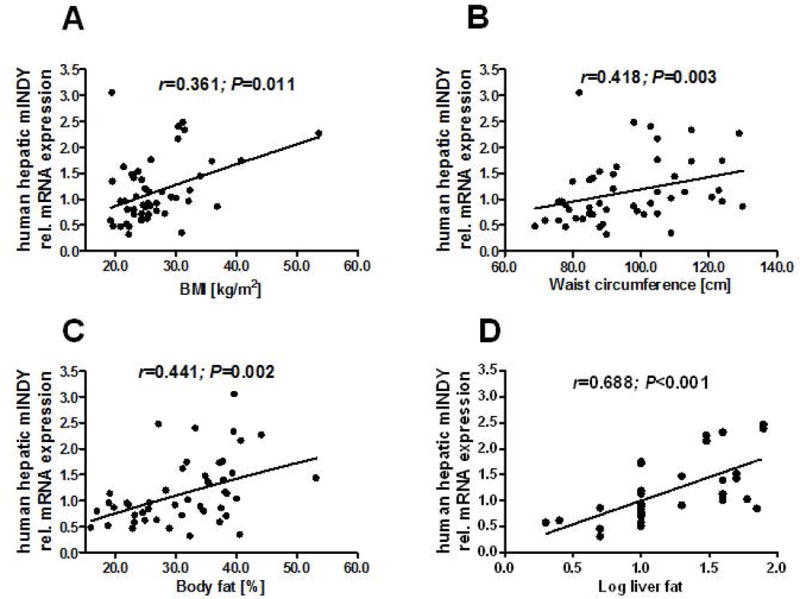
A) mIndy expression was positively associated with BMI, B) waist circumference, C) body fat, D) histologically assessed liver fat content. The degree of liver histology-determined steatosis remained significantly associated with mIndy expression after adjustment for several confounders (Model in Table 1), identifying mIndy as an independent risk factor for NAFLD in our cohort of patients.
Table 2.
Gene Ontology enrichment analysis in livers from subjects with high vs low mINDY expression.
| Gene Ontology Term | Zscore | (P_value) | (fdr) |
|---|---|---|---|
| GO0042627 CHYLOMICRON | 10.084 | 0.000 | 0.007 |
| GO0042157 LIPOPROTEIN METABOLIC PROCESS | 9.829 | 0.000 | 0.006 |
| GO0046870 CADMIUM ION BINDING | 9.253 | 3.18E‐11 | 3.37E‐09 |
| GO0005319 LIPID TRANSPORTER ACTIVITY | 8.126 | 0.002 | 0.034 |
| GO0006953 ACUTE PHASE RESPONSE | 6.519 | 0.003 | 0.042 |
| GO0006572 TYROSINE CATABOLIC PROCESS | 5.016 | 0.002 | 0.036 |
| GO0004984 OLFACTORY RECEPTOR ACTIVITY | 4.036 | 9.82E‐19 | 2.38E‐16 |
| GO0005840 RIBOSOME | 3.848 | 0.001 | 0.014 |
| GO0003735 STRUCTURAL CONSTITUENT OF RIBOSOME | 3.684 | 0.001 | 0.021 |
| GO0008270 ZINC ION BINDING | −8.407 | 3.40E‐13 | 4.44E‐11 |
| GO0003677 DNA BINDING | −8.592 | 9.26E‐13 | 1.12E‐10 |
| GO0006350 TRANSCRIPTION | −9.520 | 4.12E‐15 | 6.35E‐13 |
| GO0046872 METAL ION BINDING | −9.790 | 8.75E‐16 | 1.41E‐13 |
| GO0016020 MEMBRANE | −9.979 | 4.72E‐17 | 8.89E‐15 |
| GO0006355 REGULATION OF TRANSCRIPTION DNA DEPENDE | −10.015 | 7.29E‐17 | 1.30E‐14 |
| GO0005737 CYTOPLASM | −11.918 | 6.03E‐21 | 1.86E‐18 |
| GO0005515 PROTEIN BINDING | −14.315 | 4.65E‐29 | 1.97E‐26 |
| GO0005634 NUCLEUS | −14.608 | 1.50E‐33 | 1.02E‐30 |
Gene Ontology terms of biological processes. Positive z-score indicates increased in patients with high mIndy expression. Negative z-score indicates reduced in patients with high mIndy expression.
High fat, high sucrose diet increases hepatic mIndy expression in non-human primates
To independently confirm our human data, we used a cohort of non-human primates raised in a controlled environment. Fourteen middle-aged male rhesus monkeys were randomized into two groups and fed either a standard diet (SD, n=4) or a high-fat, high sucrose (HFS) diet (n=10) for two years, as described (27). Monkeys fed a HFS diet showed significant increases in body weight (Figure 3A), waist circumference (Figure 3B) and serum IL-6 levels (Figure 3C) when compared to SD-fed animals. Total RNA microarrays from monkey liver samples, and pairwise comparison between HFS and SD cohorts indicated a 3.5-fold increase in the z-ratio for mIndy in response to HFS. These data were confirmed by RT-PCR, with a 3.7±0.7 fold increase in hepatic mIndy expression in obese, HFS-fed monkeys as compared to SD-fed controls (Figure 3D). These data show that a high fat high sucrose diet or the resulting diet induced obesity increases mIndy expression in non human primates.
Figure 3. mIndy expression in liver tissue from non-human primates.
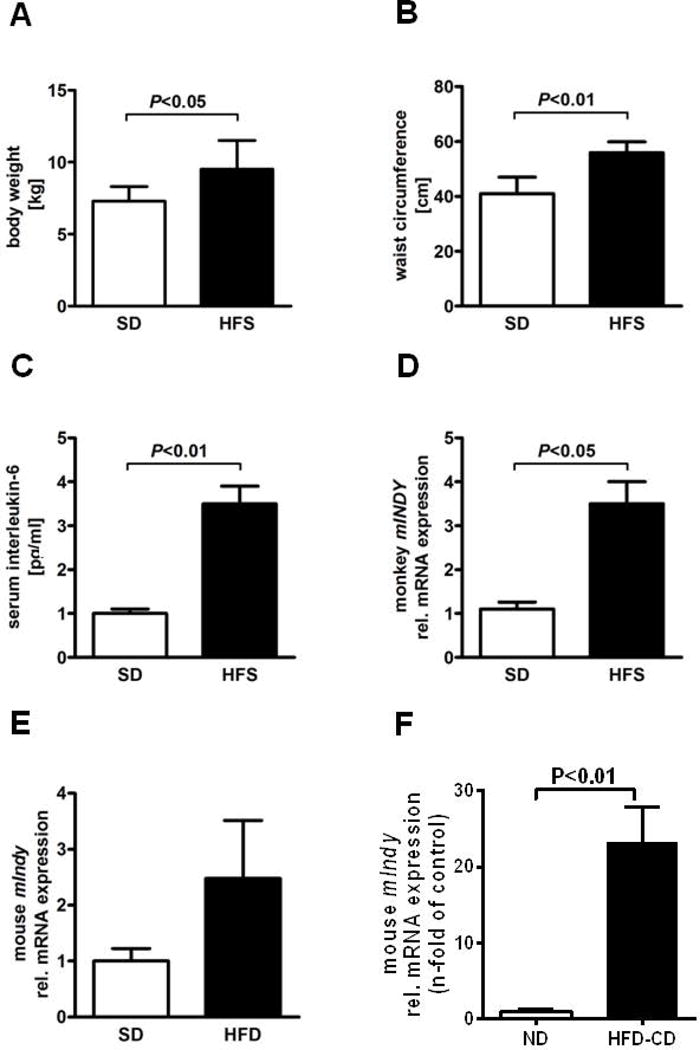
Rhesus monkeys were maintained on a standard diet (SD, n=4) or a high fat, high sucrose diet (HFS, n=10) for 24 months. Monkeys on HFS diet had A) increased BMI, B) larger waist circumference, C) elevated serum IL-6 levels, and D) increased hepatic mIndy expression compared to SD-fed animals. E) In non fasted male 16 week high fat diet fed C57BL/6 mice, hepatic mIndy expression was induced compared to the SD-fed group (n=4–5) F). In a murine NASH model (high fat/methionine low-choline deficient diet=HFD-CD), hepatic mIndy expression was markedly increased after 6 weeks of the intervention.
Similarly, in non-fasted male 16 week high fat diet fed C57BL/6 mice, hepatic mIndy expression was induced 2.5±1.0 fold in comparison to the SD-fed group (SD, n=4–5; 1.0±0.2) (Figure 3E). Interestingly, in a murine NASH model (high fat/methionine low-choline deficient diet=HFD-CD), hepatic mIndy expression was markedly increased in HFD-CD-fed mice (n=4) with > 20-fold increase (Figure 3F) after 6 weeks of the intervention. The increase of mIndy expression in the course of NASH was also time-dependent (data not shown). Since these data suggest that mIndy is regulated by an inflammatory mediator, expression levels of mIndy in murine hepatoctyes and Kupffer cells were analysed independently. In Kupffer cells, mIndy expression was very low, on a normal diet as well as on a HFD (Figure S1A).
The cytokine interleukin-6 (IL-6) is positively associated with hepatic mIndy expression
Other inflammatory mediators might also induce mIndy. Total RNA microarrays suggested that high mIndy expression was associated with activation of some acute phase response components (Table 2). The acute phase response, which serves as a core of the innate immune system, is initiated and mediated by a number of cytokines, most importantly by IL-6 (28). IL-6 independently predicted the degree of hepatic steatosis at a cut off level of 4.81pg/ml (29, 30). Therefore, patients were grouped according to serum IL-6 levels below or above this threshold. In patients with plasma IL-6 levels above this threshold, mIndy expression was twofold higher compared to patients with IL-6 levels below this threshold (P=0.008) (Figure 4A). In a cross sectional comparative analysis of serum from these patients, hepatic mIndy expression positively correlation with circulating IL-6 levels after adjustment for liver fat (r2=0.300; P=0.038, data not shown).
Figure 4. mIndy interacts with IL-6.
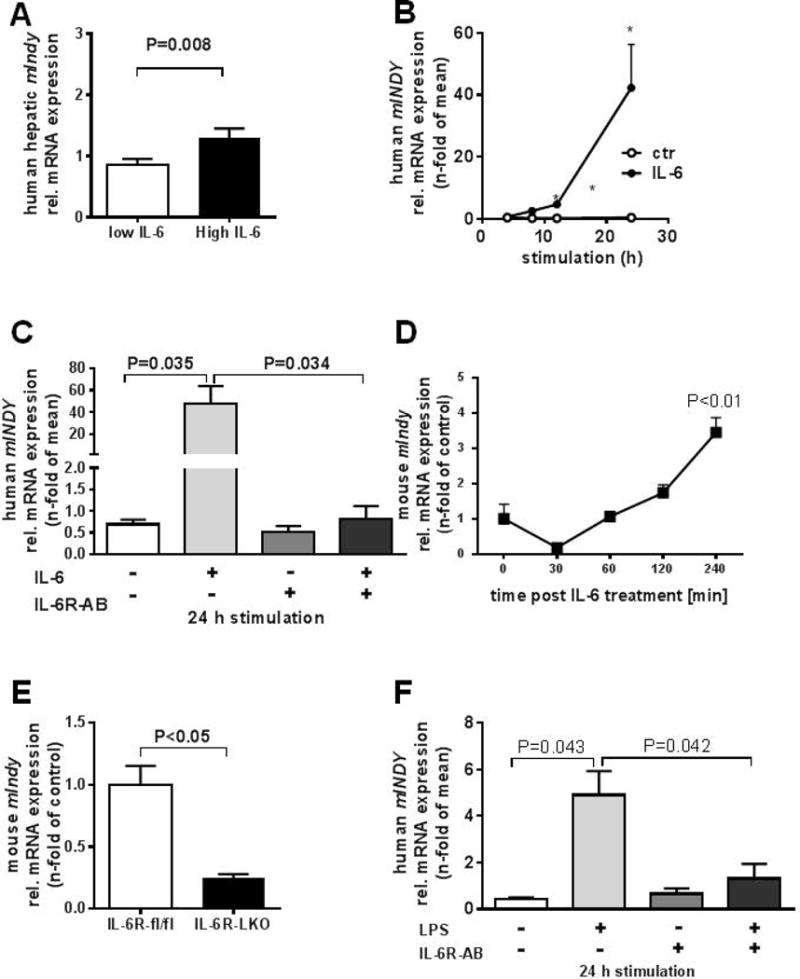
A) IL-6 is a predictor of hepatic steatosis at cut-off level 4.81pg/ml (29, 30). Patients were divided into two groups with IL-6 levels below or above this threshold and mIndy expression in liver samples were determined. B) In human primary hepatocytes, IL-6 induced mIndy expression in a time-dependent manner (n=6). C) Blockade of the IL-6R with the monoclonal IL-6R antibody tocilizumab, completely abolished IL-6-mediated induction of mIndy (n=6). D) I.v. injection of IL-6 into C57BL/6 wildtype mice leads to an increase in mIndy mRNA levels (n=4). E) Targeted, liver-specific deletion of the IL-6R (IL-6RL-KO) (n=3) in mice leads to a decrease in hepatic mIndy expression. F) Human non-parenchymal cells (including Kupffer cells) were co-cultivated with human primary hepatocytes and stimulated with lipopolysaccharide (LPS) with or without tocilizumab (n=3).
IL-6 induces hepatic mIndy expression in primary human hepatocytes and in mice
Treatment of human primary hepatocytes with IL-6 (50ng/ml) induced mIndy expression in a time-dependent manner, with a 42 ± 14 fold increase after 24 h of IL-6 (P=0.031) (Figure 4B). Moreover, blockade of IL-6 signalling with the human monoclonal IL-6 receptor antibody tocilizumab completely abolished IL-6′s ability to increase mIndy expression (Figure 4C).
To assess the ability of IL-6 to recapitulate this effect in vivo, IL-6 was injected intravenously into C57BL/6 wildtype mice. In this setting, hepatic mIndy expression was induced by 3.5±0.3 fold after 240 min (Figure 4D). To independently verify the role of the cognate IL-6 receptor in the regulation of mIndy expression, liver samples of mice with a targeted liver-specific deletion of the IL-6 receptor (IL-6RL-KO) were analyzed, which were generated by crossing IL-6R fl/fl mice to ALFP-Cre recombinase mice as described in (21). Deleting the hepatic IL-6 receptor led to a marked 68±4% reduction in hepatic mIndy expression when compared to IL-6Rfl/fl mice (Figure 4E). Total RNA microarrays from these liver samples indicated that ~900 genes where reduced in IL-6RL-KO mice compared to IL-6R fl/fl. mIndy ranked 20th among the top negatively regulated genes. Together, these data confirm that IL-6 regulates mIndy transcription in both human and mouse livers via the IL-6 receptor.
To determine whether the action of IL-6 can also be attributed to an endocrine-mediated mechanism, human non-parenchymal cells, including Kupffer cells, were co-cultivated with hepatocytes, and then stimulated with lipopolysaccharide (LPS), which provides a strong stimulus for macrophages to secrete an array of pro-inflammatory cytokines, including IL-6. In this setting, mIndy expression was induced 5-fold (Figure 4F), and pre-incubation of the co-culture with tocilizumab largely reduced the induction of mIndy by LPS stimulated cells. When hepatocytes were cultured without non-parenchymal cells, such as Kupffer cells, the LPS response was reduced but not completely presvented (Figure S1B). Together, these data suggest that IL-6, but not other cytokines, induces mIndy in an inflammatory paracrine and endocrine-related setting, because IL-6 receptor blockade largely abolished the response.
Human mINDY is induced via the transcription factor Stat3
5′-RACE experiments were performed to identify the transcription start site of the human mIndy (SLC13A5) gene, with gene-specific primers for reverse transcription in exon 6 and nested PCR primers located in the fifth and third exons. A possible transcription start site located approximately 40 bp upstream of the 5′-end of the published start ATG was found in 5 out of 7 clones that contained sequences continuing into the first exon (Figure 5A). RT-PCR was used to verify the putative transcription start site. Genomic DNA and cDNA were amplified with specific primers, whereby the forward primers were either located 26 bp upstream of the potential transcription start site or at the published ATG start codon. Normalized mINDY template numbers did not differ between cDNA and genomic DNA when using the primer directed at the ATG start codon, whereas the normalized mIndy template number in the cDNA was reduced about 95% versus genomic DNA using the primer upstream of the putative transcription start site. Hence, we tentatively assigned the 5′-upstream region of the latter transcription start site as promoter region. In silico analysis of the 618 bp human mIndy promoter fragment revealed a potential Stat responsive element sequence as shown by MatInspector software. Moreover, a second Stat responsive element was identified with the Stat consensus sequence TT(N4–6)AA (Figure 5A, 5C). Two reporter gene constructs were generated by cloning either a stretch of 618 bp containing both Stat responsive elements or a fragment of 376 bp length including one Stat binding motif upstream of the start ATG of the human mIndy gene, including the putative transcription start site, in front of luciferase in the vector pGL3-basic. Transiently transfection of mouse hepatocytes with these Stat-containing human mINDY promoter constructs increased the luciferase activity about 53 % and 138 %, when either one, or both Stat responsive elements were present, respectively (Figure 5B). To determine which of the elements is indispensible for the induction of mINDY by IL-6, mutations were introduced into both Stat3-binding elements at the 376 bp and 618 bp mIndy promotor fragments by PCR-based site-directed mutagenesis (Figure 5C) Surprisingly, none of the mutations in the potential Stat3-binding sites reduced mINDY promoter activity when stimulated with IL-6 (Figure 5D). These data suggest that other binging sites, an indirect mechanism or non-transcriptional effects contribute to the Stat3-dependent transactivation of the mIndy gene following IL-6 stimulation.
Figure 5. Characterization of the human mIndy promoter.
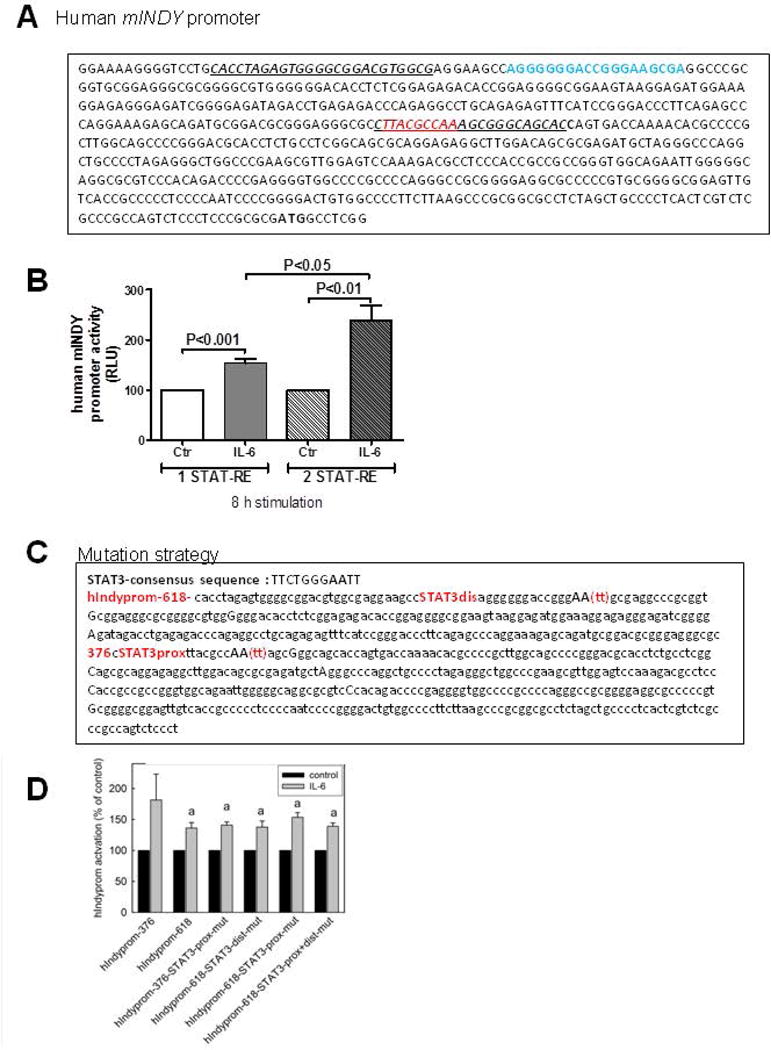
The putative start site and upstream promoter region of the human mIndy gene were determined by 5′ RACE (A) The most frequent transcription start site (bold) was located 40 bp upstream of the ATG start codon. (B) Reporter gene constructs were generated by cloning the 621 bp (prom-621, blue top) or 379 bp (prom-379, red top) sequence upstream of the ATG start codon of human mIndy in front of a luciferase reporter. C) Site directed mutagenesis strategy in 376 bp and 618 bp mIndy promoter fragments. D) Site directed mutagenesis at STAT3-elements 376 bp and 618 bp in the mIndy promoter did not reduce promoter activity. Luciferase activity was determined and compared to empty vector control.
IL-6 mediated metabolic effects via mINDY
mINDY is a transporter of citrate from the interstitial space into the cytosol. Incubation of primary human hepatocytes with IL-6 (50ng/ml) in the presence of physiological concentrations of citrate (10μmol/l) led to a significant increase both in 14C- citrate uptake (Figure 6A) and lipogenesis from citrate (Figure 6B). Deletion of mIndy in primary hepatocytes isolated from mINDY−/− mice (7) completely prevented these effects of IL-6 (Figure 6C and 6D). To determine whether this also has a role in the in vivo setting, we generated hepatocyte specific mINDY KO mice as described in the methods section. In these mice, mINDY expression was completely abolished in the liver, but not other tissues such as adipose tissue, muscle and brain (data not shown) compared to mINDY fl/fl mice. IL-6 was infused for 14 days using mini osmotic pumps as described in (31) into mINDY-L-KO and mINDY fl/fl. mINDY fl/fl mice without IL-6 infusion were used as negative control. One week prior to the experiment, an intravenous line was inserted into the right jugular vein. On the day of the experiment, mice were fasted overnight and infused with 7μCi 14C-Citrate continuously and livers were taken after 4 hours of infusion. Fatty acid synthesis from citrate was determined. Our data show that chronic infusion of IL-6 for 14 days nearly doubled fatty acid synthesis from citrate in livers of mINDY fl/fl mice. Liver specific deletion of mIndy completely prevented IL-6 induced fatty acid synthesis from citrate. These data suggest that mINDY mediates IL-6 induced hepatic lipid synthesis from citrate in mice in vivo (Figure 6E). In line with these findings, HEK293 cell lines with stable 50 fold overexpression of mIndy lead to a concentration dependent uptake of citrate (Figure 1B). Citrate induced lipogenesis increased by more than 130 fold in these cells (Figure S2). Taken together, these results suggest a novel function of IL-6, namely to stimulate hepatocyte citrate uptake and hepatic citrate induced lipogenesis in vivo. The effect of IL-6 is mediated via mINDY.
Figure 6. Cytoplasmic citrate uptake and lipid synthesis.
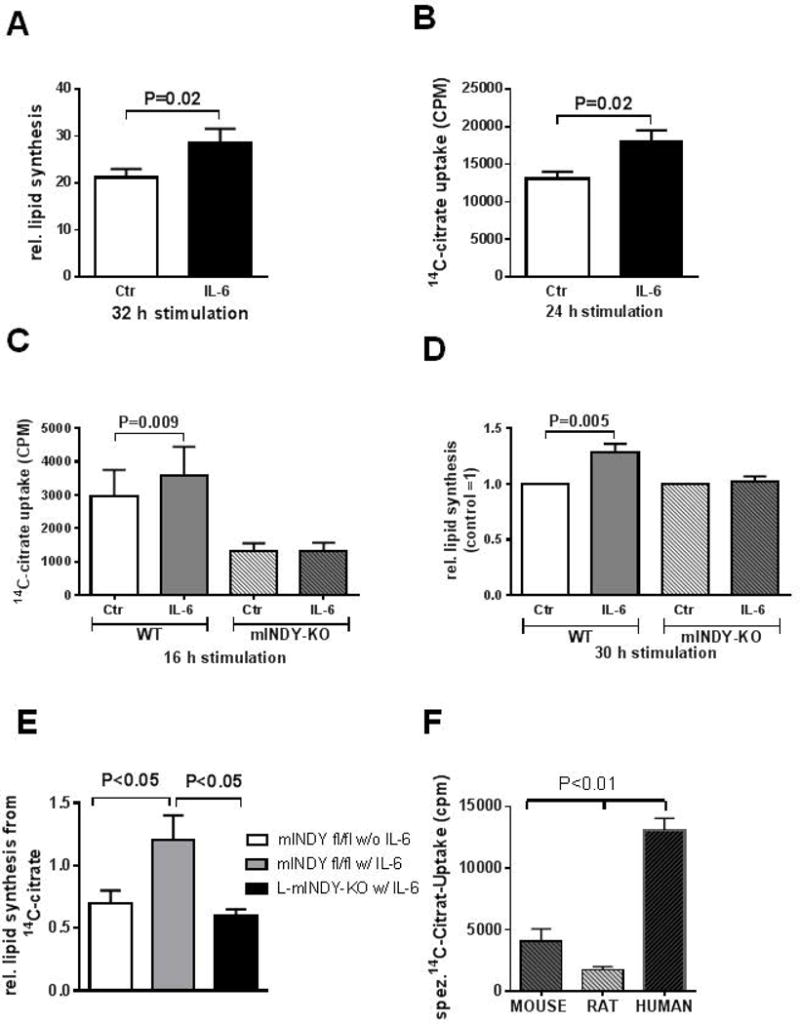
A,B) In human primary hepatocytes, IL-6 increased cytoplasmic influx of citrate (A) and lipid synthesis from citrate (B). C and D) IL-6 increased citrate uptake (C) and lipid synthesis from citrate (D) in a primary culture of hepatocytes from wildtype (WT) mice, but not from mice with deletion of mIndy (mINDY-KO). E) In liver specific mINDY KO mice (mINDY-KO) and mINDY fl/fl control mice, IL-6 was infused for 14 days via miniosmotic pumps. 14C-citrate was then administered iv for 4 hours and fatty acid synthesis was determined in liver of these mice. mINDY fl/fl mice without IL-6 infusion were used as negative controls. IL-6 increased fatty acid synthesis from citrate, an effect prevented when mIndy was deleted in the liver. F) 14C Citrate uptake into primary mouse, rat and human hepatocytes incubated for 24h hours with citrate. Data represent values+/- SEM from primary hepatocytes from independent assays from three different mice, 5 different rats and 5 different human donors.
Significance of human mINDY compared to mouse and rat mINDY
Finally, a recent publication raised questions about the translatability of mINDY as a target for the treatment of metabolic disease from the mouse to the human situation due to differences in uptake properties between species (32). Therefore, we directly measured 14C-citrate uptake into mouse, rat, and human primary hepatocytes incubated with low physiological concentrations of citrate. In this setting, citrate uptake was highest in human primary hepatocytes, followed by mouse and rats (Figure 6F). These data directly show that human mINDY takes up citrate even in low physiological doses and are rather supportive for the notion that human mINDY is a viable target for the treatment of metabolic disease.
DISCUSSION
Reduced Indy gene expression increases life span in lower organisms in most (1, 3, 7, 33) but not in all (34) studies. Moreover, deletion or knock down of Indy or its homologs lead to a lean phenotype by mechanisms akin to caloric restriction, without a reduction in food intake (1, 3, 7, 33). Here, we provide new data showing similar associations in humans. In lean subjects with low amounts of liver fat, mIndy expression was low, and mIndy expression increased with increasing BMI and liver fat content. Furthermore, our data from non-human primates indicate that mIndy is regulated in response to a ‘unhealthy’ high fat, high sucrose diet. In mice, a high fat diet but to a larger extend a high fat- NASH inducing diet (methionine low, choline deficient), also induced mIndy in the liver. Taken together, these data show for the first time that mIndy is regulated by the nutritional state and might therefore play a pathophysiological role in diet induced obesity and associated conditions, such as NAFLD.
Our data suggest that the association between mINDY, obesity and NAFLD was mediated, at least in part, by the cytokine IL-6. Moreover, we provide evidence for human mIndy being a target gene of Stat3 in response to IL-6. Both, the treatment of primary human hepatocytes with IL-6 and intravenous administration of IL-6 in mice markedly increased hepatic mIndy mRNA levels. This gene encodes the plasma membrane citrate transporter mINDY (NaCT, SLC13A5) and, thus, stimulation of human and rodent hepatocytes with IL-6 enhanced cellular uptake of citrate. Citrate, in turn, is a precursor of acetyl-CoA and fatty acid in the liver. mIndy induction in mice in vivo and in murine and human primary hepatocytes in vitro required the cognate IL-6 receptor. Indeed, the monoclonal human IL-6R antibody tocilizumab abolished the effect of IL-6 on mIndy expression and IL-6RL-KO mice displayed significantly lower hepatic mIndy levels. Our data show that two Stat responsive elements by in silico analysis of the human mIndy promoter 5′ upstream of the putative transcription start site, consistent with the notion that Stat3 transcriptional activity induces the human mIndy promoter. Indeed, reporter gene constructs containing the two Stat binding sites were activated by IL-6. However, mutating these Stat-binding sites did not result in reduced mIndy promoter activity. These data suggest that either i) other enhancer-elements, far more upstream of the transcription site mediate the effect, ii) the induction is mediated indirectly via Stat3-dependent induction of other transcription factors, iii) mIndy mRNA is regulated at the posttranscriptional level. More studies are needed to clarify these possibilities.
After stimulation of human non-parenchymal cells, including Kupffer cells, with LPS, inducing an array of cytokines, IL-6 receptor blockade attenuated mIndy induction in hepatocytes. When only hepatocytes were cultured, the LPS response was reduced. These data suggests that IL-6 likely is a cytokine contributing to the induction of mIndy in a pro-inflammatory setting and that the effect is mediated in both, a paracrine and endocrine manner from immune competent cells. Collectively, these data provide evidence for IL-6 to induce the human longevity gene homolog mIndy via Stat3 in human and rodent liver tissues.
IL-6 is increased in obesity, T2D and NAFLD. Obesity and NAFLD are characterized by a chronic inflammatory state (35). The nature of the immune response is unique as compared to an acute inflammatory responses and has a well characterized impact on metabolic regulation (35). In obesity, accumulation of immune cells such as macrophages and T-lymphocytes in the liver and white adipose tissue leads to increased secretion of IL-6 (35). The effect of IL-6 on hepatic lipid metabolism has been investigated with diverging results (20–22, 31, 36–40). IL-6 transgenic animals were protected from HFD induced obesity and fatty liver. IL-6 levels in these animals are, however, several fold higher than IL-6 concentrations in obesity (39). In studies in IL-6-deficient, and IL-6R deficient mice, the course of diet-induced hepatic steatosis support a protective effect of the cytokine in obesity and fatty liver disease (41, 42). In these animals, a compensatory increase in many other cytokines has been reported (21). In contrast, low dose application of murine IL-6 in mice lead to hepatic steatosis (43). Subchronic IL-6 administration in IL-6-deficient mice exacerbated hepatic steatosis by increasing lipogenesis (44). Importantly, neutralization of IL-6 with neutralizing antibodies in HFD fed mice improved glucose tolerance, ameliorated liver fat content (45). Longterm IL-6 incubation of rat hepatocytes increased lipid synthesis (Brass and Vetter et al., 1994), i.p. IL-6 administration in mice stimulated hepatic lipogenesis by increasing hepatic citrate (46) and acetyl-CoA concentrations (19). Taken together, these data show that IL-6 is a pleiotropic cytokine with complex roles in metabolic disease. Our data from patients and non-human primates confirm the association between IL-6 and anthropometric measures of obesity and hepatic lipid content. Moreover, our data show an association between mIndy expression and IL-6 as well as obesity and NAFLD. Our in vitro studies corroborate the observation by showing that IL-6 induced mIndy expression in primary hepatocytes from human and mice, and that IL-6 increased hepatic citrate uptake and hepatic lipogenesis from circulating citrate via mINDY in vivo.
Circulating citrate concentrations range from 50–150 μmol/L. We provide evidence that human mINDY is a high capacity transporter for circulating citrate, which is hardly saturated by circulating concentrations. Thus, mINDY allows a continuous uptake of citrate into hepatocytes. Citrate is a central metabolite, both, in cytosolic and in mitochondrial metabolism, by connecting carbohydrate catabolism and lipogenesis. Citrate is the main carbon source of fatty acid synthesis which has recently been shown to be enhanced by IL-6 (19). Moreover, citrate acts as an allosteric activator of acetyl-CoA carboxylase (ACC). Cytosolic citrate also contributes to NADPH generation via malic enzyme for lipogenesis (47). In line with this mechanism, siRNA mediated mIndy knockdown reduced total fat content in human hepatocytes (24). Knockdown of mIndy in rats using ASOs (14)and in mice using siRNA (48) reduced liver fat content when rats were fed a HFD. Most importantly, inhibition of mINDY using a novel small molecule improved glucose tolerance and ameliorated liver fat content upon high fat feeding in mice (15). Conversely, our data show that overexpression of mINDY in HEK293 cells resulted in an increase in citrate uptake and intracellular fatty acid and sterol synthesis. Moreover, enhancing the activity of citrate transport by mINDY in HepG2 cells augments citrate-induced lipid synthesis (12, 49). Inducing mIndy in obesity and NAFLD, might, thus, contribute to the pathogenic process by enhancing the uptake of citrate which is then used to fuel hepatic lipid synthesis.
In summary, our data show that the longevity gene homolog mIndy was regulated in humans according to the nutritional state and that IL-6 increased human hepatocyte mIndy expression in a paracrine and endocrine manner. Our work describes a promoter sequence of human mIndy that is located upstream of the most frequent transcription start site, which was determined by 5′-RACE. We also describe a novel function of IL-6, namely the induction of citrate uptake into human primary hepatocytes via mINDY and by this mechanism, an induction of lipid synthesis in human primary hepatocytes. Based on our data in humans, future studies are needed to address the important question whether or not the inhibition of human mINDY is beneficial in the treatment of metabolic disease, such as obesity, NAFLD and type 2 diabetes.
EXPERIMENTAL PROCEDURES
Patients
mINDY gene expression in different tissues was measured in the Human Total RNA Panel (BD Biosciences Clontech, Heidelberg, Germany), supplemented with RNA from human liver from our laboratory. For liver samples and hepatocytes, experimental procedures were performed according to the guidelines of the charitable state-controlled foundation Human Tissue and Cell Research, with the informed patient’s consent approved by the local Ethical Committee of the Charité University School of Medicine Berlin (EA2/135/08). All subjects gave written informed consent at least 24 hours prior to surgery. Forty-nine patients were enrolled between February 2009 and March 2010. Details regarding inclusion and exclusion criteria have been published (50), German Clinical Trials Register: DRKS00005450.
Preparation and cultivation of primary human hepatocytes
Tissue samples from liver resections were obtained from patients undergoing partial hepatectomy. The experimental procedures were performed according to the guidelines of the charitable state-controlled foundation Human Tissue and Cell Research, with the informed patient’s consent approved by the local Ethical Committee of the Charité University School of Medicine Berlin (EA2/007/13). Detailed information is given in the Supplemental Section.
Animals
Non-human primates
Studies were performed as described (27). Detailed information is given in the Supplemental Section.
Mice
Generation of mINDY-KO mice and IL-6RαL-KO mice have been described previously (7, 21). Detailed information is given in the Supplemental Section.
Generation of liver specific mIndy KO mice: We previously generated a targeting vector, in which exons 1 to 6 of the SLC13A5 gene were flanked by loxP sites (7). After yielding SLC13A5 (mIndy) fl/fl, these mice were crossed with Albumin-Cre mice {Kellendonk 2000}, and heterozygous animals were further intercrossed to mIndyFL/FL Alb-Cre, i.e., hepatocyte-specific mIndy knockout mice on a C57/BL6 background (mIndy L-KO).
Male C57BL/6J mice were purchased from Charles River Laboratories. First mice cohort were fed with a high fat diet or with a normal chow dor 16 weeks, starting at the age of 4 weeks. Second mice cohort were fed with a high fat-low methionine-choline deficient diet (A06071302 from Research Diets, Brunswick, NJ), used for NASH development, or with a normal fat control diet (A08051501 from Research Diets) for six weeks. Upon completion of the feeding period, a systemic perfusion of the mice with PBS was performed and livers were excised.
Chronic IL-6 treatments: Alzet osmotic pumps (Durect, Cupertino, CA) were used in mIndy L-KO and mINDy fl/fl littermate control mice. IL-6 was infused for 14 days with an infusion rate of 1 μl/h (32 μg/ml hIL-6 in 0.9% NaCl, 0.1% BSA). One week prior to the experiment, an intravenous line was inserted into the right jugular vein. On the day of the experiment, mice were fasted overnight and infused with 7μCi 14C-Citrate continuously. After 4 hours, mice were sacrificed and livers were taken and snap frozen within 1 minute and stored at −80°C for later analysis. Fatty acid synthesis from citrate was determined in these livers as described in (7).
Hepatocyte preparation and cultivation
Density gradient-purified hepatocytes were prepared without the use of collagenase, as described previously (12). Detailed information is given in the Supplemental Section.
Microarray analysis
Microarray data was analyzed using DIANE 6.0, a spreadsheet-based microarray analysis program based on JMP 7.0 from SAS system, as mentioned in our previous studies (7).
Identification of transcription initiation sites by 5′-RACE
Total RNA from two different primary human hepatocyte cultures was used with 5′-RACE system 2.0 (Life Technologies, Eggenstein, Germany) as described previously (12).
Generation of human mINDY promoter constructs
Fragments containing 618 bp or 376 bp of the putative human mIndy promoter were generated from human genomic DNA by PCR using specific primers. Fragments were cloned in the forward orientation into pGL3-basic (Promega, Mannheim, Germany). Luciferase-based reporter assays with transfected primary mouse hepatocytes were performed as described previously and compared against empty vector control transfected hepatocytes (12). Primer oligonucleotides and plasmids are given in the supplemental methods section.
Statistical Analysis
Detailed information is given in the Supplemental Section.
Supplementary Material
Acknowledgments
This work was supported by grant from the German Research Foundation (BI1292/4-2, IRTG 2251), and the German Diabetes Research Center (DZD e.V.), to ALB; and NIH/NIA grants AG16667, AG24353 and AG25277, and a Glenn Award for Research in Biological Mechanisms of Aging to SLH and NIH/NIDDK grants DK 40936, DK 49230, DK-45735, DK-059635 to GIS. ALB and JJ own shares of Eternygen GmbH.
Abbreviations
- Indy
I’m Not Dead Yet
- mIndy
mammalian homolog of Indy (gene, Slc13A5)
- mINDY
mammalian homolog of Indy (protein, NaCT)
- IL-6
Interleukin-6
- STAT-3
signal transducer and activator of transcription 3
- ASO
anti-sense oligonucleotides
- HFD
high fat diet
- PI-3
phosphoinositol 3
- MAPK
mitogen-activated protein kinases
- NAFLD
non-alcoholic fatty liver disease
- PCR
polymerase chain reaction
- Km
Michaelis constant
- Vmax
maximum rate achieved by the system
- HOMA-IR
homeostatic model assessment for insulin resistance
- SD
standard diet
- HFS
high fat high sucrose diet
- NASH
non alcoholic steatohepatitis
- IL-6RL-KO
liver specific IL-6 receptor knockout
- LPS
lipopolysaccharide
- ACC
acetyl-CoA carboxylase
- NADPH
nicotinamide adenine dinucleotide phosphate
References
- 1.Rogina B, Reenan RA, Nilsen SP, Helfand SL. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science. 2000;290:2137–2140. doi: 10.1126/science.290.5499.2137. [DOI] [PubMed] [Google Scholar]
- 2.Inoue K, Fei YJ, Zhuang L, Gopal E, Miyauchi S, Ganapathy V. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem J. 2004;378:949–957. doi: 10.1042/BJ20031261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang PY, Neretti N, Whitaker R, Hosier S, Chang C, Lu D, Rogina B, et al. Long-lived Indy and calorie restriction interact to extend life span. Proc Natl Acad Sci U S A. 2009;106:9262–9267. doi: 10.1073/pnas.0904115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz F, Karadeniz Z, Fischer-Rosinsky A, Willmes DM, Spranger J, Birkenfeld AL. Knockdown of Indy/CeNac2 extends Caenorhabditis elegans life span by inducing AMPK/aak-2. Aging (Albany NY) 2015;7:553–67. doi: 10.18632/aging.100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neretti N, Wang PY, Brodsky AS, Nyguyen HH, White KP, Rogina B, Helfand SL. Long-lived Indy induces reduced mitochondrial reactive oxygen species production and oxidative damage. Proc Natl Acad Sci U S A. 2009;106:2277–2282. doi: 10.1073/pnas.0812484106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knauf F, Rogina B, Jiang Z, Aronson PS, Helfand SL. Functional characterization and immunolocalization of the transporter encoded by the life-extending gene Indy. Proc Natl Acad Sci U S A. 2002;99:14315–14319. doi: 10.1073/pnas.222531899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR, Zhang D, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011;14:184–195. doi: 10.1016/j.cmet.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frankel S, Rogina B. Indy mutants: live long and prosper. Front Genet. 2012;3:13. doi: 10.3389/fgene.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue K, Zhuang L, Maddox DM, Smith SB, Ganapathy V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J Biol Chem. 2002;277:39469–39476. doi: 10.1074/jbc.M207072200. [DOI] [PubMed] [Google Scholar]
- 10.Inoue K, Fei YJ, Huang W, Zhuang L, Chen Z, Ganapathy V. Functional identity of Drosophila melanogaster Indy as a cation-independent, electroneutral transporter for tricarboxylic acid-cycle intermediates. Biochem J. 2002;367:313–319. doi: 10.1042/BJ20021132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gopal E, Miyauchi S, Martin PM, Ananth S, Srinivas SR, Smith SB, Prasad PD, et al. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am J Physiol Gastrointest Liver Physiol. 2007;292:G402–408. doi: 10.1152/ajpgi.00371.2006. [DOI] [PubMed] [Google Scholar]
- 12.Neuschafer-Rube F, Lieske S, Kuna M, Henkel J, Perry RJ, Erion DM, Pesta D, et al. The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes. 2014;63:1048–1057. doi: 10.2337/db13-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mancusso R, Gregorio GG, Liu Q, Wang DN. Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter. Nature. 2012;491:622–626. doi: 10.1038/nature11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pesta DH, Perry RJ, Guebre-Egziabher F, Zhang D, Jurczak M, Fischer-Rosinsky A, Daniels MA, et al. Prevention of diet-induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5) Aging (Albany NY) 2015;7:1086–93. doi: 10.18632/aging.100854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huard K, Brown J, Jones JC, Cabral S, Futatsugi K, Gorgoglione M, Lanba A, et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5) Sci Rep. 2015;5:17391. doi: 10.1038/srep17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322:1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, Mooney RA. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem. 2003;278:13740–13746. doi: 10.1074/jbc.M210689200. [DOI] [PubMed] [Google Scholar]
- 18.Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol. 2011;11:81. doi: 10.1038/nri2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perry Rachel J, Camporez J-Paulo G, Kursawe R, Titchenell Paul M, Zhang D, Perry Curtis J, Jurczak Michael J, et al. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell. 160:745–58. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. 2014;15:423–430. doi: 10.1038/ni.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wunderlich FT, Strohle P, Konner AC, Gruber S, Tovar S, Bronneke HS, Juntti-Berggren L, et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010;12:237–249. doi: 10.1016/j.cmet.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Febbraio MA, Hiscock N, Sacchetti M, Fischer CP, Pedersen BK. Interleukin-6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes. 2004;53:1643–1648. doi: 10.2337/diabetes.53.7.1643. [DOI] [PubMed] [Google Scholar]
- 23.Fuster JJ, Walsh K. The good, the bad, and the ugly of interleukin-6 signaling. EMBO J. 2014;33:1425–1427. doi: 10.15252/embj.201488856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Li H, Garzel B, Yang H, Sueyoshi T, Li Q, Shu Y, et al. SLC13A5 is A Novel Transcriptional Target of the Pregnane X Receptor and Sensitizes Drug-Induced Steatosis in Human Liver. Mol Pharmacol. 2015;87:674–82. doi: 10.1124/mol.114.097287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iacobazzi V, Infantino V. Citrate–new functions for an old metabolite. Biol Chem. 2014;395:387–399. doi: 10.1515/hsz-2013-0271. [DOI] [PubMed] [Google Scholar]
- 26.Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 Diabetes. Hepatology. 2014;59:713–723. doi: 10.1002/hep.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jimenez-Gomez Y, Mattison JA, Pearson KJ, Martin-Montalvo A, Palacios HH, Sossong AM, Ward TM, et al. Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab. 2013;18:533–545. doi: 10.1016/j.cmet.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Galiano D, Sanchez-Garrido MA, Espejo I, Montero JL, Costan G, Marchal T, Membrives A, et al. IL-6 and IGF-1 are independent prognostic factors of liver steatosis and non-alcoholic steatohepatitis in morbidly obese patients. Obes Surg. 2007;17:493–503. doi: 10.1007/s11695-007-9087-1. [DOI] [PubMed] [Google Scholar]
- 30.Pearce SG, Thosani NC, Pan JJ. Noninvasive biomarkers for the diagnosis of steatohepatitis and advanced fibrosis in NAFLD. Biomark Res. 2013;1:7. doi: 10.1186/2050-7771-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52:2784–2789. doi: 10.2337/diabetes.52.11.2784. [DOI] [PubMed] [Google Scholar]
- 32.Zwart R, Peeva PM, Rong JX, Sher E. Electrophysiological characterization of human and mouse sodium-dependent citrate transporters (NaCT/SLC13A5) reveal species differences with respect to substrate sensitivity and cation dependence. J Pharmacol Exp Ther. 2015;355:247–254. doi: 10.1124/jpet.115.226902. [DOI] [PubMed] [Google Scholar]
- 33.Fei YJ, Liu JC, Inoue K, Zhuang L, Miyake K, Miyauchi S, Ganapathy V. Relevance of NAC-2, an Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem J. 2004;379:191–198. doi: 10.1042/BJ20031807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toivonen JM, Walker GA, Martinez-Diaz P, Bjedov I, Driege Y, Jacobs HT, Gems D, et al. No influence of Indy on lifespan in Drosophila after correction for genetic and cytoplasmic background effects. PLoS Genet. 2007;3:e95. doi: 10.1371/journal.pgen.0030095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 36.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 37.Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, Kahn BB, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105:1791–1797. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthews VB, Allen TL, Risis S, Chan MH, Henstridge DC, Watson N, Zaffino LA, et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia. 2010;53:2431–2441. doi: 10.1007/s00125-010-1865-y. [DOI] [PubMed] [Google Scholar]
- 39.Sadagurski M, Norquay L, Farhang J, D’Aquino K, Copps K, White MF. Human IL6 enhances leptin action in mice. Diabetologia. 2010;53:525–535. doi: 10.1007/s00125-009-1580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henkel J, Neuschafer-Rube F, Pathe-Neuschafer-Rube A, Puschel GP. Aggravation by prostaglandin E2 of interleukin-6-dependent insulin resistance in hepatocytes. Hepatology. 2009;50:781–790. doi: 10.1002/hep.23064. [DOI] [PubMed] [Google Scholar]
- 41.El-Assal O, Hong F, Kim WH, Radaeva S, Gao B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol. 2004;1:205–211. [PubMed] [Google Scholar]
- 42.Hong F, Radaeva S, Pan HN, Tian Z, Veech R, Gao B. Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology. 2004;40:933–941. doi: 10.1002/hep.20400. [DOI] [PubMed] [Google Scholar]
- 43.Gavito AL, Bautista D, Suarez J, Badran S, Arco R, Pavon FJ, Serrano A, et al. Chronic IL-6 Administration Desensitizes IL-6 Response in Liver, Causes Hyperleptinemia and Aggravates Steatosis in Diet-Induced-Obese Mice. PLoS One. 2016;11:e0157956. doi: 10.1371/journal.pone.0157956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vida M, Gavito AL, Pavon FJ, Bautista D, Serrano A, Suarez J, Arrabal S, et al. Chronic administration of recombinant IL-6 upregulates lipogenic enzyme expression and aggravates high-fat-diet-induced steatosis in IL-6-deficient mice. Dis Model Mech. 2015;8:721–731. doi: 10.1242/dmm.019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamaguchi K, Nishimura T, Ishiba H, Seko Y, Okajima A, Fujii H, Tochiki N, et al. Blockade of interleukin 6 signalling ameliorates systemic insulin resistance through upregulation of glucose uptake in skeletal muscle and improves hepatic steatosis in high-fat diet fed mice. Liver Int. 2015;35:550–561. doi: 10.1111/liv.12645. [DOI] [PubMed] [Google Scholar]
- 46.Grunfeld C, Adi S, Soued M, Moser A, Fiers W, Feingold KR. Search for mediators of the lipogenic effects of tumor necrosis factor: potential role for interleukin 6. Cancer Res. 1990;50:4233–4238. [PubMed] [Google Scholar]
- 47.Pajor AM. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch. 2014;466:119–302013. doi: 10.1007/s00424-013-1369-y. [DOI] [PubMed] [Google Scholar]
- 48.Brachs S, Winkel AF, Tang H, Birkenfeld AL, Brunner B, Jahn-Hofmann K, Margerie D, et al. Inhibition of citrate cotransporter Slc13a5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol Metab. 2016;5:1072–1082. doi: 10.1016/j.molmet.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inoue K, Zhuang L, Maddox DM, Smith SB, Ganapathy V. Human sodium-coupled citrate transporter, the orthologue of Drosophila Indy, as a novel target for lithium action. Biochem J. 2003;374:21–26. doi: 10.1042/BJ20030827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Docke S, Lock JF, Birkenfeld AL, Hoppe S, Lieske S, Rieger A, Raschzok N, et al. Elevated hepatic chemerin mRNA expression in human non-alcoholic fatty liver disease. Eur J Endocrinol. 2013;169:547–557. doi: 10.1530/EJE-13-0112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.