

Abstract
Background
Phase I experts recommend revisiting dose-limiting toxicity (DLT) definition to include chronic and cumulative toxicities induced by new molecularly targeted therapies. Patient’s assessment of late toxicities’ tolerability is, however, unknown.
Materials and methods
A prospective survey on adverse events (AEs) tolerability on 23 National Cancer InstituteCommon Terminology Criteria for Adverse Event, Version 4 (NCI-CTCAE.v4) items was conducted at Gustave Roussy’s Phase I department. Patients’ maximum tolerability duration was recorded at baseline, during trial and at trial completion. Results were compared with the corresponding physicians’ survey.
Results
52 patients enrolled in 27 Phase I trials between May 2014 and November 2015 completed 102 forms. At baseline, the most feared G2/G3 AEs were haematuria (74%), vomiting (71%) and hyperglycemia (64%)/dry mouth (94%), hyperglycemia (92%) and vomiting (92%). At trial completion, the most feared G2/G3 AEs were personality change (83.3%), haematuria (82%) and fever (80%)/dry mouth, fever and dizziness (100% each). Tolerability score did not differ over time. More previous treatments and occurrence of severe AEs were associated with better tolerability at study completion (p=0.0234 and p=0.0153, respectively, in multivariate analysis). Patient’s tolerability differed from physician’s assessment.
Conclusion
AEs considered intolerable by patients are toxicities that directly impact their quality of life and differ from those feared by physicians or included in DLT definition. Patient-reported tolerability of AEs may help in optimising drug development.
Keywords: Dose limiting toxicity, phase 1, adverse event, tolerability, patient reported outcome
Key questions.
What is already known about this subject?
The traditional definition of dose-limiting toxicity (DLT) usually assessed during phase 1 first cycle does not suit the delayed, cumulative or chronic adverse events (AEs) induced by new molecularly targeted agents.
The DLT-TARGETT Survey highlighted that phase 1 experts recommend considering longer DLT assessment and certain G2 adverse events.
Patient-reported assessment of toxicity and tolerability in unknown.
What does this study add?
To the best of our knowledge, this is the first study to focus on patients’ reported tolerability during phase 1 trials.
There is a consensus among patients on G3 AEs that are deemed to be intolerable.
Specific grade 1, such a vomiting, personality change and confusion, is deemed intolerable when prolonged >7 days for one-third of patients, whereas grade 2 vomiting, hyperglycaemia and haematuria were not tolerable for two-thirds of patients.
Number of previous chemotherapies and occurrence of severe toxicities on trial were linked to better tolerability.
There is a gap between patient assessment of tolerability and experts’ definition of AEs to consider as DLT.
How might this impact on clinical practice?
Our study reinforces the need to readapt DLT definition in phase 1 trial design. Patient-reported assessment of tolerability and toxicity may help to better adapt this new DLT to best define the recommended phase 2 dose.
Introduction
The primary objective of phase 1 studies is to define the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) of novel agents or combinations. This determination is based on a predefined target percentage of dose-limiting toxicities (DLTs), defined traditionally as G4 haematological toxicities and G3–4 non-haematological toxicities.1 The assessment of DLTs is, therefore, critical. However, there is currently no consensus on what constitutes a DLT, and a surprising heterogeneity in DLT definition has been observed among phase 1 trials.2 A recent work from Le Tourneau et al,3 reviewing 155 phase 1 trials evaluating 111 different molecularly targeted agents (MTAs), revealed that differences in DLT definition involved toxicity type and duration, but also that the need to delay treatment, to reduce dose-intensity, or the degree of reversibility of the toxicity were very infrequently considered.
MTAs, as compared with conventional cytotoxic chemotherapy, are characterised by a different toxicity profile (they present lower haematological toxicities), a broader therapeutic window (the optimal biological dose may not always be the highest one) and a continuous administration schedule (which reinforces the importance of chronic or cumulative toxicities).4 MTD determination methods, which have been established in the era of cytotoxic agents, are therefore not valid anymore and need to be refined to better suit to these agents’ characteristics.5 6
In this context, recent initiatives have emerged from phase 1 experts, aiming at establishing novel recommendations for optimising drug development. First, the international DLT-TARGETT task force proposed updated recommendations for DLT definition and RP2D determination, based on the retrospective analysis of all toxicity data from 54 centrally reviewed phase 1 trials. They concluded that, although the DLT period should not be lengthened to keep the trials fast and efficient, more attention should be paid to dose-intensity reductions, toxicities occurring beyond cycle one and toxicity causality. A 15-question survey, conducted in parallel on 65 international phase 1 experts, revealed that 76% of them recommended incorporating selected G2 toxicities in the DLT definition as well as considering toxicity duration.7 However, there was no consensus on which G2 toxicities should be added, and for which toxicity duration these should score as DLTs. Also, toxicities’ tolerability was based on physicians’ assessment, which may differ from the patient’s one.8 In order to investigate this, we conducted a prospective phase 1 patient survey on toxicities tolerability and compared the results with the corresponding physicians’ questionnaire.
Materials and methods
Study design
A prospective patient survey on adverse events (AEs) tolerability was conducted at Gustave Roussy’s Drug Development Department. Patients had to fill the questionnaire at three time points: baseline, during study (cycle 2 day 1) and after trial completion. Results were compared with corresponding physicians’ answers (DLT-TARGETT Survey).7
Questionnaire
The questionnaire investigated 29 Common Terminology Criteria for Adverse Event, Version 4 (CTCAE.v4) items adapted in lay terms (online supplementary material. Three degrees of severity were evaluated for each item: ‘mild’, moderate’ or ‘severe’, corresponding to CTCAE grades 1, 2 and 3, respectively. The latter were defined according to their impact on the instrumental or self-care activities of daily living. For each item, we asked: (1) whether the patient already experienced the AE and (2) what was their estimate of the maximum duration (less or more than 7 days) for which this AE would be tolerable if experienced during trial. The questionnaire had to be modified two times, after an intermediary analysis that showed a low completion rate of some items. These readjustments were designed so that all information collected on previous versions could be exploited.
esmoopen-2016-000148supp001.pdf (446.6KB, pdf)
Patients’ characteristic and eligibility
The survey was distributed to patients entering phase 1 trials in DITEP (Département d’Innovation Thérapeutique et des Essais Précoces) between May 2014 and October 2015. No specific selection was made; only patients with cognitive impairment were excluded. Baseline patient characteristics as well as worst AE presented on trial were recorded.
Objectives of the study
The primary objective was to describe patients’ assessment of AEs tolerability throughout the trial. Secondary objectives were (1) to analyse toxicities tolerability according to patients’ and trials’ characteristics and (2) to compare patients’ answers with those of the corresponding physicians’ DLT-TARGETT survey.
Statistical analysis
‘Tolerability assessment’ was defined as the proportion of patients who considered specified AE tolerable for less or more than 7 days. The patient ‘Tolerability score’ variable was defined as the proportion of items assessed to be tolerable for more than 7 days; it was analysed according to demographical characteristics using linear regression models. Comparison of Tolerability scores between the different time points was done using paired Wilcoxon test.
Descriptive analyses include data from three items (other eye disorder, dysthyroidism and acute kidney injury), which were absent on the last questionnaire version, as their completion rate over the entire population was >30%. Analytical statistics excluded items absent on the last version (mucositis, peripheral sensory neuropathy, eye disorder, dysthyroidism and acute kidney injury) and were done using R software (version 3.1.3).
Results
Patients and trials
Fifty-two patients enrolled in 27 phase 1 trials participated between May 2014 and October 2015 (figure 1).
Figure 1.

Consort diagram of the study design 52 patients were enrolled and completed 102 questionnaires (median completion rate of 81%). Among the 20 patients who completed the questionnaire at study completion, four were still receiving treatment and had been on drug for >11 months.
Median age was 60 years, 28 patients (54%) were male (table 1); median time on trial was 13.2 weeks. Thirty-six patients (70%) went off-study for progressive disease, and four patients (8%) went off-study for DLT. Thirty patients completed the questionnaire at C2D1, and 20 after study completion. Four patients received >11 months of experimental treatment and were still on trial at the time of the database closure; they were censored at that time and completed a questionnaire, which was counted as ‘trial completion’ questionnaire.
Table 1.
Patient and trials characteristics (52 patients, 27 trials)
| n (%) | |
| Age (years) | |
| Median | 60 |
| Range | 25–83 |
| Sex | |
| Male | 28 (54) |
| Female | 24 (46) |
| Number of previous chemotherapy | |
| Median | 2 |
| Primary tumour site | |
| Colorectal | 10 (19) |
| Lung | 6 (12) |
| Breast | 4 (8) |
| Others | 32 (62) |
| Time on trial (weeks) | |
| Median | 13 |
| Number (%) of patients experiencing worse AE during trial | |
| Grade 1 AE | 14 (27) |
| Grade 2 AE | 15 (29) |
| Grade 3 or 4 | 10 (19) |
| Worse AE category | |
| Skin disorder | 19 (27) |
| Digestive | 18 (25) |
| General | 18 (25) |
| Metabolic | 5 (7) |
| Haematological | 4 (6) |
| Respiratory | 3 (4) |
| Eye disorder | 3 (4) |
| Cardiovascular | 1 (1) |
| Total | 71 (100) |
| Type of drug | |
| Antibody | 9 (33) |
| Small molecule | 19 (67) |
| Drug’s target | |
| Signalling pathway | 17 (63) |
| DNA repair | 1 (4) |
| Immunity | 5 (19) |
| Other | 4 (15) |
AE, adverse event.
Trials evaluated antibodies (n=9) and/or small molecules (n=19), targeting DNA repair (n=1), intracellular signalling pathways (n=17), immunity (n=5), epigenetic pathways (n=3) and others (n=1) (table 1).
Questionnaires completion
A total of 102 forms were collected: 52 at baseline, 30 during trial and 20 at trial completion, representing a total of 8874 answers (figure 1). Median time between the first and the second questionnaire was 3.1 weeks; it was 35.9 weeks between the first and the last one. Median completion rate was 5726/7038 (81.4%) questions, after exclusion of six items that were removed from the last two versions. Questions for which completion rate was the lowest were cardiac toxicity, hyperglycaemia, hypertension and personality change (with completion rates of 64.7%, 66.7%, 70% and 71.6%, respectively).
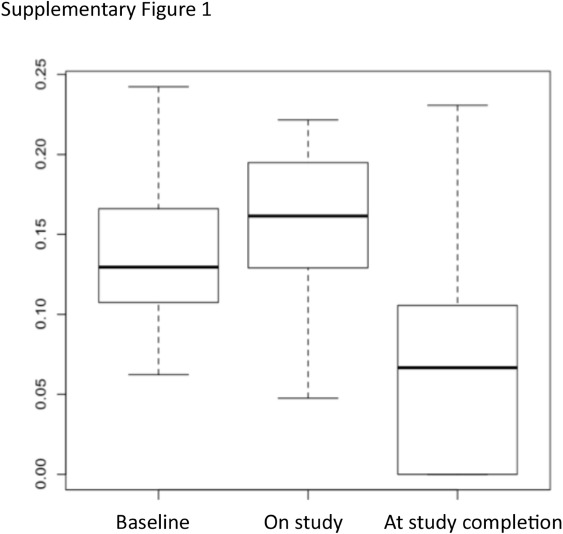
Interindividual variability in responses was significantly lower at study completion than at baseline or during the study for G3 AEs (analysis by comparison of variances: p values=0.0009199 and 0.0003622, respectively) (online supplementary figure 1).
esmoopen-2016-000148supp002.jpg (30.3KB, jpg)
{kind=link}
Comparison of toxicities tolerability at baseline, during the trial and at study completion
At baseline, G1 AEs that were deemed to be intolerable for >7 days were vomiting, personality change and confusion (33%, 30% and 24% of the patients, respectively), whereas it was vomiting (46%), nausea (33%) and haematuria (33%) during study, and personality change (58%), dizziness (44%) and vomiting (43%) at trial completion (figure 2A). The most feared G2 AEs were haematuria (74%), vomiting (71%) and hyperglycemia (64%) at baseline, whereas it was vomiting (79%), haematuria (76%) and hyperglycaemia (70%) on study, and personality change (83%), haematuria (82%) and fever (80%) at trial completion (figure 2B). For G3 AEs, dry mouth (94%), hyperglycemia (92%) and vomiting (92%) were deemed to be intolerable for >7 days at baseline, whereas haematuria (95%), other eye disorder (93%) and renal failure (93%) were the most feared AEs on study. At trial completion, seven toxicities were judged not tolerable for >7 days by all patients (100%): confusion, dry mouth, fever, personality change, headaches, dizziness and other cardiac toxicities (figure 2C).
Figure 2.

Patients’ most feared AEs reported to be intolerable when lasting more than 7 days at baseline, on study and at study completion (% patients); (A) grade 1 AEs, (B) grade 2 AEs and (C) grade 3 AEs. AE, adverse event.
When looking at each individual patient, concordance in tolerability assessment between baseline and study completion varied from 57% (cardiac toxicity) to 100% (peripheral motor neuropathy) for G3 AEs, and from 40% (constipation) to 83% (headache) for G2 AEs (figure 3). When considering the entire population, Tolerability score was not statistically different between baseline and study completion (p=0.4569), nor was the assessment of intolerable G3 AEs (p=0.1438). Interestingly, Tolerability score of G1 and G2 AEs was statistically different between baseline and study completion (p=0.0001352 and p=0.000318, respectively). Dizziness and personality change were the two G1 AEs for which difference in Tolerability score was the widest, whereas it was renal failure and dysthyroidism for G2 AEs (figure 2A,B). However, in paired analysis, the global, G1, G2 and G3 AEs Tolerability score did not significantly change between baseline and study completion (p=0.3683, p=0.5712, p=0.7841 and p=0.04967, respectively).
Figure 3.

Change in individual tolerability assessment between baseline and study completion. Change in tolerability assessment is represented for grade 2 (A) and grade 3 (B) adverse events (AEs). Each AE is represented by two columns: the first column represents the tolerability assessment at baseline, whereas the second one represents the change in tolerability assessment between baseline and study completion. In the first column, patients who assess the AE as tolerable >7 days at baseline are depicted in green; those considering it as tolerable <7 days are represented in red. In the second column, the same colour code is kept for patients who did not change their assessment at study completion; patients who changed their tolerability assessment during the study are represented in grey; patients who tolerated the AE <7 days at baseline but eventually considered it tolerable more than 7 days at study completion are represented in light grey; patients who changed their tolerability assessment in the opposite direction are represented in dark grey.
Evaluation of Tolerability score according to patients’ characteristics
At baseline, no patient characteristic was statistically associated with Tolerability score (table 2A). At study completion, a higher number of previous chemotherapies was associated with a better AEs Tolerability score (p=0.00755 and p=0.0234 in univariate and multivariate analysis, respectively) (table 2B). Surprisingly, a higher grade of the worst AE presented on trial was also associated with a better Tolerability score (p=0.0049 and p=0.0153 in univariate and multivariate analysis, respectively). The patient’s expected Tolerability score increased by 0.05241 with each additional previous line of chemotherapy; it increased by 0.11794 between G2 and G3 as the worst toxicity experienced by the patient on trial. No statistically significant association was found between tolerability and age, sex, type of primary cancer, number of previous targeted therapies, previous enrolment in phase 1 trial, trial duration or type of experimental drug.
Table 2.
Patient and trials characteristics associated with AEs Tolerability score in univariate and multivariate analyses
| Factors | Univariate analysis | Multivariate analysis |
|---|---|---|
| At baseline | ||
| Age | p=0.88379 | |
| Sex | p=0.717 | |
| Type of primary tumour | p2=0.439/p3=0.690/p4=0.166 | p2=0.6612/p3=0.7686/p4=0.100 |
| Number of previous chemotherapies | p=0.0521 | p=0.1360 |
| Number of previous targeted therapies | p=0.0532 | p=0.0781 |
| Previous phase 1 trial enrolment | p=0.0647 | p=0.6824 |
| At study completion | ||
| Age | p=0.17470 | |
| Sex | p=0.39370 | |
| Type of primary tumour | p2=0.56729/p4=0.42879 | |
| Number of previous chemotherapies | p=0.00755** | p=0.0234* |
| Number of previous targeted therapies | p=0.635 | |
| Previous phase 1 trial enrolment | p=0.374 | |
| Worst AE grade | p=0.0049** | p=0.0153* |
| Trial duration | p=0.647 | |
| Type of drug | p=0.48045 |
p2 refers to the comparison of lung cancers with CRCs, p3 refers to the comparison of breast cancers with CRCs and p4 refers to the comparison of other types with CRCs.
*p<0.05, **p<0.01.
AE, adverse event; CRC, colorectal cancer.
Comparison with physicians’ answers in DLT-TARGETT survey
Most feared mild (G1) or moderate (G2) AEs by physicians were eye disorders, confusion and blurred vision (15%, 14% and 8.6% of clinicians for G1 AEs, and 39%, 42.5% and 34% of them for G2 AEs, respectively) (figure 4). For G1 toxicities, the biggest differences in AEs tolerability between physicians and patients were vomiting, diarrhoea and fever (SD=0.212, 0.145 and 0.145, respectively). For G2 toxicities, the biggest difference in AEs tolerability between physicians and patients were haematuria, hyperglycaemia and acneiform rash (SD=0.453, 0.415 and 0.371, respectively).
Figure 4.

Comparison of tolerability assessment between patients and phase 1 experts’ tolerability of grades 2 (A) and 3 (B) AEs according to patients (left blue column) and physicians (right yellow column). AE, adverse event; DLT, dose-limiting toxicity.
Discussion
The main objective of phase 1 trial is to select the optimal drug dosage to be used in later phase trials. Once determined, the recommended dose will almost never be re-evaluated, and accurate determination is therefore key. MTAs have raised specific toxicity tolerability issues, mainly as a result from their highly diverse toxicity profile and continuous administration schedule—which associates with chronic and cumulative toxicities. Physicians have therefore recently highlighted the need to revisit the traditional assessment method of DLTs, notably by incorporating G2 toxicity items in the DLT definition. Whether toxicities that physicians anticipate as intolerable are similar to the ones that patients identify as such is unknown. Here, we report the responses of 52 phase 1 patients to a self-assessment questionnaire on toxicity tolerability and compare it with the answers of a similar survey performed in phase 1 experts. This single-centre survey shows that the patients’ most feared AEs are gastrointestinal toxicities, neurological toxicities and personality change, which differs from the physicians’ most feared toxicities. It also reveals that the most significant predictors of AEs tolerability are the number of previous treatment lines and the grade of the most severe toxicity experienced on study.
To our knowledge, this is the first study evaluating toxicity tolerability based on a self-assessment survey in phase 1 patients. Patients were representative of the usual phase 1 population and presented various cancer treatment and medical history. With a median questionnaire completion rate of 81%, we can hope that results of this survey can be applied to a broader phase 1 population. However, this was a single-centre study, and there is no doubt that other factors—such as ethnicity, family support and access to alternative treatment options—may also influence AEs tolerability. A revalidation in larger and international populations is therefore warranted.
Results of this survey revealed a relative consensus among patients regarding intolerable moderate (G2) or severe (G3) AEs. For G2 AEs, the three most feared toxicities (personality change, haematuria and vomiting) were selected by more than three out of four patients, whereas seven consensual G3 AEs were deemed not to be tolerable by all patients. This is in stark contrast with the results of the equivalent physician’s survey7 where no consensus could be found for G2 AEs that should be considered as DLTs. In the latter survey, retinopathy, confusion and blurred vision were the most frequently chosen G2 AEs (by 47%, 39% and 38% of the physicians, respectively) for being implemented in the DLT definition. This highlights that if experts seem to fear mostly AEs for which a specialised medical consulting is needed or for which no treatment can be rapidly administered, patients are more concerned by AEs affecting their daily quality of life. Interestingly, most patients reported that toxicity tolerability would highly depend on the reversibility of the AE, a point that was also raised by many physicians. If implementation of a ‘reversibility’ criteria in the DLT definition cannot be envisioned—as it would significantly lengthen the DLT period and trial duration—it would nonetheless deserve careful attention when recommending the phase 2 dose.
As almost all small molecules are developed as oral drugs and for a prolonged administration schedule (until resistance occurs), optimising patient adherence to the treatment is key. Gleevec (imatinib, Novartis) is usually a well-tolerated treatment, for which efficacy and survival benefit has been directly related to dose intensity and, therefore, adherence. However, the ADAGIO study revealed that one-third of the patients were considered to be non-adherent to the drug, and that only 15% of them were perfectly adherent with 100% of the prescribed dosage taken.9 In this example of well-tolerated drug, bothersome of symptoms explained only a very limited proportion of non-adherence. We can therefore anticipate that adherence would be even worse for drugs displaying side effects that directly impact patient’s quality of life. Adherence is indeed an important issue, which is currently the focus of an increasing number of studies.10 11 Therefore, evaluating drug tolerability using patient’s self-assessment, in parallel of the clinician’s one, may have added value for the phase 2 dose recommendation process.
Patient assessment of toxicity tolerability is a field of growing interest.12 Patient-reported outcomes (PROs) could indeed allow covering the ‘subjective’ part of AEs tolerability, thereby improving the quality of the trials data. In this purpose, the National Cancer Institute and the National Institute of Health developed the PRO-CTCAE (Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events), which is designed to be used as a companion to the CTCAE and clinician-based data reporting, in order to improve accuracy of AEs reporting in phase 3 trials.8 13 As this tool did not include any toxicity duration variable and covered all NCI-CTCAE items, we designed our own specific questionnaire that was matching the previous physician’s survey. Also, in our study, patients were asked to assess the toxicity tolerability even if they did not experience it—and thereby sometimes had to imagine the AE. This differs from the PRO studies, where patients have to report AEs when they experience it. It would however be interesting to test the PRO-CTCAE reporting system in the phase 1 setting and compare it with physicians’ reports, or to create a specific phase 1 PRO, which would notably incorporate a toxicity duration variable and quality of life assessments.14 A shared limitation between our questionnaire and PROs is the high degree of patient engagement and compliance required. In our study, 58% and 66% of enrolled patients filled in the questionnaire at C2D1 and at trial completion, respectively, and the overall completion rate was 81%. Causes of this attrition rate include mainly physical patient inability to answer (death or palliative situations), and loss in follow-up, despite several reminders.
To conclude, in a medical era where cancer is becoming a chronic disease associated with continuous therapies and where patients are more involved in their treatment, paying more attention to the impact of toxicities on patients’ quality of life is key. This subjective part of toxicities tolerability deserves being assessed and taken into account in the dose recommendation process, as a complementary information to data provided by the physician’s objective toxicity reporting. In this single-centre study, patients’ reported toxicity tolerability differed from the physician’s one. As physicians agree that drug development methods and DLT definition should be revisited, performing a similar study in wider populations and implementing a phase 1-specific PRO should be further considered.
Acknowledgments
We thank all the members of the DLT-TARGETT task force (Laurence Collette, Camilla Fowst, Percy Ivy, Stan B. Kaye, Denis Lacombe, Christophe Le Tourneau, Bernard Levy, Pierre Mancini, Christophe Massard, David Olmos, Xavier Paoletti, Elisa Rizzo, Lesley Seymour, Lillian L Siu, Jean-Charles Soria and Jaap Verweij) for having contributed to the physician’s questionnaire and for having offered to perform such study in patients during their meetings. We also thank Aicha Goubar (Biostatistics and Epidemiology Department, Villejuif, France) for her help in the statistical analysis.
Footnotes
Contributors: Conception or design of the work: SP-V. Data collection: CH, DL, CT and EL. Data analysis and interpretation: CH, XP and SP-V. Drafting the article: CH and SP-V. Critical revision of the article: CH, XP, CLT, CM, AH, J-CS and SP-V. Final approval of the version to be published: CH, DL, XP, CT, CLT, EL, AH, CM, J-CS and SP-V.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; internally peer reviewed.
References
- 1. Arbuck SG. Workshop on phase I study design. Ninth NCI/EORTC new drug development symposium, Amsterdam, March 12, 1996. Ann Oncol 1996;7:567–73. 10.1093/oxfordjournals.annonc.a010672 [DOI] [PubMed] [Google Scholar]
- 2. Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 2009;101:708–20. 10.1093/jnci/djp079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Le Tourneau C, Razak AR, Gan HK, et al. . Heterogeneity in the definition of dose-limiting toxicity in phase I cancer clinical trials of molecularly targeted agents: a review of the literature. Eur J Cancer 2011;47:1468–75. 10.1016/j.ejca.2011.03.016 [DOI] [PubMed] [Google Scholar]
- 4. Postel-Vinay S, Gomez-Roca C, Molife LR, et al. . Phase I trials of molecularly targeted agents: should we pay more attention to late toxicities? J Clin Oncol 2011;29:1728–35. 10.1200/JCO.2010.31.9236 [DOI] [PubMed] [Google Scholar]
- 5. Soria JC. Phase 1 trials of molecular targeted therapies: are we evaluating toxicities properly? Eur J Cancer 2011;47:1443–5. 10.1016/j.ejca.2011.04.009 [DOI] [PubMed] [Google Scholar]
- 6. Postel-Vinay S, Collette L, Paoletti X, et al. . Towards new methods for the determination of dose limiting toxicities and the assessment of the recommended dose for further studies of molecularly targeted agents--dose-limiting toxicity and toxicity assessment recommendation group for early trials of targeted therapies, an european organisation for research and treatment of cancer-led study. Eur J Cancer 2014;50:2040–9. 10.1016/j.ejca.2014.04.031 [DOI] [PubMed] [Google Scholar]
- 7. Paoletti X, Le Tourneau C, Verweij J, et al. . Defining dose-limiting toxicity for phase 1 trials of molecularly targeted agents: results of a DLT-TARGETT international survey. Eur J Cancer 2014;50:2050–6. 10.1016/j.ejca.2014.04.030 [DOI] [PubMed] [Google Scholar]
- 8. Dueck AC, Mendoza TR, Mitchell SA, et al. . Validity and reliability of the US national cancer institute's patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). JAMA Oncol 2015;1:1051–9. 10.1001/jamaoncol.2015.2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noens L, van Lierde MA, De Bock R, et al. . Prevalence, determinants, and outcomes of nonadherence to imatinib therapy in patients with chronic myeloid leukemia: the ADAGIO study. Blood 2009;113:5401–11. 10.1182/blood-2008-12-196543 [DOI] [PubMed] [Google Scholar]
- 10. Chirgwin JH, Giobbie-Hurder A, Coates AS, et al. . Treatment adherence and its impact on disease-free survival in the Breast International Group 1-98 trial of tamoxifen and letrozole, alone and in sequence. J Clin Oncol 2016;34:2452–9. 10.1200/JCO.2015.63.8619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hershman DL. Sticking to it: improving outcomes by increasing adherence. J Clin Oncol 2016;34:2440–2. 10.1200/JCO.2016.67.7336 [DOI] [PubMed] [Google Scholar]
- 12. Trotti A, Colevas AD, Setser A, et al. . Patient-reported outcomes and the evolution of adverse event reporting in oncology. J Clin Oncol 2007;25:5121–7. 10.1200/JCO.2007.12.4784 [DOI] [PubMed] [Google Scholar]
- 13. Basch E, Reeve BB, Mitchell SA, et al. . Development of the National Cancer Institute's patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). JNCI 2014;106:dju244 10.1093/jnci/dju244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anota A, Boulin M, Dabakuyo-Yonli S, et al. . An explorative study to assess the association between health-related quality of life and the recommended phase II dose in a phase I trial: idarubicin-loaded beads for chemoembolisation of hepatocellular carcinoma. BMJ Open 2016;6:e010696 10.1136/bmjopen-2015-010696 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
esmoopen-2016-000148supp001.pdf (446.6KB, pdf)
esmoopen-2016-000148supp002.jpg (30.3KB, jpg)