

Abstract
Objective
Deep brain stimulation (DBS) of the subthalamic nucleus (STN) is a highly effective symptomatic therapy for motor deficits in Parkinson's disease (PD). An additional, disease‐modifying effect has been suspected from studies in toxin‐based PD animal models, but these models do not reflect the molecular pathology and progressive nature of PD that would be required to evaluate a disease‐modifying action. Defining a disease‐modifying effect could radically change the way in which DBS is used in PD.
Methods
We applied STN‐DBS in an adeno‐associated virus (AAV) 1/2‐driven human mutated A53T α‐synuclein (aSyn)‐overexpressing PD rat model (AAV1/2‐A53T‐aSyn). Rats were injected unilaterally, in the substantia nigra (SN), with AAV1/2‐A53T‐aSyn or control vector. Three weeks later, after behavioral and nigrostriatal dopaminergic deficits had developed, rats underwent STN‐DBS electrode implantation ipsilateral to the vector‐injected SN. Stimulation lasted for 3 weeks. Control groups remained OFF stimulation. Animals were sacrificed at 6 weeks.
Results
Motor performance in the single pellet reaching task was impaired in the AAV1/2‐A53T‐aSyn–injected stim‐OFF group, 6 weeks after AAV1/2‐A53T‐aSyn injection, compared to preoperative levels (–82%; p < 0.01). Deficits were reversed in AAV1/2‐A53T‐aSyn, stim‐ON rats after 3 weeks of active stimulation, compared to the AAV1/2‐A53T‐aSyn stim‐OFF rats (an increase of ∼400%; p < 0.05), demonstrating a beneficial effect of DBS. This motor improvement was maintained when the stimulation was turned off and was accompanied by a higher number of tyrosine hydroxylase+ SN neurons (increase of ∼29%), compared to AAV1/2‐A53T‐aSyn stim‐OFF rats (p < 0.05).
Interpretation
Our data support the putative neuroprotective and disease‐modifying effect of STN‐DBS in a mechanistically relevant model of PD. Ann Neurol 2017;81:825–836
Deep brain stimulation (DBS) of the subthalamic nucleus (STN) is considered a standard therapy for Parkinson's disease (PD),1 the second‐most common neurodegenerative disease, for which no cure is yet available. DBS has provided robust and long‐lasting improvement of levodopa‐sensitive motor symptoms, fluctuations, dyskinesia, and quality of life in several clinical trials.2, 3, 4 Much of this benefit emerges rapidly with a “make and break” mechanism related to high‐frequency stimulation and likely represents symptomatic benefit attributed to correction of abnormal network activity. With the application of DBS earlier in the course of disease, a putative disease‐modifying effect has become of clinical interest.5, 6 Several potential mechanisms have been proposed by which DBS could act neuroprotective: STN‐DBS could reduce overactivity and excitotoxicity of glutamatergic projections from the STN to substantia nigra (SN).7, 8 Moreover, DBS may induce the expression of brain‐derived neurotrophic factor (BDNF), a neurotrophic factor that is anterogradely transported from its site of synthesis and is known to support survival of dopaminergic neurons.9 Finally, improved motor activity as an indirect effect of symptomatic improvement could enhance neuronal survival.10
To date, there is no clinical evidence for STN‐DBS‐related disease modification, but this lack of evidence could simply reflect inappropriate trial methodologies.11 In contrast, numerous preclinical studies, conducted in rodent and nonhuman primate (NHP) models of PD have demonstrated a beneficial effect of STN lesion or STN‐DBS on SN neuronal survival.8, 12, 13, 14, 15, 16, 17 However, the common drawback of these preclinical studies was the use of toxin‐mediated PD models, either by 1 methyl‐4 phenyl 1,2,3,6‐tetrahydropyridine (MPTP) in NHP14 or 6‐hydroxydopamine (6‐OHDA) in rodents.12, 15, 18, 19 These toxin models cause acute nigral lesions and do not adequately reflect the molecular pathology of human PD, specifically they do not exhibit α‐synuclein (aSyn)‐positive aggregates, a hallmark of PD, and therefore have limited translational value for studying disease‐modifying therapies.
Thus, in order to evaluate the disease‐modifying potential of DBS in a context where SN neurons are accumulating and under duress from pathological aSyn, we utilized a rat model for PD that is based on a vector driven (adeno‐associated virus [AAV] 1/2) overexpression of mutated A53T α‐synuclein (A53T‐aSyn) in dopaminergic neurons of the SN that leads to progressive and reliable neurodegeneration and motor impairment.20, 21 This model mimics the neurobiological hallmarks of PD much closer than toxin models and is thus more suitable for the investigation of DBS mechanisms of action.
Materials and Methods
Animals
Thirty‐six adult male Sprague‐Dawley rats were purchased from Charles River Laboratories (Sulzfeld, Germany) and kept under standard conditions (21°C, 12‐hour light/dark cycle). Twenty‐five rats were included in the main study; 11 rats were used for analyses of the STN. All applicable international, national, and/or institutional guidelines for care and use of animals were followed. The local authorities at the Regierung von Unterfranken (Würzburg, Germany) approved all animal experiments.
AAV1/2 Serotype Injection
Rats with an average weight of 280 g were stereotactically injected (as described by Koprich et al20) unilaterally into the SN contralateral to the dominant forepaw (determined by the single pellet reaching task pretesting) with either 2.0μl of empty AAV1/2 (empty vector; EV) or 2.0μl of AAV1/2‐expressing human mutated A53T‐aSyn both at a concentration of 2.55 × 1012 genomic particles (gp)/ml by using coordinates from bregma: –5.2mm anteroposterior (AP); either –2.0mm (right) or +2.0mm (left) mediolateral (ML); –7.4mm dorsoventral (DV; rat brain atlas of Paxinos and Watson22).
DBS
Twenty‐two animals underwent ipsilateral microelectrode‐guided implantation of monopolar DBS electrodes (UE‐PSEGSECN1M; FHC Inc., Bowdoin, ME) into the STN. Platinum/iridium electrodes with an impedance between 0.8 and 1.0MOhm were implanted ipsilateral to the AAV1/2 injection using the following coordinates: –3.6mm AP, either –2.5mm (right) or +2.5mm (left) ML, and –7.7mm DV. A custom‐made plug (GT‐Labortechnik, Arnstein, Germany) was connected to the electrode that was fixed to the skull by dental cement. High‐frequency stimulation of rats was performed by using a cable‐bound multichannel systems stimulus generator (Type STG 4004; MultichannelSystems, Reutlingen, Germany) with rectangular pulse shape, 60‐μs pulse length, and 130‐Hz frequency. Amplitude was set at 20% below the occurrence of side effects (ie, orofacial or forelimb dyskinesia). In the interventional groups, stimulation was activated the same day as DBS implantation lasting 21 days whereas the control groups remained OFF stimulation.
Single Pellet Reaching Task
Animals were placed in a Plexiglas box (34 × 14cm) with an opening on the side filled with a food pellet and trained to grasp and pass the pellet to their mouth as described by Lindau et al23 The first 20 reaching attempts were scored per session. The first 3 days of training were used to assess handedness of the rat. Rats were trained with their dominant hand daily for 13 days before injection of AAV1/2. Motor performance was assessed with the investigator blinded to experimental groups and treatment conditions pre‐OP (operative), 3 weeks after STN‐DBS electrode implantation and after turning off the DBS. To maintain reproducibility, training sessions were performed every 5 days between data collection sessions. Three animals were excluded from the study because of insufficient pre‐OP performance in the single pellet reaching task.
Tissue Processing and Immunohistochemistry
After transcardial perfusion with 0.1M of phosphate‐buffered saline (PBS) brains were dissected coronally at the region of –0.26mm from bregma. The ventral part, including the ventral striatum, was snap frozen in liquid dry ice‐cooled isopentane for high‐performance liquid chromatography (HPLC). The dorsal part, including the dorsal striatum, STN, and the SN, was immersion‐fixed in 4% paraformaldehyde in 0.1M of PBS for 2 days and cryo‐protected in 30% sucrose/0.1M of PBS solution for another 4 days followed by freezing of the tissue in liquid dry ice‐cooled isopentane.
Next, 40‐μm coronal cryo‐sections of the brain at the region of –0.8, –3.12 to –4.20, and –4.36 to –6.72mm relative to the bregma to represent the dorsal part of the striatum, the STN and SN were serially cut (six series). Sections were preincubated for 1 hour in 10% natural goat serum (NGS)/2% bovine serum albumin (BSA)/0.5% Triton X‐100 in 0.1M of PBS and then incubated with either rabbit anti‐rat‐tyrosine hydroxylase (TH)‐antibody (striatum, SN; Abcam, Cambridge, UK) diluted in 2% NGS/2% BSA/0.5% Triton X‐100 in PBS or anti‐NeuN (neuron nucleus; SN; Merck, Millipore, CA) antibody diluted in 2% NGS/2% BSA/0.5% Triton X‐100 in 0.1M of PBS for overnight at room temperature (RT; TH) or 4°C (NeuN). A biotinylated secondary goat‐anti‐rabbit‐antibody was applied for 2 hours at RT, followed by avidin/biotin reagent (Dako, High Wycombe, UK) before incubation and staining with diaminobenzidine‐HCl and H2O2.
Nissl Staining
Sections were incubated in cresyl violet solution (1g of cresyl violet + 10ml of 100% acetic acid ad 1l of distilled water) for 30 minutes at RT. After washing in distilled water, sections were dehydrated in an ascending of ethanol series (70%, 96%, and 100%) and then incubated in xylene solution.
TH/aSyn Immunofluorescence Double Staining
Sections were blocked with 10% NGS and 2% BSA for 1 hour at RT and simultaneously incubated overnight at 4°C with rabbit‐anti‐mouse TH and mouse‐anti‐human aSyn (Invitrogen, Frederick, MD) antibodies followed by incubation with fluorescence labelled goat anti‐mouse cyanine (Cy) 2 and goat‐anti‐rabbit Cy3 secondary antibodies (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) for 2 hours at RT. 4′,6‐diamidino‐2‐phenylindole (DAPI) nuclear staining (Sigma‐Aldrich, St. Louis, MO) was performed at RT for 20 minutes.
NeuN/Terminal Deoxynucleotidyl Transferase dUTP Nick End Immunofluorescence
STN sections were blocked with 5% BSA/5% NGS/0.3% Triton X‐100 in 0.1M of PBS for 1 hour at RT, incubated with mouse‐anti‐NeuN antibodies (Merck, Millipore, CA, USA) for overnight at 4°C followed by incubation with fluorescence‐labeled goat‐anti‐mouse Alexa Fluor 488 secondary antibodies (Dianova, Hamburg, Germany) for 2 hours at RT. The In Situ Cell Death Detection Kit, TMR Red (Roche, Mannheim, Germany) was applied for 1 hour at RT according to the manufacturer's description. Cell nuclei staining was then performed with DAPI (1:500,000) for 20 minutes at RT. Rat brain sections with infarction by photochemically initiated thrombosis, as described by Watson et al,24 were used as positive controls. Analysis of this staining was performed with the investigator blinded to experimental groups and treatment conditions.
Unbiased Stereology of SN and STN Neurons
Stereology was performed with the Stereo Investigator software package (version 11.07; MicroBrightField Biosciences, Williston, VT). The investigator was blinded to experimental groups by using coded object slides. Nine SN and four STN sections in average separated by 240μm (one of six series) were used for counting. TH+ or NeuN+ neurons in both SN pars compacta and reticulata and Nissl+ STN neurons were included within each selected region. Counting parameters were: TH, Nissl: grid size 130 × 130μm, counting frame 60 × 60μm, and 1.5‐μm guard zone; NeuN: grid size 200 × 200μm, counting frame 60 × 60μm, and 1.5‐μm guard zone. Actual mounted thickness was determined by randomly selecting sections and determining thickness at every counting site. Sections were viewed under a 100×/1.25 numerical aperture objective (Olympus, Tokyo, Japan) on a BX53 microscope. Gundersen coefficients of error for m = 1 were all less than or equal to 0.09 for each section counted.
Catecholamine quantification by HPLC
HPLC was performed as described.21 The investigator was blinded to experimental groups and treatment conditions. Brain sections were homogenized followed by centrifuging at 10,000g, 20 minutes. Catecholamines were determined from the supernatant. Values of catecholamines are expressed as ng analyte/mg total protein.
Statistical Analysis
Normal distribution was confirmed for each set of data by Q‐Q‐plots, thus parametric methods were used. For the single pellet reaching task, the analysis of HPLC catecholamine quantification and for stereological estimation of cell counts comparing the four groups, the one‐way analysis of variance (ANOVA) test with Tukey's multiple comparison post‐test was used. For analysis of changes after turning off the STN‐DBS in AAV1/2‐A53T‐aSyn rats, the paired Student t test was implemented. Effect size measured by eta‐squared and power analyses were performed by using the software G*Power 3.1. An eta‐squared value ≥0.14 was considered as a strong effect, a power value >80% was considered as sufficient.
* p < 0.05 and ** p < 0.01 were considered as significant p values.
Experimental Sequence
The experimental sequence is summarized in Figure 1A. Starting 16 days before stereotactic AAV1/2 injection, all animals were trained for the behavioral studies and baseline motor performance was assessed. Starting with the vector injection at day 0, the implantation of the DBS electrodes in the STN took place 3 weeks later, on day 21. Rats were divided into four groups: activated stimulation (stim‐ON: AAV1/2‐A53T‐aSyn, n = 10 and AAV1/2‐EV, n = 4) and unactivated stimulation (stim‐OFF: AAV1/2‐A53T‐aSyn, n = 5 and AAV1/2‐EV, n = 3). Continuous DBS treatment started immediately lasting for the next 3 weeks in the stim‐ON interventional groups until day 42. DBS was then switched off for 24 hours in the AAV1/2‐A53T‐aSyn stim‐ON group for another behavioral testing after washing out the symptomatic neurostimulation effect. Finally, on day 43, all animals were killed for further investigations.
Figure 1.
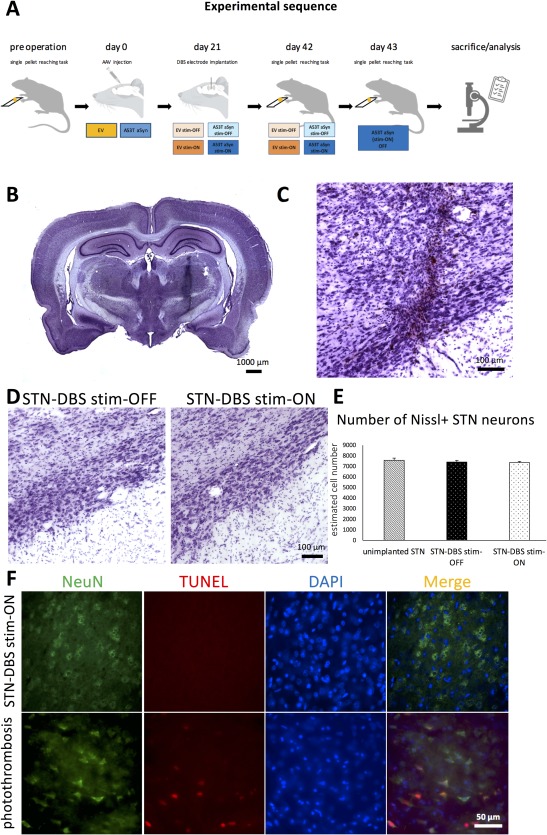
Experimental sequence and control of successful STN microelectrode implantation. (A) Time course of the experimental sequence in a schematic overview depicting the relevant procedural hallmarks. (B) Nissl staining of an exemplary 40‐μm coronal whole‐brain section showing the insertion channel of the unilateral STN‐DBS microelectrode in the right hemisphere and (C) higher magnification of the targeted subthalamic area. (D) Representative images of the STN of nonstimulated (STN‐DBS stim‐OFF) and stimulated (STN‐DBS stim‐ON) rats, showing comparable cell numbers after quantification (E; p = 0.6785; F(2, 12) = 0.4006; unimplanted STN/stim‐OFF/stim‐ON, n = 4/4/7). (F) Upper panel: TUNEL+ profiles (red) are not detected in NeuN+ neurons (green) within the stimulated STN. In contrast, in rat brain (cortex) damaged by photochemically initiated thrombosis, that is used as positive control, TUNEL+ profiles (red) that colocalize with cell nuclei (DAPI staining, blue) are visible within NeuN+ neurons (green; lower panel). DAPI = 4′,6‐diamidino‐2‐phenylindole; DBS = deep brain stimulation; NeuN = neuron nucleus; STN = subthalamic nucleus; TUNEL = terminal deoxynucleotidyl transferase dUTP nick end.
Results
Verification of Continuous DBS, STN Electrode Placement, and SN AAV1/2‐A53T‐aSyn Injection
After 3 weeks of active STN‐DBS, the integrity of the wiring deployed in each rat was tested. Three animals of the AAV1/2‐A53T‐aSyn stim‐ON group had broken stimulation circuits and were excluded from further analysis. To verify correct positioning of the STN‐DBS electrodes in the remaining rats, we generated postmortem Nissl staining of the subthalamic area to demonstrate that the trajectory of the implanted electrode ended in the region of the STN without disruption of the nucleus itself (Fig 1B,C). One further rat of the AAV1/2‐A53T‐aSyn stim‐ON group was excluded from analysis because STN positioning was off target. Stereological quantification of STN Nissl+ neurons did not reveal any significant differences comparing STN stim‐ON with stim‐OFF rats (Fig 1D,E; p = 0.6785; F (2, 12) = 0.4006). Additionally, terminal deoxynucleotidyl transferase dUTP nick end (TUNEL) staining in the STN after 3 weeks of STN stimulation (STN‐DBS stim‐ON) did not show any signs for TUNEL‐positive profiles as an indicator for DNA fragmentation and cell apoptosis (Fig 1F). Therefore, a delayed lesion of the STN induced by neurostimulation itself could be excluded. Double‐immunohistochemical staining with antibodies against human aSyn and rat TH in sections of the SN after injection of the AAV1/2‐A53T‐aSyn vector showed a homogenous and widespread staining pattern of pathologic A53T‐aSyn in the injected hemisphere and an intracellular localization of aSyn in TH+ neurons in the merged pictures of both stim‐ON and stim‐OFF rats (Fig 2A,B), whereas no aSyn deposition could be observed in the untreated hemisphere serving as control (Fig 2C). Moreover, transport of A53T‐aSyn to the striatum along the nigrostriatal tract was observed 6 weeks after AAV1/2‐A53T‐aSyn injection (Fig 2D). A single animal was excluded from the study because of unsuccessful injection of AAV1/2‐A53T‐aSyn into the SN (stim‐OFF group).
Figure 2.
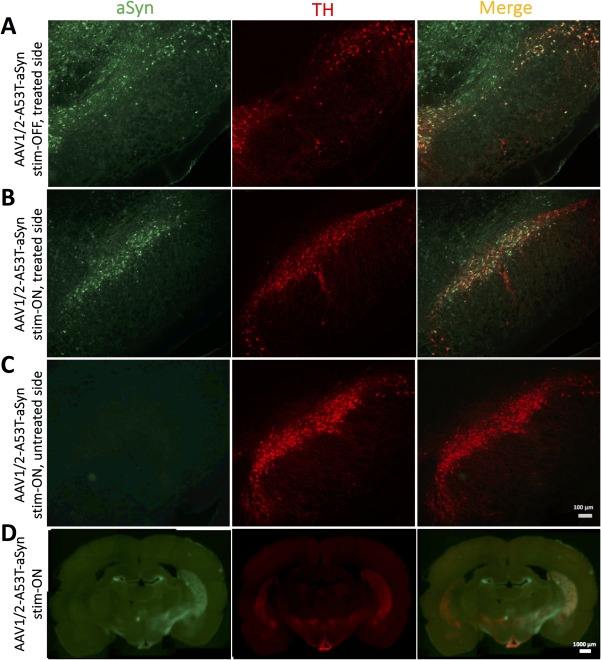
AAV1/2‐A53T‐aSyn injection into the SN leads to widespread expression of pathological A53T‐aSyn that projects into the striatum. (A,B) Representative immunofluorescence double stainings showing expression of A53T‐aSyn (green) and TH+ dopaminergic neurons (red) in the SN after 43 days of AAV injection in a (A) stim‐OFF rat of the AAV1/2‐A53T‐aSyn‐treated hemisphere and a (B) stim‐ON AAV1/2‐A53T‐aSyn‐treated rat revealing a widespread A53T‐aSyn expression in the SN of both rats and more surviving TH+ cells in the AAV1/2‐A53T‐aSyn stim‐ON brain. (C) No detectable aSyn aggregates in the SN of the untreated side of the AAV1/2‐A53T‐aSyn stim‐ON rat are visible. (D) Coronal whole‐brain slice of a AAV1/2‐A53T‐aSyn stim‐ON rat shows the deposition of aSyn (green) and nigrostriatal dopaminergic projections (red) in the dorsal striatum. AAV = adeno‐associated virus; aSyn = α‐synuclein; SN = substantia nigra; TH = tyrosine hydroxylase.
STN‐DBS Improves Motor Performance in AAV1/2‐A53T‐aSyn Rats
Assessment of the single pellet reaching task on day 42 (6 weeks after AAV1/2 injection and 3 weeks after STN electrode implantation) demonstrated a significant reduction of task performance, to 18.3 ± 10.7% of preoperative levels in AAV1/2‐A53T‐aSyn stim‐OFF animals (p < 0.01). This impairment could be restored in AAV1/2‐A53T‐aSyn rats by active STN‐DBS (stim‐ON), to 73.6 ± 12.8% of preoperative levels (p < 0.05), a level of performance that was not significantly different to AAV1/2‐EV‐injected, stim‐ON animals (p > 0.05). In EV controls, performance was not significantly altered, compared to preoperative levels, whether stimulation was on or off, in the AAV1/2‐EV‐injected stim‐OFF group (118.9 ± 10.7%, mean ± standard error of the mean [SEM]) and AAV1/2‐EV stim‐ON rats (88.3 ± 19.2%; Fig 3A; p = 0.0025; F (3, 11) = 9.197; eta‐squared = 0.715; power = 99%). After 24 hours of stimulation‐washout in the AAV1/2‐A53T‐aSyn stim‐ON group, motor performance was still 66.6 ± 9.5% of baseline level, which was not significantly different from stim‐ON in the same animals (p > 0.05) and still significantly increased compared to AAV1/2‐A53T‐aSyn stim‐OFF level (p < 0.05; Fig 3B).
Figure 3.
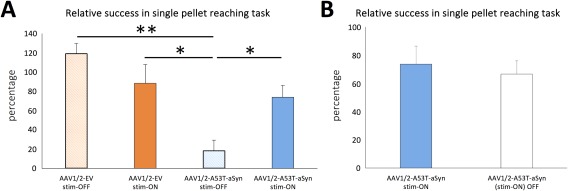
Long‐term STN‐DBS improves motor outcome in AAV1/2‐A53T‐aSyn‐injected rats. (A) Diagram shows a significantly worse motor performance in the single pellet reaching task of the AAV1/2‐A53T‐aSyn‐injected stim‐OFF animals (light blue bar) compared to the AAV1/2‐EV control groups (light orange bar, dark orange bar) and a significant restoration of this behavioral impairment in the AAV1/2‐A53T‐aSyn DBS stim‐ON group (dark blue bar). Motor performance was calculated and reported as percentage of the individual preoperative baseline performance (pre‐AAV injection; p = 0.0025; F(3, 11) = 9.197; EV stim‐OFF/EV stim‐ON/A53T stim‐OFF/A53T stim‐ON, n = 3/3/4/5). (B) After turning off the DBS for 24 hours, the performance in the single pellet reaching task decreased nonsignificantly around 7% in the AAV1/2‐A53T‐aSyn stim‐ON group (white bar; t(4) = 1.364). Data are presented as mean ± SEM. *p < 0.05; **p < 0.01. AAV = adeno‐associated virus; aSyn = α‐synuclein; DBS = deep brain stimulation; EV = empty vector; SEM = standard error of the mean. STN = subthalamic nucleus.
STN‐DBS Diminishes Nigral Loss of TH+ Neurons After AAV1/2‐A53T‐aSyn Injection
Stereological estimation of cell numbers within the SN demonstrated a significant decrease in TH+ neurons in stim‐OFF, AAV1/2‐A53T‐aSyn rats, by ∼25%, compared to either AAV1/2‐EV group (AAV1/2‐EV stim‐OFF 10,113 ± 105, AAV1/2‐EV stim‐ON 10,670 ± 459, AAV1/2‐A53T‐aSyn stim‐OFF 7,295 ± 761; Fig 4A–C,E). However, rats that received unilateral active STN‐DBS for 3 weeks after AAV1/2‐A53T‐aSyn injection had significantly more TH+ SN cells (increase of ∼29%), compared to AAV1/2‐A53T‐aSyn stim‐OFF rats (9,455 ± 267; p < 0.05; Fig 4D,E). The level of TH+ cells in AAV1/2‐A53T‐aSyn stim‐ON rats was not significantly different to that in AAV1/2‐EV vehicle stim‐ON or stim‐OFF (both p > 0.05) animals (p = 0.0014; F (3, 13) = 9.516; eta‐squared = 0.687; power = 99%).
Figure 4.
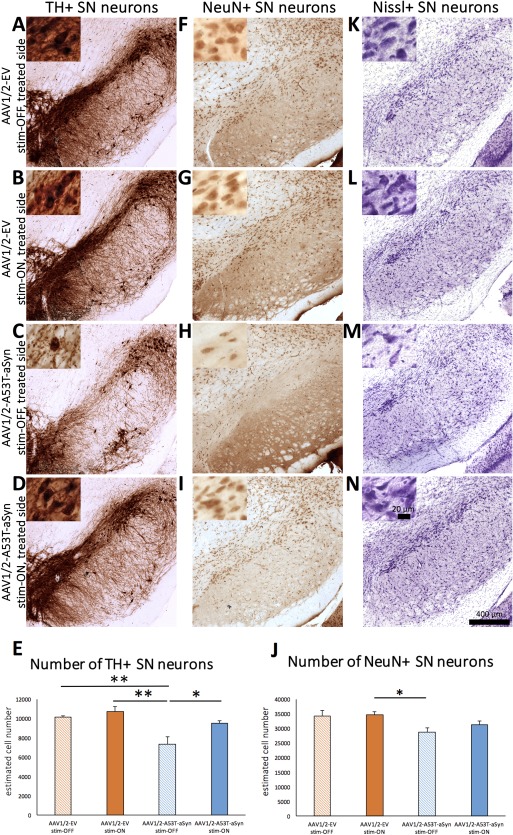
Long‐term STN‐DBS leads to rescue of TH+ SN neurons in AAV1/2‐A53T‐aSyn rats. (A–D) Images of representative immunohistochemical‐stained sections for TH+ dopaminergic neurons and staining with neuronal marker NeuN (F–I) and Nissl (K–N) of the rat SN in all four experimental groups. Insets show higher magnification of SN pars compacta neurons. (E) Diagram shows the estimated cell numbers of quantified TH+ neurons in the SN analyzed by unbiased stereology. A significant reduction of TH+ cell number could be observed in AAV1/2‐A53T‐aSyn DBS stim‐OFF animals (light blue bar) compared to control AAV1/2‐EV rats that was not present in the AAV1/2‐A53T‐aSyn DBS stim‐ON treatment group, indicating a possible neuroprotective effect of STN‐DBS (p = 0.0014; F(3, 13) = 9.516; EV stim‐OFF/EV stim‐ON/A53T stim‐OFF/A53T stim‐ON, n = 3/4/4/6). Stereological estimation of NeuN+ total neuron cell numbers using the one‐way ANOVA demonstrated overall statistically significant differences among the four groups of rats (p = 0.0198; F(3, 13) = 4.684; EV stim‐OFF/EV stim‐ON/A53T stim‐OFF/A53T stim‐ON, n = 3/4/4/6). Tukey's multiple comparison post‐test showed a significant reduction of NeuN+ neuron number in AAV1/2‐A53T‐aSyn stim‐OFF rats compared to the AAV1/2‐EV stim‐ON group. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01. AAV = adeno‐associated virus; aSyn = α‐synuclein; ANOVA = analysis of variance; DBS = deep brain stimulation; EV = empty vector; NeuN = neuron nucleus; SEM = standard error of the mean; STN = subthalamic nucleus; TH = tyrosine hydroxylase.
Additionally, total neuronal number in the SN was analyzed by labeling with NeuN (Fig 4F–J) and Nissl staining (Fig 4K–N). The greatest number of NeuN+ cells was observed in the EV control groups (AAV1/2‐EV stim‐OFF 34,172 ± 1,884, AAV1/2‐EV stim‐ON 34,781 ± 743; Fig 4F,G,J). Statistical analysis by one‐way ANOVA demonstrated an overall significance of treatment (p = 0.0198; F (3, 13) = 4.684; eta‐squared = 0.519; power = 88%); therefore, the differences among the means of all four treatment groups were compared using Tukey's multiple comparison post‐test (Fig 4J). This analysis revealed a statistically significant reduction of NeuN+ SN neurons in the AAV1/2‐A53T‐aSyn stim‐OFF group (28,691 ± 1,529) compared to the AAV1/2‐EV stim‐ON rats (Fig 4H,J). By contrast, there was no significant difference in comparing AAV1/2‐EV rats with AAV1/2‐A53T‐aSyn rats after 3 weeks of STN stimulation (31,221 ± 993; Fig 4I,J). Qualitative analysis of Nissl‐stained SN demonstrated comparable results (Fig 4K–N).
For examination of dopaminergic neurotransmitter level, striatal HPLC was performed and analyzed for dopamine (DA) and the metabolite homovanilic acid (HVA) 6 weeks after AAV1/2 administration. AAV1/2‐EV injection did not reduce striatal DA, whether STN stimulation was on or off (98.5 ± 2.7% stim‐OFF, 103.9 ± 2.1% stim‐ON). In contrast, AAV1/2‐A53T‐aSyn injection resulted in a significant decrease of DA in the ipsilateral striatum, whether stimulation was on or off (79.7 ± 3.2%), relative to the uninjected side, in stim‐OFF rats and 81.4 ± 6.8% in stim‐ON animals (Fig 5A; p = 0.0157; F (3, 13) = 5.033; eta‐squared = 0.537; power = 91%). HVA/DA ratio as an indicator for DA turnover showed a nonsignificant increase in the AAV1/2‐A53T‐aSyn‐injected stim‐OFF group compared to both EV groups (132.1 ± 8.8% vs ∼99%; p = 0.051; F (3, 13) = 3.384; eta‐squared = 0.439; power = 75%). STN‐DBS did not lead to significant changes in the HVA/DA ratio in AAV1/2‐A53T‐aSyn‐rats (109.7 ± 6.9%; Fig 5B).
Figure 5.
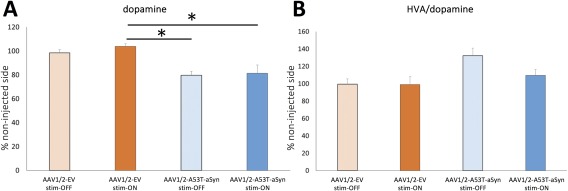
Long‐term STN‐DBS does not rescue DA deficit in AAV1/2‐A53T‐aSyn rats. (A) Measurement of striatal DA levels by HPLC also indicates a reduction in the AAV1/2‐A53T‐aSyn DBS stim‐OFF group (light blue bar), but also in the AAV1/2‐A53T‐aSyn stim‐ON rats compared to AAV1/2‐EV rats (p = 0.0157; F(3, 13) = 5.033; EV stim‐OFF/EV stim‐ON/A53T stim‐OFF/A53T stim‐ON, n = 3/4/4/6). (B) DA turnover measured by HVA/DA ratio by HPLC shows a nonsignificant increase in the AAV1/2‐A53T‐aSyn DBS stim‐OFF group compared to AAV1/2‐A53T‐aSyn DBS stim‐ON rats and EV controls (p = 0.051; F(3, 13) = 3.384; EV stim‐OFF/EV stim‐ON/A53T stim‐OFF/A53T stim‐ON, n = 3/4/4/6). Data are presented as mean ± SEM. AAV = adeno‐associated virus; aSyn = α‐synuclein; DA = dopamine; DBS = deep brain stimulation; EV = empty vector; HPLC = high‐performance liquid chromatography; HVA = homovanilic acid; SEM = standard error of the mean.
Discussion
We present results of chronic high‐frequency STN neurostimulation in a PD rat model that provides chronic stress on SN neurons by AAV1/2‐A53T‐aSyn expression, progressive neurodegeneration, and the presence of insoluble aSyn aggregates as opposed to the more acute MPTP‐ or 6‐OHDA‐mediated toxicity without aSyn pathology that have been used in previous reports investigating the effects of DBS in PD animal models.9, 12, 13, 14, 15, 16, 20 We could demonstrate that unilateral injection of AAV1/2‐A53T‐aSyn into the SN, followed by ipsilateral STN‐DBS electrode implantation 3 weeks later, can be accomplished with low dropout rates attributed to mistargeting the SN (6%) or STN (5%). However, higher dropout rates were found attributed to connection uncoupling with our experimental DBS apparatus (21% in the stim‐ON groups). To be translationally relevant, we implanted STN‐DBS electrodes 3 weeks after AAV1/2 injection, when damage to the nigrostriatal tract and motor deficits are established.21 To control for a possible subthalamotomy‐like effect of electrode insertion,25 we included an AAV1/2‐A53T‐aSyn stim‐OFF group. Neuronal damage in the STN by stimulation itself was excluded by demonstration of comparable STN neuron cell counts in stimulated versus unstimulated rats and lack of TUNEL+ profiles in the STN after stimulation. For assessment of behavioral deficits, we used the single pellet reaching task, which was most sensitive to detect unilateral motor impairment in rats. Sensitivity of motor testing was further increased by performing the surgical procedure on the dominant hemisphere as identified by previous screening for paw preference.
Injection of AAV1/2‐A53T‐aSyn into the SN induced a significant worsening of pellet reaching in the stim‐OFF group compared to AAV1/2‐EV rats after 6 weeks, which was in keeping with the previously reported motor impairment in this model assessed using the cylinder test.21 Chronic neurostimulation of the STN restored motor performance to near baseline level in AAV1/2‐A53T‐aSyn rats, which corroborates the symptomatic benefit of DBS previously established by various motor tasks in the 6‐OHDA PD rat model.15, 26 The magnitude of improvement was comparable with the high efficacy of STN‐DBS in human PD, where average motor symptom reductions of 40% to 60% have been described.27 However, after turning off stimulation for 1 day in the DBS‐treated AAV1/2‐A53T‐aSyn group, we observed sustained benefit. In contrast, in human PD, a stimulation pause of 30 to 60 minutes is usually sufficient to wash out most of the symptomatic DBS effect28; therefore, we considered 24 hours as a sufficiently long washout period in our experiment. Thus, despite a mild deterioration of motor symptoms in AAV1/2‐A53T‐aSyn rats 24 hours after stimulation was terminated, the motor performance in this group remained significantly better compared to unstimulated AAV1/2‐A53T‐aSyn‐treated rats, suggesting a disease‐modifying effect in this model. We propose that the lack of a persistent beneficial effect of STN‐DBS after turning off stimulation in human PD patients is a result of DBS implantation being at too late a disease stage for neuronal rescue. Thus, in human PD, the onset of nigrostriatal degeneration precedes the appearance of motor impairment with estimated loss of SN neurons by already 30% at this time point (for review, see Cheng et al29). The likely basis of sustained motor improvement after chronic STN‐DBS in AAV1/2‐A53T‐aSyn rats was a reduced loss of SN TH+ neurons in DBS‐treated compared to unstimulated AAV1/2‐A53T‐aSyn rats. Additionally, we observed a significant reduction of NeuN+ SN neurons in AAV1/2‐A53T‐aSyn stim‐OFF rats compared to AAV1/2‐EV stim‐ON animals. However, the differences between the various groups were not as pronounced as for TH expression. A similar dissociation between TH+ dopaminergic and total SN neurons is very common in toxic PD rodent models induced by MPTP30 or 6‐OHDA,12 because damage to the dopaminergic neurons might lead to functional loss of TH phenotype before cell death occurs. Both a rescue of TH expression only31 and in addition total SN neuron number8, 13, 14 have been described in PD models after STN lesioning/DBS. However, more studies reported the number of TH+ neurons without determining total SN neuron counts.12, 15, 16, 17, 25, 32 The inability to synthesize DA by loss of TH enzymatic function is considered a pivotal step in PD disease progression and a hallmark of PD,33 underlining the translational importance of our findings. We consider the positive effect of DBS on TH expression in our AAV1/2‐A53T‐aSyn model as protective on neuronal function, and hence “neuroprotective,” even though it may not entirely prevent cell death.
Various reports exist on a beneficial impact of STN silencing on neuronal survival in toxin‐based rodent models of PD. After nigrostriatal lesion by 3‐nitropropionic acid (3‐NP) injection, a preventive ablation of the STN, by quinolinic acid 2 weeks before 3‐NP administration, led to an attenuation of dopaminergic SN neuron loss.8 In a comparable approach using the 6‐OHDA model, prevention of dopaminergic SN neuronal degeneration by 1 week before STN lesioning, using kainic acid injections, was demonstrated.17 Additionally, ibotenic acid lesioning of STN neurons (loss of 50–60%), 1 week before 6‐OHDA delivery, rescued 23% of dopaminergic SN neurons.32 Although the exact physiological mechanisms of DBS are still debated, in particular whether high‐frequency stimulation drives or inhibits axons,34, 35 the behavioral effects of DBS are lesioning‐like and similar observations have been made in settings using experimental DBS to study neuroprotection: Improved motor function and less SN neurodegeneration was observed in 6‐OHDA rats that received STN‐DBS for 2 weeks starting 2 days before 6‐OHDA administration.15 The time point of STN neuromodulation within the course of disease may be crucial for the magnitude of neuronal rescue in the SN. Whereas kainic acid injection into the ipsilateral STN 1 hour after 6‐OHDA injection rescued 72.9% of dopaminergic neurons compared to the contralateral hemisphere, treatment at 7 days after 6‐OHDA administration did not prevent an almost complete loss, to 5.6% of control.16 A common drawback of these studies is that treatment effects of STN neuromodulation were generally studied in a preventive mode (eg, before administration of the toxin), and not after degeneration has commenced, as reflecting the clinical PD situation. Only a few preclinical studies in toxin‐based models with a more clinically relevant design have been conducted. STN‐DBS commencing 6 days after MPTP administration in monkeys led to survival of dopaminergic neurons in the SN.14 A beneficial effect on SN dopaminergic neuron survival was observed by bilateral striatal 6‐OHDA injections in rats and at the same time bilateral STN electrode implantations that were activated 1 week later, and stimulated for 1 hour per day, over a period of 3 months, although this stimulation protocol does not reflect the chronic stimulation in PD patients.13 Another group demonstrated that unilateral 6‐OHDA administration and STN‐DBS electrode implantation at the same time, followed by chronic stimulation for 2 weeks that commenced 2 weeks after surgery, also reduced dopaminergic SN cell loss.12 The drawback of the two latter studies was that subthalamic electrodes were implanted at the same time point as 6‐OHDA injection, and although stimulation was delayed, this experimental design did not control for a possible microlesioning effect of electrode insertion and again did not reflect the sequence of events in the human disease. The design of our study mimics the clinical application of DBS in this respect. Delivery of the AAV1/2 at a slightly lower concentration of 1.7 × 1012 gp/ml than in our current study led to significant motor deficits and striatal dopaminergic fiber loss already after 3 weeks whereas dopaminergic neuron loss in the SN was milder and not significant.21 By explicitly choosing this time point for STN‐DBS electrode implantation, we aimed to mimic a moderate stage of human PD. Focusing on the consequences of AAV1/2‐A53T‐aSyn‐induced neurodegeneration and subsequent DBS treatment in the nigrostriatal pathway, we observed a reduction of striatal DA levels in both AAV1/2‐A53T‐aSyn stim‐OFF and stim‐ON groups. Additionally, a nonsignificant increase of DA turnover in unstimulated compared to stimulated AAV1/2‐A53T‐aSyn rats was observed. These findings suggest that striatal dopaminergic terminals are most sensitive to A53T‐aSyn overexpression, and that DBS does not rescue these terminals while preventing the functional impairment of their corresponding nigral cell bodies. DBS, however, may reduce metabolic stress and contain function in SN neurons. In this context, high‐frequency stimulation of the STN has been shown to increase striatal DA release in naïve and 6‐OHDA rats.36, 37 Moreover, as in PD, the STN is overactive and may facilitate glutamatergic exitocitoxicity of the SN pars compacta; several studies propose a reduction of STN‐mediated SN damage as the main mechanism of the neuroprotective effect of STN‐DBS/STN lesioning, but experiments proving this concept are still lacking.8, 14, 15, 16, 17 Additionally, an increase in BDNF in the nigrostriatal tract and M1 cortex by STN‐DBS has been described in 6‐OHDA rats, pointing toward a more complex mechanism of neuroprotection by DBS.9
Our study provides the first evidence of a neuroprotective effect of STN‐DBS in a progressive, aSyn‐related model of PD. The experimental methodology implemented will now allow us to study the underlying neurobiological mechanisms and the best time point of DBS in order to maximize the disease‐modifying effect.
Table 1.
Summary of Data and Statistical Analyses
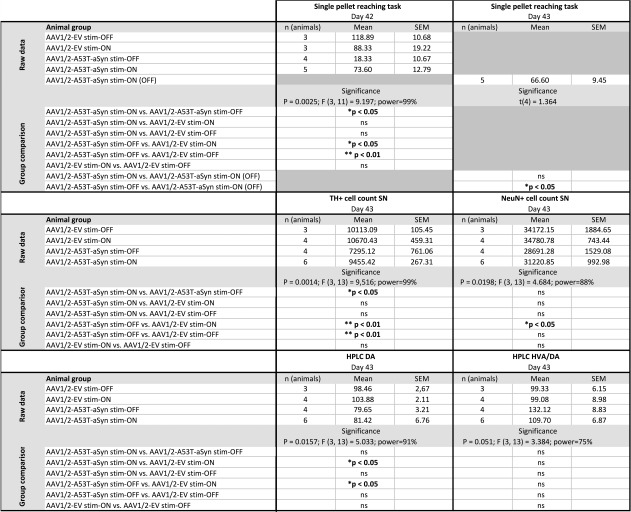
|
For each experiment, the n number per group, mean ± SEM, time point of analysis, p value, F/t value, group comparison using Tukey's multiple comparison post‐test, and statistical power is given.
AAV = adeno‐associated virus; aSyn = α‐synuclein; DA = dopamine; EV = empty vector; HPLC = high‐performance liquid chromatography; HVA = homovanilic acid; ns = not significant; SEM = standard error of the mean; SN = substantia nigra; TH = tyrosine hydroxylase.
Author Contributions
C.W.I., J.B.K., J.V., and J.M.B. developed the concept of this study; C.W.I. and J.B.K. designed this study. C.W.I., M.R., T.M., J.B.K., and F.F. acquired and analyzed data. C.W.I. and T.M. drafted the manuscript and figures. T.M., M.R., J.B.K., and C.W.I. contributed equally to this work.
Potential Conflicts of Interest
Nothing to report.
Acknowledgment
The work was supported by the University Research Funds by the State of Bavaria, Germany, and by a personal funding from family Thorsten Fischer, Würzburg, Germany.
We are grateful to Keali Röhm, Louisa Frieß, and Heike Menzel for their expert technical assistance and to Helga Brünner for the animal care.
References
- 1. Volkmann J. Deep brain stimulation for Parkinson's disease. Parkinsonism Relat Disord 2007;13(suppl 3):S462–S465. [DOI] [PubMed] [Google Scholar]
- 2. Krack P, Batir A, Van Blercom N, et al. Five‐year follow‐up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med 2003;349:1925–1934. [DOI] [PubMed] [Google Scholar]
- 3. Schupbach WM, Chastan N, Welter ML, et al. Stimulation of the subthalamic nucleus in Parkinson's disease: a 5 year follow up. J Neurol Neurosurg Psychiatry 2005;76:1640–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weaver FM, Follett K, Stern M, et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA 2009;301:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schuepbach WM, Rau J, Knudsen K, et al. Neurostimulation for Parkinson's disease with early motor complications. N Engl J Med 2013;368:610–622. [DOI] [PubMed] [Google Scholar]
- 6. Charles D, Konrad PE, Neimat JS, et al. Subthalamic nucleus deep brain stimulation in early stage Parkinson's disease. Parkinsonism Relat Disord 2014;20:731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus‐mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol 1998;44(3 suppl 1):S175–S188. [DOI] [PubMed] [Google Scholar]
- 8. Nakao N, Nakai E, Nakai K, et al. Ablation of the subthalamic nucleus supports the survival of nigral dopaminergic neurons after nigrostriatal lesions induced by the mitochondrial toxin 3‐nitropropionic acid. Ann Neurol 1999;4:640–651. [PubMed] [Google Scholar]
- 9. Spieles‐Engemann AL, Steece‐Collier K, Behbehani MM, et al. Subthalamic nucleus stimulation increases brain derived neurotrophic factor in the nigrostriatal system and primary motor cortex. J Parkinsons Dis 2011;1:123–136. [PMC free article] [PubMed] [Google Scholar]
- 10. Zigmond MJ, Smeyne RJ. Exercise: is it a neuroprotective and if so, how does it work? Parkinsonism Relat Disord 2014;20(suppl 1):S123–S127. [DOI] [PubMed] [Google Scholar]
- 11. Harnack D, Kupsch A. The impact of subthalamic deep brain stimulation on nigral neuroprotection‐myth or reality? Neuromodulation 2010;13:160–167. [DOI] [PubMed] [Google Scholar]
- 12. Spieles‐Engemann AL, Behbehani MM, Collier TJ, et al. Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis 2010;39:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Temel Y, Visser‐Vandewalle V, Kaplan S, et al. Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res 2006;1120:100–105. [DOI] [PubMed] [Google Scholar]
- 14. Wallace BA, Ashkan K, Heise CE, et al. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP‐treated monkeys. Brain 2007;130(pt 8):2129–2145. [DOI] [PubMed] [Google Scholar]
- 15. Maesawa S, Kaneoke Y, Kajita Y, et al. Long‐term stimulation of the subthalamic nucleus in hemiparkinsonian rats: neuroprotection of dopaminergic neurons. J Neurosurg 2004;100:679–687. [DOI] [PubMed] [Google Scholar]
- 16. Chen L, Liu Z, Tian Z, et al. Prevention of neurotoxin damage of 6‐OHDA to dopaminergic nigral neuron by subthalamic nucleus lesions. Stereotact Funct Neurosurg 2000;75:66–75. [DOI] [PubMed] [Google Scholar]
- 17. Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6‐OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci 1996;8:1408–1414. [DOI] [PubMed] [Google Scholar]
- 18. Meredith GE, Kang UJ. Behavioral models of Parkinson's disease in rodents: a new look at an old problem. Mov Disord 2006;21:1595–1606. [DOI] [PubMed] [Google Scholar]
- 19. Spieles‐Engemann AL, Collier TJ, Sortwell CE. A functionally relevant and long‐term model of deep brain stimulation of the rat subthalamic nucleus: advantages and considerations. Eur J Neurosci 2010;32:1092–1099. [DOI] [PubMed] [Google Scholar]
- 20. Koprich JB, Johnston TH, Reyes MG, et al. Expression of human A53T alpha‐synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson's disease. Mol Neurodegener 2010;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koprich JB, Johnston TH, Huot P, et al. Progressive neurodegeneration or endogenous compensation in an animal model of Parkinson's disease produced by decreasing doses of alpha‐synuclein. PLoS One 2011;6:e17698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates, 5th ed Amsterdam; Boston, MA: Elsevier Academic; 2005. [Google Scholar]
- 23. Lindau NT, Banninger BJ, Gullo M, et al. Rewiring of the corticospinal tract in the adult rat after unilateral stroke and anti‐Nogo‐A therapy. Brain 2014;137(pt 3):739–756. [DOI] [PubMed] [Google Scholar]
- 24. Watson BD, Dietrich WD, Busto R, et al. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol 1985;17:497–504. [DOI] [PubMed] [Google Scholar]
- 25. Harnack D, Meissner W, Jira JA, et al. Placebo‐controlled chronic high‐frequency stimulation of the subthalamic nucleus preserves dopaminergic nigral neurons in a rat model of progressive Parkinsonism. Exp Neurol 2008;210:257–260. [DOI] [PubMed] [Google Scholar]
- 26. Fang X, Sugiyama K, Akamine S, et al. Improvements in motor behavioral tests during deep brain stimulation of the subthalamic nucleus in rats with different degrees of unilateral parkinsonism. Brain Res 2006;1120:202–210. [DOI] [PubMed] [Google Scholar]
- 27. Deuschl G, Schade‐Brittinger C, Krack P, et al. A randomized trial of deep‐brain stimulation for Parkinson's disease. N Engl J Med 2006;355:896–908. [DOI] [PubMed] [Google Scholar]
- 28. Temperli P, Ghika J, Villemure JG, et al. How do parkinsonian signs return after discontinuation of subthalamic DBS? Neurology 2003;60:78–81. [DOI] [PubMed] [Google Scholar]
- 29. Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jackson‐Lewis V, Jakowec M, Burke RE, et al. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine. Neurodegeneration 1995;4:257–269. [DOI] [PubMed] [Google Scholar]
- 31. Paul G, Meissner W, Rein S, et al. Ablation of the subthalamic nucleus protects dopaminergic phenotype but not cell survival in a rat model of Parkinson's disease. Exp Neurol 2004;185:272–280. [DOI] [PubMed] [Google Scholar]
- 32. Carvalho GA, Nikkhah G. Subthalamic nucleus lesions are neuroprotective against terminal 6‐OHDA‐induced striatal lesions and restore postural balancing reactions. Exp Neurol 2001;171:405–417. [DOI] [PubMed] [Google Scholar]
- 33. Tabrez S, Jabir NR, Shakil S, et al. A synopsis on the role of tyrosine hydroxylase in Parkinson's disease. CNS Neurol Disord Drug Targets 2012;11:395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng F, Lammert K, Nixdorf‐Bergweiler BE, et al. Axonal failure during high frequency stimulation of rat subthalamic nucleus. J Physiol 2011;589(pt 11):2781–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McIntyre CC, Hahn PJ. Network perspectives on the mechanisms of deep brain stimulation. Neurobiol Dis 2010;38:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meissner W, Reum T, Paul G, et al. Striatal dopaminergic metabolism is increased by deep brain stimulation of the subthalamic nucleus in 6‐hydroxydopamine lesioned rats. Neurosci Lett 2001;303:165–168. [DOI] [PubMed] [Google Scholar]
- 37. Meissner W, Harnack D, Reese R, et al. High‐frequency stimulation of the subthalamic nucleus enhances striatal dopamine release and metabolism in rats. J Neurochem 2003;85:601–609. [DOI] [PubMed] [Google Scholar]