

Abstract
Protofibrils of the 42 amino acids long amyloid-β peptide are transient pre-fibrillar intermediates in the process of peptide aggregation into amyloid plaques and are thought to play a critical role in the pathology of Alzheimer’s disease. Hence, there is a need for research reagents and potential diagnostic reagents for detection and imaging of such aggregates. Here we describe an in vitro selection of Affibody molecules that bind to protofibrils of Aβ42cc, which is a stable engineered mimic of wild type Aβ42 protofibrils. Several binders were identified that bind Aβ42cc protofibrils with low nanomolar affinities, and which also recognize wild type Aβ42 protofibrils. Dimeric head-to-tail fusion proteins with subnanomolar binding affinities, and very slow dissociation off-rates, were also constructed. A mapping of the chemical properties of the side chains onto the Affibody scaffold surface reveals three distinct adjacent surface areas of positively charged surface, nonpolar surface and a polar surface, which presumably match a corresponding surface epitope on the protofibrils. The results demonstrate that the engineered Aβ42cc is a suitable antigen for directed evolution of affinity reagents with specificity for wild type Aβ42 protofibrils.
Introduction
Aggregation of the amyloid-β peptide (Aβ) in the brain is a hallmark of Alzheimer’s disease1. The end-state of the aggregation is amyloid fibrils, which become deposited into senile plaques. However, Aβ aggregation involves a number of intermediate aggregation states, collectively called soluble oligomers, and evidence suggests direct causative links between soluble Aβ oligomers and synapse dysfunction2, 3. The aggregation path of the 42-residue Aβ42 peptide to amyloid fibrils is believed to involve the formation of pentameric or hexameric oligomers (paranuclei) that associate into larger protofibrils, which eventually undergo a structural interconversion into amyloid fibrils4–6. Please note that we refer to protofibrils as a class of rod-like pre-fibrillar aggregates that are distinct from amyloid fibrils6.
However, smaller dimeric or trimeric (low-n) oligomers7, and various aggregates that form in the presence of membranes or detergents, have also been described (and reviewed)6. In fact, much of the details surrounding oligomer formation and interconversion in vivo as well as in vitro remain elusive. New binding agents that specifically recognize intermediate aggregates, and which can be used to detect the presence of such aggregates, would represent valuable tools in the research and such agents might also be used within diagnostic or even therapeutic applications.
In this work, we describe the selection of Affibody molecules8, 9 that selectively recognize protofibrils of Aβ. There are several advantages and potential applications of in vitro selected binders. One apparent strength is that the state of target is under full control during the selection, which is not normally the case with IgG antibodies that are often generated by immunization. Another advantage, compared to IgG antibodies, is that Affibody molecules, in which the 58-residue small three-helix Z-domain is used as a scaffold for variation, are easily produced in large amounts in bacterial cultures or even by peptide synthesis. A third potential advantage relates to possible applications for therapy or brain imaging, as a smaller protein domain presumably will undergo a more rapid transfer through the blood-brain-barrier than large IgG antibodies. Finally, we wanted to address a more basic question in protein engineering: can an in-vitro selected protein binder be identified that can discriminate a certain protein aggregate from other aggregated and monomeric forms of the same peptide?
Protofibrils of wild type Aβ42 are instable and therefore not optimal as targets for binding protein selection using for instance phage display. However, we have previously engineered an Aβ42 cysteine variant (Aβ42 cc) that forms protofibrils, which do not convert into amyloid fibrils10–12. Briefly, in Aβ42 cc, alanine residues 21 and 30 are replaced with cysteine residues, allowing an intramolecular disulfide bond to form. The disulfide locks the peptide in a hairpin conformation, which is compatible with the conformation of Aβ42wt in protofibrils, but incompatible with the conformation observed in fibrils13–15. Aggregation of Aβ42 cc is therefore halted at the protofibril stage. In previous studies, size exclusion chromatography profiles, circular dichroism spectra and electron microscopy images showed a clear resemblance between the Aβ42cc protofibrils and what has previously been reported for Aβ42wt protofibrils10. We also demonstrated that the Aβ42cc protofibrils are recognized by a protofibril-selective antibody (mAb158). In a follow up study11, we characterized the biophysical properties of Aβ42cc protofibrils in detail and compared them with Aβ42wt protofibrils, using atomic force microscopy, analytical ultracentrifugation, nanoparticle tracking analysis, binding of the dye ANS, binding to oligomer-specific antibodies (A11 and OC) and a synaptotoxicity assay. In summary, none of the techniques revealed any significant differences between Aβ42cc protofibrils and Aβ42wt protofibrils, except the fact that Aβ42cc do not undergo structural conversion into amyloid fibrils.
Ideally, a comparison of the atomic structures of Aβ42cc and Aβ42wt protofibrils should be carried out to clarify the degree of structural similarity. However, structural biology of protein aggregates is extremely challenging because of sample heterogeneities. We have presented a structural model for the Aβ42cc protofibrils based on solid-state NMR data12, which is essentially different compared to the structure of Aβ42wt amyloid fibrils. Moreover, by a combination of magic angle spinning NMR and solution state NMR, we also showed that the N-terminal part of the peptide is disordered in the protofibrils16.
From previous studies, we can thus conclude that Aβ42 cc protofibrils are a good mimic of wild type Aβ42 protofibrils. Moreover, Aβ42 cc protofibrils are stable and thereby suitable as target for selection of binding proteins.
Results
Phage display selections
Affibody molecules were selected using phage display with Aβ42 cc protofibrils as the target molecule. The protofibrils were biotinylated and attached to streptavidin coated magnetic beads. Binding proteins were selected from a phage library containing 1.4 × 1010 Affibody variants17, 18. A total of 13 surface-located amino acid residues in helices 1 and 2 of the Z-domain from Staphylococcus aureus protein A were randomized to construct the phage library19. The randomized positions can be found in Fig. 1a,b. This library was designed to exclude cysteines and should thus not allow for the selection of cysteine-containing Affibody molecules that can form dimers, such as the Aβ-monomer binding Affibody molecule that was selected and studied by us previously17, 20. The selection was carried out in two tracks, in which one involved a pre-selection against fibrils of Aβ42wt to remove fibril binders. Six rounds of selection and phage amplification were performed while gradually decreasing the target concentration from 2 µM (on a monomer basis) to 0.2 nM in the final round. Affibody variants in phage clones that remained after the fourth and final round were expressed as fusions to an albumin binding domain (ABD) and screened for protofibril binding performance using an enzyme linked immunosorbent assay (ELISA). In addition, it was vital for the study that none of the selected protofibril binders recognize mature amyloid fibrils. Therefore, the fibril binding properties were also assayed (see Supplementary Fig. S1). A total of 56 protofibril binders, identified in a single-point ELISA of 744 randomly selected Affibody variants, were ranked according to the apparent equilibrium dissociation constants (EC50 values; see Supplementary Fig. S2). It should be noted that these values are indicative of relative binding affinities in the ELISA format and not equilibrium dissociation constants. The sequences of 25 Affibody variants with the lowest ELISA EC50 values are shown in Fig. 1a. A comparison of sequence similarity in the form of a phylogenetic tree is shown in Supplementary Fig. S3.
Figure 1.
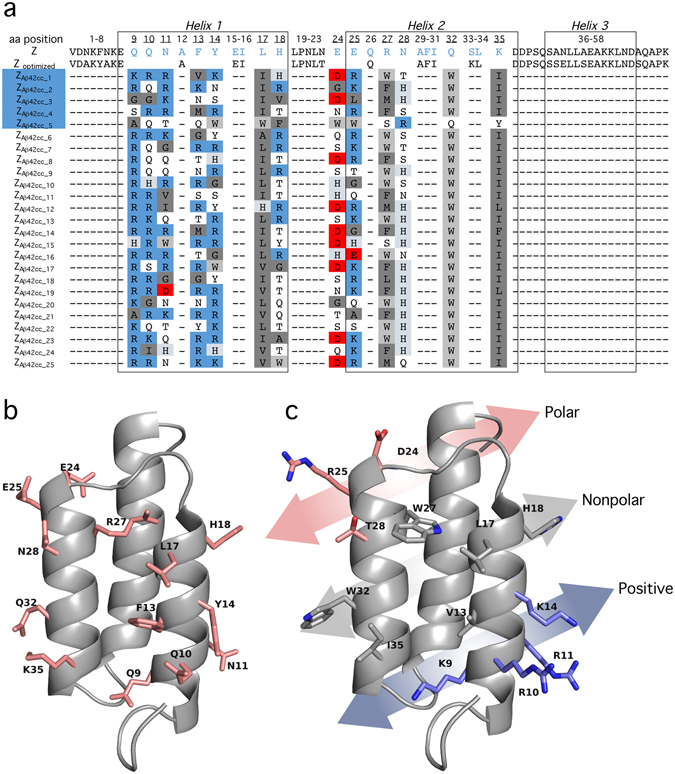
Sequence and cartoon representation of Affibody molecules selected as Aβ42cc protofibril binders. (a) Sequences of the top 25 selected Affibody molecules. The underlined amino acid positions are randomized in the phage display selection. The helical secondary structures are represented in boxes. The Affibody molecules chosen for further characterization are highlighted in blue. From 744 randomly picked clones, the sequence of ZAβ42cc_1 was identified eight times, ZAβ42cc_2 and ZAβ42cc_3 one time, ZAβ42cc_4 eight times and ZAβ42cc_5 three times. (b) The original Z-domain scaffold. Side chains of residues that are subjected to variation in the phage library are indicated. (c) Representation of side chains that are mutated in binder ZA β 42cc_1. The chemical properties of the side chains of the selected binders group into three chemically distinct regions of the surface, which identifies a ‘positive-nonpolar-polar’ recognition surface pattern.
Five strong Aβ42 cc protofibril binders, denoted ZAβ42cc_1, ZAβ42cc_2, ZAβ42cc_3, ZAβ42cc_4, and ZAβ42cc_5, were selected for further characterization. These molecules were subcloned, expressed and purified. Three of these (ZAβ42cc_1, ZAβ42cc_2 and ZAβ42cc_4) were selected because they appeared as strong binders in the ELISA. ZAβ42cc_3 and ZAβ42cc_5 were selected because their sequences differ from most other selected binders (Fig. 1a). At least ZAβ42cc_5 most likely displays a different topology when binding to Aβ42cc protofibrils.
Selectivity of binders to different Aβ aggregates
We then profiled the binding selectivity of the five selected binders (as mentioned above) to different Aβ aggregates using a second type of ELISA. This assay involved immobilization of Affibody-ABD fusions via an anti-ABD antibody followed by incubation with Aβ42cc protofibrils, Aβ42wt protofibrils (wt = wild type), Aβ42wt fibrils or Aβ42wt monomer. A non Aβ-binder Affibody molecule and PBS buffer were used as controls. The presence of bound Aβ remaining after washing was then detected using an HRP-conjugated antibody (6E10) that recognizes an N-terminal sequence of Aβ21 and which therefore most likely is insensitive to the Aβ aggregation state. A recent study suggests that the N-terminus of the peptide is disordered and more dynamic than the core structure of the protofibrils16.
In another assay, we used the antibody mAb1C3, a monoclonal mouse anti-Aβ IgG, which displays some specificity for Aβ42 protofibrils, compared to monomeric and low-molecular weight Aβ aggregates21. The Aβ42wt protofibrils used for this assay were made by either incubating Aβ42 monomer at 4 °C overnight or for 10 minutes at room temperature, followed by protofibril isolation by size exclusion chromatography22.
The five selected Affibody molecules all bound well to Aβ42cc protofibrils, as expected (Fig. 2). More important, three binders, and in particular ZAβ42cc_1, also recognized Aβ42wt protofibrils, which was the intended outcome of the present selection. None of the five binders recognized monomer and fibrils of Aβ42wt.
Figure 2.
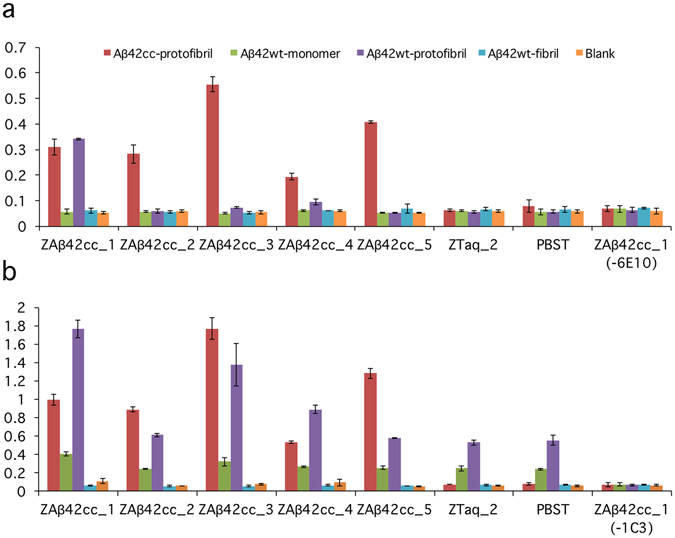
Binding profiling of five Affibody molecules to different Aβ aggregates, analyzed by ELISA. (a) Aβ42wt and Aβ42cc aggregates (50 nM assay concentration), bound to Affibody molecules, were detected by 6E10-HRP. 6E1021 recognizes the N-terminus of Aβ. As expected, all five Affibody molecules show binding to Aβ42cc protofibrils. The Affibody molecule ZAβ42cc_1 also binds to Aβ42wt protofibrils. No binding could be observed to either wild type monomer or fibrils. (b) Same as in (a), except that the assay concentration of Aβ42 was 1 µM and mAb1C3 was used for detection (weakly specific for protofibrils21). The binding profile has the same pattern as in (a) for protofibrils (both wt and cc) and Aβ42wt fibrils, but with a higher background for Aβ42wt protofibrils. (a,b) Two controls involve replacing the Affibody molecule with either an irrelevant Affibody molecule (ZTaq_2) or by PBS-T. In a third control experiment PBS-T was added instead of Aβ-specific antibody (6E10 or mAb1C3). Values are means of duplicate experiments.
We used an affinity based capturing approach to verify selective binding of Affibody molecules to protofibrils. For this experiment, we chose ZAβ42cc_1 and ZAβ42cc_4 since these two molecules, according to the selectivity profiling ELISA (Fig. 2a), recognize wild type protofibrils with an affinity that is close to that of Aβ42cc protofibrils. The Affibody molecules were recombinantly produced in E. coli and purified as described in the Methods section. They were immobilized on a nickel charged resin employing the His6 tag. The resin was briefly incubated with either Aβ42cc protofibrils, Aβ42wt protofibrils, Aβ42wt fibrils or Aβ42wt monomer. The resin was then pelleted and non-bound Aβ was collected in the supernatant, while bound Aβ was eluted together with Affibody molecules using a buffer containing imidazole. An SDS-PAGE analysis of the samples obtained from the affinity capture assay verifies that the two Affibody molecules ZAβ42cc_1 and ZAβ42cc_4 bind protofibrillar aggregates of Aβ42cc and Aβ42wt with similar selectivities, but do not recognize the monomer and fibrils of Aβ42wt (Fig. 3).
Figure 3.
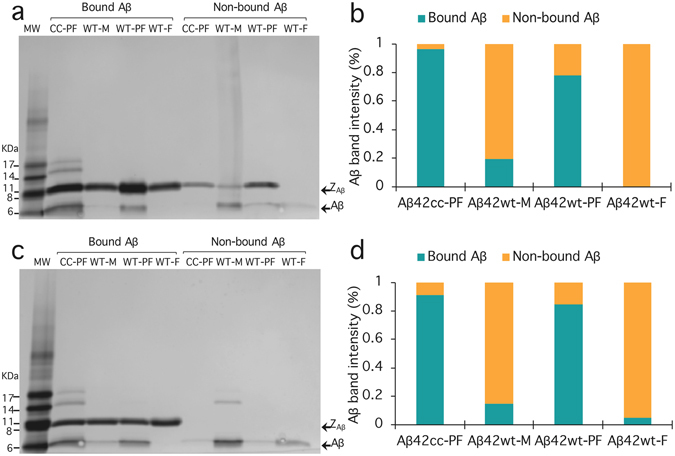
Binding of Affibody molecules to various Aβ aggregates and monomer in a batch mode affinity experiment. The SDS-PAGE analysis shows that Affibody ZA β 42cc_1 (a) and ZA β 42cc_4 (c) recognize protofibrillar aggregates of Aβ42cc and Aβ42wt (bound Aβ). Wild type monomer and fibrils did not bind to the Affibody molecules and were recovered in the supernatant (non-bound Aβ). The designations MW, CC-PF, WT-M, WT-PF and WT-F refer to molecular weight marker (GE Healthcare), Aβ42cc protofibrils, Aβ42wt monomer, Aβ42wt protofibrils and Aβ42wt fibrils, respectively. (b) and (d), Aβ band intensity for ZA β 42cc_1 and ZA β 42cc_4, respectively. The band intensities were obtained with the graphic program ImageJ32. The data was normalized with respect to each Aβ-species, i.e. the sum of the intensities for bands originating from one Aβ-species is equal to 100%.
Binding kinetics of Affibody molecules
Next, we used surface plasmon resonance (SPR) to study the kinetics for association and dissociation of the selected Affibody molecules to Aβ42cc protofibrils. For this we immobilized Aβ42cc protofibrils on a Biacore CM5 sensor chip using amine coupling chemistry. Affibody binding kinetics to this surface (Fig. 4 and Supplementary Table S1) was in all cases characterized by non-exponential association and non-exponential and saturation dependent dissociation. Collected data did fit well to a global heterogeneous binding model with two binding sites, if local maximum response (Rmax value) was assumed. The monomeric Affibody molecules, also in ABD-fused format, all bound with similar affinities and kinetics with average dissociation constants for the high-affinity sites of k d = 5 (±2) × 10−4 s−1 and K D = 1.7 (±0.6) nM. A non Aβ-binding Affibody molecule (ZTaq_1) and a ZTaq_1-ABD fusion protein were used as controls. Of these, ZTaq_1 showed no binding, and ZTaq_1-ABD showed very weak binding to Aβ42cc protofibrils (results not shown).
Figure 4.
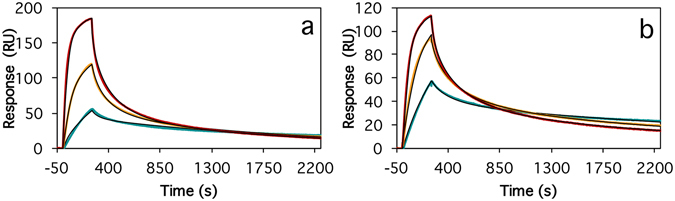
Association and dissociation kinetics for binding of two representative monomeric Affibody constructs to immobilized Aβ42cc protofibrils. Affibody concentrations are 10 (green), 20 (yellow) and 40 nM (red). The data was fitted to a heterogeneous ligand binding model with local maximum response (Rmax) values. The kinetics of the slow association and dissociation phases are k a = 2.8 (±0.1) × 105 s−1 M−1 and k a = 2.5 (±0.1) × 105 s−1 M−1 and k d = 4.7 (±0.05) × 10−4 s−1 and k d = 6.2 (±0.05) × 10−4 s−1 for ZAβ42cc_1 (a) and ZAβ42cc_4 (b), respectively, which correspond to dissociation constants for the stronger binding site of K D = 1.6 (±0.1) and K D = 2.5 (±0.2) nM, respectively.
Binding of dimeric Affibody constructs
Finally, we investigated if the affinity of the selected Affibody molecules could be improved even further by linking the binders into head-to-tail dimers. For this, we constructed a number of dimeric fusion proteins with (GGGS)n or (GGGGS)n linkers of variable lengths and studied their binding to Aβ42cc protofibrils by SPR (Fig. 5 and Supplementary Table S1). In total, we studied 14 dimeric constructs of four Affibody molecules with or without ABD-fusions. All dimers, except one for which the slow binding off-rate could not be determined, bound with affinities that are either equal to those of the corresponding monomers, or up to one order of magnitude stronger compared to the monomers. There was, in two of four cases, an apparent effect of linker length on binding affinity. For instance, dimeric ZAβ42cc_4 without linker (ZAβ42cc_4–ZAβ42cc_4) bound with a K D = 0.26 nM, intermediate length linkers showed a K D = 0.6 to 0.7 nM, and the ZAβ42cc_4 dimer with the longest linker (ZAβ42cc_4–(GGGS)4–ZAβ42cc_4) appeared to bind somewhat weaker (K D = 1.1 nM). This trend was however not common to the whole dimer binding data set.
Figure 5.
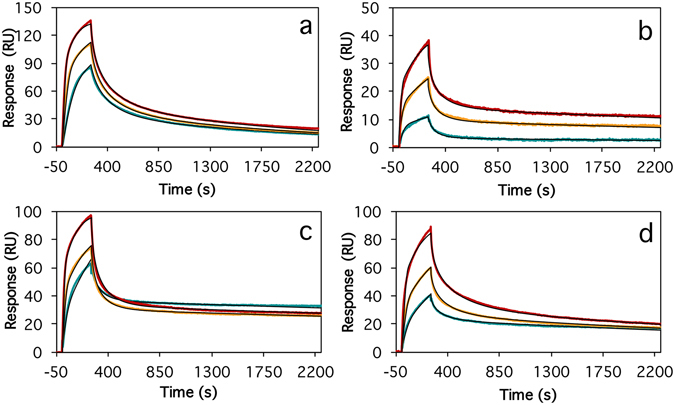
Association and dissociation kinetics for binding of some of the head-to-tail Affibody dimers to immobilized Aβ42cc protofibrils. Data were fit to a heterogeneous ligand binding model with local maximum response (Rmax) values. (a) ZAβ42cc_1-(GGGGS)- ZAβ42cc_1-ABD with Affibody dimer concentrations of 10 (green), 20 (yellow) and 40 nM (red). The kinetics of the slow association and dissociation phases are k a = 3.1 (±0.1) × 105 s−1 M−1 and k d = 5.2 (±0.03) × 10−4 s−1 corresponding to a dissociation constant for the stronger binding site of K D = 1.7 (±0.1) nM. (b) ZAβ42cc_1-(GGGGS)4- ZAβ42cc_1-ABD; 10, 20 and 40 nM; k a = 1.4 (±0.1) × 105 s−1 M−1, k d = 1.6 (±0.01) × 10−4 s−1, K D = 1.1 (±0.1) nM. (c) ZAβ42cc_4- ZAβ42cc_4 (no linker); 7.8, 15.5 and 31.2 nM; k a = 3.4 (±0.1) × 105 s−1 M−1, k d = 9.1 (±0.01) × 10−5 s−1, K D = 0.27 (±0.01) nM. (d) ZAβ42cc_4-(GGGS)4- ZAβ42cc_4; 7.8, 15.5 and 31.2 nM; k a = 2.2 (±0.3) × 105 s−1 M−1, k d = 2.5 (±0.01) × 10−4 s−1, K D = 1.1 (±0.05) nM.
Discussion
Research in the field of Alzheimer’s disease is in need of new good methods to detect and distinguish various forms of aggregated amyloid-β. Conformation specific monoclonal antibodies have previously been developed for this purpose21, 23, 24. Here, we explore alternatives to IgG antibodies by the in vitro selection of Affibody molecules binding to protofibrillar aggregates of Aβ42. Such binders would potentially have a number of technological advantages, as compared to using antibodies, in applications including tissue imaging, use of PET ligands, diagnostic biomarker profiling and basic research8.
We used protofibrils formed by Aβ42cc as the target for binding protein selection. Aβ42cc contains an intramolecular disulfide that locks the peptide in a hairpin conformation that is incompatible with the conformation of Aβ in amyloid fibrils. Aβ42cc aggregation is therefore halted at the protofibrillar state. We have in previous studies shown that Aβ42cc protofibrils are good mimics of protofibrils formed by wild type Aβ42 10, 11.
However, it was essential that the selected binding proteins also recognized wild type Aβ42 aggregates. Therefore, we performed a profiling assay in an ELISA format to assess this aspect (Fig. 2). The results with conformation independent 6E10 antibody detection indicate that one, or possibly three, of the five tested Affibody molecules bound wild type protofibrils with an affinity that, at least in the case of ZAβ42cc_1, was close to that for binding of Aβ42cc. Detection using the weakly protofibril conformation-selective mAb1C3 monoclonal antibody indicated that wild type protofibrils remained bound to ZAβ42cc_1, ZAβ42cc_3 and possibly ZAβ42cc_4 at the time of detection. It cannot be excluded that all Affibody molecules indeed recognize wild type protofibrillar aggregates with high affinity. This is because of the rapid interconversion of wild type protofibrils into mature fibrils that occurs within hours25. Hence, with the incubation times for Affibody and antibody binding in the ELISA experiment results, shown in Fig. 2 (one hour each at room temperature), wild type protofibrils, except those most strongly bound, might have converted into fibrils, which would have been washed off before detection. Hence, the ELISA in Fig. 2 is a very stringent assay and repeating these experiments with shorter incubation times might reveal that all selected binders recognize wild type protofibrillar aggregates.
We employed an affinity capture assay to confirm the aggregate selectivity of the Affibody binders. Shorter incubation times were chosen here in order to minimize the intricacy originating from the maturation of the protofibrils to fibrils25, and thus improve the resolution for detection of selective binding. The results confirm selective binding of ZA β 42cc_1 and ZA β 42cc_4 to protofibrils. However, in contrast to the ELISA selectivity profiling, where the selectivity of ZAβ42cc_1 to Aβ42cc protofibrils was higher than ZAβ42cc_4, both these Affibody molecules bind Aβ42cc protofibrils to a similar extent. Interestingly, the results suggest that these two Affibody molecules also recognized Aβ42wt protofibrils and Aβ42cc protofibrils with similar selectivity. A possible explanation for this “selectivity improvement” compared to the ELISA profiling is the reduced incubation time, which would be in line with the intention of this experiment.
The SPR data indicated that the best selected Affibody molecules interacted with Aβ42cc with slow off-rates in the order of k d = 1 to 7 × 10−4 s−1, and equilibrium dissociation constants in the range K D = 1 to 3 nM. The (total) concentrations of soluble Aβ42 in body fluids of healthy humans range from ca. 200 pM in the cerebrospinal fluid to ca. 20 pM in blood plasma. The concentration of Aβ42 protofibrils in brains of patients with Alzheimer’s disease has not been determined, but it should be lower, and perhaps much lower, than the total soluble Aβ42 concentration. Hence, it would be desirable to develop even stronger binding Affibody molecules to ensure the detection of protofibrillar species. To reach this, we explored the possibility to link Affibody molecules into dimers to achieve even slower off-rates and thereby higher affinity. The rational for this strategy was that protofibrillar protein aggregates should have a modular morphology in which Affibody binding sites are repeated in a regular manner. Hence, simultaneous engagement of two closely located sites by a single molecule (a dimer) should increase the affinity. In fact, we recently demonstrated that Affibody dimers can be improved 1000-fold compared to monomers as binders of a protein containing multiple similar binding surfaces26.
With dimers of Affibody molecules we also achieved slower off-rates, as expected, and in some cases also significantly higher affinities (KD of around 300 pM) compared to monomer binding, but the affinity gain was not quite as great as one would have hoped based on earlier studies. We do not completely understand why higher affinities are not achieved and further studies, including structural comparisons of monomeric and dimeric constructs, are necessary to address this issue.
Sequences of selected Affibody molecules are, with a few exceptions, homologous (Fig. 1a). In fact, 23 of 25 of the listed sequences in Fig. 1a appear to reflect the same binding surface (Fig. 1c). One of these sequence (ZAβ42cc_3) appears remotely homologous, and one (ZAβ42cc_5) appears to reflect a different binding surface. The 23 homologous sequences are characterized by the selection of positively charged residues, and in particular arginines, at positions 9, 10, 11, and to large extent also at positions 13, 14 and 25. Similarly, nonpolar side chains are selected at positions 17, 27, 32 and 35. Interestingly, tryptophan occurs at position 32 in all conserved sequences. The homology at positions 24 and 28 are predominantly polar, perhaps with a tendency for negative and positive charge, respectively. Position 18 does not clearly appear to have been subjected to any selection pressure.
The chemical properties of selected side chains define three chemically distinct regions of the Z-domain scaffold surface as illustrated in Fig. 1c. Residues 9, 10, 11, 13, 14, which are rich in basic residues, form an area of strong positive electrostatic potential. Adjacent to this surface is a non-polar surface (residues 17, 27 and 35) with a nonpolar and bulky tryptophan side chain (residue 32) at the edge. Finally, the nonpolar surface is flanked by a polar surface. The surface pattern positive-nonpolar-polar presumably matches the binding epitope on Aβ42cc protofibrils. In fact, the structural model of a hexameric protofibril building block that recently was derived by our group, using solid-state nuclear magnetic resonance data and Rosetta modeling12, indeed contains surfaces that appear to match the selected Affibody molecules. However, further structural studies are needed to confirm the binding site at the protofibrils.
To conclude, amyloid peptides are difficult to use as antigens in directed evolution. These peptides rapidly form different forms of aggregates and the antigen hence tends to be a mix of different molecular species, hampering reproducibility, specificity for the intended form and success rate. Here, we show that using the engineered Aβ42cc, we prevent formation of higher order aggregates and trap the peptide in the protofibril state to obtain a more defined antigen for successful selection of specific binders, which importantly also recognise Aβ42wt protofibrils. We believe that the approach could facilitate future efforts on development of affinity reagents (including monoclonal antibodies) for specific forms of Aβ aggregates and in particular for Aβ protofibrils.
Methods
Aβ production and preparation of aggregates
Aβ42cc and Aβ42wt were produced by co-expression with the ZA β 3 Affibody molecule and purified as described previously10, 11, 27. Aβ peptide was separated from the ZA β 3 by denaturation in 7 M guanidinium hydrochloride followed by immobilized metal ion affinity chromatography (IMAC) under denaturating conditions.
Aβ42cc protofibrils were obtained by overnight dialysis of monomeric fractions against 20 mM Na-phosphate, pH 7.4, 50 mM NaCl, 1 mM EDTA at room temperature. The dialysis was continued for another 7 h in the same buffer without EDTA, followed by heating to 60 °C for 10 min.
Protofibrils of Aβ42wt were freshly prepared for each experiment, essentially as described in ref. 25, and used immediately. Aβ42wt monomer was diluted to a concentration of 100 µM and the pH was adjusted to 7.4 (with 1 M HCl). Protofibrils were allowed to form either overnight at 4 °C or for 10 minutes at room temperature. The protofibrils fraction (elution volume, Ve = 8 mL) was isolated from monomeric fraction (Ve = 13 mL) by size exclusion chromatography (SEC) using a Superdex 75 10/300 GL column (GE Healthcare) with 20 mM NaPi, pH 7.4, 150 mM NaCl buffer.
To assemble Aβ42wt fibrils, the monomer was centrifuged to pellet any existing insoluble aggregate and the soluble fraction was diluted to 25 µM with phosphate buffered saline (PBS: 2.68 mM KCl, 1.47 mM KH2PO4, 137 mM NaCl, 8.1 mM Na2HPO4, pH 7.4) and incubated at 28 °C with 110 rpm shaking for 80 h22. The fibrils were spun down and washed twice with PBS.
Phage display selections
A combinatorial phage library of the Z domain, with randomized positions 9, 10, 11, 13, 14, 17, 18, 24, 25, 27, 28, 32 and 35, was prepared as described in9, 17, 19, 28. Aβ42cc protofibrils and Aβ42wt fibrils were biotinylated as described in ref. 18, and the unreacted biotin was removed by SEC on a Superdex 200 10/300 GL column (GE Healthcare). The selection and amplification were performed in PBS-T (0.1% Tween20 added to PBS) at room temperature as described previously26. Prior to the first cycle of protofibril biopanning, fibril binders were removed from the library by incubation of phages with pre-adsorbed streptavidin beads. The remaining phage particles in the supernatant were panned for 1.5 − 2.0 h against Aβ42cc protofibrils in six cycles. The concentration of Aβ42cc protofibrils was 2 µM (monomer subunit concentration) in the first cycle, and decreased five times in subsequent selection rounds. In the last cycle, the library was panned in three parallel tracks using a 4–0.2 nM target concentration. DNA sequencing of the generated binders was performed as previously described26.
Screening ELISA
Phages from randomly selected clones after the fourth and sixth selection rounds were produced and screened for Aβ42cc protofibril binding activity by an ELISA. The expression was performed directly from the phagemid vector, which yielded the Affibody variants as albumin binding domain (ABD) fusion proteins that became secreted in the E. coli periplasm, as described previously18. For ELISA, 50 μL periplasmic fraction was transferred to Costar high binding half area 96 well plates (Corning), previously coated with 2 µg/mL of a goat anti-ABD IgG HP001, and blocked with PBSC (0.5% Casein (Sigma) in PBS), for 1.5 h incubation. The plates were washed four times with PBS-T, prior to addition of 42 nM biotinylated Aβ42cc protofibrils per well and incubated for 1 h. After washing the wells four times, streptavidin-HRP (Dako) diluted 1:30,000 in PBSC was added to the wells and incubated for 1 h. TMB substrates A and B were mixed 1:1 and added to washed wells and incubated for 7 min according to the manufacturer’s instructions (ImmunoPure TMB Substrate Kit; Thermo Scientific). Stop solution (2 M H2SO4) was added and the absorbance at 450 nm was measured in an ELISA reader (Victor 3, Perkin Elmer).
Binding profiling to different Aβ aggregates
The five Affibody variants (Fig.1,a) as ABD-fusion proteins were analyzed for ability to bind different aggregated forms of Aβ42cc and Aβ42wt using an ELISA assay at room temperature. Briefly, 50 μL periplasmic fraction was transferred to Costar high binding half area 96 well plates (Corning), previously coated with 2 µg/mL of HP001 and blocked with 0.5% Casein (Sigma) in PBS for 1.5 h incubation. The plates were washed four times with PBS-T prior to addition of 50 nM or 1 µM Aβ42 peptide solutions and incubation for 1 h. After washing, plates were incubated with mAb1C3, monoclonal mouse anti-Aβ IgG, (2 µg/mL)21, or mAb(6E10)-HRP, mouse anti-Aβ IgG1 (33 ng/mL) (SIG-39345, BioSite) for 1 h. Prior to washing, wells with mAb1C3, were incubated with an anti-mouse-HRP antibody (G21040, Invitrogen) for 1 h. TMB substrates A and B were mixed at a ratio of 1:1, and added to washed wells and incubated for 5 to 30 min according to the manufacturer’s instructions (ImmunoPure TMB Substrate Kit; Thermo Scientific). Stop solution (2 M H2SO4) was added to the wells, and the absorbance was measured at 450 nm in an ELISA plate reader (Victor 3, Perkin Elmer). An Affibody molecule specific for Taq-polymerase (ZTaq_2) was used as negative control.
The selectivity of binders to different Aβ aggregates was also tested using Affibody molecules as ligands in a batch mode experiment. For this experiment, the Affibody molecules were essentially produced as described below. ZAβ42cc_1 or ZAβ42cc_4 (65 µg) were immobilized on 75 µL Ni2+ charged IMAC Sepharose 6 fast flow (GE Healthcare) by incubation for 20 min at room temperature. The beads were pelleted by centrifugation and washed with buffer A (10 mM Tris-HCl, pH 7.4) to remove unbound Affibody molecules. In total 1.7 µg of Aβ42cc protofibrils, Aβ42 protofibrils, Aβ42 fibrils and Aβ42 monomer were separately incubated with Affibody bound IMAC Sepharose for 5 min at room temperature. Aβ42wt protofibrils were freshly prepared by SEC as explained above and used immediately. The IMAC-Affibody-Aβ complex was pelleted and the supernatant was transferred into new tube. The complex was washed once with buffer A. The Affibody-bound Aβ was eluted with 20 mM NaPi pH 7.4, 50 mM NaCl, 300 mM imidazole. Eluate and supernatant were analyzed using SDS-PAGE. The samples were prepared in SDS-PAGE sample buffer (Bio-Rad) containing 15 mM TCEP in final concentration (for fibril sample 8 M urea was included). Samples were heated to 90 °C for 5 min prior to loading on Criterion precast 4–20% gradient gels (Bio-Rad). Protein bands were visualized by silver staining29. The gels were scanned using Gel Doc EZ Imager (Bio-Rad).
Affibody molecule production
The Affibody molecules selected for further characterization were produced with N-terminal His6 tags or C-terminal ABD fusion molecules for purification.
For N-terminal His6 tag constructs, the Affibody molecules were cloned into pET28b(+) expression vectors, yielding the final constructs GSSHHHHHHLQ[ZAβ42cc_X]VD, where ZAβ42cc_X denotes the selected Affibody molecule. E. coli BL21STAR (DE3) cells (Invitrogen) were transformed with the expression plasmids and cultivated at 37 °C in TB medium (50 µg/L kanamycin). At an OD600nm of approximately 1, protein expression was induced by addition of IPTG to a final concentration of 0.2 mM. The temperature was lowered to 16 °C and the culture was incubated for 18 h prior harvest. The protein purification was done by an IMAC purification using a 5 mL HiTrap Chelating HP column (GE Healthcare) with 20 mM Tris-HCl, pH 8.0, 0.5 M NaCl as running buffer, and 20 mM Tris-HCl, pH 8.0, 0.5 M NaCl, 400 mM imidazole for elution of bound protein. The IMAC purification was followed by a SEC purification using a Superdex 75 10/300 GL column (GE Healthcare), with 20 mM NaPi, pH 7.4, 150 mM NaCl as running buffer.
The genes for the Affibody molecules with a C-terminal ABD035 tag30 were cloned into the expression vector pET26b(+). The obtained constructs were [ZAβ42cc_X]-ABD035, where ZAβ42cc_X denotes the selected Affibody molecule. The encoded proteins were essentially expressed as described above, however using a TSB + Y as cultivation medium and 1 mM IPTG for induction of the protein expression. The Affibody molecules were purified by affinity chromatography on a 2 mL anti-ABD agarose column (Affibody AB) using tris-buffered saline (TST: 25 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA, 0.5% (w/v) Tween 20, pH 8.0) as running buffer, 5 mM NH4Ac (pH 5.5) for washing and 0.5 M HAc (pH 2.8) for elution. The eluted proteins were buffer-exchanged to PBS on a NAP-10 columns (GE Healthcare).
Head-to-tail dimeric constructs were designed with (GGGS)n or (GGGGS)n linkers, and with a N-terminal His6 tags or C-terminal ABD-tags for purification, respectively. The proteins obtained were GSSHHHHHHLQ[[ZAβ42cc_X]-(GGGS)n-[ZAβ42cc_X]], or [[ZAβ42cc_X]-(GGGGS)n-[ZAβ42cc_X]]-ABD035, where ZAβ42cc_X denotes the selected Affibody sequence. Dimeric Affibody molecules were expressed and purified as described for the monomer. The molecular weight of the purified proteins was verified by LC/MS (Agilent Technologies 6520 ESI-Q-TOF).
Surface plasmon resonance analysis
Surface plasmon resonance (SPR) studies were performed on a Biacore X100 instrument (GE Healthcare). The Aβ42cc protofibrils were immobilized onto a Biacore CM5-sensor chip (GE Healthcare), as described previously31.
Five or six concentrations of each analyte were prepared in HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM ETDA, 0.005% Tween-20, pH 7.4) and injected over the immobilized chip surface for 250 s to record analyte binding to the surface. Dissociation was observed for 2,000 s in running buffer. The sensor surface was regenerated after each injection with 20 mM NaOH with 90 s contact times. All experiments were carried out at 25 °C with a flow rate of 10 μL/min.
SPR data sets were analyzed using Biacore X100 Evaluation 2.0.1 software and curve fitting was performed with a heterogeneous binding site model using global kinetic fitting, but with local adjustment of the parameter Rmax.
Electronic supplementary material
Acknowledgements
This work was supported by grants 521-2010-2517 and 621-2011-5812 from the Swedish Research Council (V.R.) to T.H., and by grant FO2015-0174 from the Swedish Brain Foundation to S.S., as well as the Wallenberg Center for Protein Research. The authors would like to thank Dr. Anatoly Dubnovitsky for expertise and assistance.
Author Contributions
T.H. conceived the project; E.W., M.M.R., H.L., and C.L. designed and performed experiments, and analyzed the results; E.W., M.M.R., H.L., E.G., B.S., C.L., M.S., J.L., S.S., and T.H. contributed to writing the manuscript.
Competing Interests
E.W. and E.G. are employees of Affibody AB, Stockholm, Sweden, and T.H. is a shareholder and serves on the Board of Directors of Alzinova AB, Gothenburg, Sweden.
Footnotes
Elisabet Wahlberg and M. Mahafuzur Rahman contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06377-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Kuperstein I, et al. Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benilova I, Strooper Bd. Promiscuous Alzheimer’s amyloid: yet another partner. Science. 2013;341:1354–1355. doi: 10.1126/science.1244166. [DOI] [PubMed] [Google Scholar]
- 4.Harper J, Wong S, Lieber C, Lansbury P. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/S1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 5.Walsh D, Lomakin A, Benedek G, Condron M, Teplow D. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 6.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-protein assembly and Alzheimer disease. J Biol Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shankar G, et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Löfblom J, et al. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Letters. 2010;584:2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 9.Nord K, et al. Binding proteins selected from combinatorial libraries of an α-helical bacterial receptor domain. Nature Biotech. 1997;15:772–777. doi: 10.1038/nbt0897-772. [DOI] [PubMed] [Google Scholar]
- 10.Sandberg A, et al. Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc Natl Acad Sci USA. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubnovitsky A, et al. Amyloid-β protofibrils: size, morphology and synaptotoxicity of an engineered mimic. PLoS ONE. 2013;8:e66101. doi: 10.1371/journal.pone.0066101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lendel C, et al. A Hexameric Peptide Barrel as Building Block of Amyloid-β Protofibrils. Angew Chem Int Ed Engl. 2014;53:12756–12760. doi: 10.1002/anie.201406357. [DOI] [PubMed] [Google Scholar]
- 13.Xiao, Y. et al. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol (2015). [DOI] [PMC free article] [PubMed]
- 14.Wälti MA, et al. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc Natl Acad Sci USA. 2016;113:E4976–E4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colvin MT, et al. Atomic Resolution Structure of Monomorphic Abeta42 Amyloid Fibrils. J Am Chem Soc. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lendel C, et al. Combined Solution- and Magic Angle Spinning NMR Reveals Regions of Distinct Dynamics in Amyloid β Protofibrils. ChemistrySelect. 2016;1:5850–5853. doi: 10.1002/slct.201601468. [DOI] [Google Scholar]
- 17.Grönwall C, et al. Selection and characterization of Affibody ligands binding to Alzheimer amyloid β peptides. J Biotechnol. 2007;128:162–183. doi: 10.1016/j.jbiotec.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Lindborg M, et al. Engineered high-affinity affibody molecules targeting platelet-derived growth factor receptor β in vivo. J Mol Biol. 2011;407:298–315. doi: 10.1016/j.jmb.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 19.Feldwisch J, et al. Design of an optimized scaffold for affibody molecules. J Mol Biol. 2010;398:232–247. doi: 10.1016/j.jmb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Hoyer W, Grönwall C, Jonsson A, Ståhl S, Härd T. Stabilization of a β-hairpin in monomeric Alzheimer’s amyloid-β peptide inhibits amyloid formation. Proc Natl Acad Sci USA. 2008;105:5099–5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Englund H, et al. Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J Neurochem. 2007;103:334–345. doi: 10.1111/j.1471-4159.2007.04759.x. [DOI] [PubMed] [Google Scholar]
- 22.Jan A, Hartley DM, Lashuel HA. Preparation and characterization of toxic Aβ aggregates for structural and functional studies in Alzheimer’s disease research. Nat Protoc. 2010;5:1186–1209. doi: 10.1038/nprot.2010.72. [DOI] [PubMed] [Google Scholar]
- 23.Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 24.Kayed R, et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luheshi LM, et al. Sequestration of the Aβ peptide prevents toxicity and promotes degradation in vivo. PLoS Biology. 2010;8:e1000334. doi: 10.1371/journal.pbio.1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindborg M, et al. High-affinity binding to staphylococcal protein A by an engineered dimeric Affibody molecule. Protein Eng Des Sel. 2013;26:635–644. doi: 10.1093/protein/gzt038. [DOI] [PubMed] [Google Scholar]
- 27.Macao B, et al. Recombinant amyloid beta-peptide production by coexpression with an affibody ligand. BMC Biotechnol. 2008;8:82. doi: 10.1186/1472-6750-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nord K, Nilsson J, Nilsson B, Uhlén M, Nygren P-Å. A combinatorial library of an α-helical bacterial receptor domain. Protein Eng. 1995;8:601–608. doi: 10.1093/protein/8.6.601. [DOI] [PubMed] [Google Scholar]
- 29.Gromova, I.a.C., J.E. in Cell Biology: A Laboratory Handbook, Vol. 4, 219–223 (Elsevier, San Diego; 2006).
- 30.Jonsson A, Dogan J, Herne N, Abrahmsén L, Nygren PÅ. Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng Des Sel. 2008;21:515–527. doi: 10.1093/protein/gzn028. [DOI] [PubMed] [Google Scholar]
- 31.Rahman MM, Zetterberg H, Lendel C, Härd T. Binding of Human Proteins to Amyloid-β Protofibrils. ACS Chem Biol. 2015;10:766–774. doi: 10.1021/cb5008663. [DOI] [PubMed] [Google Scholar]
- 32.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.