

Abstract
Renal proximal tubules are targets for toxicity due in part to the expression of transporters that mediate the secretion and reabsorption of xenobiotics. Alterations in transporter expression and/or function can enhance the accumulation of toxicants and sensitize the kidneys to injury. This can be observed when xenobiotic uptake by carrier proteins is increased or efflux of toxicants and their metabolites is reduced. Nephrotoxic chemicals include environmental contaminants (halogenated hydrocarbon solvents, the herbicide paraquat, the fungal toxin ochratoxin, and heavy metals) as well as pharmaceuticals (certain beta-lactam antibiotics, antiviral drugs, and chemotherapeutic drugs). This review explores the mechanisms by which transporters mediate the entry and exit of toxicants from renal tubule cells and influence the degree of kidney injury. Delineating how transport proteins regulate the renal accumulation of toxicants is critical for understanding the likelihood of nephrotoxicity resulting from competition for excretion or genetic polymorphisms that affect transporter function.
Keywords: Transporters, OAT, OCT2, OCTN2, MRP2, MDR1, BCRP, MATE1, MATE2-K
Graphical Abstract
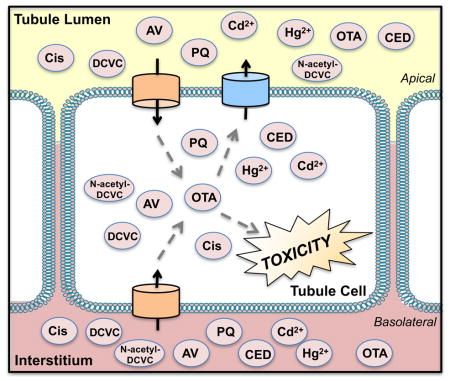
1. Introduction
An important function of the kidney is to excrete metabolic waste products and xenobiotics from the circulation. During this process, the kidney may become vulnerable to toxicity. The pathways for xenobiotic elimination include glomerular filtration and tubular secretion. Drugs smaller than 50 to 65 kDa pass through the glomerulus in the unbound form [1]. Large or charged chemicals are often cleared from the circulation by transporter-mediated renal secretion. Much of the xenobiotic secretion in the kidney occurs in proximal tubules. The uptake of organic anions and cations as well as zwitterions from the blood is the first step in renal secretion. Subsequently, chemicals can undergo active efflux by transporters on the apical brush border membrane where they are added to the ultrafiltrate or by transporters on the basolateral membrane where they re-enter the blood. Chemicals in the ultrafiltrate are either removed to the urine or reabsorbed by apical transporters and returned to the circulation. Depending on the physicochemical properties, reactivity, and propensity for binding to intracellular components, chemicals can also be retained inside tubule cells. Alterations in the rate and/or extent of uptake versus efflux can influence the net intracellular concentrations of drugs, environmental contaminants, and the formation of metabolites. The focus of this review will be primarily on the transport mechanisms and subsequent toxicities of chemicals in proximal tubules.
2. Renal Transporters
2.1. Uptake Transporters
Proximal tubules contain several uptake proteins that are part of the solute carrier (SLC) drug transporter family, including the organic anion transporters (OATs), the organic cation transporters (OCTs), the organic cation/carnitine transporters (OCTNs), and the organic anion transporting polypeptides (OATP). Human OAT1-3 (SLC22A6-8) proteins are largely found on the basolateral membrane of proximal tubules [2–8] although OAT2/Oat2 may also localize to the apical surface of human and rat proximal tubules [9]. OAT4 (SLC22A11) and Oat5 (SLC22A10/19) proteins have been detected on the apical membrane of human and rat renal proximal tubules, respectively [10, 11]. OATs are responsible for the uptake of organic anions, such as beta-lactam antibiotics and nonsteroidal anti-inflammatory drugs [12, 13]. Endogenous dicarboxylate anions (such as alpha-ketoglutarate) are considered to be the driving force for organic anion exchange [14–18]. Among the OCTs, human kidneys are abundantly enriched with OCT2 (SLC22A2) protein on the basolateral membrane of proximal tubules [19, 20]. By comparison, rodent kidneys have two isoforms (Oct1 and 2, Slc22a1 and 2, respectively) with overlapping functions on the basolateral membrane [19]. Substrates for OCT2 include the hypoglycemic drug metformin and the cancer drug cisplatin [21–23]. OCT3 (SLC22A3) is also present in human kidneys and transports cations; however, its role in nephrotoxicity remains largely unknown. OCTN1/Octn1 and OCTN2/Octn2 (SLC22A4 and 5) are both localized to the apical side of rodent and human proximal tubules [24–26]. OCTN1/Octn1 and OCTN2/Octn2 transport carnitine and cation drugs including the antiseizure drug valproic acid and the antibiotic cephaloridine [27, 28]. Among OATPs, OATP4C1 (SLCO4C1) is detected at high levels on the basolateral membranes of proximal tubules [29, reviewed in 30]. Typical OATP substrates include cardiac glycosides and anti-cholesterol statin drugs [29, 31].
2.2. Efflux Transporters
Renal efflux transporters include ATP-binding cassette (ABC) and SLC transporters. Multidrug resistance protein 1 (MDR1, ABCB1), multidrug resistance-associated protein 2, 4 (MRP2, 4, ABCC2, 4), and breast cancer resistance protein (BCRP, ABCG2) belong to the ABC transporter family. Multidrug and toxin extrusion 1 and 2-K (MATE1, 2-K, SLC47A1, 2) are efflux transporters in the SLC transporter family.
MDR1/P-glycoprotein (P-gp) is localized to the apical membrane of rodent and human proximal tubules [32, 33] and actively exports drugs with diverse structures including chemotherapeutic agents, steroids, cardiac glycosides, and immunosuppressant drugs [reviewed in 34, 35]. MRPs function as extrusion pumps for organic anions, including beta-lactam antibiotics, as well as glucuronide and glutathione (GSH) conjugates such as acetaminophen-glucuronide and arsenic-GSH [36–38]. Two isoforms found on the apical side of proximal tubules are MRP2 [37, 39] and 4 [40]. In the kidneys, human MRP4 has 5-fold higher mRNA expression than human MRP2 as well as greater affinity for uptake of the organic anion, para-aminohippurate (PAH) in membrane vesicles (Km values: 160 μM for MRP4 and 5 mM MRP2) [41]. BCRP accepts a wide variety of organic anions as substrates including anticancer drugs, sulfated conjugates, and the Hoechst 33342 dye [42–49]. Human BCRP protein is localized to the apical membrane of proximal tubules but at levels lower than observed in rodent kidneys [50]. MATE transporters are members of the SLC family and function as cation exchangers. Rodent Mate1 as well as human MATE1 and MATE2-K are found on apical membranes and efflux organic cations in exchange for protons [51–54]. In in vitro models, MATE proteins typically act as uptake carriers, in particular, when non-polarized cells are acidified to pH 6.5 using ammonium chloride [55, 56]. Typical substrates for MATE1 include the histamine H2 receptor antagonist, cimetidine, the antidiabetic agent, metformin, and the herbicide, paraquat [55–57]. MATE2-K shares overlapping substrates with MATE1 including cimetidine and metformin [52]. Many cationic drugs are eliminated using renal secretion by the OCT2/MATE pathway. Imbalances in OCT2-mediated uptake or MATE-mediated efflux can result in accumulation of cationic toxins in proximal tubules. More recently, the Na+-dependent phosphate transporter 4 (NPT4, SLC17A3) was identified as an efflux transporter of urate and important for the pathogenesis of gout [58]. NPT4 protein was shown to localize to the apical side of human proximal tubules and transports various organic anions including PAH and ochratoxin A [59].
3. Prototypical Nephrotoxicants
This section provides an overview of chemicals that injure proximal tubules using renal transport mechanisms.
3. 1. Environmental Chemicals
A wide range of naturally-occurring and synthetic chemicals are introduced into the environment daily. These include nephrotoxicants such as halogenated hydrocarbons, herbicides, mycotoxins, and heavy metals that contaminate food and water sources. Regulatory efforts have largely limited human exposure to these chemicals. Nonetheless, understanding the renal absorption and secretion of each chemical provides mechanistic insight into how transporters influence their propensity to cause proximal tubule damage.
3. 1.1. Trichloroethylene
Solvents, such as trichloroethylene (TCE), have contaminated food, water, and air through widespread industrial use. Epidemiological studies have attributed renal cell carcinomas to the long-term exposure of industrial workers to high levels of TCE in Germany [60, 61]. In rats and mice, exposure to oral and inhaled TCE caused renal tubular cytomegaly and karyomegaly as well as the formation of tumors, most notably in male rats due to their higher rate of metabolism [62, 63]. In support of this, metabolism studies identified the reactive and toxic metabolites of TCE responsible for DNA mutations and protein alkylation [64]. In the liver, the majority of TCE is oxidized by cytochrome P450 enzymes [65–68], while a small portion is conjugated to GSH by glutathione S-transferases to form S-1,2,-dichlorovinyl-L-cysteine (DCVC) and subsequently N-acetyl-DCVC [66, 68–73]. Following removal from the liver back to the circulation, DCVC and N-acetyl-DCVC accumulate in proximal tubules where N-acetyl-DCVC can be deacetylated [74]. This is important because DCVC is a substrate for renal beta lyases, which further metabolize cysteine (Cys)-containing compounds into highly reactive species that bind DNA, proteins, and lipids [75–78]. Thus, GSH-conjugated metabolites of TCE are important mediators of renal injury [71]. Rabbits administered DCVC developed dose-dependent morphological necrosis of the proximal tubules and proteinuria [79]. Primary human proximal tubules exhibit apoptosis, necrosis, and proliferation following exposure to DCVC at a range of concentrations from 10 to 1000 μM [80].
Since DCVC is charged at physiological pH, it was predicted that transport proteins would be required for entry into cells [81]. To determine the role of transport in the tubular damage caused by TCE metabolites, rabbit renal cortical slices were directly exposed to DCVC and N-acetyl DCVC [82]. Probenecid, a prototypical Oat inhibitor, prevented N-acetyl DCVC uptake by 80% and reduced cellular damage, indicating the involvement of Oats in basolateral influx. Further studies using rabbit proximal tubules demonstrated that unlabeled DCVC, PAH, and probenecid inhibited the basolateral uptake of radiolabeled DCVC by 70–85% (Figure 1) [83]. Further, PAH and probenecid also attenuated the damage produced by DCVC. Similar results have revealed a role for human OAT-mediated uptake of DCVC. Chinese hamster ovary (CHO) cells stably transfected with hOAT1 exhibited uptake of DCVC that was blocked by PAH (IC50: 208 μM) [84]. Conversely, interference of organic cation transporters using tetraethylammonium (TEA) had no effect on DCVC uptake in rat renal proximal tubules [85], suggesting little interaction with basolateral OCT transporters.
Figure 1. Transport of S-1,2,-dichlorovinyl-L-cysteine (DCVC) and N-acetyl-DCVC.

Organic anion transporter 1 (OAT1) transports DCVC and N-acetyl-DCVC across the basolateral membrane of proximal tubules while amino acid transporters, such as system ASC, A, and L, reabsorb the metabolites of trichlorethylene on the apical surface. N-acetyl-DCVC is removed from renal cells by multidrug resistance-associated protein 2 (MRP2), whereas DCVC is not.
Multiple amino acid transporters have also been implicated in the influx of DCVC using isolated rat proximal tubule cells and porcine kidney LLC-PK1 cell lines [85–87]. In isolated rat proximal tubules, DCVC uptake was prevented using substrates of the system A, ASC, and L transporters, which are proteins responsible for the uptake of amino acids such as alanine, serine, and cysteine [85]. Amino acid transporters along the apical membrane are important for DCVC reabsorption. Rabbit and rat brush-border membrane vesicles were used to assess the apical uptake of DCVC [86, 88]. In rabbit brush-border membrane vesicles, [35S]DCVC was transported by a saturable and Na+-dependent process. This transport could be inhibited up to 80% by several amino acids including L-phenylalanine, L-leucine, and L-cysteine. Based on kinetic transport studies, the authors concluded that inhibition of [35S]DCVC uptake by L-phenylalanine was competitive, which suggested the two chemicals shared a common transport pathway. Basolateral and apical uptake pathways allow entry of DCVC and N-acetyl DCVC into renal cells, which may contribute to the acute and chronic toxicity seen with TCE exposures.
Few studies have explored the role of efflux transporters in the disposition of TCE metabolites. Recently, membrane vesicles generated from mouse proximal tubules were used to demonstrate that Mrp2 transported N-acetyl DCVC, but not DCVC, in an ATP-dependent manner (Km 36.6 μM) [89]. Further, mouse proximal tubule-derived cells endogenously expressing Mrp2 showed marked inhibition (40–50%) of N-acetyl DCVC transport from the basolateral-to-apical direction after transfection with antisense mouse Mrp2 RNA. Transcellular transport of DCVC was not affected by transfection of anti-Mrp2 RNA, consistent with the membrane vesicle data. The authors suggest that N-acetyl DCVC may be consecutively taken up by the basolateral Oat1 transporter and excreted by Mrp2 into the ultrafiltrate. Additional studies are necessary to understand the exit pathway for other metabolites, such as DCVC, from proximal tubule cells.
3.1.2. Paraquat
Paraquat (PQ), a 1,1′-dimethyl-4,4′-bipyridinium cation, was discovered in 1955 and has been widely used as an herbicide since 1962. It is an effective weed killer that becomes inactivated once in contact with the soil leading to relatively low bioaccumulation in the environment. The ability of PQ to elicit toxicity extends to mammalian species as clinical cases of pulmonary and renal injury were reported as early as the 1960s. As PQ is not absorbed through intact skin, the majority of the poisoning cases are due to accidental or intentional ingestions. In 1966, Bullivant reported two of the initial human cases of accidental PQ poisoning by ingestion in New Zealand [90]. The peak concentration of PQ in humans was observed within 2 to 4 hours after ingestion [91]. PQ rapidly distributes into tissues such as the lungs, kidneys, and liver. Pneumotoxicity is the primary cause of mortality because PQ concentrates in the lungs using the polyamine uptake system [92]. Animal studies have also demonstrated that repeated, low doses of PQ can cause degeneration of the dopaminergic neurons in the substantia nigra pars compacta [93].
The kidneys are responsible for eliminating the majority of PQ that is absorbed into the systemic circulation, and are therefore also susceptible to injury [94]. PQ is minimally metabolized and largely excreted in the urine unchanged. Elimination of PQ occurs through a combination of glomerular filtration and active tubular secretion such that the total renal clearance exceeds the glomerular filtration rate (GFR) [95]. Secretion of PQ was found to be concentration- and time-dependent, and inhibited by organic cations in rats [95]. The mechanism of damage to the lungs involves redox cycling which leads to the formation of a PQ radical and superoxide [96]; a similar mechanism has been proposed for renal cells. Administration of PQ to rodents causes acute tubular degeneration, necrosis, and hemorrhage in the kidneys within 17 to 24 h [97–99]. These histopathological changes eventually lead to acute renal failure, which impairs the ability of the kidneys to further excrete PQ [100–102]. Clinically, PQ toxicity presents as reduced GFR, albuminuria, and glycosuria [103]. Ingestion of PQ at doses greater than 20 mg/kg leads to acute tubular necrosis and renal failure in humans [102].
The proximal tubules are the primary site of PQ damage, which suggests that this portion of the nephron is responsible for facilitating its secretion. In vitro studies have investigated the transport of PQ in LLC-PK1 cells. These studies pointed to basolateral OCT transporters as mediators of the uptake of PQ from the blood (Figure 2). In LLC-PK1 cells, the transport of PQ concentration in the basolateral-to-apical direction was saturable with time [104]. Additionally, transport was inhibited by organic cations, including quinine, cimetidine, and TEA, with no change observed in the presence of organic anions. Similar results were demonstrated in primary rat proximal tubule cells [105]. Mechanistic transport studies using HEK293 cells overexpressing human OCT1, 2, or 3 revealed that overexpression of OCT2, but not OCT1 or OCT3, enhanced the accumulation (12-fold) and cytotoxicity (18-fold) of PQ [57]. The cytotoxicity (LC50) of PQ in HEK293-OCT2 cells was observed at 23 μM (compared to 417 μM in control cells). Additionally, uptake of PQ in cells transfected with a common variant of the OCT2/SLC22A2 gene (rs316019; A270S) increased accumulation 3-fold compared to the wild-type reference allele [57]. The SLC22A2 A270S variant has an allele frequency between 10% and 20% in some populations [106]. While this variant is associated with decreased metformin transport in vitro [107], the interaction of PQ with SLC22A2 A270S enhanced PQ transport. Further investigation of this variant is needed to understand the transport differences between substrates.
Figure 2. Transport of Paraquat.

Organic cation transporter 2 (OCT2) is primarily responsible for the accumulation of paraquat (PQ) in proximal tubules whereby redox cycling results in toxicity. Efflux of PQ takes place through the multidrug and toxin extrusion protein 1 (MATE1) and multidrug resistance protein 1 (MDR1).
Apical secretion of PQ in the kidneys declines in the presence of certain cations including TEA and 1-methyl-4-phenylpyridinium (MPP+), suggesting the involvement of the organic cation/H+ exchangers, MATE1 and MATE2-K [105, 108]. In HEK293 cells overexpressing human MATE1 or rat Mate1, uptake of PQ was saturable and time-dependent while empty vector controls showed no activity [57]. The maximal rate of PQ transport in rat Mate1 expressing cells was 2.5-fold higher than human MATE1 but the two orthologs shared similar affinity (Km values). The cytotoxicity of PQ in HEK293 cells overexpressing human MATE1 revealed an LC50 value of 125 μM compared to 717 μM for vector controls [57]. Infusion of 50 mg/kg PQ intravenously in Mate1-deficient mice increased renal PQ concentrations by 147% at 90 min compared to wild-type mice [109]. Additionally, the plasma area-under-the-curve and maximal concentration of PQ in Mate1-null mice were increased 64% and 57%, respectively, compared to wild-type controls. Mate1-null mice administered PQ (20 mg/kg, intraperitoneally) had significantly higher mRNA levels of two kidney injury biomarkers, N-acetyl-Beta-D-glucosaminidase (NAG) and kidney injury molecule-1 (Kim-1) as well as enhanced necrosis and tubular degeneration compared to wild-type mice [109]. A direct role for MATE2-K still needs to be elucidated.
Interestingly, Mate1-null mice had reduced accumulation of PQ in their livers which correlated with an induction of liver Mdr1a mRNA expression [109]. However, this compensatory increase was not seen in the kidneys. Furthermore, administration of dexamethasone (a Mdr1 inducer) to rats reduced the severity of PQ-induced pulmonary damage, providing further anecdotal evidence supporting Mdr1 as an efflux transporter for PQ [110]. While the authors of this study did not evaluate the effect of dexamethasone on renal accumulation of PQ, the urinary excretion of PQ was reportedly unchanged. Subsequent studies have further analyzed PQ transport and cytotoxicity in vitro in human proximal tubule cells and HEK293 cells overexpressing MDR1 as well as in Mdr1a/1b-null mice [111]. Intracellular accumulation of PQ was reduced by 60% in MDR1-overexpressing HEK293 cells. Intracellular accumulation of PQ was increased by 50% in proximal tubule cells treated with PSC833, a specific inhibitor of MDR1 or transfected with siRNA targeted against the MDR1/ABCB1 [111]. Enhanced PQ accumulation after PSC833 treatment corresponded with a 2-fold decrease in cell viability compared to non-treated human proximal tubule cells. Likewise, accumulation of PQ was 750% higher in the kidneys of Mdr1a/1b-null mice after 4 h [111]. Histopathology of kidney sections from Mdr1a/1b-null mice showed increased susceptibility to PQ nephrotoxicity, as manifested by epithelial cell swelling, vacuolation, apoptosis, and necrosis compared to wild-type mice.
3.1.3. Ochratoxin A
A number of fungal by-products, called mycotoxins, cause nephrotoxicity. These include citrinin, aflatoxin B1, ochratoxins and others. In this review, we will focus on ochratoxin due to the abundance of existing data regarding its renal transport. Ochratoxin A (OTA) is a mycotoxin produced by Aspergillus ochraceous, Aspergillus carbonarius, and Pencillium verrucosum [112]. OTA is structurally different from ochratoxin B and C and represents the most prevalent form [113]. Human exposure to OTA results from the consumption of contaminated small grains (barley, wheat, corn), coffee beans, and grapes. OTA can cause various chronic tubule-interstitial syndromes including Balkan nephropathy [114, 115]. Initial reports suggesting that mycotoxins cause nephrotoxicity were based on observations in pigs [115, 116]. Pigs fed mycotoxins consumed large amounts of water, urinated continuously, and had pain near the kidneys. Although the kidneys are an important target for OTA-induced injury, OTA can also cause neurotoxicity, teratogenicity, and immunotoxicity in rodents [117, 118].
OTA has a long circulating half-life (103 h in rats) due to extensive binding to albumin (99%) in the serum [119]. As a result, the glomerular filtration of OTA is minimal [120, 121]. OTA toxicity is observed in proximal and distal tubules due to reabsorption in these portions of the nephron following secretion and to a much lesser extent, filtration [122–124]. Male Wistar rats treated with 0.5 mg/kg of OTA had impaired proximal tubule functioning, glycosuria, and enzymuria [119]. OTA is carcinogenic in mice, with renal tumors particularly evident in male mice [125]. The mechanism of OTA toxicity has been characterized. OTA inhibits protein synthesis in mice by competing with phenylalanine [126]. In addition, OTA can deplete ATP production [127], covalently bind to DNA [128], as well as produce free radicals [129], which collectively contribute to renal cell death.
In vitro studies have shown that OTA interacts with the same transport system as other organic anions (Figure 3) [130]. In vesicles from the renal brush border and basolateral membranes of canine kidneys, OTA preferentially inhibited OAT-mediated transport of PAH, without affecting organic cation transport [131]. OTA produced a dose-dependent depletion of ATP in nephron segments, most significantly in the S2 and S3 segments of the proximal tubules [130]. Probenecid, an organic anion inhibitor, protected against the depletion of ATP by OTA, further supporting the involvement of the organic anion transport pathway. Similar results have been reported in isolated renal proximal tubules from rabbits [132]. OAT1 was identified early as a key transporter of OTA. Rat OAT1 expressed in Xenopus oocytes mediated the sodium-independent uptake of OTA with a Km value of 2 μM [133]. S2 segment proximal tubule cells isolated from transgenic mice stably expressing human OAT1, OAT3, or OAT4 exhibited OTA uptake, which could be competitively blocked by OAT inhibitors (PAH, probenecid, piroxicam) [130, 134]. OAT4 is typically found on the apical membrane of human renal proximal tubules and functions as an organic anion exchanger with the dicarboxylate ion, glutarate [10]. Stable expression of the human OAT4/SLC22A11 gene in mouse proximal tubules increased uptake of OTA [134]. The estimated Km for OAT4-mediated uptake of OTA was 23 μM, suggesting high affinity transport. Organic anions, such as probenecid, penicillin G, aspartame and others, significantly inhibited the influx of OTA by OAT4 [134]. One study has explored the interaction of organic cations with peritubular uptake of OTA in single rabbit renal proximal tubules [135]. Inhibition of OTA was blocked by the organic cation TEA by only 7%. Based on these studies, the basolateral and apical uptake of OTA largely involves OAT proteins.
Figure 3. Transport of Ochratoxin A.

Various organic anion transporters (OATs) are involved in the uptake of ochratoxin A (OTA) into proximal tubules including OAT1, 3, and 4. Other uptake transporters include the organic anion transporting polypeptide 1a1 (Oatp1a1). Efflux of OTA likely occurs through the multidrug resistance-associated protein 2 (Mrp2), breast cancer resistance protein (BCRP), and Na+-dependent phosphate transporter (NPT4).
Other organic anion transporters that participate in the renal uptake of OTA include rat Oatp1a1 (formerly known as Oatp1) and rat Oat5. Oatp1a1 is located on the apical surface of the rat proximal tubules [136]. Rat Oatp1a1-mediated uptake of OTA in stably transfected CHO cells yielded a Km of 29 μM [137]. Rat Oat5 expressed in Xenopus laevis oocytes enabled the sodium-independent uptake of OTA (Km 0.34 μM, Vmax 0.26 pmol/oocyte/h) [138]. Immunohistochemical staining showed that Oat5 was localized to the apical membrane of proximal tubules in the corticomedullary regions as well as the S2 and S3 segments. Oat5 may have a counter ion specificity that is unique when compared to other Oats since its function was unaffected by addition of dicarboxylate-type ions and urate to the transport media for Xenopus oocytes expressing mouse Oat5 [138]. Oat-K1 is a kidney-specific, organic anion carrier found on the apical side of the proximal straight tubules of the rat kidney, which enables the pH-independent reabsorption of OTA [139]. OTA was found to be reabsorbed in all nephron segments to varying degrees and was independent of pH in male Wistar rats [140]. Moreover, the reabsorption of OTA was inhibited by sulfobromopthalein, a substrate of Oat-K1 in microinfused rat proximal tubules.
Multiple efflux transporters have been investigated for their ability to remove OTA from cells. Membrane vesicles isolated from HEK cells expressing recombinant human MRP efflux transporters were used for indirect and direct measurement of OTA transport. OTA inhibited MRP1- and MRP2-mediated transport of PAH with IC50 values of 53 μM and 58 μM, respectively [141]. Additionally, OTA was found to be a direct substrate of MRP2 in HEK-MRP2 cells with a net ATP-dependent transport rate of 1.2 nmol/mg protein/min at 200 μM. In another study, MDR1, MRP2, and BCRP were studied as potential efflux transporters of OTA using intestinal Caco-2 cells [142]. Caco-2 cells secreted OTA to the apical compartment in a concentration-dependent manner while no effect on the absorptive ability was seen. Treatment with MRP2 and BCRP inhibitors, MK571 (35% inhibition) and Ko143 (50% inhibition), respectively, reduced OTA secretion. The MDR1 inhibitor, PSC833, did not affect OTA transport. These studies suggest the involvement of MRP2 and BCRP in OTA efflux. OTA has also been shown to be a substrate for other ABC-transporters such as ABCA8, an anion pump and transporter of lipids [143]. However, the importance of this transporter is uncertain as only faint expression has been detected in the kidneys of mice and its localization in polarized epithelial cells is not currently known [144]. Nonetheless, further studies in kidney cells are needed to confirm the ability of MRP2, BCRP, and ABCA8 to export OTA and reduce the extent of toxicity.
NPT4 is a Na+-dependent phosphate transporter of urate that is also found on the apical side of human renal proximal tubules. Xenopus oocytes expressing human NPT4 demonstrated time- and concentration-dependent transport of OTA (Km 802.8 ± 137.3 μM, Vmax 518.7± 76.4 fmol/h per oocyte) [145]. The effect of different ionic conditions on NPT4-mediated transport was also studied. While a lack of extracellular Na+ did not affect OTA transport, NPT4-mediated OTA transport was sensitive to changes in plasma membrane potential and elevation of extracellular K+ concentrations. The presence of an outward Cl− gradient also did not influence OTA transport.
3.1.4. Heavy Metals
Many heavy metals including lead, mercury, cadmium, copper, and uranium can be toxic to the kidneys. Studies evaluating the transport of heavy metals and their metabolites have shed some light on the mechanism of toxicity. Many of the heavy metals specifically target and damage the proximal tubules, which suggests a role for active transport processes unique to this region of the nephron. In this section, we highlight two of the more commonly studied heavy metal nephrotoxicants, cadmium and mercury.
3.1.4.1. Cadmium
Cadmium (Cd2+) is a divalent heavy metal pollutant to which people are exposed from various sources including cigarettes, occupational dust and fumes, food, and water [reviewed in 146]. Much of the Cd2+ absorbed into the body is through inhalation (20–50%), whereas only 5% is absorbed by ingestion [147, 148]. Cd2+ accumulates in the body due to the absence of an efficient excretory mechanism from storage sites and a long half-life of 10 to 30 years [148]. Cd2+ exposure can lead to numerous conditions including neurological diseases, renal dysfunction, pulmonary complications, diabetes, cancer, immunosuppression, bone disorders, and cardiovascular diseases [149–156]. The mechanism(s) of Cd2+ toxicity are still unclear but are thought to involve the indirect generation of superoxide and hydroxyl radicals inside cells. Such oxidative stress subsequently stimulates cellular proliferation and alters apoptotic and DNA repair mechanisms [157].
The kidney is one of the main targets for Cd2+ toxicity. Injury to proximal tubular cells leads to tubular necrosis, interstitial fibrosis, and glomerular epithelial cell hypertrophy in rats [158]. With chronic exposure, tubular damage can progress to overt renal Fanconi syndrome, denoted by glycosuria, aminoaciduria, and phosphaturia, that may culminate in renal failure [159, 160]. An interplay between the liver and kidneys has been implicated in the toxicity of Cd2+. In studies using rats and chickens, the majority of circulating Cd2+ was bound to albumin and transported to the liver where low molecular weight (less than 7 kDa), high-affinity metal-binding proteins called metallothioneins (MTs) were induced [161, 162]. In the liver, Cd2+ can dissociate from albumin and bind to MT to form Cd2+-MT complexes that are returned to the systemic circulation. Circulating Cd2+-MT complexes are freely filtered through glomeruli and reabsorbed by tubular cells. Alternatively, Cd2+ can bind to other thiol-containing molecules such as Cys or GSH and be excreted into the bile [163–165].
There is some evidence that basolateral transporters participate in the proximal tubule uptake of Cd2+. Studies in Chinese hamster ovary (CHO-K1) cells transfected with rabbit Slc22a1(Oct1) or Slc22a2 (Oct2) genes have shown that Cd2+ dose-dependently inhibits the uptake of the OCT substrate [3H]-TEA (IC50 values: Oct1 96 μM and Oct2 207 μM) (Figure 4) [166]. In this study, TEA (10 mM) also reduced Cd2+ (3 μM) cytotoxicity in Oct1- and Oct2-expressing CHO-K1 cells. Furthermore, administration of TEA to rats was found to decrease Cd2+ accumulation by 7-fold in the kidneys. These data indirectly suggest that the OCT substrate TEA competes with Cd2+ for transport by Oct1 and 2. Similarly, human OCT2 has been shown to transport Cd2+ [167]. The possibility of other basolateral transporters to participate in Cd2+ uptake has yet to be explored. Although there are no direct studies exploring OAT uptake of Cd2+, studies have shown that increased basolateral uptake of cadmium (60–70%) in rat kidneys occurred after co-administration of cadmium with Cys or GSH [165]. It is conceivable that similar to other metals such as mercury, Cys- and GSH-conjugated Cd2+ can be transported across the basolateral plasma membrane by OAT/Oats.
Figure 4. Transport of Cadmium.

Cadmium (Cd2+) is bound to albumin in the circulation after absorption. In the liver, Cd2+ forms a complex with metallothioneins (MT) that are released into the circulation. Cd2+ also binds to thiol-containing groups such as L-cysteine (Cys) and glutathione (GSH). After filtration, the Cd2+-MT complexes are reabsorbed by megalin and cubulin into the proximal tubules on the apical side and undergo degradation to ionic Cd2+. Cd2+ also displays “molecular mimicry” and uses divalent metal transporters (DMT1, divalent metal ion transporter-1) and zinc/iron proteins (ZIP8/14, zinc/iron-regulated transporter 8/14) for reabsorption. Amino acid transporters, such as system B0,+, may also reabsorb Cys-conjugates of Cd2+ on the apical plasma membrane. Organic cation transporter 2 (OCT2) (and possibly Oct1 in rodents) may also influx Cd2+ across the basolateral side of tubule cells. Multidrug resistance protein 1 (MDR1) and multidrug and toxin extrusion protein 1 and 2-K (MATE1/2-K) contribute to the limited Cd2+ that is secreted.
There is a significant evidence suggesting that Cd2+ enters proximal tubules across the apical surface of the plasma membrane. Following glomerular filtration, the Cd2+-MT complex is taken up by megalin and cubilin, which are multiligand, endocytic receptors [168]. Megalin and cubilin are highly enriched on the brush border of proximal tubules and mediate various physiological functions such as the reabsorption of proteins and vitamins and regulation of calcium homeostasis [reviewed in 169, reviewed in 170]. Upon binding, these proteins are transferred to lysosomes where they are degraded to release ionic Cd2+, thereby initiating cellular damage processes [168, 171]. The ability of amino acid transporters to influx Cys-conjugates of Cd2+ was also recently studied [164]. Amino acids that are substrates of system B0,+, including L-cystine, and L-arginine, as well as L-methionine reduced the uptake of Cys-conjugated Cd2+ (Cys-S-Cd-S-Cys) by 50%. Amino acids that are not substrates of system B0,+, such as L-aspartic acid and L-glutamate, did not modulate the uptake of Cys-conjugated Cd2+. Given these data, the authors suggested that Cys-conjugates of Cd2+ may mimic entry of amino acids through transporters systems such as system B0,+, in order to gain access across the apical membrane following filtration.
The apical transport of Cd2+ may also occur through a process of “ionic and molecular mimicry” due to size and charge similarity with essential divalent elements including iron (Fe2+), zinc (Zn2+) and calcium (Ca2+) [reviewed in 172, 173]. Transporters for these metals also recognize Cd2+ and consequently, the systemic levels of some essential metals affect the extent of Cd2+ accumulation and distribution. Cd2+ can be reabsorbed from the tubule lumen through zinc/iron-regulated transporters (ZRT/IRT) also called ZIP proteins. ZIP8, ZIP14, and divalent metal transporter 1 (DMT1) proteins mediate the absorption of Cd2+ across the apical side of mouse proximal tubule cells [174, 175]. Studies have shown that silencing ZIP8, ZIP14, and DMT1 genes significantly reduced the uptake of Cd2+ [174]. Deficiencies in Fe2+, Zn2+, or Ca2+ can increase Cd2+ accumulation as well as susceptibility to kidney toxicity [176]. One study revealed that low stores of Fe2+ enhanced the intestinal absorption of dietary Cd2+ in both mice and humans [177]. In mice fed low calcium diets, more Cd2+ accumulated in the kidneys following oral CdCl2 compared to normal diet fed controls. Additionally, enhanced levels of intestinal MT-1 mRNA and calcium transporter 1 mRNA were observed [178]. This suggests that more Cd2+ is reabsorbed intestinally through the deficient essential metal transporter and bound to MT, which then preferentially accumulates in the kidneys. Wang et al. (2010) studied the acute effects of Fe2+, Zn2+, or Ca2+ on apical Cd2+ transport in isolated perfused rabbit proximal tubules [164]. The presence of Fe2+, Zn2+, and Ca2+ in the lumen inhibited uptake of Cd2+ by 42%, 48%, and 27–69%, respectively. These studies showed that essential metals play several roles in modulating Cd2+-induced cellular injury. They may competitively inhibit renal Cd2+ uptake and during periods of deficiency, there is enhancement of Cd2+ absorption from the intestine, which leads to accumulation in the kidney.
Due to the high protein binding of Cd2+ in the liver and kidneys (approximately 75% in humans) [179], the rate of excretion into urine is low. Despite the limited renal excretion of Cd2+, research has demonstrated a potential role for active transporters in its removal. Initially, it was shown that Cd2+ increased levels of Mdr1 protein and function in the brush border membrane of the S1 and S2 segments of rat proximal tubules [180]. Incubation of proximal tubule cells cultured from rats with 10 μM cadmium chloride (CdCl2) led to the time-dependent increase in the levels of Mdr1 protein over 72 h that protected against Cd2+-induced apoptosis [180]. The Mdr1 inhibitors, verapamil, cyclosporine A, and nifedipine, enhanced susceptibility to Cd2+ toxicity in rat, LLC-PK1 (Porcine Kidney) and OK (Opossum Kidney) proximal tubule cell lines [180–182]. Additionally, MDR1 inhibitors decreased the basolateral-to-apical transport and increased the apical-to-basolateral transport of Cd2+ in LLC-PK1 cells overexpressing human MDR1 [182]. These results supported the participation of rat and human Mdr1/MDR1 in the apical efflux of Cd2+from the kidney. More recent data support an alternate mechanism for MDR1’s ability to protect against Cd2+toxicity. Using MDR1-MDCK cells, it was demonstrated that MDR1 could efflux ceramide, a lipid by-product of cellular damage that contributes to renal cell apoptosis [183].
The ability of a Cd2+/H+ antiporter to efflux Cd2+ has been revealed in LLC-PK1 cells and brush border membrane vesicles from rat kidneys [184, 185]. A reduction in the pH of the culture media (from 7.4 to 5.5) decreased Cd2+ accumulation by 60% and increased the efflux of Cd2+ by 15% in LLC-PK1 cells treated with CdCl2. An organic cation/H+ antiport system, which may be the same as the Cd2+/H+ antiporter, has been further investigated for its ability to transport Cd2+ in the presence of other cations [186]. In LLC-PK1 cells, treatment with N′-methylnicotinamide (NMN), cisplatin, and TEA (substrates of H+ antiport system) increased intracellular Cd2+ accumulation in a concentration-dependent manner and decreased transcellular Cd2+ transport. Enhanced accumulation of Cd2+ in the presence of cations was largely due to a modest inhibition of apical Cd2+ efflux presumably due to competition. The accumulation of Cd2+ was only elevated by 25% at the highest cation concentration tested (100 μM). However, the simultaneous inhibition of basolateral transport systems, such as Oct2, was not examined which could have contributed to the limited Cd2+ accumulation. While not demonstrated experimentally, it is likely that the above mentioned organic cation/H+ antiport system is Mate1/2-K, which was shown to efflux the same substrates, NMN, cisplatin, and TEA in a pH-dependent manner [52, 54, 187]. Recently, it was shown that mouse Mate1 as well as human MATE1 and MATE2-K could transport Cd2+ and influence its cytotoxicity in HEK kidney cells [188].
3.1.4.2. Mercury
Mercury is a heavy metal pollutant that enters the environment through natural or industrial processes and is found in air, soil, and water [189]. The different forms of mercury in the environment encountered by humans include elemental mercury, organic mercury (methylmercury; CH3Hg+) and inorganic mercury (Hg2+). Hg2+ is most likely to accumulate in the kidneys [190–194] and is a major contributor to mercury-induced renal damage, whereas methylmercury is more likely to cross the blood-brain barrier and cause significant adverse effects to the brain [189]. However, both exposures are important to nephrotoxicity as methylmercury can also accumulate in the kidney and/or be converted to Hg2+ within the body as evidenced by the time-dependent accumulation of Hg2+ in subcellular fractions of the kidneys, livers, and brains of rats exposed to methylmercury [195–198]. The renal uptake of Hg2+ is rapid since as much as 50% of a nontoxic dose can be found in rat kidneys within a few hours after exposure [199]. Both Hg2+ and methylmercury target the proximal tubules [196–198, 200–203]. Mercury has high affinity for sulfhydryl groups [204]; therefore in the plasma and tissues, Hg2+ ions are found conjugated to thiol-containing molecules such as GSH, Cys, homocysteine (Hcy), N-acetylcysteine (NAC), and albumin. This strong affinity also contributes to the mechanism of toxicity where binding of mercuric ions to critical proteins can initiate cell injury pathways. Binding of mercuric ions to thiol-rich proteins, such as metallothionein, also increases intracellular retention [205–207]. The Cys-conjugated species of both inorganic and organic mercury seem to be the primary form found in proximal tubular cells. A number of conjugated mercury species are substrates for uptake and efflux transporters, playing a role in susceptibility to mercury toxicity.
The basolateral uptake of Hg2+ and methylmercury into proximal tubules occurs primarily by OATs (Figure 5). Torres et al. (2011) showed that Oat1-null mice exposed to Hg2+ exhibit less renal injury as assessed by histology and biochemical endpoints including blood urea nitrogen (BUN) and serum creatinine (SCr) compared to wild-type mice [208]. Furthermore, differences in the enrichment of Oat1 and Oat3 proteins in male and female rats resulted in sex-dependent susceptibility to mercury-induced kidney damage [209]. Female rats have 5- and 3-fold lower levels of renal Oat1 and Oat3 protein, respectively. In turn, mercury chloride (HgCl2)-exposed female rats exhibited less renal impairment than male counterparts. Human OAT1 has also been implicated in Hg2+ uptake. Transfection of Madin-Darby canine kidney (MDCK) cells with human OAT1 decreased cell viability after exposure to mercuric conjugates of Cys, NAC, or GSH (Cys-Hg2+, NAC-Hg2+, GSH-Hg2+) compared to control, nontransfected cells [210, 211]. Interestingly, in this cellular system, the most toxic conjugate was found to be NAC-Hg2+ (LC50 40 μM), followed by Cys-Hg2+ (LC50 80 μM) while the least toxic was GSH-Hg2+ and did not reach an LC50 at the concentrations tested (0–100 μM). For comparison, it should be noted that the LC50 of HgCl2 was 16 μM. Additionally, oocytes from Xenopus laevis injected with cRNA for human OAT1 showed preferential uptake of Cys-Hg2+, but not the GSH-Hg2+ [210]. PAH inhibited the uptake of Cys-Hg2+ in a dose-dependent manner. Methylmercury has also been shown to be transported by OATs. Treatment of male Swiss OF1 mice with the OAT inhibitor, probenecid, reduced tubular damage from methylmercury by 80–90% [212]. Xenopus laevis oocytes expressing rat Oat1 showed increased uptake of NAC-conjugated methylmercury (Km: 31 μM) but not Cys or GSH-conjugates. However, rat Oat3-expressing oocytes did not transport NAC, GSH-, or Cys conjugates of methylmercury [213]. Additional studies in MDCK cells expressing human OAT1 showed that the human isoform could mediate the uptake of Cys-, Hcy, and NAC-S-conjugates of methylmercury [214–216]. The series of in vivo and in vitro studies point to the involvement of basolaterally localized OATs/Oats in the uptake and renal toxicity of Hg2+, methylmercury, and their NAC, Cys, and GSH conjugates.
Figure 5. Transport of Mercury.

Mercury (Hg2+) and methylmercury (CH3Hg2+) bind to sulfhydryl groups including thiol-containing molecules such as glutathione (GSH), cysteine (Cys), homocysteine (Hcy), and N-acetylcysteine (NAC). These conjugated mercury species are taken up basolaterally by organic anion transporters (OAT) and apically by amino acid transporters including L -cysteine, system B0,+, and Na+-dependent low-affinity L -systems. Efflux transporters on the brush border membrane include multidrug resistance-associated protein 2 (MRP2) and breast cancer resistance protein (BCRP).
After GSH-Hg2+ is broken down by γ-glutamyltransferase on the apical brush border membrane to form the Cys-Hg2+conjugate, amino acid transporters mediate reabsorption into proximal tubule cells. This was evidenced by the reduced rate of apical disappearance flux (JD) of mercuric conjugates following inhibition of γ-glutamyltransferase in perfused S2 segments of rabbit proximal tubules [217]. Additionally, when L-lysine or cycloleucine were added to isolated, perfused S2 segments of rabbit proximal tubules, the transport rate was reduced by 50%. Most likely, multiple amino acid transporters participated in the apical influx of dicysteinylmercury including the L-cysteine, system B0,+, system B0, system ASC, and other Na+-dependent and independent systems [218]. System B0,+, a transporter of various amino acids including methionine, is localized to the apical membrane of proximal tubule cells. Similarly, Cys and Hcy conjugates of methylmercury are structurally similar to the amino acid methionine. It was proposed that conjugates of methylmercury might mimic the entry of methionine into cells [219]. The uptake of Cys, GSH, and NAC conjugates of methylmercury in perfused rabbit proximal tubule segments was inhibited by the amino acids L-methionine and L-cysteine. Compared to control Xenopus laevis oocytes, transport of the Cys-methylmercury conjugate was 10-fold greater in oocytes expressing the mouse system B0,+ [220]. Further studies showed that the amino acid transporter mediated uptake of Cys-methylmercury across the apical side of the plasma membrane. In rat proximal tubule-derived NRK-52E cells grown in monolayers, uptake of Cys-methylmercury was attributed to a Na+-independent amino acid transporter inhibited by L-cysteine, L-leucine, L-isoleucine, and L-phenylalanine on the apical side. PAH, probenecid, and glutarate did not affect uptake, suggesting apical uptake was not mediated by Oats [214].
Mercury is known to be a substrate of the apical efflux transporters, Mrp2 and Bcrp. Exposure of killifish proximal tubule cells to HgCl2 reduced Mrp2 transport of fluorescein methotrexate (a substrate for MRP2) [221]. Since Hg2+ accumulation was not measured in this study, this effect could be secondary to cellular toxicity. However, other results suggest that reduced Mrp2 transport by HgCl2 may be the direct result of inhibited transporter function. Treatment of MDCK cells with HgCl2 showed a dose-dependent induction of Mrp1 and Mrp2 mRNA and protein levels compared to vehicle-treated MDCK cells [222]. HgCl2 inhibited the growth of MDCK cells, which was further enhanced in the presence of the Mrp inhibitor, MK-571 [222]. Moreover, MK-571 increased the intracellular accumulation of Hg2+ (30% higher), pointing to a role for Mrps in the efflux of this metal [222]. Compared to control vesicles, there was 1.5- to 2-fold greater transport of Cys-S-conjugates of Hg2+ and methylmercury in membrane vesicles expressing MRP2. In vivo, TR− (transport deficient) rats treated with HgCl2, Cys-S-Hg-S-Cys, and Cys-methylmercury, but not methylmercury, demonstrated slightly higher renal burden and lower mercury concentrations in the urine (4- to 10-fold) relative to control rats [223]. Recent studies examined the role of Mrp2 in the site-specificity of Hg2+-induced nephropathy [224]. TR− rats lacking Abcc2/Mrp2 expression and Mrp2-null mice exhibited different patterns of nephropathy compared to control Wistar rats and FVB mice, respectively. TR− rats treated with 1.5 and 2.25 μM/kg Hg2+ and Mrp2-null mice treated with 18.5–19.5 μM/kg Hg2+ displayed cellular injury and death in the S1 and S2 segments of proximal tubules, whereas Wistar rats and wild-type mice exhibited S3 segment injury. Evidence of the differential pattern of injury and accumulation of Hg2+ is indicative of Mrp2 heterogeneity and heavy metal handling along the nephron in rodents. It was proposed that proximal portions of tubules may secrete Hg2+ into the lumen via Mrp2 for subsequent reabsorption in the distal S3 segment where the toxic effects are exerted [224].
Interestingly, polymorphisms in the ABCC2 gene that encodes MRP2 were associated with the differential excretion of mercury in subjects from Indonesia, the Philippines, Tanzania, and Zimbabwe [225]. In the subgroups with the highest exposures (found in Zimbabwe), subjects with the MRP2/ABCC2 variant (rs1885301; G/A) variant (N=146) had 166% higher concentrations of mercury in the urine than those with the wild-type allele (p=0.027). The G > A polymorphism (rs1885301) results in loss of a transcription-factor binding site (Fetal Alz-50 clone 1, FAC1) and gain of another site (FAST-1 SMAD interacting protein) in the 5′ UTR. Although the functional role of these changes is unknown, they may enhance MRP2 activity. By comparison, carriers of a non-synonymous polymorphism in MRP2/ABCC2 (rs2273697; G/A) had 2.2-fold lower urinary concentrations of mercury, suggesting decreased function. However, the influence of these SNPs on MRP2 mRNA and protein levels in kidneys as well as susceptibility to Hg2+-induced nephrotoxicity remains unclear. Nonetheless, these data further support a role for MRP2 in the excretion of mercury conjugates from the kidneys.
In addition to Mrp2/MRP2, the Bcrp transporter can participate in the efflux of mercuric species from proximal tubules. Uptake of the Cys-conjugate of Hg2+ in inside-out membrane vesicles expressing mouse Bcrp was 5-fold higher than control vesicles [226]. Furthermore, Bcrp-null rats treated with HgCl2 have 1.5-fold higher renal concentrations of Hg2+ than wild-type rats. Hematologic, hepatic, and fecal burden of Hg2+ were also greater in Bcrp-null rats compared to wild-type rats. Histopathologic analysis, SCr, and BUN indicated increased severity of renal injury in Bcrp-null rats at the doses (1.5 and 2 μM/kg HgCl2) tested. At lower doses (0.5 and 1.5 μM/kg), the decrease in urinary excretion of Hg2+ in Bcrp-null rats correlated well with greater renal accumulation. However, Bcrp-null rats treated with a higher dose (2 μM/kg) showed an increase in urinary excretion of Hg2+ compared to corresponding wild-type rats. This may be due to enhanced kidney damage or compensation by other efflux proteins such as Mrp2. Differences in the pattern of mercury-induced nephropathy in transport deficient rodent models pointed to distinct roles for Bcrp and Mrp2 in Hg2+ efflux. Unlike Mrp2-deficient mice and rats, which exhibit mercury-induced necrosis on the outer cortex (where S1 and S2 segments are located), Bcrp-null rats exposed to nephrotoxic doses of mercury chloride exhibited necrosis on the inner cortex and the outer strip of the outer medulla (S3 segments). These disparate findings are likely due to the segmental differences in the distribution of the Bcrp and Mrp2 proteins as well as their intrinsic ability to transport conjugated forms of mercury. Collectively, these studies point to multiple mechanisms for the elimination of Hg2+ from proximal tubule cells.
The uptake of mercury across the brush border membrane can be inhibited leading to a lower extent of tubular injury. Application of the heavy metal chelator, DMPS, on the apical surface of isolated rabbit proximal tubules enhanced the urinary secretion of methylmercury [219]. DMPS diminished the apical influx of Cys-methylmercury suggesting that DMPS conjugates of mercury formed in the tubule lumen are not reabsorbed. Others have demonstrated that DMPS also plays a role in reducing the proximal tubular uptake and toxicity mediated by Hg2+. In isolated perfused rabbit proximal tubule cells, addition of DMPS to Hg2+at the basolateral or apical surface resulted in very little to no uptake of Hg2+ or toxicity [227]. These studies are evidence that Hg2+ or methylmercury bound to DMPS are poor substrates for proximal tubule transporters.
In vivo, Mrp2 plays a significant role in the secretion of Hg2+ and methylmercury from proximal tubule cells after administration with DMPS [228–230]. TR− rats that lack Abcc2/Mrp2 expression exhibited less Hg2+ in urine and feces and greater renal burden of Hg2+ than control Wistar rats following injection with HgCl2, conjugated Hg2+ (Cys-Hg2+ or Hcy-Hg2+), or methylmercury. Once treated with DMPS, the renal content of Hg2+ was reduced and urinary excretion was increased, however to a lesser extent in TR- rats (7-fold higher Hg2+ in total renal mass, 2.5-fold lower Hg2+ excretion) [228–230]. To further understand the role of other human MRP isoforms in the disposition of DMPS-S-conjugates of Hg2+, inside-out membrane vesicles expressing human MRP4 were used [231]. Although human MRP2-containing vesicles transported DMPS-S-conjugates of Hg2+ (1.7-fold), uptake into human MRP4-containing vesicles did not differ from controls. These data indicate that at least a portion of DMPS-induced elimination of Hg2+ and methylmercury occurs through MRP2/Mrp2. In addition, Bcrp may also play a role in the elimination of DMPS-Hg2+. Compared to control vesicles, the DMPS-conjugate of Hg2+ accumulated to 5-fold greater concentrations in inside-out membrane vesicles expressing mouse Bcrp [226]. Further studies are needed to determine the affinity of human BCRP for the DMPS-conjugates of methylmercury and to assess the in vivo ability of BCRP to facilitate urinary secretion.
3.2. Pharmaceuticals
An important dose-limiting adverse effect of a number of pharmaceuticals is nephrotoxicity, which in turn reduces their overall utility as therapeutic agents. This section will highlight several examples of therapeutic classes and/or specific drugs that are associated with direct tubular injury due to transporter-mediated accumulation.
3.2.1. Beta-lactam antibiotics
Beta-lactam antibiotics include penicillins, cephalosporins, and carbapenems. Structurally, these medications are cyclic dipeptides containing lactam rings that react with bacterial carboxypeptidases to prevent cell wall synthesis [232]. The antibacterial spectrum of naturally-occurring beta-lactams was broadened by modifying the main structure to enhance reactivity [233]. However, increased reactivity in the early development of cephalosporins and carbapenems resulted in a greater propensity for nephrotoxicity [234]. Nephrotoxic beta-lactams can produce structural damage and proximal tubule necrosis in rabbits and various other laboratory species within 24 hours [235–237]. The mechanism(s) underlying tubular injury include the ability of beta-lactams to actively accumulate in renal proximal tubular cells, acylate proteins [238], and/or induce lipid peroxidation (for cephaloridine) [239–241]. The side group substitutions of beta-lactams greatly affect transport rates and protein reactivity and hence contribute to the varying degrees of nephrotoxicity [242, 243]. Very few cephalosporins cause tubular damage at therapeutic doses. Cephaloridine, a first generation cephalosporin which is nephrotoxic at therapeutic doses, is no longer used. However, cephaloridine remains a classic example of how transport can be a critical determinant of drug-induced renal injury.
Single, high doses of cephaloridine lead to proximal tubule necrosis in rats, dogs, and other various laboratory species [244–246]. This injury was prevented by administration of probenecid along with cephaloridine [247]. Further studies revealed that a lower concentration of cephaloridine was found in cortical regions of rabbit kidneys after probenecid treatment compared to animals treated with cephaloridine alone [246]. These data indicated that cephaloridine interacted with Oat transporters on the basolateral membrane. Mouse S3 proximal straight tubule cells stably expressing rat Oat1 were used to study the uptake of radiolabeled PAH and cephaloridine. Oat1-expressing cells treated with cephaloridine exhibited dose-dependent inhibition of [14C]-PAH uptake, 2-fold increase in [14C]-cephaloridine uptake, decreased viability, and increased lipid peroxidation compared to vector-expressing cells (Figure 6). Probenecid treatment diminished the effect of cephaloridine on cell viability as well as lipid peroxidation by 1.5- to 2-fold [248]. Other studies have also shown involvement of rat Oat3 in cephaloridine-induced nephrotoxicity [249]. Proximal tubule cells stably expressing rat Oat3 (both basolaterally and apically) exhibited higher [14C]-cephaloridine uptake and cytotoxicity that was reversed by probenecid treatment. More recent studies showed that cephaloridine inhibited organic anion uptake in proximal tubule cells stably expressing human OAT1-3 in a competitive manner (Ki OAT1: 0.74 mM, OAT2: 2.09 mM, OAT3: 2.46 mM) [12, 250]. Cephaloridine (5 mM) decreased the viability of proximal tubule cells expressing OAT1, OAT2, or OAT3 (30–50%), which could be reversed by probenecid in OAT1- and OAT3-expressing cells. These studies indicated that both rat and human OATs were involved in the uptake of cephaloridine, and possibly other cephalosporins, across the basolateral plasma membrane.
Figure 6. Transport of Cephaloridine.

On the basolateral side of the plasma membrane, cephaloridine (CED) is taken into tubule cells by the organic anion transporters 1, 2, and 3 (OAT 1, 2, 3) whereas OAT4 and the organic cation/carnitine transporter 2 (OCTN2) mediate uptake on the apical side. Efflux transporters for cephaloridine are largely unknown and may involve multidrug and toxin extrusion protein 1 and 2-K (MATE1/2-K).
In studies by Takeda et. al., (2002) exploring the interaction of human OATs with cephalosporins, cephaloridine competitively inhibited organic anion uptake 31-fold in isolated proximal tubule cells expressing the human OAT4 transporter (Ki OAT4: 3.63 mM) [12]. Cephaloridine also decreased the viability of OAT4-expressing proximal tubule cells by 40%, which was modestly reversed by probenecid. OAT4 and additional transporters, such as the organic cation/carnitine transporter OCTN2, present on the apical surface likely reabsorb cephaloridine from the tubular lumen. Human OCTN2-expressing HeLa cells exhibited cephaloridine uptake compared to vector-transfected cells where cephaloridine accumulation was undetectable [28]. While other cephalosporins were reported to interact with the human PEPT1 and rat Pept2 peptide transporters, cephaloridine does not. More recently, mouse Octn2 was also shown to play a role in the apical uptake of cephaloridine. Experiments with juvenile visceral steatosis mice, which have a functional deficiency in the Octn2 gene, showed increased renal clearance of cephaloridine [251]. Furthermore, direct uptake of cephaloridine was shown in Xenopus laevis oocytes expressing mouse Octn2 (Km: 3 mM). This accumulation of cephaloridine could be inhibited by the Octn2 substrates, carnitine (IC50: 14.8 μM) and TEA (0.68 mM).
Early studies suggest that cephaloridine might also interact with an apical cation transport system. Administration of a cationic transport inhibitor, cyanine 863, before or after cephaloridine administration to rabbits resulted in 2- to 3-fold greater susceptibility to nephrotoxicity [252]. PAH and TEA accumulation were measured in slices from rabbit renal cortical tissue and accumulation of each chemical was reduced 2-fold by pretreatment with cyanine 863. However, the cationic substrates quinine and NMN showed a small or no effect, respectively, on the extent of nephrotoxicity or PAH/TEA accumulation. The authors suggested that the efflux of cephaloridine was mediated by a cationic transporter, and that inhibition of this system increased susceptibility to toxicity. However, since direct measurements of intracellular concentrations of cephaloridine were not made in this study, it was hard to conclude that a definite interaction exists. Additional studies using cells expressing specific cationic transporters, such as the MATEs, are needed to understand the efflux of cephaloridine and related beta-lactams from tubular cells.
It is important to note apparent species differences that have been reported [253, 254]. Cephaloridine inhibited NMN transport across basolateral membrane vesicles isolated from rabbits; however, no differences were observed in basolateral membrane vesicles isolated from rats or dogs. Conversely, cephaloridine did inhibit NMN transport in brush-border membrane vesicles from rat and dogs (56%), suggesting similar interactions with apical cation transporters in these species.
There are very few known efflux mechanisms for cephaloridine. Future studies are unlikely given the infrequent clinical use of cephaloridine and increased availability of minimally nephrotoxic beta-lactam alternatives. Cephaloridine did not affect MRP4-mediated ATP-dependent uptake of [3H]dehydroepiandrosterone in membrane vesicles [255]. However, other cephalosporins tested in this study did show inhibitory effects. Uptake studies of membrane vesicles from Sf9 cells expressing rat Mrp2, rat Bcrp, and human MRP2 revealed increased uptake of high molecular weight cephalosporins by all three transporters [36]. These apical efflux transport pathways (Bcrp and MRP2/Mrp2) may be important for the toxicity of other renally-cleared cephalosporins which warrants further study in proximal tubule cells.
3.2.2. Antiviral Drugs
Several potent medications are being used successfully to treat viral infections caused by cytomegalovirus, herpes virus, and retroviruses. Antiviral drugs differ in their mechanism of action. Nucleotide analogs, including cidofovir, adefovir, and tenofovir, are structurally-related to endogenous nucleotides. This structure allows the drugs to be incorporated into viral DNA by reverse transcriptase [256]. Nucleotides, unlike nucleosides, contain phosphonate groups, allowing them to skip the phosphorylation step before incorporation into viral DNA [reviewed in 257]. Despite their efficacy, the clinical use of nucleotide analogs can be limited by their propensity to cause nephrotoxicity. Thirty-five and sixty percent of the intravenously administered doses of cidofovir and adefovir, respectively, are renally secreted and have been shown to accumulate in proximal tubules at higher concentrations than in other organs [258, 259]. Dose-limiting nephrotoxicity can be observed in nearly 15% of treated patients [260]. Renal-related side effects reported in patients prescribed antiviral agents include Fanconi syndrome, progressive declines in kidney function, diabetes insipidus, and tubular dysfunction [260]. Most often, nephrotoxicity tends to occur in patients with pre-existing renal disease and those administered antiviral drugs for prolonged periods or prescribed concomitant nephrotoxic agents [260]. The pathophysiological changes are multifactorial and include acute tubular necrosis possibly through direct mitochondrial damage [261].
Cidofovir, adefovir, and tenofovir are anionic drugs efficiently transported by human OAT1 (Figure 7) [262–264]. Overexpression of human OAT1 in CHO cells caused a 500-fold increase in the extent of cytotoxicity for these drugs [262–264]. Likewise, human OAT3 also mediated the time-dependent uptake for adefovir, cidofovir, and tenofovir [265]. It should be noted that OAT1 was found to be a more efficient transporter of adefovir, cidofovir, and tenofovir compared to OAT3. Other antiviral drugs including acyclovir and ganciclovir were also revealed as substrates of OAT1, OAT2, and OCT1 [266, 267]. By comparison, OCT2 did not mediate the uptake of adefovir, cidofovir, tenofovir, acyclovir, ganciclovir, or penciclovir [267]. Further studies compared the potency of adefovir, cidofovir, and tenofovir for hOAT1-mediated transport. From most potent to least potent are: adefovir (Km: 23.8 μM), tenofovir (Km: 33.8 μM), cidofovir (Km: 58.0 μM) [268]. Cytotoxicity was also compared in CHO cells expressing hOAT1: adefovir (IC50: 1.4 μM), cidofovir (IC50: 3.0 μM), and tenofovir (IC50: 21.0 μM). These data may partially explain the lower incidence of nephrotoxicity associated with tenofovir, compared to adefovir [269]. However, other factors such as reactivity to intracellular components, likely also play a role in susceptibility to injury.
Figure 7. Transport of Antiviral Drugs.

Anionic antiviral (AV) drugs are transported into renal cells by the organic anion transporters 1 and 3 (OAT1, 3) and removed from proximal tubules using the multidrug resistance-associated proteins 2 and 4 (MRP2, 4).
The ability of tubular cells to efficiently efflux nephrotoxic antiviral drugs is a critical determinant of the extent of toxicity. To date, a variety of renal transporters have been investigated for their ability to efflux nucleotide phosphonates. Using intact renal proximal tubules from killifish, Miller et al. (2001) tested the interaction of adefovir and cidofovir with fluorescent substrates of efflux transporters [270]. Fluorescein-methotrexate was used as the substrate of Mrp2 whereas a fluorescent cyclosporine A analog was used as the substrate of Mdr1. Both adefovir and cidofovir reduced the apical accumulation of fluorescein-methotrexate. Notably, cidofovir inhibited transport activity more potently than adefovir (adefovir IC50: 50 μM; cidofovir IC50: 10 μM). However, even at high concentrations (250 μM), adefovir and cidofovir failed to alter the transport of the fluorescent cyclosporine A analog, possibly indicating no interaction with Mdr1. Although these studies indicate a potential interaction of these chemicals for the same efflux transport pathway, direct measurements or transport kinetics were not assessed. Further studies pointed to a limited role for the human MRP2 transporter in adefovir and cidofovir efflux. Inverted membrane vesicles expressing human MRP2 and BCRP showed no ATP-dependent uptake of [3H]-adefovir or [3H]-cidofovir [271].
The MRP4 transporter is likely more relevant than MRP2 for the apical efflux of nucleotide phosphonate drugs in humans. Initial evidence revealed that MRP4 was overexpressed in an adefovir-resistant human T-lymphoblast CEM cell line [272]. Indeed, the overexpression of the human MRP4 and MRP5 transporters in HEK293 cells conferred resistance to cytotoxicity for a range of acyclic nucleotide phosphonates including adefovir, but with the exception of cidofovir [273]. Inverted membrane vesicles prepared from HEK293-MRP4 cells accumulated [3H]adefovir and [3H]tenofovir in a concentration-dependent manner (Km > 1 mM) which was inhibited by the MRP4 substrate, dehydroepiandrosterone sulfate [271]. By contrast, no transport of [3H]cidofovir was observed. Imaoka et al. (2007) also examined the transport of antiviral agents by Mrp4 in vivo [271]. Mrp4-null mice had reduced tubular secretion and exhibited a 2-fold increase in the kidney accumulation of adefovir and tenofovir compared to wild-type mice. Consistent with data obtained from membrane vesicles, the kidney concentration of [3H]cidofovir did not differ between Mrp4-null and wild-type mice. Taken together, these data point to MRP4 as a low affinity transporter for the secretion of adefovir and tenofovir, but not cidofovir. Although nucleotide phosphonates have similar structures, it is evident from these studies that transporter affinities among this class of medications may contribute to the varying degrees of toxicity reported.
3.2.3. Cisplatin
Cisplatin (cis-diamminedichloroplatinum II) is an antineoplastic drug used to treat solid tumors in the lung, testis, ovary, cervix, bladder, and colon. The primary mechanism for cisplatin’s efficacy is the ability to bind DNA and form inter- and intrastrand adducts, which prevent cancer cell replication. Nephrotoxicity is a major dose-limiting side-effect of cisplatin along with peripheral neuropathy, ototoxicity, vomiting, and diarrhea. Up to a third of patients treated with high doses (100 mg/m2) of cisplatin suffer from nephrotoxicity [274]. Upon entry inside proximal tubule cells, cisplatin undergoes biotransformation to aquated intermediates which leads to intercalation of DNA and adduct formation on proteins. Early work demonstrated that cisplatin was actively transported into renal tubules [275], but it was not until recently that researchers identified the transporters responsible for this process.
Early in vitro studies highlighted the existence of a transport system for cisplatin on the basolateral side of proximal tubules (Figure 8) [276–278]. Application of cisplatin to the basolateral side of MDCK cells resulted in a loss of epithelial monolayer integrity, as measured by transepithelial electrical resistance [279]. Furthermore, the OCT2 inhibitor, cimetidine, was able to partially reverse the drop in electrical resistance in cisplatin-treated cells. Other studies demonstrated that cisplatin inhibited the uptake of a fluorescent OCT2 substrate, 4-[4-(dimethylamino)styryl]-N-methylpyridinium (ASP+) into HEK293 cells stably transfected with the human OCT2 gene and in freshly isolated human proximal tubules [22]. Increased platinum accumulation and injury in HEK293 cells expressing OCT2 was inhibited by cimetidine in a concentration-dependent manner [22, 280]. Assessment of additional basolateral organic ion transporters, including OCT1, OCT3, OAT1, and OAT3, showed that only OCT1 had the potential to increase cisplatin cytotoxicity; however, to a lesser degree than OCT2. Uptake transporters on the apical side of proximal tubules, such as OCTN1 and OCTN2, did not increase transport of cisplatin suggesting they are not involved in reabsorption [280].
Figure 8. Transport of Cisplatin.

Cisplatin is taken up into the proximal tubules by the organic cation transporter 2 (OCT2) and also possibly by copper transporter 1 (CTR1). Cisplatin is removed by efflux using the multidrug and toxin extrusion protein 1 (MATE1). Cisplatin also further conjugates with glutathione (GSH) in the proximal tubules and has been proposed to undergo secretion by the multidrug resistance-associated protein 2 (MRP2).
Building on prior in vitro studies, Filipski et al. (2009) utilized double Oct1/2-null mice and demonstrated a 50% reduction in the urinary excretion of cisplatin [281]. Reduced secretion of cisplatin in the Oct1/2-null mice correlated with less acute renal tubular necrosis as well as minimal changes in BUN and SCr. There are conflicting reports regarding the importance of genetic polymorphisms in OCT2/SLC22A2 on cisplatin disposition and acute kidney injury in patients. Several studies have observed an association between a loss-of-function genetic variant at position 808G>T (rs316019) and protection from kidney injury in cisplatin-treated cancer patients (n=78 Filipski et al.; n=53 Iwata et al.; n=123 Zhang et al.) [281–283]. The rs316019 variant was not associated with changes in the systemic clearance or plasma concentrations of cisplatin. However, a separate study of patients with esophageal cancer (n=95) treated with cisplatin and 5-fluorouracil revealed no relationship between changes in eGFR in individuals who were homozygous wild-type (808GG) compared to individuals carrying the variant allele (808GT+TT) [284]. Further studies are needed to validate this polymorphism as a predictor of likelihood for cisplatin kidney injury.
Recent research has also suggested that the copper transporter 1 (CTR1) is an additional basolateral uptake carrier for cisplatin. Down-regulation of endogenous CTR1 protein levels in HEK293 cells using copper chloride (CuCl2) (100 μM) or siRNA significantly lowered cisplatin uptake [285]. Cellular apoptosis and necrosis were decreased in both HEK293 cells and rat proximal tubule cells treated with cisplatin after CTR1/Ctr1 knockdown. Finally, CTR1 knockdown and concomitant treatment with cimetidine resulted in the additive reduction of cisplatin uptake in HEK293 cells [285], suggesting a combined nephroprotection due to inhibition of OCT2 and CTR1 function. However, there are conflicting studies which dispute a role for CTR1 in cisplatin uptake. In a separate study, HEK cells overexpressing human CTR1 did not increase the rate of cisplatin influx [286]. The cisplatin-sensitive ovarian cancer cell line A2780 has 2-fold higher levels of human CTR1 and greater cisplatin influx compared to the resistant cell line A2780CP. However, siRNA knockdown of CTR1 in A2780 cells did not affect cisplatin uptake; it should be noted that OCT2 levels following CTR1 knockdown were not quantified since induction of this alternate uptake carrier could shift the uptake pathway for cisplatin [286]. While evidence suggests a potential interaction between cisplatin and CTR1, in vivo studies are needed to determine its role in the drug’s renal secretion.
As described above, the transepithelial transport of organic cations is often mediated by uptake through OCT2 that is coupled to secretion by the MATE1 and 2-K transporters across the brush border membrane. There are conflicting data regarding the use of intracellular acidification to demonstrate MATE1 uptake of cisplatin. In HEK293 cells overexpressing rat Mate1, human MATE1, or human MATE2-K, there was no difference in cisplatin uptake compared to empty vector cells under acidified intracellular conditions using 30 mM ammonium chloride for 20 min [23]. Subsequent studies showed, however, that treatment of mouse Mate1-HEK293 cells with 30 mM ammonium chloride did markedly increase transport and accumulation of platinum 4-fold compared to empty vector cells [187]. Differences in pH conditions, which were not always reported, could have led to the discrepant findings.
The involvement of Mate1 in cisplatin secretion has also been confirmed in vivo. Mate1-null mice treated with 15 mg/kg cisplatin intraperitoneally exhibited higher cisplatin plasma and renal concentrations and greater susceptibility to nephrotoxicity compared to wild-type mice [187]. Additionally, the Mate1 inhibitor pyrimethamine enhanced renal injury in wild-type mice. Another study was conducted to assess the association between a genetic polymorphism in MATE1 (rs2289669; G>A) along with the systemic exposure and toxicity of cisplatin (doses of 60 to 80 mg/m2) in patients (n=53) with advanced carcinomas [282]. The results showed no significant differences in SCr, eGFR, BUN, or plasma concentration of cisplatin among patients with and without this variant. Although there is clear evidence that cisplatin is a substrate for MATE1, the extent of MATE1 involvement in cisplatin secretion in humans requires further study.
Another important transporter associated with cisplatin extrusion from proximal tubules is Mrp2. Early studies demonstrated that Mrp2 could efflux glutathione conjugates of cisplatin from mouse cancer cells [287–289]. Overexpression of the human MRP2 transporter was shown to increase cisplatin resistance 10-fold in HEK293 cells [287] and reduce cisplatin accumulation by 30% in LLC-PK1 cells [288]. An initial in vivo report suggested that the disposition and extent of toxicity of cisplatin were similar in Mrp2-null and wild-type mice, as demonstrated by the urinary excretion of platinum and kidney histology [290]. However, in the study by Sprowl et al. (2012), accumulation of platinum in mouse kidneys was not assessed [290]. More recently, Wen et al. (2014) found that cisplatin inhibited transport of the MRP2/Mrp2 substrate 5(6)-carboxy-2′,7′-dichlorofluorescein by 70 to 80% in vesicles expressing mouse Mrp2 and human MRP2 [291]. Furthermore, Mrp2-null mice treated with cisplatin exhibited 2-fold higher BUN levels, a greater increase in SCr concentration, and more severe proximal tubule degeneration and necrosis compared to wild-type mice. Kidney concentrations of platinum in Mrp2-null mice were 30% higher than those observed in wild-type mice. Transgenic expression of the human MRP2/ABCC2 gene in Mrp2-null mice reduced the accumulation of platinum in the kidneys to levels observed in wild-type mice without altering plasma platinum levels. Nephrotoxicity was also reduced in humanized MRP2 mice compared to Mrp2-null mice [291]. These data show direct in vivo evidence of the involvement of MRP2 in the efflux of platinum species and nephroprotection.
4. Summary
Transporter proteins in the kidneys coordinate the secretion and/or reabsorption of structurally-diverse chemicals including heavy metals, herbicides, mycotoxins, and drugs. Because of their critical role in the overall disposition of xenobiotics, transporters regulate the renal exposure and subsequently the toxicity of xenobiotics. Beyond the chemicals described in this review, other commonly prescribed nephrotoxic drugs as well as environmental toxins share the same renal transport pathways responsible for clearance. These include additional antibiotics, immunosuppressant drugs, and other heavy metals. The use of in vitro models of transporter-stably expressing cell lines and renal proximal tubule cells have been a routine experimental approach thus far. These experimental tools have been instrumental in studying both transporter-mediated toxicities as well as drug-drug interactions and have been helpful alternatives to using in vivo models. Scientists in the field have a growing understanding of the potential for different carriers and pumps to contribute to chemical secretion. However, more research is needed to assess the redundancy of transporters, the influence of genetic variants on the disposition and toxicity of nephrotoxicants, the intracellular unbound concentrations of nephrotoxicants as well as the molecular mechanisms that regulate transporter expression and function in the kidneys. New renal tubule models, including a growing availability of primary human proximal tubule cells, cells transfected with multiple transporters, and kidneys-on-a-chip [292], hold great promise to address these critical gaps in the literature and provide novel approaches to screen for drug interactions and nephrotoxicity.
Acknowledgments
Funding.
This work was supported by the National Institutes of Health Institute of Diabetes and Digestive and Kidney Diseases [Grant DK080774, DK093903], National Institute of Environmental Health Sciences [Grants ES020522, ES005022, ES007148], and a Predoctoral Fellowship in Pharmaceutical Science from the American Foundation for Pharmaceutical Education.
Abbreviations
- ABC
ATP-binding cassette
- ASP+
4-[4-(dimethylamino)styryl]-N-methylpyridinium
- BCRP
breast cancer resistance protein
- BUN
blood urea nitrogen
- CdCl2
cadmium chloride
- CHO
Chinese hamster ovary
- CTR1
copper transporter 1
- Cys
cysteine
- DMT1
divalent metal transporter 1
- DMPS
2,3-dimercaptopropane-1-sulfonic acid
- GFR
glomerular filtration rate
- GSH
glutathione
- Hcy
homocysteine
- IRT
iron-regulated transporters
- JD
disappearance flux
- KIM-1
kidney injury molecule-1
- MDCK
Madin-Darby canine kidney
- MT
metallothioneins
- MATE
multidrug and toxin extrusion protein
- MRP
multidrug resistance-associated protein
- MDR1
multidrug resistance protein 1
- NAG
N-acetyl-Beta-D-glucosaminidase
- NAC
N-acetylcysteine
- NMN
N′-methylnicotinamide
- NPT
Na+-dependent phosphate transporter
- OAT
organic anion transporter
- OATP
organic anion transporting polypeptide
- OTA
ochratoxin A
- OK
opossum kidney
- OCT
organic cation transporter
- OCTN
organic cation/carnitine transporter
- PQ
paraquat
- PAH
p-aminohippurate
- P-gp
P-glycoprotein
- PK
porcine kidney
- SCr
serum creatinine
- SLC
solute carrier
- TCE
trichloroethylene
- TEA
tetraethylammonium
- ZRT
zinc-regulated transporters
References
- 1.Brater DHS. Disposition and dose requirements of drugs in renal insufficiency. In: Seldinand DGG, editor. The Kidney. Lippincott Williams & Wilkins; Philadelphia: 2000. pp. 2923–2942. [Google Scholar]
- 2.Hosoyamada M, Sekine T, Kanai Y, Endou H. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. Am J Physiol. 1999;276:F122–128. doi: 10.1152/ajprenal.1999.276.1.F122. [DOI] [PubMed] [Google Scholar]
- 3.Tojo A, Sekine T, Nakajima N, Hosoyamada M, Kanai Y, Kimura K, Endou H. Immunohistochemical localization of multispecific renal organic anion transporter 1 in rat kidney. J Am Soc Nephrol. 1999;10:464–471. doi: 10.1681/ASN.V103464. [DOI] [PubMed] [Google Scholar]
- 4.Eraly SA, Vallon V, Vaughn DA, Gangoiti JA, Richter K, Nagle M, Monte JC, Rieg T, Truong DM, Long JM, Barshop BA, Kaler G, Nigam SK. Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. J Biol Chem. 2006;281:5072–5083. doi: 10.1074/jbc.M508050200. [DOI] [PubMed] [Google Scholar]
- 5.Enomoto A, Takeda M, Shimoda M, Narikawa S, Kobayashi Y, Kobayashi Y, Yamamoto T, Sekine T, Cha SH, Niwa T, Endou H. Interaction of human organic anion transporters 2 and 4 with organic anion transport inhibitors. J Pharmacol Exp Ther. 2002;301:797–802. doi: 10.1124/jpet.301.3.797. [DOI] [PubMed] [Google Scholar]
- 6.Cha SH, Sekine T, Fukushima JI, Kanai Y, Kobayashi Y, Goya T, Endou H. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol Pharmacol. 2001;59:1277–1286. doi: 10.1124/mol.59.5.1277. [DOI] [PubMed] [Google Scholar]
- 7.Kojima R, Sekine T, Kawachi M, Cha SH, Suzuki Y, Endou H. Immunolocalization of multispecific organic anion transporters, OAT1, OAT2, and OAT3, in rat kidney. J Am Soc Nephrol. 2002;13:848–857. doi: 10.1681/ASN.V134848. [DOI] [PubMed] [Google Scholar]
- 8.Bahn A, Ljubojevic M, Lorenz H, Schultz C, Ghebremedhin E, Ugele B, Sabolic I, Burckhardt G, Hagos Y. Murine renal organic anion transporters mOAT1 and mOAT3 facilitate the transport of neuroactive tryptophan metabolites. Am J Physiol Cell Physiol. 2005;289:C1075–1084. doi: 10.1152/ajpcell.00619.2004. [DOI] [PubMed] [Google Scholar]
- 9.Shen H, Liu T, Morse BL, Zhao Y, Zhang Y, Qiu X, Chen C, Lewin AC, Wang XT, Liu G, Christopher LJ, Marathe P, Lai Y. Characterization of Organic Anion Transporter 2 (SLC22A7): A Highly Efficient Transporter for Creatinine and Species-Dependent Renal Tubular Expression. Drug Metab Dispos. 2015;43:984–993. doi: 10.1124/dmd.114.062364. [DOI] [PubMed] [Google Scholar]
- 10.Ekaratanawong S, Anzai N, Jutabha P, Miyazaki H, Noshiro R, Takeda M, Kanai Y, Sophasan S, Endou H. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J Pharmacol Sci. 2004;94:297–304. doi: 10.1254/jphs.94.297. [DOI] [PubMed] [Google Scholar]
- 11.Anzai N, Jutabha P, Enomoto A, Yokoyama H, Nonoguchi H, Hirata T, Shiraya K, He X, Cha SH, Takeda M, Miyazaki H, Sakata T, Tomita K, Igarashi T, Kanai Y, Endou H. Functional characterization of rat organic anion transporter 5 (Slc22a19) at the apical membrane of renal proximal tubules. J Pharmacol Exp Ther. 2005;315:534–544. doi: 10.1124/jpet.105.088583. [DOI] [PubMed] [Google Scholar]
- 12.Takeda M, Babu E, Narikawa S, Endou H. Interaction of human organic anion transporters with various cephalosporin antibiotics. Eur J Pharmacol. 2002;438:137–142. doi: 10.1016/s0014-2999(02)01306-7. [DOI] [PubMed] [Google Scholar]
- 13.Apiwattanakul N, Sekine T, Chairoungdua A, Kanai Y, Nakajima N, Sophasan S, Endou H. Transport properties of nonsteroidal anti-inflammatory drugs by organic anion transporter 1 expressed in Xenopus laevis oocytes. Mol Pharmacol. 1999;55:847–854. [PubMed] [Google Scholar]
- 14.Wolff NA, Philpot RM, Miller DS, Pritchard JB. Functional expression of renal organic anion transport in Xenopus laevis oocytes. Mol Cell Biochem. 1992;114:35–41. doi: 10.1007/BF00240295. [DOI] [PubMed] [Google Scholar]
- 15.Sekine T, Watanabe N, Hosoyamada M, Kanai Y, Endou H. Expression cloning and characterization of a novel multispecific organic anion transporter. J Biol Chem. 1997;272:18526–18529. doi: 10.1074/jbc.272.30.18526. [DOI] [PubMed] [Google Scholar]
- 16.Sweet DH, Wolff NA, Pritchard JB. Expression cloning and characterization of ROAT1. The basolateral organic anion transporter in rat kidney. J Biol Chem. 1997;272:30088–30095. doi: 10.1074/jbc.272.48.30088. [DOI] [PubMed] [Google Scholar]
- 17.Bakhiya A, Bahn A, Burckhardt G, Wolff N. Human organic anion transporter 3 (hOAT3) can operate as an exchanger and mediate secretory urate flux. Cell Physiol Biochem. 2003;13:249–256. doi: 10.1159/000074539. [DOI] [PubMed] [Google Scholar]
- 18.Koepsell H, Endou H. The SLC22 drug transporter family. Pflugers Arch. 2004;447:666–676. doi: 10.1007/s00424-003-1089-9. [DOI] [PubMed] [Google Scholar]
- 19.Karbach U, Kricke J, Meyer-Wentrup F, Gorboulev V, Volk C, Loffing-Cueni D, Kaissling B, Bachmann S, Koepsell H. Localization of organic cation transporters OCT1 and OCT2 in rat kidney. Am J Physiol Renal Physiol. 2000;279:F679–687. doi: 10.1152/ajprenal.2000.279.4.F679. [DOI] [PubMed] [Google Scholar]
- 20.Motohashi H, Sakurai Y, Saito H, Masuda S, Urakami Y, Goto M, Fukatsu A, Ogawa O, Inui K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol. 2002;13:866–874. doi: 10.1681/ASN.V134866. [DOI] [PubMed] [Google Scholar]
- 21.Kimura N, Okuda M, Inui K. Metformin transport by renal basolateral organic cation transporter hOCT2. Pharm Res. 2005;22:255–259. doi: 10.1007/s11095-004-1193-3. [DOI] [PubMed] [Google Scholar]
- 22.Ciarimboli G, Ludwig T, Lang D, Pavenstadt H, Koepsell H, Piechota HJ, Haier J, Jaehde U, Zisowsky J, Schlatter E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol. 2005;167:1477–1484. doi: 10.1016/S0002-9440(10)61234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol. 2007;74:477–487. doi: 10.1016/j.bcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Tamai I, Yabuuchi H, Nezu J, Sai Y, Oku A, Shimane M, Tsuji A. Cloning and characterization of a novel human pH-dependent organic cation transporter, OCTN1. FEBS Lett. 1997;419:107–111. doi: 10.1016/s0014-5793(97)01441-5. [DOI] [PubMed] [Google Scholar]
- 25.Tamai I, China K, Sai Y, Kobayashi D, Nezu J, Kawahara E, Tsuji A. Na(+)-coupled transport of L-carnitine via high-affinity carnitine transporter OCTN2 and its subcellular localization in kidney. Biochim Biophys Acta. 2001;1512:273–284. doi: 10.1016/s0005-2736(01)00328-5. [DOI] [PubMed] [Google Scholar]
- 26.Tamai I, Nakanishi T, Kobayashi D, China K, Kosugi Y, Nezu J, Sai Y, Tsuji A. Involvement of OCTN1 (SLC22A4) in pH-dependent transport of organic cations. Mol Pharm. 2004;1:57–66. doi: 10.1021/mp0340082. [DOI] [PubMed] [Google Scholar]
- 27.Ohashi R, Tamai I, Yabuuchi H, Nezu JI, Oku A, Sai Y, Shimane M, Tsuji A. Na(+)-dependent carnitine transport by organic cation transporter (OCTN2): its pharmacological and toxicological relevance. J Pharmacol Exp Ther. 1999;291:778–784. [PubMed] [Google Scholar]
- 28.Ganapathy ME, Huang W, Rajan DP, Carter AL, Sugawara M, Iseki K, Leibach FH, Ganapathy V. beta-lactam antibiotics as substrates for OCTN2, an organic cation/carnitine transporter. J Biol Chem. 2000;275:1699–1707. doi: 10.1074/jbc.275.3.1699. [DOI] [PubMed] [Google Scholar]
- 29.Mikkaichi T, Suzuki T, Onogawa T, Tanemoto M, Mizutamari H, Okada M, Chaki T, Masuda S, Tokui T, Eto N, Abe M, Satoh F, Unno M, Hishinuma T, Inui K, Ito S, Goto J, Abe T. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci U S A. 2004;101:3569–3574. doi: 10.1073/pnas.0304987101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikkaichi T, Suzuki T, Tanemoto M, Ito S, Abe T. The organic anion transporter (OATP) family. Drug Metab Pharmacokinet. 2004;19:171–179. doi: 10.2133/dmpk.19.171. [DOI] [PubMed] [Google Scholar]
- 31.Nakai D, Nakagomi R, Furuta Y, Tokui T, Abe T, Ikeda T, Nishimura K. Human liver-specific organic anion transporter, LST-1, mediates uptake of pravastatin by human hepatocytes. J Pharmacol Exp Ther. 2001;297:861–867. [PubMed] [Google Scholar]
- 32.Jette L, Beaulieu E, Leclerc JM, Beliveau R. Cyclosporin A treatment induces overexpression of P-glycoprotein in the kidney and other tissues. Am J Physiol. 1996;270:F756–765. doi: 10.1152/ajprenal.1996.270.5.F756. [DOI] [PubMed] [Google Scholar]
- 33.Ernest S, Rajaraman S, Megyesi J, Bello-Reuss EN. Expression of MDR1 (multidrug resistance) gene and its protein in normal human kidney. Nephron. 1997;77:284–289. doi: 10.1159/000190289. [DOI] [PubMed] [Google Scholar]
- 34.Schinkel AH, Borst P. Multidrug resistance mediated by P-glycoproteins. Semin Cancer Biol. 1991;2:213–226. [PubMed] [Google Scholar]
- 35.Tang-Wai DF, Kajiji S, DiCapua F, de Graaf D, Roninson IB, Gros P. Human (MDR1) and mouse (mdr1, mdr3) P-glycoproteins can be distinguished by their respective drug resistance profiles and sensitivity to modulators. Biochemistry. 1995;34:32–39. doi: 10.1021/bi00001a005. [DOI] [PubMed] [Google Scholar]
- 36.Kato Y, Takahara S, Kato S, Kubo Y, Sai Y, Tamai I, Yabuuchi H, Tsuji A. Involvement of multidrug resistance-associated protein 2 (Abcc2) in molecular weight-dependent biliary excretion of beta-lactam antibiotics. Drug Metab Dispos. 2008;36:1088–1096. doi: 10.1124/dmd.107.019125. [DOI] [PubMed] [Google Scholar]
- 37.Schaub TP, Kartenbeck J, Konig J, Vogel O, Witzgall R, Kriz W, Keppler D. Expression of the conjugate export pump encoded by the mrp2 gene in the apical membrane of kidney proximal tubules. J Am Soc Nephrol. 1997;8:1213–1221. doi: 10.1681/ASN.V881213. [DOI] [PubMed] [Google Scholar]
- 38.Ghanem CI, Ruiz ML, Villanueva SS, Luquita MG, Catania VA, Jones B, Bengochea LA, Vore M, Mottino AD. Shift from biliary to urinary elimination of acetaminophen-glucuronide in acetaminophen-pretreated rats. J Pharmacol Exp Ther. 2005;315:987–995. doi: 10.1124/jpet.105.090613. [DOI] [PubMed] [Google Scholar]
- 39.Scheffer GL, Kool M, Heijn M, de Haas M, Pijnenborg AC, Wijnholds J, van Helvoort A, de Jong MC, Hooijberg JH, Mol CA, van der Linden M, de Vree JM, van der Valk P, Elferink RP, Borst P, Scheper RJ. Specific detection of multidrug resistance proteins MRP1, MRP2, MRP3, MRP5, and MDR3 P-glycoprotein with a panel of monoclonal antibodies. Cancer Res. 2000;60:5269–5277. [PubMed] [Google Scholar]
- 40.van Aubel RA, Smeets PH, Peters JG, Bindels RJ, Russel FG. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J Am Soc Nephrol. 2002;13:595–603. doi: 10.1681/ASN.V133595. [DOI] [PubMed] [Google Scholar]
- 41.Smeets PH, van Aubel RA, Wouterse AC, van den Heuvel JJ, Russel FG. Contribution of multidrug resistance protein 2 (MRP2/ABCC2) to the renal excretion of p-aminohippurate (PAH) and identification of MRP4 (ABCC4) as a novel PAH transporter. J Am Soc Nephrol. 2004;15:2828–2835. doi: 10.1097/01.ASN.0000143473.64430.AC. [DOI] [PubMed] [Google Scholar]
- 42.Merino G, Jonker JW, Wagenaar E, Pulido MM, Molina AJ, Alvarez AI, Schinkel AH. Transport of anthelmintic benzimidazole drugs by breast cancer resistance protein (BCRP/ABCG2) Drug Metab Dispos. 2005;33:614–618. doi: 10.1124/dmd.104.003319. [DOI] [PubMed] [Google Scholar]
- 43.Robey RW, Steadman K, Polgar O, Bates SE. ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther. 2005;4:187–194. [PubMed] [Google Scholar]
- 44.Huang Y, Sadee W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Lett. 2006;239:168–182. doi: 10.1016/j.canlet.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 45.Ando T, Kusuhara H, Merino G, Alvarez AI, Schinkel AH, Sugiyama Y. Involvement of breast cancer resistance protein (ABCG2) in the biliary excretion mechanism of fluoroquinolones. Drug Metab Dispos. 2007;35:1873–1879. doi: 10.1124/dmd.107.014969. [DOI] [PubMed] [Google Scholar]
- 46.Enokizono J, Kusuhara H, Sugiyama Y. Effect of breast cancer resistance protein (Bcrp/Abcg2) on the disposition of phytoestrogens. Mol Pharmacol. 2007;72:967–975. doi: 10.1124/mol.107.034751. [DOI] [PubMed] [Google Scholar]
- 47.Pan G, Giri N, Elmquist WF. Abcg2/Bcrp1 mediates the polarized transport of antiretroviral nucleosides abacavir and zidovudine. Drug Metab Dispos. 2007;35:1165–1173. doi: 10.1124/dmd.106.014274. [DOI] [PubMed] [Google Scholar]
- 48.Myllynen P, Kummu M, Kangas T, Ilves M, Immonen E, Rysa J, Pirila R, Lastumaki A, Vahakangas KH. ABCG2/BCRP decreases the transfer of a food-born chemical carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in perfused term human placenta. Toxicol Appl Pharmacol. 2008;232:210–217. doi: 10.1016/j.taap.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 49.Kim M, Turnquist H, Jackson J, Sgagias M, Yan Y, Gong M, Dean M, Sharp JG, Cowan K. The multidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin Cancer Res. 2002;8:22–28. [PubMed] [Google Scholar]
- 50.Huls M, Brown CD, Windass AS, Sayer R, van den Heuvel JJ, Heemskerk S, Russel FG, Masereeuw R. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008;73:220–225. doi: 10.1038/sj.ki.5002645. [DOI] [PubMed] [Google Scholar]
- 51.Otsuka M, Yasuda M, Morita Y, Otsuka C, Tsuchiya T, Omote H, Moriyama Y. Identification of essential amino acid residues of the NorM Na+/multidrug antiporter in Vibrio parahaemolyticus. J Bacteriol. 2005;187:1552–1558. doi: 10.1128/JB.187.5.1552-1558.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol. 2006;17:2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- 53.Nishihara K, Masuda S, Ji L, Katsura T, Inui K. Pharmacokinetic significance of luminal multidrug and toxin extrusion 1 in chronic renal failure rats. Biochem Pharmacol. 2007;73:1482–1490. doi: 10.1016/j.bcp.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 54.Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem Pharmacol. 2007;74:359–371. doi: 10.1016/j.bcp.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 55.Ohta KY, Inoue K, Hayashi Y, Yuasa H. Molecular identification and functional characterization of rat multidrug and toxin extrusion type transporter 1 as an organic cation/H+ antiporter in the kidney. Drug Metab Dispos. 2006;34:1868–1874. doi: 10.1124/dmd.106.010876. [DOI] [PubMed] [Google Scholar]
- 56.Terada T, Masuda S, Asaka J, Tsuda M, Katsura T, Inui K. Molecular cloning, functional characterization and tissue distribution of rat H+/organic cation antiporter MATE1. Pharm Res. 2006;23:1696–1701. doi: 10.1007/s11095-006-9016-3. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y, Zhang S, Sorani M, Giacomini KM. Transport of paraquat by human organic cation transporters and multidrug and toxic compound extrusion family. J Pharmacol Exp Ther. 2007;322:695–700. doi: 10.1124/jpet.107.123554. [DOI] [PubMed] [Google Scholar]
- 58.Dehghan A, Kottgen A, Yang Q, Hwang SJ, Kao WL, Rivadeneira F, Boerwinkle E, Levy D, Hofman A, Astor BC, Benjamin EJ, van Duijn CM, Witteman JC, Coresh J, Fox CS. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008;372:1953–1961. doi: 10.1016/S0140-6736(08)61343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jutabha P, Anzai N, Kitamura K, Taniguchi A, Kaneko S, Yan K, Yamada H, Shimada H, Kimura T, Katada T, Fukutomi T, Tomita K, Urano W, Yamanaka H, Seki G, Fujita T, Moriyama Y, Yamada A, Uchida S, Wempe MF, Endou H, Sakurai H. Human sodium phosphate transporter 4 (hNPT4/SLC17A3) as a common renal secretory pathway for drugs and urate. J Biol Chem. 2010;285:35123–35132. doi: 10.1074/jbc.M110.121301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vamvakas S, Bruning T, Thomasson B, Lammert M, Baumuller A, Bolt HM, Dekant W, Birner G, Henschler D, Ulm K. Renal cell cancer correlated with occupational exposure to trichloroethene. J Cancer Res Clin Oncol. 1998;124:374–382. doi: 10.1007/s004320050186. [DOI] [PubMed] [Google Scholar]
- 61.Charbotel B, Fevotte J, Hours M, Martin JL, Bergeret A. Case-control study on renal cell cancer and occupational exposure to trichloroethylene. Part II: Epidemiological aspects. Ann Occup Hyg. 2006;50:777–787. doi: 10.1093/annhyg/mel039. [DOI] [PubMed] [Google Scholar]
- 62.National Toxicology Program. 1990. Carcinogenesis studies of trichloroethylene (without epichlorhydrin) (CAS No. 79–01–6) in F344/N rats and B6C3F1 mice gavage studies. [PubMed] [Google Scholar]
- 63.National Toxicology Program. 1988. Toxicology and carcinogenesis studies of trichloroethylene (CAS No. 79–01–6) in four strains of rats (ACI, August, Marshall, Osborne-Mendel) (gavage studies) [PubMed] [Google Scholar]
- 64.Anders MW, Lash L, Dekant W, Elfarra AA, Dohn DR. Biosynthesis and biotransformation of glutathione S-conjugates to toxic metabolites. Crit Rev Toxicol. 1988;18:311–341. doi: 10.3109/10408448809037470. [DOI] [PubMed] [Google Scholar]
- 65.Miller RE, Guengerich FP. Oxidation of trichloroethylene by liver microsomal cytochrome P-450: evidence for chlorine migration in a transition state not involving trichloroethylene oxide. Biochemistry. 1982;21:1090–1097. doi: 10.1021/bi00534a041. [DOI] [PubMed] [Google Scholar]
- 66.Lash LH, Lipscomb JC, Putt DA, Parker JC. Glutathione conjugation of trichloroethylene in human liver and kidney: kinetics and individual variation. Drug Metab Dispos. 1999;27:351–359. [PubMed] [Google Scholar]
- 67.Lash LH, Putt DA, Huang P, Hueni SE, Parker JC. Modulation of hepatic and renal metabolism and toxicity of trichloroethylene and perchloroethylene by alterations in status of cytochrome P450 and glutathione. Toxicology. 2007;235:11–26. doi: 10.1016/j.tox.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lash LH, Putt DA, Parker JC. Metabolism and tissue distribution of orally administered trichloroethylene in male and female rats: identification of glutathione- and cytochrome P-450-derived metabolites in liver, kidney, blood, and urine. J Toxicol Environ Health A. 2006;69:1285–1309. doi: 10.1080/15287390500360133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lock EA, Sani Y, Moore RB, Finkelstein MB, Anders MW, Seawright AA. Bone marrow and renal injury associated with haloalkene cysteine conjugates in calves. Arch Toxicol. 1996;70:607–619. doi: 10.1007/s002040050319. [DOI] [PubMed] [Google Scholar]
- 70.Birner G, Bernauer U, Werner M, Dekant W. Biotransformation, excretion and nephrotoxicity of haloalkene-derived cysteine S-conjugates. Arch Toxicol. 1997;72:1–8. doi: 10.1007/s002040050461. [DOI] [PubMed] [Google Scholar]
- 71.Lash LH, Xu Y, Elfarra AA, Duescher RJ, Parker JC. Glutathione-dependent metabolism of trichloroethylene in isolated liver and kidney cells of rats and its role in mitochondrial and cellular toxicity. Drug Metab Dispos. 1995;23:846–853. [PubMed] [Google Scholar]
- 72.Lash LH, Putt DA, Brashear WT, Abbas R, Parker JC, Fisher JW. Identification of S-(1,2-dichlorovinyl)glutathione in the blood of human volunteers exposed to trichloroethylene. J Toxicol Environ Health A. 1999;56:1–21. doi: 10.1080/009841099158204. [DOI] [PubMed] [Google Scholar]
- 73.Lash LH, Qian W, Putt DA, Hueni SE, Elfarra AA, Krause RJ, Parker JC. Renal and hepatic toxicity of trichloroethylene and its glutathione-derived metabolites in rats and mice: sex-, species-, and tissue-dependent differences. J Pharmacol Exp Ther. 2001;297:155–164. [PubMed] [Google Scholar]
- 74.Uttamsingh V, Anders MW. Acylase-catalyzed deacetylation of haloalkene-derived mercapturates. Chem Res Toxicol. 1999;12:937–942. doi: 10.1021/tx990090p. [DOI] [PubMed] [Google Scholar]
- 75.Volkel W, Dekant W. Chlorothioketene, the ultimate reactive intermediate formed by cysteine conjugate beta-lyase-mediated cleavage of the trichloroethene metabolite S-(1,2-Dichlorovinyl)-L-cysteine, forms cytosine adducts in organic solvents, but not in aqueous solution. Chem Res Toxicol. 1998;11:1082–1088. doi: 10.1021/tx980084d. [DOI] [PubMed] [Google Scholar]
- 76.Dekant W, Berthold K, Vamvakas S, Henschler D. Thioacylating agents as ultimate intermediates in the beta-lyase catalysed metabolism of S-(pentachloro-butadienyl)-L-cysteine. Chem Biol Interact. 1988;67:139–148. doi: 10.1016/0009-2797(88)90093-2. [DOI] [PubMed] [Google Scholar]
- 77.Muller M, Birner G, Dekant W. Reactivity of haloketenes and halothioketenes with nucleobases: chemical characterization of reaction products. Chem Res Toxicol. 1998;11:454–463. doi: 10.1021/tx9701438. [DOI] [PubMed] [Google Scholar]
- 78.Hayden PJ, Ichimura T, McCann DJ, Pohl LR, Stevens JL. Detection of cysteine conjugate metabolite adduct formation with specific mitochondrial proteins using antibodies raised against halothane metabolite adducts. J Biol Chem. 1991;266:18415–18418. [PubMed] [Google Scholar]
- 79.Silber PM, Gandolfi AJ, Brendel K. Early biological indicators of S-(1,2-dichlorovinyl)-L-cysteine nephrotoxicity in the rabbit. Drug Chem Toxicol. 1986;9:285–303. doi: 10.3109/01480548608998281. [DOI] [PubMed] [Google Scholar]
- 80.Lash LH, Hueni SE, Putt DA. Apoptosis, necrosis, and cell proliferation induced by S-(1,2-dichlorovinyl)-L-cysteine in primary cultures of human proximal tubular cells. Toxicol Appl Pharmacol. 2001;177:1–16. doi: 10.1006/taap.2001.9295. [DOI] [PubMed] [Google Scholar]
- 81.Birner G, Vamvakas S, Dekant W, Henschler D. Nephrotoxic and genotoxic N-acetyl-S-dichlorovinyl-L-cysteine is a urinary metabolite after occupational 1,1,2-trichloroethene exposure in humans: implications for the risk of trichloroethene exposure. Environ Health Perspect. 1993;99:281–284. doi: 10.1289/ehp.9399281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolfgang GH, Gandolfi AJ, Stevens JL, Brendel K. N-acetyl S-(1,2-dichlorovinyl)-L-cysteine produces a similar toxicity to S-(1,2-dichlorovinyl)-L-cysteine in rabbit renal slices: differential transport and metabolism. Toxicol Appl Pharmacol. 1989;101:205–219. doi: 10.1016/0041-008x(89)90270-6. [DOI] [PubMed] [Google Scholar]
- 83.Dantzler WH, Evans KK, Groves CE, Welborn JR, North J, Stevens JL, Wright SH. Relation of cysteine conjugate nephrotoxicity to transport by the basolateral organic anion transport system in isolated S2 segments of rabbit proximal renal tubules. J Pharmacol Exp Ther. 1998;286:52–60. [PubMed] [Google Scholar]
- 84.Groves CE, Munoz L, Bahn A, Burckhardt G, Wright SH. Interaction of cysteine conjugates with human and rabbit organic anion transporter 1. J Pharmacol Exp Ther. 2003;304:560–566. doi: 10.1124/jpet.102.043455. [DOI] [PubMed] [Google Scholar]
- 85.Lash LH, Anders MW. Uptake of nephrotoxic S-conjugates by isolated rat renal proximal tubular cells. J Pharmacol Exp Ther. 1989;248:531–537. [PubMed] [Google Scholar]
- 86.Schaeffer VH, Stevens JL. The transport of S-cysteine conjugates in LLC-PK1 cells and its role in toxicity. Mol Pharmacol. 1987;31:506–512. [PubMed] [Google Scholar]
- 87.Schaeffer VH, Stevens JL. Mechanism of transport for toxic cysteine conjugates in rat kidney cortex membrane vesicles. Mol Pharmacol. 1987;32:293–298. [PubMed] [Google Scholar]
- 88.Wright SH, Wunz TM, North J, Stevens JL. Na-dependent transport of S-(1,2-dichlorovinyl)-L-cysteine by renal brush-border membrane vesicles. J Pharmacol Exp Ther. 1998;285:162–169. [PubMed] [Google Scholar]
- 89.Tsirulnikov K, Abuladze N, Koag MC, Newman D, Scholz K, Bondar G, Zhu Q, Avliyakulov NK, Dekant W, Faull K, Kurtz I, Pushkin A. Transport of N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine, a metabolite of trichloroethylene, by mouse multidrug resistance associated protein 2 (Mrp2) Toxicol Appl Pharmacol. 2010;244:218–225. doi: 10.1016/j.taap.2009.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bullivant CM. Accidental poisoning by paraquat: Report of two cases in man. Br Med J. 1966;1:1272–1273. doi: 10.1136/bmj.1.5498.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Houze P, Baud FJ, Mouy R, Bismuth C, Bourdon R, Scherrmann JM. Toxicokinetics of paraquat in humans. Hum Exp Toxicol. 1990;9:5–12. doi: 10.1177/096032719000900103. [DOI] [PubMed] [Google Scholar]
- 92.Rose MS, Smith LL, Wyatt I. Evidence for energy-dependent accumulation of paraquat into rat lung. Nature. 1974;252:314–315. doi: 10.1038/252314b0. [DOI] [PubMed] [Google Scholar]
- 93.McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 94.Daniel JW, Gage JC. Absorption and excretion of diquat and paraquat in rats. Br J Ind Med. 1966;23:133–136. doi: 10.1136/oem.23.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chan BS, Seale JP, Duggin GG. The mechanism of excretion of paraquat in rats. Toxicol Lett. 1997;90:1–9. doi: 10.1016/s0378-4274(96)03820-9. [DOI] [PubMed] [Google Scholar]
- 96.Smith LL. Mechanism of paraquat toxicity in lung and its relevance to treatment. Hum Toxicol. 1987;6:31–36. doi: 10.1177/096032718700600105. [DOI] [PubMed] [Google Scholar]
- 97.Ecker JL, Hook JB, Gibson JE. Nephrotoxicity of paraquat in mice. Toxicol Appl Pharmacol. 1975;34:178–186. doi: 10.1016/0041-008x(75)90185-4. [DOI] [PubMed] [Google Scholar]
- 98.Lock EA. The effect of paraquat and diquat on renal function in the rat. Toxicol Appl Pharmacol. 1979;48:327–336. doi: 10.1016/0041-008x(79)90039-5. [DOI] [PubMed] [Google Scholar]
- 99.Lock EA, Ishmael J. The acute toxic effects of paraquat and diquat on the rat kidney. Toxicol Appl Pharmacol. 1979;50:67–76. doi: 10.1016/0041-008x(79)90493-9. [DOI] [PubMed] [Google Scholar]
- 100.Beebeejaun AR, Beevers G, Rogers WN. Paraquat poisoning-prolonged excretion. Clin Toxicol. 1971;4:397–407. doi: 10.3109/15563657108990492. [DOI] [PubMed] [Google Scholar]
- 101.Kodagoda N, Jayewardene RP, Attygalle D. Poisoning with paraquat. Forensic Sci. 1973;2:107–111. doi: 10.1016/0300-9432(73)90018-6. [DOI] [PubMed] [Google Scholar]
- 102.Vale JA, Meredith TJ, Buckley BM. Paraquat poisoning: clinical features and immediate general management. Hum Toxicol. 1987;6:41–47. doi: 10.1177/096032718700600107. [DOI] [PubMed] [Google Scholar]
- 103.Vaziri ND, Ness RL, Fairshter RD, Smith WR, Rosen SM. Nephrotoxicity of paraquat in man. Arch Intern Med. 1979;139:172–174. [PubMed] [Google Scholar]
- 104.Chan BS, Lazzaro VA, Seale JP, Duggin GG. Transport of paraquat in a renal epithelial cell line LLC-PK1. J Pharmacol Exp Ther. 1996;279:625–632. [PubMed] [Google Scholar]
- 105.Chan BS, Lazzaro VA, Seale JP, Duggin GG. Characterisation and uptake of paraquat by rat renal proximal tubular cells in primary culture. Hum Exp Toxicol. 1996;15:949–956. doi: 10.1177/096032719601501202. [DOI] [PubMed] [Google Scholar]
- 106.Kang HJ, Song IS, Shin HJ, Kim WY, Lee CH, Shim JC, Zhou HH, Lee SS, Shin JG. Identification and functional characterization of genetic variants of human organic cation transporters in a Korean population. Drug Metab Dispos. 2007;35:667–675. doi: 10.1124/dmd.106.013581. [DOI] [PubMed] [Google Scholar]
- 107.Song IS, Shin HJ, Shin JG. Genetic variants of organic cation transporter 2 (OCT2) significantly reduce metformin uptake in oocytes. Xenobiotica. 2008;38:1252–1262. doi: 10.1080/00498250802130039. [DOI] [PubMed] [Google Scholar]
- 108.Wright SH, Wunz TM. Paraquat2+/H+ exchange in isolated renal brush-border membrane vesicles. Biochim Biophys Acta. 1995;1240:18–24. doi: 10.1016/0005-2736(95)00166-0. [DOI] [PubMed] [Google Scholar]
- 109.Li Q, Peng X, Yang H, Wang H, Shu Y. Deficiency of multidrug and toxin extrusion 1 enhances renal accumulation of paraquat and deteriorates kidney injury in mice. Mol Pharm. 2011;8:2476–2483. doi: 10.1021/mp200395f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dinis-Oliveira RJ, Remiao F, Duarte JA, Ferreira R, Sanchez Navarro A, Bastos ML, Carvalho F. P-glycoprotein induction: an antidotal pathway for paraquat-induced lung toxicity. Free Radic Biol Med. 2006;41:1213–1224. doi: 10.1016/j.freeradbiomed.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 111.Wen X, Gibson CJ, Yang I, Buckley B, Goedken MJ, Richardson JR, Aleksunes LM. MDR1 transporter protects against paraquat-induced toxicity in human and mouse proximal tubule cells. Toxicol Sci. 2014;141:475–483. doi: 10.1093/toxsci/kfu141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.JK-G, Harwig T, Scott PM, Rechcigl M. Handbook of Foodborne Diseases of Biological Origin. CRC Press; Boca Raton, FL, USA: 1983. pp. 193–238. [Google Scholar]
- 113.O’Brien E, Dietrich D. Mycotoxins affecting the kidney. In: Hook J, editor. Toxicology of Kidney. CRC Press; Boca Raton, FL, USA: 2004. pp. 895–936. [Google Scholar]
- 114.Pavlovic M, Plestina R, Krogh P. Ochratoxin A contamination of foodstuffs in an area with Balkan (endemic) nephropathy. Acta Pathol Microbiol Scand B. 1979;87:243–246. doi: 10.1111/j.1699-0463.1979.tb02433.x. [DOI] [PubMed] [Google Scholar]
- 115.Tapia MO, Seawright AA. Experimental ochratoxicosis A in pigs. Aust Vet J. 1984;61:219–222. doi: 10.1111/j.1751-0813.1984.tb05993.x. [DOI] [PubMed] [Google Scholar]
- 116.C.C.f.A.S.a. Technology. Mycotoxins: Risks in Plant, Animal, and Human Systems, Task Force Report No. 139. Ames, Iowa: 2003. p. 199. [Google Scholar]
- 117.Mayura K, Stein AF, Berndt WO, Phillips TD. Teratogenic effects of Ochratoxin A in rats with impaired renal function. Toxicology. 1984;32:277–285. doi: 10.1016/0300-483x(84)90080-5. [DOI] [PubMed] [Google Scholar]
- 118.Haubeck HD, Lorkowski G, Kolsch E, Roschenthaler R. Immunosuppression by ochratoxin A and its prevention by phenylalanine. Appl Environ Microbiol. 1981;41:1040–1042. doi: 10.1128/aem.41.4.1040-1042.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kumagai S. Ochratoxin A: plasma concentration and excretion into bile and urine in albumin-deficient rats. Food Chem Toxicol. 1985;23:941–943. doi: 10.1016/0278-6915(85)90112-7. [DOI] [PubMed] [Google Scholar]
- 120.Stojkovic R, Hult K, Gamulin S, Plestina R. High affinity binding of ochratoxin A to plasma constituents. Biochem Int. 1984;9:33–38. [PubMed] [Google Scholar]
- 121.Hagelberg S, Hult K, Fuchs R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J Appl Toxicol. 1989;9:91–96. doi: 10.1002/jat.2550090204. [DOI] [PubMed] [Google Scholar]
- 122.Ringot D, Chango A, Schneider YJ, Larondelle Y. Toxicokinetics and toxicodynamics of ochratoxin A, an update. Chem Biol Interact. 2006;159:18–46. doi: 10.1016/j.cbi.2005.10.106. [DOI] [PubMed] [Google Scholar]
- 123.Stein AF, Phillips TD, Kubena LF, Harvey RB. Renal tubular secretion and reabsorption as factors in ochratoxicosis: effects of probenecid on nephrotoxicity. J Toxicol Environ Health. 1985;16:593–605. doi: 10.1080/15287398509530766. [DOI] [PubMed] [Google Scholar]
- 124.Cooper P. The kidney and ochratoxin A. Food Cosmet Toxicol. 1979;17:406–408. doi: 10.1016/0015-6264(79)90347-x. [DOI] [PubMed] [Google Scholar]
- 125.Bendele AM, Carlton WW, Krogh P, Lillehoj EB. Ochratoxin A carcinogenesis in the (C57BL/6J X C3H)F1 mouse. J Natl Cancer Inst. 1985;75:733–742. [PubMed] [Google Scholar]
- 126.Creppy EE, Roschenthaler R, Dirheimer G. Inhibition of protein synthesis in mice by ochratoxin A and its prevention by phenylalanine. Food Chem Toxicol. 1984;22:883–886. doi: 10.1016/0278-6915(84)90170-4. [DOI] [PubMed] [Google Scholar]
- 127.Poor M, Veres B, Jakus PB, Antus C, Montsko G, Zrinyi Z, Vladimir-Knezevic S, Petrik J, Koszegi T. Flavonoid diosmetin increases ATP levels in kidney cells and relieves ATP depleting effect of ochratoxin A. J Photochem Photobiol B. 2014;132:1–9. doi: 10.1016/j.jphotobiol.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 128.Pfohl-Leszkowicz A, Manderville RA. An update on direct genotoxicity as a molecular mechanism of ochratoxin a carcinogenicity. Chem Res Toxicol. 2012;25:252–262. doi: 10.1021/tx200430f. [DOI] [PubMed] [Google Scholar]
- 129.Sorrenti V, Di Giacomo C, Acquaviva R, Barbagallo I, Bognanno M, Galvano F. Toxicity of ochratoxin a and its modulation by antioxidants: a review. Toxins (Basel) 2013;5:1742–1766. doi: 10.3390/toxins5101742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jung KY, Takeda M, Kim DK, Tojo A, Narikawa S, Yoo BS, Hosoyamada M, Cha SH, Sekine T, Endou H. Characterization of ochratoxin A transport by human organic anion transporters. Life Sci. 2001;69:2123–2135. doi: 10.1016/s0024-3205(01)01296-6. [DOI] [PubMed] [Google Scholar]
- 131.Sokol PP, Ripich G, Holohan PD, Ross CR. Mechanism of ochratoxin A transport in kidney. J Pharmacol Exp Ther. 1988;246:460–465. [PubMed] [Google Scholar]
- 132.Groves CE, Morales M, Wright SH. Peritubular transport of ochratoxin A in rabbit renal proximal tubules. J Pharmacol Exp Ther. 1998;284:943–948. [PubMed] [Google Scholar]
- 133.Tsuda M, Sekine T, Takeda M, Cha SH, Kanai Y, Kimura M, Endou H. Transport of ochratoxin A by renal multispecific organic anion transporter 1. J Pharmacol Exp Ther. 1999;289:1301–1305. [PubMed] [Google Scholar]
- 134.Babu E, Takeda M, Narikawa S, Kobayashi Y, Enomoto A, Tojo A, Cha SH, Sekine T, Sakthisekaran D, Endou H. Role of human organic anion transporter 4 in the transport of ochratoxin A. Biochim Biophys Acta. 2002;1590:64–75. doi: 10.1016/s0167-4889(02)00187-8. [DOI] [PubMed] [Google Scholar]
- 135.Welborn JR, Groves CE, Wright SH. Peritubular transport of ochratoxin A by single rabbit renal proximal tubules. J Am Soc Nephrol. 1998;9:1973–1982. doi: 10.1681/ASN.V9111973. [DOI] [PubMed] [Google Scholar]
- 136.Bergwerk AJ, Shi X, Ford AC, Kanai N, Jacquemin E, Burk RD, Bai S, Novikoff PM, Stieger B, Meier PJ, Schuster VL, Wolkoff AW. Immunologic distribution of an organic anion transport protein in rat liver and kidney. Am J Physiol. 1996;271:G231–238. doi: 10.1152/ajpgi.1996.271.2.G231. [DOI] [PubMed] [Google Scholar]
- 137.Eckhardt U, Schroeder A, Stieger B, Hochli M, Landmann L, Tynes R, Meier PJ, Hagenbuch B. Polyspecific substrate uptake by the hepatic organic anion transporter Oatp1 in stably transfected CHO cells. Am J Physiol. 1999;276:G1037–1042. doi: 10.1152/ajpgi.1999.276.4.G1037. [DOI] [PubMed] [Google Scholar]
- 138.Youngblood GL, Sweet DH. Identification and functional assessment of the novel murine organic anion transporter Oat5 (Slc22a19) expressed in kidney. Am J Physiol Renal Physiol. 2004;287:F236–244. doi: 10.1152/ajprenal.00012.2004. [DOI] [PubMed] [Google Scholar]
- 139.Takeuchi A, Masuda S, Saito H, Abe T, Inui K. Multispecific substrate recognition of kidney-specific organic anion transporters OAT-K1 and OAT-K2. J Pharmacol Exp Ther. 2001;299:261–267. [PubMed] [Google Scholar]
- 140.Dahlmann A, Dantzler WH, Silbernagl S, Gekle M. Detailed mapping of ochratoxin A reabsorption along the rat nephron in vivo: the nephrotoxin can be reabsorbed in all nephron segments by different mechanisms. J Pharmacol Exp Ther. 1998;286:157–162. [PubMed] [Google Scholar]
- 141.Leier I, Hummel-Eisenbeiss J, Cui Y, Keppler D. ATP-dependent para-aminohippurate transport by apical multidrug resistance protein MRP2. Kidney Int. 2000;57:1636–1642. doi: 10.1046/j.1523-1755.2000.00007.x. [DOI] [PubMed] [Google Scholar]
- 142.Schrickx J, Lektarau Y, Fink-Gremmels J. Ochratoxin A secretion by ATP-dependent membrane transporters in Caco-2 cells. Arch Toxicol. 2006;80:243–249. doi: 10.1007/s00204-005-0041-5. [DOI] [PubMed] [Google Scholar]
- 143.Tsuruoka S, Ishibashi K, Yamamoto H, Wakaumi M, Suzuki M, Schwartz GJ, Imai M, Fujimura A. Functional analysis of ABCA8, a new drug transporter. Biochem Biophys Res Commun. 2002;298:41–45. doi: 10.1016/s0006-291x(02)02389-6. [DOI] [PubMed] [Google Scholar]
- 144.Wakaumi M, Ishibashi K, Ando H, Kasanuki H, Tsuruoka S. Acute digoxin loading reduces ABCA8A mRNA expression in the mouse liver. Clin Exp Pharmacol Physiol. 2005;32:1034–1041. doi: 10.1111/j.1440-1681.2005.04301.x. [DOI] [PubMed] [Google Scholar]
- 145.Jutabha P, Anzai N, Hayashi K, Domae M, Uchida K, Endou H, Sakurai H. A novel human organic anion transporter NPT4 mediates the transport of ochratoxin A. J Pharmacol Sci. 2011;116:392–396. doi: 10.1254/jphs.10227sc. [DOI] [PubMed] [Google Scholar]
- 146.Jarup L, Berglund M, Elinder CG, Nordberg G, Vahter M. Health effects of cadmium exposure--a review of the literature and a risk estimate. Scand J Work Environ Health. 1998;24(Suppl 1):1–51. [PubMed] [Google Scholar]
- 147.Berglund M, Akesson A, Nermell B, Vahter M. Intestinal absorption of dietary cadmium in women depends on body iron stores and fiber intake. Environ Health Perspect. 1994;102:1058–1066. doi: 10.1289/ehp.941021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect. 2004;112:1099–1103. doi: 10.1289/ehp.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Descotes J. Immunotoxicology of cadmium. IARC Sci Publ. 1992:385–390. [PubMed] [Google Scholar]
- 150.Kazantzis G. Cadmium nephropathy. Contrib Nephrol. 1979;16:161–166. doi: 10.1159/000402891. [DOI] [PubMed] [Google Scholar]
- 151.Kjellstrom T. Mechanism and epidemiology of bone effects of cadmium. IARC Sci Publ. 1992:301–310. [PubMed] [Google Scholar]
- 152.Kjellstrom T, Friberg L, Rahnster B. Mortality and cancer morbidity among cadmium-exposed workers. Environ Health Perspect. 1979;28:199–204. doi: 10.1289/ehp.28-1637490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Messner B, Knoflach M, Seubert A, Ritsch A, Pfaller K, Henderson B, Shen YH, Zeller I, Willeit J, Laufer G, Wick G, Kiechl S, Bernhard D. Cadmium is a novel and independent risk factor for early atherosclerosis mechanisms and in vivo relevance. Arterioscler Thromb Vasc Biol. 2009;29:1392–1398. doi: 10.1161/ATVBAHA.109.190082. [DOI] [PubMed] [Google Scholar]
- 154.Schwartz GG, Il’yasova D, Ivanova A. Urinary cadmium, impaired fasting glucose, and diabetes in the NHANES III. Diabetes Care. 2003;26:468–470. doi: 10.2337/diacare.26.2.468. [DOI] [PubMed] [Google Scholar]
- 155.Smith TJ, Petty TL, Reading JC, Lakshminarayan S. Pulmonary effects of chronic exposure to airborne cadmium. Am Rev Respir Dis. 1976;114:161–169. doi: 10.1164/arrd.1976.114.1.161. [DOI] [PubMed] [Google Scholar]
- 156.Viaene MK, Masschelein R, Leenders J, De Groof M, Swerts LJ, Roels HA. Neurobehavioural effects of occupational exposure to cadmium: a cross sectional epidemiological study. Occup Environ Med. 2000;57:19–27. doi: 10.1136/oem.57.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Galan A, Garcia-Bermejo L, Troyano A, Vilaboa NE, Fernandez C, de Blas E, Aller P. The role of intracellular oxidation in death induction (apoptosis and necrosis) in human promonocytic cells treated with stress inducers (cadmium, heat, X-rays) Eur J Cell Biol. 2001;80:312–320. doi: 10.1078/0171-9335-00159. [DOI] [PubMed] [Google Scholar]
- 158.Aughey E, Fell GS, Scott R, Black M. Histopathology of early effects of oral cadmium in the rat kidney. Environ Health Perspect. 1984;54:153–161. doi: 10.1289/ehp.8454153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Johri N, Jacquillet G, Unwin R. Heavy metal poisoning: the effects of cadmium on the kidney. Biometals. 2010;23:783–792. doi: 10.1007/s10534-010-9328-y. [DOI] [PubMed] [Google Scholar]
- 160.Piscator M. Long-term observations on tubular and glomerular function in cadmium-exposed persons. Environ Health Perspect. 1984;54:175–179. doi: 10.1289/ehp.8454175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shaikh ZA, Smith JC. The biosynthesis of metallothionein rat liver and kidney after administration of cadmium. Chem Biol Interact. 1976;15:327–336. doi: 10.1016/0009-2797(76)90138-1. [DOI] [PubMed] [Google Scholar]
- 162.Weser U, Donay F, Rupp H. Cadmium-induced synthesis of hepatic metallothionein in chicken and rats. FEBS Lett. 1973;32:171–174. doi: 10.1016/0014-5793(73)80764-1. [DOI] [PubMed] [Google Scholar]
- 163.Cherian MG, Vostal JJ. Biliary excretion of cadmium in rat. I. Dose-dependent biliary excretion and the form of cadmium in the bile. J Toxicol Environ Health. 1977;2:945–954. doi: 10.1080/15287397709529493. [DOI] [PubMed] [Google Scholar]
- 164.Wang Y, Zalups RK, Barfuss DW. Potential mechanisms involved in the absorptive transport of cadmium in isolated perfused rabbit renal proximal tubules. Toxicol Lett. 2010;193:61–68. doi: 10.1016/j.toxlet.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zalups RK. Evidence for basolateral uptake of cadmium in the kidneys of rats. Toxicol Appl Pharmacol. 2000;164:15–23. doi: 10.1006/taap.1999.8854. [DOI] [PubMed] [Google Scholar]
- 166.Soodvilai S, Nantavishit J, Muanprasat C, Chatsudthipong V. Renal organic cation transporters mediated cadmium-induced nephrotoxicity. Toxicol Lett. 2011;204:38–42. doi: 10.1016/j.toxlet.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 167.Thevenod F, Ciarimboli G, Leistner M, Wolff NA, Lee WK, Schatz I, Keller T, Al-Monajjed R, Gorboulev V, Koepsell H. Substrate- and cell contact-dependent inhibitor affinity of human organic cation transporter 2: studies with two classical organic cation substrates and the novel substrate cd2+ Mol Pharm. 2013;10:3045–3056. doi: 10.1021/mp400113d. [DOI] [PubMed] [Google Scholar]
- 168.Klassen RB, Crenshaw K, Kozyraki R, Verroust PJ, Tio L, Atrian S, Allen PL, Hammond TG. Megalin mediates renal uptake of heavy metal metallothionein complexes. Am J Physiol Renal Physiol. 2004;287:F393–403. doi: 10.1152/ajprenal.00233.2003. [DOI] [PubMed] [Google Scholar]
- 169.Thevenod F. Catch me if you can! Novel aspects of cadmium transport in mammalian cells. Biometals. 2010;23:857–875. doi: 10.1007/s10534-010-9309-1. [DOI] [PubMed] [Google Scholar]
- 170.Christensen EI, Birn H. Megalin and cubilin: synergistic endocytic receptors in renal proximal tubule. Am J Physiol Renal Physiol. 2001;280:F562–573. doi: 10.1152/ajprenal.2001.280.4.F562. [DOI] [PubMed] [Google Scholar]
- 171.Wolff NA, Abouhamed M, Verroust PJ, Thevenod F. Megalin-dependent internalization of cadmium-metallothionein and cytotoxicity in cultured renal proximal tubule cells. J Pharmacol Exp Ther. 2006;318:782–791. doi: 10.1124/jpet.106.102574. [DOI] [PubMed] [Google Scholar]
- 172.Bridges CC, Zalups RK. Molecular and ionic mimicry and the transport of toxic metals. Toxicol Appl Pharmacol. 2005;204:274–308. doi: 10.1016/j.taap.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Clarkson TW. Molecular and ionic mimicry of toxic metals. Annu Rev Pharmacol Toxicol. 1993;33:545–571. doi: 10.1146/annurev.pa.33.040193.002553. [DOI] [PubMed] [Google Scholar]
- 174.Fujishiro H, Yano Y, Takada Y, Tanihara M, Himeno S. Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics. 2012;4:700–708. doi: 10.1039/c2mt20024d. [DOI] [PubMed] [Google Scholar]
- 175.He L, Wang B, Hay EB, Nebert DW. Discovery of ZIP transporters that participate in cadmium damage to testis and kidney. Toxicol Appl Pharmacol. 2009;238:250–257. doi: 10.1016/j.taap.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Waalkes MP, Kovatch R, Rehm S. Effect of chronic dietary zinc deficiency on cadmium toxicity and carcinogenesis in the male Wistar [Hsd: (WI)BR] rat. Toxicol Appl Pharmacol. 1991;108:448–456. doi: 10.1016/0041-008x(91)90091-r. [DOI] [PubMed] [Google Scholar]
- 177.Flanagan PR, McLellan JS, Haist J, Cherian G, Chamberlain MJ, Valberg LS. Increased dietary cadmium absorption in mice and human subjects with iron deficiency. Gastroenterology. 1978;74:841–846. [PubMed] [Google Scholar]
- 178.Min KS, Ueda H, Tanaka K. Involvement of intestinal calcium transporter 1 and metallothionein in cadmium accumulation in the liver and kidney of mice fed a low-calcium diet. Toxicol Lett. 2008;176:85–92. doi: 10.1016/j.toxlet.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 179.Post C, Johansson B, Allenmark S. Organ distribution and protein binding of cadmium in autopsy material from heavy smokers. Environ Res. 1984;34:29–37. doi: 10.1016/0013-9351(84)90073-2. [DOI] [PubMed] [Google Scholar]
- 180.Thevenod F, Friedmann JM, Katsen AD, Hauser IA. Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem. 2000;275:1887–1896. doi: 10.1074/jbc.275.3.1887. [DOI] [PubMed] [Google Scholar]
- 181.Endo T, Kimura O, Sakata M. Effects of P-glycoprotein inhibitors on cadmium accumulation in cultured renal epithelial cells, LLC-PK1, and OK. Toxicol Appl Pharmacol. 2002;185:166–171. doi: 10.1006/taap.2002.9533. [DOI] [PubMed] [Google Scholar]
- 182.Kimura O, Endo T, Hotta Y, Sakata M. Effects of P-glycoprotein inhibitors on transepithelial transport of cadmium in cultured renal epithelial cells, LLC-PK1 and LLC-GA5-COL 150. Toxicology. 2005;208:123–132. doi: 10.1016/j.tox.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 183.Lee WK, Torchalski B, Kohistani N, Thevenod F. ABCB1 protects kidney proximal tubule cells against cadmium-induced apoptosis: roles of cadmium and ceramide transport. Toxicol Sci. 2011;121:343–356. doi: 10.1093/toxsci/kfr071. [DOI] [PubMed] [Google Scholar]
- 184.Endo T, Kimura O, Sakata M. Comparative studies of cadmium and mercury accumulation by LLC-PK1 cells: effects of pH on uptake and efflux. Toxicol Lett. 1996;87:77–83. doi: 10.1016/0378-4274(96)03707-1. [DOI] [PubMed] [Google Scholar]
- 185.Endo T, Kimura O, Sakata M. pH-dependent transport of cadmium in rat renal brush border membrane vesicles: cadmium efflux via H+-antiport. Toxicol Lett. 1998;99:99–107. doi: 10.1016/s0378-4274(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 186.Endo T, Kimura O, Sakata M. Bidirectional transport of cadmium across apical membrane of renal epithelial cell lines via H+-antiporter and inorganic anion exchanger. Toxicology. 1998;131:183–192. doi: 10.1016/s0300-483x(98)00133-4. [DOI] [PubMed] [Google Scholar]
- 187.Nakamura T, Yonezawa A, Hashimoto S, Katsura T, Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80:1762–1767. doi: 10.1016/j.bcp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 188.Yang H, Guo D, Obianom ON, Su T, Polli JE, Shu Y. Multidrug and toxin extrusion proteins mediate cellular transport of cadmium. Toxicol Appl Pharmacol. 2017;314:55–62. doi: 10.1016/j.taap.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.ATSDR. Toxicological profile for mercury. U.S. Department of Health and Human Services, Centers for Disease Control; Atlanta, GA: 2008. [Google Scholar]
- 190.Ashe WF, Largent EJ, Dutra FR, Hubbard DM, Blackstone M. Behavior of mercury in the animal organism following inhalation. AMA Arch Ind Hyg Occup Med. 1953;7:19–43. [PubMed] [Google Scholar]
- 191.Clarkson TW, Magos L. Studies on the binding of mercury in tissue homogenates. Biochem J. 1966;99:62–70. doi: 10.1042/bj0990062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Friberg L. On the value of measuring mercury and cadmium concentration in urine. Prac Lek. 1959;11:162. [PubMed] [Google Scholar]
- 193.Friberg L, Odeblad E, Forssman S. Distribution of two mercury compounds in rabbits after a single subcutaneous injection; a radiometric and autoradiographic study of the distribution of mercuric chloride and phenylmercuric acetate. AMA Arch Ind Health. 1957;16:163–168. [PubMed] [Google Scholar]
- 194.Hahn LJ, Kloiber R, Leininger RW, Vimy MJ, Lorscheider FL. Whole-body imaging of the distribution of mercury released from dental fillings into monkey tissues. FASEB J. 1990;4:3256–3260. doi: 10.1096/fasebj.4.14.2227216. [DOI] [PubMed] [Google Scholar]
- 195.Omata S, Sato M, Sakimura K, Sugano H. Time-dependent accumulation of inorganic mercury in subcellular fractions of kidney, liver, and brain of rats exposed to methylmercury. Arch Toxicol. 1980;44:231–241. doi: 10.1007/BF00278031. [DOI] [PubMed] [Google Scholar]
- 196.Magos L, Butler WH. The kinetics of methylmercury administered repeatedly to rats. Arch Toxicol. 1976;35:25–39. doi: 10.1007/BF00333983. [DOI] [PubMed] [Google Scholar]
- 197.Norseth T, Clarkson TW. Studies on the biotransformation of 203Hg-labeled methyl mercury chloride in rats. Arch Environ Health. 1970;21:717–727. doi: 10.1080/00039896.1970.10667325. [DOI] [PubMed] [Google Scholar]
- 198.Norseth T, Clarkson TW. Biotransformation of methylmercury salts in the rat studied by specific determination of inorganic mercury. Biochem Pharmacol. 1970;19:2775–2783. doi: 10.1016/0006-2952(70)90104-8. [DOI] [PubMed] [Google Scholar]
- 199.Zalups RK. Early aspects of the intrarenal distribution of mercury after the intravenous administration of mercuric chloride. Toxicology. 1993;79:215–228. doi: 10.1016/0300-483x(93)90213-c. [DOI] [PubMed] [Google Scholar]
- 200.Berlin M, Gibson S. Renal uptake, excretion, and retention of mercury. I. A study in the rabbit during infusion of mercuric chloride. Arch Environ Health. 1963;6:617–625. doi: 10.1080/00039896.1963.10663450. [DOI] [PubMed] [Google Scholar]
- 201.Berlin M, Ullberg S. Accumulation and retention of mercury in the mouse. I. An autoradiographic study after a single intravenous injection of mercuric chloride. Arch Environ Health. 1963;6:589–601. doi: 10.1080/00039896.1963.10663447. [DOI] [PubMed] [Google Scholar]
- 202.Berlin M, Ullberg S. Accumulation and retention of mercury in the mouse. III. An autoradiographic comparison of methylmercuric dicyandiamide with inorganic mercury. Arch Environ Health. 1963;6:610–616. doi: 10.1080/00039896.1963.10663449. [DOI] [PubMed] [Google Scholar]
- 203.Taugner R, Winkel Kz, Iravani J. On the localization of mercuric chloride concentration in the rat kidney. Virchows Arch Pathol Anat Physiol Klin Med. 1966;340:369–383. [PubMed] [Google Scholar]
- 204.Hughes WL. A physicochemical rationale for the biological activity of mercury and its compounds. Ann N Y Acad Sci. 1957;65:454–460. doi: 10.1111/j.1749-6632.1956.tb36650.x. [DOI] [PubMed] [Google Scholar]
- 205.Zalups RK, Cherian MG. Renal metallothionein metabolism after a reduction of renal mass. I. Effect of unilateral nephrectomy and compensatory renal growth on basal and metal-induced renal metallothionein metabolism. Toxicology. 1992;71:83–102. doi: 10.1016/0300-483x(92)90056-k. [DOI] [PubMed] [Google Scholar]
- 206.Zalups RK, Cherian MG, Barfuss DW. Mercury-metallothionein and the renal accumulation and handling of mercury. Toxicology. 1993;83:61–78. doi: 10.1016/0300-483x(93)90092-7. [DOI] [PubMed] [Google Scholar]
- 207.Zalups RK, Koropatnick J. Temporal changes in metallothionein gene transcription in rat kidney and liver: relationship to content of mercury and metallothionein protein. J Pharmacol Exp Ther. 2000;295:74–82. [PubMed] [Google Scholar]
- 208.Torres AM, Dnyanmote AV, Bush KT, Wu W, Nigam SK. Deletion of multispecific organic anion transporter Oat1/Slc22a6 protects against mercury-induced kidney injury. J Biol Chem. 2011;286:26391–26395. doi: 10.1074/jbc.M111.249292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hazelhoff MH, Bulacio RP, Torres AM. Gender related differences in kidney injury induced by mercury. Int J Mol Sci. 2012;13:10523–10536. doi: 10.3390/ijms130810523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Aslamkhan AG, Han YH, Yang XP, Zalups RK, Pritchard JB. Human renal organic anion transporter 1-dependent uptake and toxicity of mercuric-thiol conjugates in Madin-Darby canine kidney cells. Mol Pharmacol. 2003;63:590–596. doi: 10.1124/mol.63.3.590. [DOI] [PubMed] [Google Scholar]
- 211.Zalups RK, Aslamkhan AG, Ahmad S. Human organic anion transporter 1 mediates cellular uptake of cysteine-S conjugates of inorganic mercury. Kidney Int. 2004;66:251–261. doi: 10.1111/j.1523-1755.2004.00726.x. [DOI] [PubMed] [Google Scholar]
- 212.Ban M, de Ceaurriz J. Probenecid-induced protection against acute hexachloro-1,3-butadiene and methyl mercury toxicity to the mouse kidney. Toxicol Lett. 1988;40:71–76. doi: 10.1016/0378-4274(88)90184-1. [DOI] [PubMed] [Google Scholar]
- 213.Koh AS, Simmons-Willis TA, Pritchard JB, Grassl SM, Ballatori N. Identification of a mechanism by which the methylmercury antidotes N-acetylcysteine and dimercaptopropanesulfonate enhance urinary metal excretion: transport by the renal organic anion transporter-1. Mol Pharmacol. 2002;62:921–926. doi: 10.1124/mol.62.4.921. [DOI] [PubMed] [Google Scholar]
- 214.Zalups RK, Ahmad S. Handling of cysteine S-conjugates of methylmercury in MDCK cells expressing human OAT1. Kidney Int. 2005;68:1684–1699. doi: 10.1111/j.1523-1755.2005.00585.x. [DOI] [PubMed] [Google Scholar]
- 215.Zalups RK, Ahmad S. Transport of N-acetylcysteine s-conjugates of methylmercury in Madin-Darby canine kidney cells stably transfected with human isoform of organic anion transporter 1. J Pharmacol Exp Ther. 2005;314:1158–1168. doi: 10.1124/jpet.105.086645. [DOI] [PubMed] [Google Scholar]
- 216.Zalups RK, Ahmad S. Handling of the homocysteine S-conjugate of methylmercury by renal epithelial cells: role of organic anion transporter 1 and amino acid transporters. J Pharmacol Exp Ther. 2005;315:896–904. doi: 10.1124/jpet.105.090530. [DOI] [PubMed] [Google Scholar]
- 217.Cannon VT, Barfuss DW, Zalups RK. Molecular homology and the luminal transport of Hg2+ in the renal proximal tubule. J Am Soc Nephrol. 2000;11:394–402. doi: 10.1681/ASN.V113394. [DOI] [PubMed] [Google Scholar]
- 218.Cannon VT, Zalups RK, Barfuss DW. Amino acid transporters involved in luminal transport of mercuric conjugates of cysteine in rabbit proximal tubule. J Pharmacol Exp Ther. 2001;298:780–789. [PubMed] [Google Scholar]
- 219.Wang Y, Zalups RK, Barfuss DW. Luminal transport of thiol S-conjugates of methylmercury in isolated perfused rabbit renal proximal tubules. Toxicol Lett. 2012;213:203–210. doi: 10.1016/j.toxlet.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bridges CC, Zalups RK. System b0,+ and the transport of thiol-s-conjugates of methylmercury. J Pharmacol Exp Ther. 2006;319:948–956. doi: 10.1124/jpet.106.109371. [DOI] [PubMed] [Google Scholar]
- 221.Terlouw SA, Graeff C, Smeets PH, Fricker G, Russel FG, Masereeuw R, Miller DS. Short- and long-term influences of heavy metals on anionic drug efflux from renal proximal tubule. J Pharmacol Exp Ther. 2002;301:578–585. doi: 10.1124/jpet.301.2.578. [DOI] [PubMed] [Google Scholar]
- 222.Aleo MF, Morandini F, Bettoni F, Giuliani R, Rovetta F, Steimberg N, Apostoli P, Parrinello G, Mazzoleni G. Endogenous thiols and MRP transporters contribute to Hg2+ efflux in HgCl2-treated tubular MDCK cells. Toxicology. 2005;206:137–151. doi: 10.1016/j.tox.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 223.Bridges CC, Joshee L, Zalups RK. MRP2 and the handling of mercuric ions in rats exposed acutely to inorganic and organic species of mercury. Toxicol Appl Pharmacol. 2011;251:50–58. doi: 10.1016/j.taap.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zalups RK, Joshee L, Bridges CC. Novel Hg2+-induced nephropathy in rats and mice lacking Mrp2: evidence of axial heterogeneity in the handling of Hg2+ along the proximal tubule. Toxicol Sci. 2014;142:250–260. doi: 10.1093/toxsci/kfu171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Engstrom K, Ameer S, Bernaudat L, Drasch G, Baeuml J, Skerfving S, Bose-O’Reilly S, Broberg K. Polymorphisms in genes encoding potential mercury transporters and urine mercury concentrations in populations exposed to mercury vapor from gold mining. Environ Health Perspect. 2013;121:85–91. doi: 10.1289/ehp.1204951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bridges CC, Zalups RK, Joshee L. Toxicological significance of renal Bcrp: Another potential transporter in the elimination of mercuric ions from proximal tubular cells. Toxicol Appl Pharmacol. 2015;285:110–117. doi: 10.1016/j.taap.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zalups RK, Parks LD, Cannon VT, Barfuss DW. Mechanisms of action of 2,3-dimercaptopropane-1-sulfonate and the transport, disposition, and toxicity of inorganic mercury in isolated perfused segments of rabbit proximal tubules. Mol Pharmacol. 1998;54:353–363. doi: 10.1124/mol.54.2.353. [DOI] [PubMed] [Google Scholar]
- 228.Bridges CC, Joshee L, Zalups RK. Multidrug resistance proteins and the renal elimination of inorganic mercury mediated by 2,3-dimercaptopropane-1-sulfonic acid and meso-2,3-dimercaptosuccinic acid. J Pharmacol Exp Ther. 2008;324:383–390. doi: 10.1124/jpet.107.130708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bridges CC, Joshee L, Zalups RK. MRP2 and the DMPS- and DMSA-mediated elimination of mercury in TR(-) and control rats exposed to thiol S-conjugates of inorganic mercury. Toxicol Sci. 2008;105:211–220. doi: 10.1093/toxsci/kfn107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zalups RK, Bridges CC. MRP2 involvement in renal proximal tubular elimination of methylmercury mediated by DMPS or DMSA. Toxicol Appl Pharmacol. 2009;235:10–17. doi: 10.1016/j.taap.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 231.Bridges CC, Joshee L, van den Heuvel JJ, Russel FG, Zalups RK. Glutathione status and the renal elimination of inorganic mercury in the Mrp2(-/-) mouse. PLoS One. 2013;8:e73559. doi: 10.1371/journal.pone.0073559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Waxman DJ, Strominger JL. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu Rev Biochem. 1983;52:825–869. doi: 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- 233.Hoover JR, Dunn GL, Jakas DR, Lam LL, Taggart JJ, Guarini JR, Phillips L. Semisynthetic cephalosporins. Synthesis and structure-activity relationships of 7-mandelamido-3-cephem-4-carboxylic acids. J Med Chem. 1974;17:34–41. doi: 10.1021/jm00247a008. [DOI] [PubMed] [Google Scholar]
- 234.Tune BM, Hsu CY, Fravert D. Cephalosporin and carbacephem nephrotoxicity. Roles of tubular cell uptake and acylating potential. Biochem Pharmacol. 1996;51:557–561. doi: 10.1016/0006-2952(95)02237-6. [DOI] [PubMed] [Google Scholar]
- 235.Atkinson RM, Currie JP, Davis B, Pratt DA, Sharpe HM, Tomich EG. Acute toxicity of cephaloridine, an antibiotic derived from cephalosporin C. Toxicol Appl Pharmacol. 1966;8:398–406. doi: 10.1016/0041-008x(66)90050-0. [DOI] [PubMed] [Google Scholar]
- 236.Birnbaum J, Kahan FM, Kropp H, MacDonald JS. Carbapenems, a new class of beta-lactam antibiotics. Discovery and development of imipenem/cilastatin. Am J Med. 1985;78:3–21. doi: 10.1016/0002-9343(85)90097-x. [DOI] [PubMed] [Google Scholar]
- 237.Tune BM, Fravert D. Mechanisms of cephalosporin nephrotoxicity: a comparison of cephaloridine and cephaloglycin. Kidney Int. 1980;18:591–600. doi: 10.1038/ki.1980.177. [DOI] [PubMed] [Google Scholar]
- 238.Tune BM, Fravert D. Cephalosporin nephrotoxicity. Transport, cytotoxicity and mitochondrial toxicity of cephaloglycin. J Pharmacol Exp Ther. 1980;215:186–190. [PubMed] [Google Scholar]
- 239.Kuo CH, Maita K, Sleight SD, Hook JB. Lipid peroxidation: a possible mechanism of cephaloridine-induced nephrotoxicity. Toxicol Appl Pharmacol. 1983;67:78–88. doi: 10.1016/0041-008x(83)90246-6. [DOI] [PubMed] [Google Scholar]
- 240.Tune BM, Fravert D, Hsu CY. Oxidative and mitochondrial toxic effects of cephalosporin antibiotics in the kidney. A comparative study of cephaloridine and cephaloglycin. Biochem Pharmacol. 1989;38:795–802. doi: 10.1016/0006-2952(89)90233-5. [DOI] [PubMed] [Google Scholar]
- 241.Cojocel C, Hannemann J, Baumann K. Cephaloridine-induced lipid peroxidation initiated by reactive oxygen species as a possible mechanism of cephaloridine nephrotoxicity. Biochim Biophys Acta. 1985;834:402–410. doi: 10.1016/0005-2760(85)90014-1. [DOI] [PubMed] [Google Scholar]
- 242.Inui K, Okano T, Takano M, Kitazawa S, Hori R. Carrier-mediated transport of amino-cephalosporins by brush border membrane vesicles isolated from rat kidney cortex. Biochem Pharmacol. 1983;32:621–626. doi: 10.1016/0006-2952(83)90485-9. [DOI] [PubMed] [Google Scholar]
- 243.Indelicato JM, Dinner A, Peters LR, Wilham WL. Hydrolysis of 3-chloro-3-cephems. Intramolecular nucleophilic attack in cefaclor. J Med Chem. 1977;20:961–963. doi: 10.1021/jm00217a021. [DOI] [PubMed] [Google Scholar]
- 244.Atkinson RM, Caisey JD, Currie JP, Middleton TR, Pratt DA, Sharpe HM, Tomich EG. Subacute toxicity of cephaloridine to various species. Toxicol Appl Pharmacol. 1966;8:407–428. doi: 10.1016/0041-008x(66)90051-2. [DOI] [PubMed] [Google Scholar]
- 245.Welles JS, Gibson WR, Harris PN, Small PM, Anderson RC. Toxicity, distribution, and excretion of cephaloridine in laboratory animals. Antimicrob Agents Chemother (Bethesda) 1965;5:863–869. [PubMed] [Google Scholar]
- 246.Tune BM. Effect of organic acid transport inhibitors on renal cortical uptake and proximal tubular toxicity of cephaloridine. J Pharmacol Exp Ther. 1972;181:250–256. [PubMed] [Google Scholar]
- 247.Child KJ, Dodds MG. Nephron transport and renal tubular effects of cephaloridine in animals. Br J Pharmacol Chemother. 1967;30:354–370. doi: 10.1111/j.1476-5381.1967.tb02142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Takeda M, Tojo A, Sekine T, Hosoyamada M, Kanai Y, Endou H. Role of organic anion transporter 1 (OAT1) in cephaloridine (CER)-induced nephrotoxicity. Kidney Int. 1999;56:2128–2136. doi: 10.1046/j.1523-1755.1999.00789.x. [DOI] [PubMed] [Google Scholar]
- 249.Jung KY, Takeda M, Shimoda M, Narikawa S, Tojo A, Kim DK, Chairoungdua A, Choi BK, Kusuhara H, Sugiyama Y, Sekine T, Endou H. Involvement of rat organic anion transporter 3 (rOAT3) in cephaloridine-induced nephrotoxicity: in comparison with rOAT1. Life Sci. 2002;70:1861–1874. doi: 10.1016/s0024-3205(02)01500-x. [DOI] [PubMed] [Google Scholar]
- 250.Khamdang S, Takeda M, Babu E, Noshiro R, Onozato ML, Tojo A, Enomoto A, Huang XL, Narikawa S, Anzai N, Piyachaturawat P, Endou H. Interaction of human and rat organic anion transporter 2 with various cephalosporin antibiotics. Eur J Pharmacol. 2003;465:1–7. doi: 10.1016/s0014-2999(03)01381-5. [DOI] [PubMed] [Google Scholar]
- 251.Kano T, Kato Y, Ito K, Ogihara T, Kubo Y, Tsuji A. Carnitine/organic cation transporter OCTN2 (Slc22a5) is responsible for renal secretion of cephaloridine in mice. Drug Metab Dispos. 2009;37:1009–1016. doi: 10.1124/dmd.108.025015. [DOI] [PubMed] [Google Scholar]
- 252.Wold JS, Turnipseed SA, Miller BL. The effect of renal cation transport inhibition on cephaloridine nephrotoxicity. Toxicol Appl Pharmacol. 1979;47:115–122. doi: 10.1016/0041-008x(79)90078-4. [DOI] [PubMed] [Google Scholar]
- 253.Kasher JS, Holohan PD, Ross CR. Effect of cephaloridine on the transport of organic ions in dog kidney plasma membrane vesicles. J Pharmacol Exp Ther. 1983;225:606–610. [PubMed] [Google Scholar]
- 254.Williams PD, Hitchcock MJ, Hottendorf GH. Effect of cephalosporins on organic ion transport in renal membrane vesicles from rat and rabbit kidney cortex. Res Commun Chem Pathol Pharmacol. 1985;47:357–371. [PubMed] [Google Scholar]
- 255.Ci L, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. Involvement of MRP4 (ABCC4) in the luminal efflux of ceftizoxime and cefazolin in the kidney. Mol Pharmacol. 2007;71:1591–1597. doi: 10.1124/mol.106.031823. [DOI] [PubMed] [Google Scholar]
- 256.Furman PA, Fyfe JA, St Clair MH, Weinhold K, Rideout JL, Freeman GA, Lehrman SN, Bolognesi DP, Broder S, Mitsuya H, et al. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci U S A. 1986;83:8333–8337. doi: 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cihlar T, Ray AS. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res. 2010;85:39–58. doi: 10.1016/j.antiviral.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 258.Cundy KC, Barditch-Crovo P, Walker RE, Collier AC, Ebeling D, Toole J, Jaffe HS. Clinical pharmacokinetics of adefovir in human immunodeficiency virus type 1-infected patients. Antimicrob Agents Chemother. 1995;39:2401–2405. doi: 10.1128/aac.39.11.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Cundy KC, Petty BG, Flaherty J, Fisher PE, Polis MA, Wachsman M, Lietman PS, Lalezari JP, Hitchcock MJ, Jaffe HS. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 1995;39:1247–1252. doi: 10.1128/aac.39.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Shimizu M, Furusyo N, Ikezaki H, Ogawa E, Hayashi T, Ihara T, Harada Y, Toyoda K, Murata M, Hayashi J. Predictors of kidney tubular dysfunction induced by adefovir treatment for chronic hepatitis B. World J Gastroenterol. 2015;21:2116–2123. doi: 10.3748/wjg.v21.i7.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Tanji N, Tanji K, Kambham N, Markowitz GS, Bell A, D’Agati DV. Adefovir nephrotoxicity: possible role of mitochondrial DNA depletion. Hum Pathol. 2001;32:734–740. doi: 10.1053/hupa.2001.25586. [DOI] [PubMed] [Google Scholar]
- 262.Cihlar T, Lin DC, Pritchard JB, Fuller MD, Mendel DB, Sweet DH. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol Pharmacol. 1999;56:570–580. doi: 10.1124/mol.56.3.570. [DOI] [PubMed] [Google Scholar]
- 263.Ho ES, Lin DC, Mendel DB, Cihlar T. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J Am Soc Nephrol. 2000;11:383–393. doi: 10.1681/ASN.V113383. [DOI] [PubMed] [Google Scholar]
- 264.Cihlar T, Ho ES, Lin DC, Mulato AS. Human renal organic anion transporter 1 (hOAT1) and its role in the nephrotoxicity of antiviral nucleotide analogs. Nucleosides Nucleotides Nucleic Acids. 2001;20:641–648. doi: 10.1081/NCN-100002341. [DOI] [PubMed] [Google Scholar]
- 265.Uwai Y, Ida H, Tsuji Y, Katsura T, Inui K. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2) Pharm Res. 2007;24:811–815. doi: 10.1007/s11095-006-9196-x. [DOI] [PubMed] [Google Scholar]
- 266.Takeda M, Khamdang S, Narikawa S, Kimura H, Kobayashi Y, Yamamoto T, Cha SH, Sekine T, Endou H. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J Pharmacol Exp Ther. 2002;300:918–924. doi: 10.1124/jpet.300.3.918. [DOI] [PubMed] [Google Scholar]
- 267.Cheng Y, Vapurcuyan A, Shahidullah M, Aleksunes LM, Pelis RM. Expression of organic anion transporter 2 in the human kidney and its potential role in the tubular secretion of guanine-containing antiviral drugs. Drug Metab Dispos. 2012;40:617–624. doi: 10.1124/dmd.111.042036. [DOI] [PubMed] [Google Scholar]
- 268.Cihlar T, Laflamme G, Fisher R, Carey AC, Vela JE, Mackman R, Ray AS. Novel nucleotide human immunodeficiency virus reverse transcriptase inhibitor GS-9148 with a low nephrotoxic potential: characterization of renal transport and accumulation. Antimicrob Agents Chemother. 2009;53:150–156. doi: 10.1128/AAC.01183-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Dkhil MA, Al-Quraishy S, Diab MM, Othman MS, Aref AM, Abdel Moneim AE. The potential protective role of Physalis peruviana L. fruit in cadmium-induced hepatotoxicity and nephrotoxicity. Food Chem Toxicol. 2014;74:98–106. doi: 10.1016/j.fct.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 270.Miller DS. Nucleoside phosphonate interactions with multiple organic anion transporters in renal proximal tubule. J Pharmacol Exp Ther. 2001;299:567–574. [PubMed] [Google Scholar]
- 271.Imaoka T, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol Pharmacol. 2007;71:619–627. doi: 10.1124/mol.106.028233. [DOI] [PubMed] [Google Scholar]
- 272.Schuetz JD, Connelly MC, Sun D, Paibir SG, Flynn PM, Srinivas RV, Kumar A, Fridland A. MRP4: A previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat Med. 1999;5:1048–1051. doi: 10.1038/12487. [DOI] [PubMed] [Google Scholar]
- 273.Reid G, Wielinga P, Zelcer N, De Haas M, Van Deemter L, Wijnholds J, Balzarini J, Borst P. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol Pharmacol. 2003;63:1094–1103. doi: 10.1124/mol.63.5.1094. [DOI] [PubMed] [Google Scholar]
- 274.Shord SS, Thompson DM, Krempl GA, Hanigan MH. Effect of concurrent medications on cisplatin-induced nephrotoxicity in patients with head and neck cancer. Anticancer Drugs. 2006;17:207–215. doi: 10.1097/00001813-200602000-00013. [DOI] [PubMed] [Google Scholar]
- 275.Reece PA, Stafford I, Russell J, Gill PG. Nonlinear renal clearance of ultrafilterable platinum in patients treated with cis-dichlorodiammineplatinum (II) Cancer Chemother Pharmacol. 1985;15:295–299. doi: 10.1007/BF00263904. [DOI] [PubMed] [Google Scholar]
- 276.Pan BF, Sweet DH, Pritchard JB, Chen R, Nelson JA. A transfected cell model for the renal toxin transporter, rOCT2. Toxicol Sci. 1999;47:181–186. doi: 10.1093/toxsci/47.2.181. [DOI] [PubMed] [Google Scholar]
- 277.Endo T, Kimura O, Sakata M. Carrier-mediated uptake of cisplatin by the OK renal epithelial cell line. Toxicology. 2000;146:187–195. doi: 10.1016/s0300-483x(00)00176-1. [DOI] [PubMed] [Google Scholar]
- 278.Kolb RJ, Ghazi AM, Barfuss DW. Inhibition of basolateral transport and cellular accumulation of cDDP and N-acetyl- L-cysteine-cDDP by TEA and PAH in the renal proximal tubule. Cancer Chemother Pharmacol. 2003;51:132–138. doi: 10.1007/s00280-002-0537-0. [DOI] [PubMed] [Google Scholar]
- 279.Ludwig T, Riethmuller C, Gekle M, Schwerdt G, Oberleithner H. Nephrotoxicity of platinum complexes is related to basolateral organic cation transport. Kidney Int. 2004;66:196–202. doi: 10.1111/j.1523-1755.2004.00720.x. [DOI] [PubMed] [Google Scholar]
- 280.Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1–3 and multidrug and toxin extrusion family) J Pharmacol Exp Ther. 2006;319:879–886. doi: 10.1124/jpet.106.110346. [DOI] [PubMed] [Google Scholar]
- 281.Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86:396–402. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Iwata K, Aizawa K, Kamitsu S, Jingami S, Fukunaga E, Yoshida M, Yoshimura M, Hamada A, Saito H. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol. 2012;16:843–851. doi: 10.1007/s10157-012-0638-y. [DOI] [PubMed] [Google Scholar]
- 283.Zhang J, Zhou W. Ameliorative effects of SLC22A2 gene polymorphism 808 G/T and cimetidine on cisplatin-induced nephrotoxicity in Chinese cancer patients. Food Chem Toxicol. 2012;50:2289–2293. doi: 10.1016/j.fct.2012.03.077. [DOI] [PubMed] [Google Scholar]
- 284.Hinai Y, Motoyama S, Niioka T, Miura M. Absence of effect of SLC22A2 genotype on cisplatin-induced nephrotoxicity in oesophageal cancer patients receiving cisplatin and 5-fluorouracil: report of results discordant with those of earlier studies. J Clin Pharm Ther. 2013;38:498–503. doi: 10.1111/jcpt.12097. [DOI] [PubMed] [Google Scholar]
- 285.Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2009;296:F505–511. doi: 10.1152/ajprenal.90545.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Ivy KD, Kaplan JH. A re-evaluation of the role of hCTR1, the human high-affinity copper transporter, in platinum-drug entry into human cells. Mol Pharmacol. 2013;83:1237–1246. doi: 10.1124/mol.113.085068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Cui Y, Konig J, Buchholz JK, Spring H, Leier I, Keppler D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol Pharmacol. 1999;55:929–937. [PubMed] [Google Scholar]
- 288.Kawabe T, Chen ZS, Wada M, Uchiumi T, Ono M, Akiyama S, Kuwano M. Enhanced transport of anticancer agents and leukotriene C4 by the human canalicular multispecific organic anion transporter (cMOAT/MRP2) FEBS Lett. 1999;456:327–331. doi: 10.1016/s0014-5793(99)00979-5. [DOI] [PubMed] [Google Scholar]
- 289.Ishikawa T, Ali-Osman F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem. 1993;268:20116–20125. [PubMed] [Google Scholar]
- 290.Sprowl JA, Gregorc V, Lazzari C, Mathijssen RH, Loos WJ, Sparreboom A. Associations between ABCC2 polymorphisms and cisplatin disposition and efficacy. Clin Pharmacol Ther. 2012;91:1022–1026. doi: 10.1038/clpt.2011.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wen X, Buckley B, McCandlish E, Goedken MJ, Syed S, Pelis R, Manautou JE, Aleksunes LM. Transgenic expression of the human MRP2 transporter reduces cisplatin accumulation and nephrotoxicity in Mrp2-null mice. Am J Pathol. 2014;184:1299–1308. doi: 10.1016/j.ajpath.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wilmer MJ, Ng CP, Lanz HL, Vulto P, Suter-Dick L, Masereeuw R. Kidney-on-a-Chip Technology for Drug-Induced Nephrotoxicity Screening. Trends Biotechnol. 2016;34:156–170. doi: 10.1016/j.tibtech.2015.11.001. [DOI] [PubMed] [Google Scholar]